

Summary
In Parkinson’s disease (PD), α-Synuclein (αS) pathologically impacts the brain, a highly lipid-rich organ. We investigated how alterations in αS or lipid/fatty acid homeostasis affect each other. Lipidomic profiling of human αS-expressing yeast revealed increases in oleic acid (OA, 18:1), diglycerides and triglycerides. These findings were recapitulated in rodent and human neuronal models of αS dyshomeostasis (overexpression; patient-derived triplication or E46K mutation; E46K mice). Preventing lipid droplet formation or augmenting OA increased αS yeast toxicity; suppressing the OA-generating enzyme stearoyl-CoA-desaturase (SCD) was protective. Genetic or pharmacological SCD inhibition ameliorated toxicity in αS-overexpressing rat neurons. In a C. elegans model, SCD knockout prevented αS-induced dopaminergic degeneration. Conversely, we observed detrimental effects of OA on αS homeostasis: in human neural cells, excess OA caused αS inclusion formation, which was reversed by SCD inhibition. Thus, monounsaturated fatty acid metabolism is pivotal for αS-induced neurotoxicity, and inhibiting SCD represents a novel PD therapeutic approach.
eTOC Bulb
α-Synuclein is an abundant nerve cell component that forms abnormal aggregates in Parkinson’s disease and other fatal brain disorders. No disease-modifying drugs are available. Here, we identify new drug targets in lipid pathways and describe how cellular lipid alterations drive α-synuclein toxicity.
Graphical Abstract
Introduction
Lipids contribute to many cellular processes, including energy storage, membrane synthesis, signaling and protein modification. The brain is the second most lipid-rich organ (Sastry, 1985). Lipid and FA homeostasis are essential determinants of neural development, neurotransmission and receptor activation. Many CNS disorders and neurodegenerative diseases are associated with lipid dyshomeostasis, including epilepsy (Trimbuch et al., 2009), schizophrenia (Adibhatla and Hatcher, 2007), Huntington’s disease (Epand et al., 2016), Alzheimer’s disease (Foley, 2010), motor neuron diseases (Schmitt et al., 2014) and PD (Klemann et al., 2017). Hence, cells tightly regulate lipid synthesis, uptake and subcellular distribution of precursors, especially FAs, to maintain normal function. One fundamental equilibrating mechanism is storage of FAs as triglycerides (TGs) in cytosolic lipid droplets (LDs) to prevent FA accumulation having cytotoxic consequences (Listenberger et al., 2003).
The strongly PD-associated protein αS is a 14 kDa polypeptide highly expressed in brain. It interacts with phospholipids (Bodner et al., 2009; Stockl et al., 2008) and FAs (Lucke et al., 2006; Sharon et al., 2001). Expression of αS promotes LD formation (Outeiro and Lindquist, 2003), and genome-wide association studies (GWAS) and postmortem brain analyses have identified proteins related to lipid metabolism and LD biology as associated with PD. Seipin, localized at ER/LD contact sites and involved in LD biogenesis (Chen and Goodman, 2017; Walther et al., 2017), may be differentially expressed in PD vs. control brains (Licker et al., 2014; van Dijk et al., 2012). DGKQ, a diacylglycerol kinase controlling cellular diglyceride (DG) (Chen et al., 2013), and FA elongase 7 (Chang et al., 2017), a determinant of acyl-chain length and hence lipid composition/membrane fluidity, are designated PD risk factors. A global analysis of PD association studies highlighted lipid metabolism as the common link among four key processes involved in pathogenesis (Klemann et al., 2017).
Formation of αS-rich cytoplasmic inclusions, a hallmark of PD, is likely to be triggered by changes in αS folding and assembly. Reduction in physiological α-helical tetramers/multimers relative to monomers leads to αS-rich cytoplasmic inclusions and neurotoxicity (Bartels et al., 2011; Burre et al., 2014; Dettmer et al., 2013; Dettmer et al., 2015a; Dettmer et al., 2015b; Wang et al., 2011). Despite clear links between αS, PD and lipid pathways, the impact of lipid metabolism on αS conformation/assembly state and associated phenotypes (e.g., vesicle trafficking defects) has not been well-defined. The neutral lipids pathway stores excess FAs ultimately in LDs to prevent FA-induced lipotoxicity (Listenberger et al., 2003). The role of LDs in protecting against or exacerbating αS pathology, e.g., by sequestering excess FAs or providing a structural platform for αS deposition, is unclear. FAs of varying chain lengths and degrees of saturation influence multiple biological processes when incorporated into membrane lipids via membrane thickness, curvature, fluidity and bending flexibility. There is a relationship between membrane curvature, membrane composition and αS binding (Davidson et al., 1998; Pranke et al., 2011; Westphal and Chandra, 2013).
Here, we define the impact of αS expression on the lipidome from yeast to human neurons. We show TGs are protective against αS cytotoxicity and associated ER trafficking defects by preventing the accumulation of oleic acid (OA, 18:1) and DG. Importantly, we identify stearoyl-CoA desaturase (SCD) inhibitors that potently rescue αS cytotoxicity and inclusion formation. SCD is rate-limiting in the production of OA. We define a mechanism of this rescue through preservation of the protective tetrameric form of αS by saturated lipids and a reduction of the inclusion-prone αS monomer. A high degree of conservation of lipid pathways between species (Nielsen, 2009) enabled us to validate yeast genetic and biochemical results in rat cortical neurons, a C. elegans model of dopaminergic neuron degeneration, iPSC (induced pluripotent stem cell)-derived human neurons, PD patient neurons and a mouse model of familial PD (fPD).
Results
αS expression impairs lipid homeostasis in yeast
To assess αS-related alterations of cellular lipid homeostasis in an unbiased fashion, human αS was expressed in Saccharomyces cerevisiae under an estradiol-regulated promoter (Aranda-Diaz et al., 2017) that allowed tight control of αS expression and resulting proteotoxicity (Fig S1A–D). We induced αS expression for 12 h to achieve different degrees of cytotoxicity and assessed the effects on lipid profiles via unbiased liquid chromatography/mass spectrometry (LC/MS).
The most prominent lipid class changes were increases in DG and TG, dependent on αS expression levels (Fig 1A, Fig S1E, Fig S5A) and detectable as early as 6 h after αS induction (Fig S1F). DG and TG are components of the neutral lipid pathway (Fig 1B). DGs are precursors and TGs are key components of LDs; hence, one might expect accumulation of TG to be accompanied by enrichment of LD. Indeed, αS expression correlated with LD accumulation (Fig 1C), in keeping with a previous observation (Outeiro and Lindquist, 2003).
Fig 1– αS expression alters lipid metabolism in yeast. LDs protect against αS toxicity in yeast.

A) Lipid profiles of vector and human αS expression in yeast 12 h post induction. Lipid species (116) indicated by color and in the order of the key. B) Primary pathway for LD formation. DG and TG metabolic pathways are highly conserved between mammals (enzymes-red) and yeast (enzymes-blue). ACC1, cytosolic acetyl-CoA carboxylase; ATGL, adipose triglyceride lipase; DGAT1, 2: diacylglycerol acyltransferases; DGK, diglyceride kinase (multiple isoforms in mammals); OA, oleic acid; HSL, hormone-sensitive lipase; LCAT, lecithin:cholesterol acyltransferase; Seipin, integral membrane protein; SCD1, stearoyl-coA-desaturase; LPIN, lipid phosphatases; ER, endoplasmic reticulum; LD, lipid droplet. Whether lipolysis-derived DG enters the ER is currently unknown. C) αS expression increases LD formation in yeast. Green: BODIPY (LDs). Bar chart: integrated density (ImageJ) fold difference of uninduced vs induced (12h post induction). n = 22/condition. p<0.0005, t-test. D) LDs (TG) protect against αS toxicity. Differences in αS toxicity in 4 different strain backgrounds: (i) wt; (ii) dga1Δ lro1Δ; (iii) are1Δ are2Δ; (iv) LDΔ = dga1Δ lro1Δ are1Δ are2Δ. Samples induced at 5 nM estradiol. Table S2: statistical analysis of all yeast growth curves.
To assess the role of LDs in αS toxicity, we analyzed yeast strains lacking genes in the two branches of LD biosynthesis: the diacylglycerol acyltransferases DGA1 and LRO1 (TG biosynthesis) and the sterol acyltransferases ARE1 and ARE2 (sterol ester biosynthesis) (Fig 1B). The combined blocking of LD biosynthesis (dga1Δ lro1Δ are1Δ are2Δ - designated LDΔ) enhanced αS-related cytotoxicity, suggesting a protective role for LD formation in αS toxicity (Fig 1D and Fig S1G). Combined lack of Dga1 and Lro1 enhanced αS toxicity, suggesting TGs protect against αS toxicity. However, deletion of the sterol branch (are1Δ are2Δ) did not appreciably impact αS toxicity (Fig 1D).
We hypothesized that deleting the TG branch of LD synthesis causes a buildup of DG and FA (Fig 1B) that enhances αS toxicity. Indeed, neutral lipids analyses of wt vs. dga1Δ lro1Δ yeast strains expressing αS revealed increased DG in the mutant (Fig 2A and Fig S1H). Given the known ER localization of DGs, we asked whether DG accumulation exacerbates αS-mediated ER trafficking defects (Tardiff et al., 2013). We analyzed the trafficking of carboxypeptidase Y (CPY) in wt vs. dga1Δ lro1Δ strains and found increased αS-mediated ER accumulation of CPY in the mutant that lacks TG synthesis and accumulates DG (Fig 2B).
Fig 2– DG accumulation induced by αS expression is toxic and causes a trafficking defect.

A) Neutral lipid profiles of human αS expression in wt and dga1Δ lro1Δ yeast 6 h post induction. B) αS-induced ER accumulation of CPY is exacerbated in dga1Δ lro1Δ vs wt. CPY Immunoblot (ImageJ quantified). n=3. p=0.01, t-test. C) Deletion of lipases TGL3 and TGL4 rescues αS toxicity. D) Neutral lipid profiles of human αS expression in wt and tgl3Δ tgl4Δ yeast 12 h post induction. E) αS-induced ER accumulation of CPY is reduced in the tgl3Δ tgl4Δ strain (ImageJ quantified). n=3.p=0.007,t-test. F) Choline addition rescues αS toxicity. G) αS-induced ER accumulation of CPY is alleviated upon 0.5 mM choline addition (ImageJ quantified). n=3. p=0.002, t-test.
In a tgl3Δ/tgl4Δ mutant strain that lacks the two major TG lipases and accumulates about three-fold elevated levels of TG and LDs (Kurat et al., 2006; Wagner and Daum, 2005), we tested whether preventing TG degradation to DG and FA reduces αS toxicity. Indeed, deletion of the lipases efficiently rescued αS toxicity (Fig 2C, S1I), and lipid profiling confirmed DG decreases in the deletion mutants (Fig 2D), accompanied by relief of the CPY trafficking defect (Fig 2E). To further test the relevance of DG accumulation in αS toxicity, we added choline to cells expressing αS; choline enters the Kennedy pathway of PC synthesis and consumes DG in the ER (Fig 1B) as an alternative pathway to dissipate DG (McMaster, 2017). αS toxicity was suppressed by choline addition (Fig 2F, S1J) and the CPY trafficking defect was ameliorated (Fig 2G). Given the intermediary nature of DG in the neutral lipids pathway in processing FAs into TG, we next assessed FA levels in αS-expressing cells and established the role of FA in αS toxicity.
OA enhances αS toxicity
After observing increases in DG, TG and LD, we next asked whether αS expression impacted cellular FA content and composition. FA profiling revealed that αS expression causes multi-fold elevated levels of unsaturated FAs (UFA), most prominently OA (Fig 3A, Fig S2A–B). This increase was αS dose-dependent and evident as early as 6 h post αS induction (Fig S2B). Even greater monounsaturated FA increases were observed in the DG-accumulating dga1Δ lro1Δ strain (Fig 3B). Deletion of lipases Tgl3 and Tgl4 was associated with decreased OA (Fig 3C).
Fig 3-. OA exacerbates αS toxicity.

A) OA is increased upon αS expression. Intracellular FA analysis of αS-expressing wt yeast 12 h post induction. ** p<0.005; ****p<0.0001 (one way Anova). B) The αS-associated OA phenotype is more pronounced in dga1Δ lro1Δ vs. wt. Intracellular FA analysis of αS-expressing wt yeast 6 h post induction. ****p<0.0001 (one way Anova).C) The αS-associated OA phenotype is reduced in a tgl3Δ tgl4Δ strain vs. wt. Intracellular FA analysis of αS-expressing wt yeast 12 h post induction. ****p<0.0001 (one way Anova). D) Treatment with exogenous OA enhances αS toxicity. E) Dampening of OLE1 expression suppresses αS toxicity. F) Dampening OLE1 expression mitigates the αS-induced neutral lipid profile phenotype 12 h post induction. G) αS-induced ER accumulation of CPY is ameliorated in an OLE1 damp strain. (ImageJ quantified). n=3.p=0.0018, t-test.
To test whether increased cellular OA content enhanced αS toxicity, we exposed yeast to exogenous OA: this treatment strongly aggravated αS cytotoxicity (Fig 3D, Fig S2C) and was specific to OA (S2D). To verify this genetically, a yeast strain was assessed that expresses a mutant, hyperactive cytosolic acetyl-CoA carboxylase, catalyzing the initial step in de novo long-chain FA synthesis. This mutant strain, which produces >3-fold TG and >10-fold OA compared to wt (Hofbauer et al., 2014), exhibited enhanced αS toxicity (Fig S2E–F). Conversely, genetically down-regulating Ole1, an essential Δ9 FA desaturase, rescued αS toxicity (Fig 3E, Fig S2G). Dampening Ole1 also correlated with decreased DG (Fig 3F, Fig S2H) and rescue of the CPY trafficking defect (Fig 3G).
We next analyzed a Sei1 mutant that makes fewer but larger (‘super-sized’) LDs (Cartwright et al., 2015; Fei et al., 2011). Sei1 regulates LD morphology from ER initiation to maturation (Cartwright et al., 2015; Szymanski et al., 2007; Wang et al., 2016) and forms a complex with Ldb16, an LD assembly protein (Grippa et al., 2015; Wang et al., 2014a). We found Sei1 or Ldb16 deletion led to αS toxicity resistance (Fig S2I–J), prevented OA increase (Fig S2K) and fully suppressed the αS-associated lipid profile (Fig S2L). Sei1/Ldb16 deletion also prevented the CPY trafficking defect (Fig S2M).
Our genetic and biochemical analyses in the αS cytotoxicity yeast model suggest increased OA production and DG accumulation in the ER enhance αS toxicity if OA and DG are not converted to TG and ultimately deposited in LD.
αS expression alters lipid homeostasis in rat cortical neurons
Given the significant cytopathology of cortical neurons in PD and dementia with Lewy Bodies(DLB) (Braak et al., 2006; Dickson, 2012), we compared αS-induced lipid homeostasis changes in yeast to αS overexpression in primary neurons from rat embryonic cortex. Rat cortical neurons were transduced with lentivirus expressing human wt αS under the synapsin promoter. To obtain a quantitative understanding of the impact of αS on cellular lipids, neurons expressing two different αS levels (MOI-1, MOI-5) were profiled for lipid content by LC/MS at 14d and 20d post transduction.
Rat cortical neuron lipid profiles were more extensive than yeast profiles, in accordance with the larger number and greater degree of complexity of mammalian lipid species. Expression of human αS altered both neutral and phospholipids in a time- and dose-dependent manner. As with the yeast profiles, changes in neutral lipids represented the most pronounced phenotype (Fig 4A; Fig S3A–C; Fig S5B). Greatest TG fold increases were observed at MOI5 at 20 d. MOI1 and 14 d time-point effects were less pronounced but trended similarly.
Fig 4-. αS expression alters lipid metabolism in rat and C. elegans synucleinopathy models.

A) Lipid profiles of human αS expression in rat cortical neurons. Lipid species (516) are indicated by color and in the order of the key on the right of the map. B) αS expression increases LD formation in rat cortical neurons. Microscopy-Green: BODIPY (LDs); Red: αS. Blue: Hoechst (nucleus). Neurons were imaged at 14d (ImageJ quantified). Bar chart: integrated density signal fold difference for MOI1 vs MOI5. n=16 cells. p<0.0001,t-test. C) LDs protect against αS toxicity in rat cortical neurons. Neuronal survival was measured following expression of αS in control rat cortical neurons and in neurons with knockdown of DGAT1 and DGAT2 (D1+D2). Fig S3E, RTPCR knockdown data. % Viability (Resazurin to Resorufin conversion). n=6. ****p<0.0001 *p=0.02 (one way Anova). D) Reduction in LPIN expression suppresses αS toxicity in rat cortical neurons. Neuronal survival was measured following expression of αS in control rat cortical neurons and in neurons with knockdown of LPIN1, LPIN2, LPIN3. Fig S3F, RTPCR knockdown data. %Viability (Resazurin to Resorufin conversion). n=6 ****p<0.0001 **p=0.007 (one way Anova). E) OA content is increased upon human αs expression in rat cortical neurons. Intracellular FA analysis was performed in control and human αS expressing rat cortical neurons. *p=0.02, t-test. F) Reduction in SCD1 rescues αS toxicity. Neuronal survival was measured following expression of human αS in control rat cortical neurons and in neurons with SCD1 knockdown. Fig S4E, RT-PCR knockdown data. % Viability (Resazurin to Resorufin conversion). n=6. ****p<0.0001 (one way Anova). G) Inhibition of SCD1 rescues αS toxicity (% ATP). Survival of neurons was measured following treatment with SCD1 inhibitor in control and human αS-expressing rat cortical neurons. ****p<0.0001 ***p≤0.0005 (one way Anova). H) SCD1 inhibition rescues DGAT1+DGAT2+αS-associated toxicity in rat cortical neurons. DGAT1 and DGAT2 (D1+D2) were knocked down in control vs human αS-expressing rat cortical neurons + DMSO or SCD inhibitor. %Viability (Resazurin to Resorufin conversion). n=6. ***p<0.0005 (one way Anova). I) SCD knockdown in a C. elegans model of dopaminergic neuron degeneration rescued an αS-induced dopaminergic neuron degeneration phenotype. Open arrowheads-CEP dendrites (white), ADE dendrites (black);closed arrowheads-CEP cell bodies (white), ADE cell bodies (black); *p<0.05.
We also saw increased LD formation (Fig 4B), in agreement with our observations in yeast. To investigate whether LDs are protective against αS toxicity in neurons, we depleted diacylglycerol acyltransferases DGAT1 and DGAT2 (similar functions to yeast DGA1 and LRO1; Fig 1B). This depletion enhanced αS toxicity, corroborating a protective role for TG and LDs (Fig 4C and Fig S3D–E).
The phosphatidate phosphatase lipin genes (LPIN1–3) are responsible for conversion of PA to DG (Zhang and Reue, 2017) (Fig 1B). We knocked down LPIN1, LPIN2 and LPIN3; this suppressed αS toxicity in the cortical neurons, indicating DG depletion reduces αS toxicity, as in yeast (Fig 4D, Fig S3F).
In yeast, the deletion of seipin rescued αS toxicity by suppressing OA overproduction and DG accumulation. Similarly, knockdown of seipin rescued αS toxicity in the neurons (Fig S3G–H).
Inhibition of OA synthesis suppresses αS toxicity
We sought to establish whether OA plays a key role in the mechanism of αS toxicity in rat cortical neurons. FA profiling revealed increased OA levels in αS-expressing neurons relative to a vector control (Fig 4E; Fig S4A). Addition of exogenous OA exacerbated αS neurotoxicity (Fig S4B–C), while additions of stearic (18:0), palmitic (16:0) and palmitoleic (16:1) acids did not impact toxicity (Fig S4C–D). Our findings in yeast prompted us to investigate whether knockdown of SCD1, the rat homolog of OLE1, could rescue rodent neurons from αS toxicity. Reduction of SCD1 fully suppressed αS toxicity (Fig 4F; Fig S4E). Profiling of these neurons confirmed decreased OA and DG (Fig S4I–J) upon SCD1 knockdown. Pharmacological SCD1 inhibition also rescued αS toxicity in this model (Fig 4G, S4F–G). Given the connection between DGAT, OA metabolism and TG synthesis (Fig 1B), we next asked whether SCD1 inhibition could overcome enhanced αS toxicity from DGAT loss of function. Pharmacological inhibition of SCD1 fully rescued the increased αS toxicity associated with DGAT knockdown (Fig 4H, S4H). Both OA and DG levels were decreased in αS-expressing neurons with a seipin or SCD1 knockdown relative to control cells (Fig S4I–J).
To establish PD relevance, we asked whether decreasing OA levels could rescue an αS phenotype in intact dopaminergic (DAergic) neurons in vivo, namely in a C. elegans model that expresses αS in DAergic neurons. In this species, fat-6 and fat-7 desaturases (orthologs of SCD1/OLE1) convert the saturated stearic acid (18:0) to the monounsaturated OA (18:1). Degeneration of DAergic neurons in wt vs. fat-6&fat-7-deficient animals expressing αS was measured over for 10 d beginning with the first day of adulthood. All animals showed little DAergic neurodegeneration on day 1, but significant DAergic neurodegeneration was observed for UA44 animals on day 10. In contrast, fat-6&fat-7-deficient mutants were significantly protected from αS neurotoxicity (Fig 4I). Thus, genetic inhibition of monounsaturated FA synthesis can suppress αS-induced degeneration of dopaminergic neurons in vivo.
αS excess or fPD-linked point mutations alter neutral lipid homeostasis in human neurons
To establish whether the observations of excess wt αS in yeast and rat cortical neurons extended to human cells, we used human iPSC-derived neurons. Cells were transduced with lentivirus expressing wt human αS or vector control under the synapsin promoter and LC/MS profiled. Overexpression of αS altered neutral and phospholipids (Fig 5A, Fig S5C–E), with the most notable change being increased TG. The TG buildup resulted in increased LD formation (Fig 5B), conforming with similar observations in yeast and rat cortical neurons. FA profiling 14 d post transduction revealed increased OA in the wt αS overexpressing neurons relative to vector control (Fig 5C, Fig S5F). In keeping with our findings, addition of 1 μM exogenous OA significantly enhanced αS neurotoxicity (Fig 5D, S5G).
Fig 5– αS-associated lipid metabolism phenotypes in PD-relevant human neuron models.

A)Lipid profiles of αS overexpression in human iPSCs-derived neurons. Lipid species indicated by color and in the order of the key. B)αS expression increases LD formation in human iPSC-derived neurons. Microscopy-Green:BODIPY(LDs); Blue:Hoechst (nucleus);Red:αS (ImageJ quantified). Bar chart:fold difference in integrated density signal MOI1 vs MOI5. n=7 cells.p<0.0005,t-test. C)OA is increased upon αS overexpression in human iPS neurons. Intracellular FA analysis of αS overexpressing human iPSC-derived neurons.**p0.008, t-test. D)Treatment with exogenous OA enhances αS toxicity in human iPS neurons. Treatment of αS-expressing human iPS neurons expressing vector or αS with 1μM OA, ST (stearic), POA (palmitoleic), PA (palmitic). % Viability (Resazurin to Resorufin conversion). n=6. ***p<0.0005, **p<0.01 (one way Anova). E) Neutral lipid profiles of patient triplication and isogenic corrected neurons identify increased DG in the triplication line. Lipid species indicated by color and in the order of the key. Lipid profiling performed 23 d after differentiation to neurons. F) Neutral lipid profiles of human neurons, wt vs. E46K αS. Lipid species indicated by color and in the order of the key. Lipid profiling performed 36 d after differentiation to neurons. WB confirmed an equal amount of wt and E46K αS.
Increased levels of wt αS via triplication of the αS locus can cause early-onset, severe PD (Singleton et al., 2003). To further assess the relevance of our lipid findings to PD, we differentiated human αS triplication and isogenic genetically-corrected control lines to neurons and profiled at DIV 23. Profiling further supported the neutral lipid pathway as being altered by αS excess: triplication neurons exhibited increased DG relative to their genetically corrected controls. (Fig 5E and Fig S5H).
To probe for further relevance to PD, we compared the human embryonic stem cell line (BGO1) to its isogenic genetically-engineered BGO1-SNCAE46K line carrying the PD-causing E46K mutation (Soldner et al., 2011). Lines were differentiated to neurons and profiled 36 d post terminal differentiation. The BGO1-SNCAE46K neurons substantiated a role for the neutral lipid pathway in PD: they contained more DG and TG relative to the isogenic BGO1 wt αS neurons (Fig 5F, Fig S5I). Amounts of wt and E46K αS protein were similar (Fig 5F).
An fPD hu E46K αS mutant mouse model that displays motor deficits has altered brain neutral lipid homeostasis in vivo
To further support the PD relevance of our αS-mediated neutral lipid alterations, in particular, increased UFA, DG and TG, we compared these analytes in 12 mo old wt αS mice (WT) vs. human αSE46K-expressing mice (E46K). Total cortical lipid extracts were assayed by colorimetric enzymatic assays. In the fPD E46K αS mice, brain levels of UFA, DG and TG were significantly elevated vs. WT (Fig 6A-6C). Importantly, the E46K-mediated brain lipid accumulation was accompanied by progressive motor deficits, shown by increased time in the classical pole-climbing test and decreased endurance in the wire-hanging test (Fig 6D-6E). Amounts of wt and E46K hu αS protein were indistinguishable (Fig 6F). These in vivo data suggest that E46K αS accumulation influences neutral lipid regulation and is associated with PD-relevant motor phenotypes.
Fig 6-. Expression of fPD αS E46K alters brain FA composition in mice displaying motor deficits.

12 mo male WT and E46K αS mouse cortices were analyzed: A) UFAs; B) DG; C) TG levels; n=3; N=2 independent expts. Evaluation of motor behavior: D) quantification of time to descend a pole; n=6; E) endurance in wire hanging; n=5–6. F) WB shows equal αS expression. * p<0.02 **p<0.01,t-test.
SCD inhibition decreases αS-positive inclusions, decreases pSer129 αS and increases αS tetramer:monomer ratio
We specifically investigated the impact of OA on αS homeostasis in human cells. We previously reported multiple lines of evidence that a native form of αS in intact neurons and other cells is an α-helical homo-tetramer and this neuronal species is physiological, resists pathological aggregation and occurs in equilibrium with αS monomers, which are prone to form cytotoxic oligomers if present in excess (Bartels et al., 2011; Dettmer et al., 2013; Dettmer et al., 2015a; Dettmer et al., 2015b; Dettmer et al., 2017). These and related studies have addressed the relationship of normal αS helical conformation and native tetrameric assembly state on αS cytotoxicity (Burre et al., 2014; Gould et al., 2014; Gurry et al., 2013; Wang et al., 2014b; Wang et al., 2011; Westphal and Chandra, 2013). αS has also been found to bind and be altered by OA (Sharon et al., 2003; Sharon et al., 2001). We therefore sought to test whether cellular OA accumulation impacts physiological αS assembly state and whether this could represent a mechanism of αS neurotoxicity. Overexpression of wt or fPD mutant αS in neural cells does not readily cause inclusion formation (Dettmer et al., 2015b). However, amplifying the fPD E46K mutation (where the KTKEGV repeat #4 becomes KTKKGV) by expressing analogous E→K mutations in the two adjacent KTKEGV repeat motifs (i.e., E35K+E46K+E61K, designated αS 3K) can induce multiple, round cytoplasmic inclusions whose formation can be monitored in neural lines that express αS3K::YFP (Dettmer et al., 2015a). This αS 3K inclusion formation is sensitive to known modulators of wt αS toxicity e.g., the new potential PD drug nortriptyline (Collier et al., 2017). We ‘FA loaded’ αS3K::YFP-expressing human neuroblastoma cells by conditioning them in increasing (non-toxic) concentrations of FAs. We observed a dose-dependent increase in αS3K::YFP-positive cytoplasmic inclusions (Fig 7A, S5J), specific to OA. Treatment with SCD1-directed DsiRNAs (Fig 7B) and pharmacological SCD1 inhibition reduced αS-positive cytoplasmic inclusions (Fig 7C, 7D; Fig S5K–L), the latter dose-dependently. The αS 3K protein has reduced cytosolic solubility vs. wt αS, a marked decrease in the native αS tetrameric assembly form (αS60) and excess monomers (αS14) (Dettmer et al., 2015a). Treatment of αS3K M17D cells with an SCD1 inhibitor increased αS60 (tetramers) and decreased αS14 monomers (Fig 7E). αS was shifted from excessively membrane-associated/PBS-insoluble (TX-soluble) state to the more physiological cytosolic (PBS-soluble) protein (Fig 7F). Abnormally phosphorylated αS (pSer129) has been observed in many PD models, including mouse models overexpressing the A53T or A30P αS mutants (Schell et al., 2009; Wakamatsu et al., 2007). In humans, increased αS phosphorylation is associated with fPD (Lesage et al., 2013), DLB (Obi et al., 2008) and increased pathological severity of idiopathic PD (Anderson et al., 2006; Walker et al., 2013). Pharmacological SCD1 inhibition dramatically decreased the amount of phosphorylated αS, while total αS did not change (Fig 7G). To further establish PD relevance, we analyzed the impact of SCD1 inhibition on tetramer:monomer ratio and αS phosphorylation state in cells expressing the fPD E46K αS mutation we found earlier (Figs. 5F, 6A–C) to impact the neutral lipids pathway. Importantly, our previous work had identified the E46K mutation as significantly decreasing the αS tetramer:monomer ratio (Dettmer et al., 2015a). SCD1 inhibition increased the αS tetramer:monomer ratio (Fig 7H, S5M) and decreased pSer129 αS without changing total αS levels (Fig 7I). Both genetic and pharmacological SCD1 inhibition suggest that lowering OA levels returns αS to its normal assembly state.
Fig 7– OA impacts αS inclusion formation, tetramer:monomer ratio and pS129.

A) Exogenous OA increases αS inclusions in αS3K-expressing neuroblastoma cells. *p<0.05 ****p<0.0001 (one way Anova). B) SCD1 knockdown decreases αS inclusions in αS3K-expressing neuroblastoma cells. C: control (scrambled DsiRNA). ***p<0.0005; ****p<0.0001. C) SCD inhibition decreases αS inclusions. *p<0.05 ****p<0.0001 (one way Anova). D) SCD inhibition decreases αS inclusions (microscopy) in αS3K-expressing neuroblastoma cells. E&F) SCD1 inhibition increases 60kDa αS:14kDa αS in αS3K-expressing neuroblastoma cells. *non-specific band (Perrin et al., 2003).*p<0.05 **p<0.01. G) SCD inhibition decreases pS129 αS: total αS. n=6. *p<0.05 ***p<0.005,t-test. H) SCD1 inhibition increases 60kDa αS:14kDa αS and 80kDa αS: 14kDa αS in fPD E46K-expressing neuroblastoma cells. N=2, n=6. I) SCD1 inhibition decreases pS129 αS: total αS in E46K-expressing cells. N=2, n=10. **p<0.01, t-test.
Discussion
We performed unbiased lipidomic analyses and a series of related genetic and biochemical experiments to provide new insights into the complex interplay between αS and lipid metabolic pathways. Our findings have direct implications for αS cytotoxicity mechanisms and for several PD relevant phenotypes. We observed pronounced effects of excess human αS on lipid homeostasis, which correlated across multiple cellular systems. These diverse model systems converged on neutral lipid pathway alterations being caused by αS dyshomeostasis. Each cellular model system emphasized a role for neutral lipid pathway components, culminating in our in vivo fPD mouse model implicating all principal pathway components. Perhaps our most important disease-relevant observation was identifying a key role for OA in the cellular response to excess/mutant αS. This lead to identifying SCD inhibitors capable of dose-dependently suppressing αS cytotoxicity, inclusion formation, hyperphosphorylation and an abnormal decrease in tetramer:monomer equilibrium. These beneficial effects strongly recommend SCD as a new target for treating synucleinopathy.
We find αS principally increases monounsaturated FA species, specifically OA. UFAs have been reported to promote αS membrane binding and pathological aggregation (Lucke et al., 2006; Sharon et al., 2003; Sharon et al., 2001) and increase αS cytotoxicity (Jo et al., 2002; Snead and Eliezer, 2014). Similar chain length saturated FAs did not impact αS-expressing cells, and a comparable UFA, POA, did not exert the same level of toxicity. Evidence for some specificity of the pathobiological impact of OA and POA has been reported (Lockshon et al., 2012; Petschnigg et al., 2009).
Our lipidomic analyses led us to the discovery that SCD downregulation strongly ameliorates αS cytotoxicity. The origin of increased OA by excess αS is currently under investigation. Our preliminary findings suggest ACC1 and OLE1 desaturase gene upregulation upon αS expression in yeast. The degree of fatty acyl side chain unsaturation of phospholipids is a major determinant of membrane fluidity, which in yeast is sensed by membrane-bound transcriptional activators, Mga2 and Spt23. Mga2 is an activator of OLE1. Mga2 transmembrane helices sense lipid packing in the membrane (Covino et al., 2016). Upregulation of OLE1, despite increased OA, indicates a disruption of this regulatory circuit by αS.
We find Seipin mutants suppress αS toxicity. Seipin plays a role in LD biogenesis and may act as a scaffolding protein coordinating multiple lipid metabolic pathways (Qi et al., 2017). It was found to interact with and inhibit GPAT, the initial enzyme of glycerolipid synthesis (Pagac et al., 2016). This is of particular interest because GPAT2 and the desaturase OLE1 may compete for a common acyl-CoA pool (De Smet et al., 2012). A direct regulatory impact of seipin on FA desaturation was also identified in seipin mutant cell lines derived from a Berardinelli-Seip congenital lipodystrophy patient that displayed decreased SCD activity (Boutet et al., 2009).
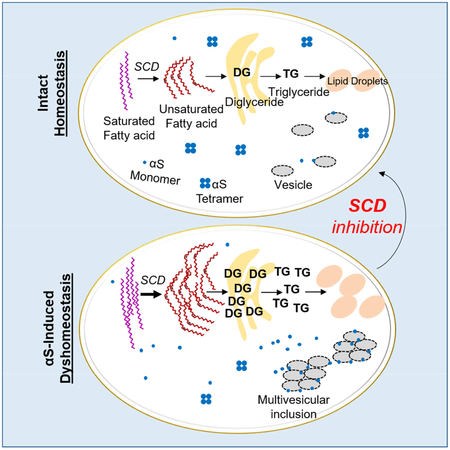
We propose the following model (Fig S6). (A) Under conditions of normal αS and lipid homeostasis, αS occurs in equilibrium between monomers and α-helically-folded physiological tetramers. Tetramers are principally soluble (cytosolic) while monomers occur in both membrane-associated and cytosolic pools (Chandra et al., 2005; Dettmer et al., 2015b; Dettmer et al., 2017; Westphal and Chandra, 2013). FA/DG/TG production and degradation are normal, as are LD size and number. (B) For mild αS accumulation unaccompanied by a genetic lipid abnormality, αS triggers changes in lipid homeostasis. This is initially compensated: increased FA production (Fig 3) results in some DG accumulation and TG production (Fig 1, 4, 5, 6). Cells store excess FAs as TG in LDs. DG accumulation in the ER is modest and tolerated. (C) Greater αS accumulation over a longer time can exceed compensatory mechanisms, particularly if there is also a genetic variant in lipid biosynthesis or metabolism. TG synthesis and LD biogenesis pathways are overwhelmed, and DG accumulates in the ER, causing membrane trafficking defects, possibly aggravating αS trafficking defects. Thus, genetic risk factors may influence the cell’s ability to achieve compensatory mechanisms to αS accumulation proposed in scenario B (Fig 1, 2, 4). State B could move towards state C with aging, the major PD risk factor.
Increased OA appears to play a key role in αS toxicity, raising the question of how this toxicity is mediated. Our data suggest when OA levels are high, a larger fraction of αS is bound to membranes. Increased αS membrane binding is associated with vesicle-rich inclusions and excess fPD-mutant αS monomers at vesicle membranes (Dettmer et al., 2017; Soper et al., 2008). Abrogating normal tetramers and shifting them to excess, aggregation-prone monomers leads to αS inclusions and neurotoxicity(Dettmer et al., 2015a). αS overexpression increases OA, and this makes inclusions more prominent. This connection may explain the observed increased cytotoxicity, since increased αS membrane localization can be toxic (Chandra et al., 2005; Choi et al., 2004; Rochet et al., 2004). We propose two mutually non-exclusive mechanisms for increased αS membrane binding: direct binding of αS to OA that is incorporated as fatty acyl side chains into membrane lipids (Sharon et al., 2003; Sharon et al., 2001); and increased membrane fluidity due to a greater degree of unsaturation (Fig S6). We hypothesize that high OA levels promote αS membrane binding, enhancing membrane-associated toxicity (Pranke et al., 2011; Volles and Lansbury, 2007). Increased membrane association of monomers may enable their gradual local sequestration into αS aggregates, ultimately including fibrils (Galvagnion, 2017). We expect increased FA saturation or decreases in unsaturated FAs can rescue αS toxicity through changes in membrane fluidity.
We observe striking changes in neutral lipids downstream of αS. We cannot conclude that all toxicity is directly caused by these lipid alterations, nor are these alterations necessarily the sole cause of phenotypes such as trafficking defects. They do, however, indicate an important drug target for PD research. Our approach to delineating the effects of αS accumulation in cells began with unbiased lipidomic analyses in yeast and moved on to genetic and pharmacological validation of the implicated neutral lipid pathways in mammalian neurons. These data, derived from multiple cell sources and mutation scenarios in vitro and in vivo, all point to a common underlying mechanism of αS toxicity: interfering with FA desaturation and the deregulated metabolic flux of UFAs into various cellular lipids. Our approach has identified αS-lipid interactions that suggest a promising new target for pharmacological intervention, SCD. αS toxicity and relative reduction of physiological αS tetramers was efficiently prevented by SCD genetic down-regulation or pharmacological inhibition. This decreased the elevated αS association with membranes and αS phosphorylation, and it increased the levels of native αS tetramers, reestablishing a physiological tetramer:monomer ratio. Our findings thus indicate that partial inhibition of SCD would be a rationally based therapeutic approach to αS neurotoxicity, an approach with therapeutic implications for PD, DLB and other human synucleinopathies.
Star Methods
All abbreviations are listed in Supplementary Table 1.
Growing and inducing yeast cultures
All experiments were performed in the Saccharomyces cerevisiae BY4741 background. Deletion strains were transformed with the estradiol transcriptional regulator (nat+) and a copy of human αS regulated by the estradiol promoter (leu+). The standard lithium acetate transformation protocol was used for all yeast transformations. Yeast cells were routinely cultured in complete synthetic medium (CSM). Yeast cells were uninduced (0 nm nanoMolar) estradiol or induced at varying estradiol concentrations at different time points indicated in figure legends. For induction, cells were grown in CSM.Raff overnight, diluted in CSM.Gal for 6–8 hours and then log phase cells were induced in CSM.Gal containing estradiol. Uninduced cultures were included as controls to assess the impact of gene deletion on growth rate independent of αS expression. Yeast growth curves were performed in triplicate in an Epoch2 Microplate Spectrophotometer (Biotek) at 30°C with intermittent shaking. Average and standard deviations are reported in figures. OD600nm readings were taken every 15 min.
Lipidomic and FA profiling and Analysis
LCMS was employed to examine lipid content changes in yeast, rat cortical neurons, human iPSC-derived neurons expressing human αS and patient derived neurons. Samples were extracted using a chloroform/methanol extraction. For yeast strains, uninduced and induced yeast cultures were washed once in cold LCMS grade water. Cells were pelleted and resuspended in 600 μl of methanol. 300 μl of LCMS water and 400 μl chloroform were added to the cells. Cells were disrupted by bead beating for 12 mins at 4 °C using the Qiagen TissuLyserII at a frequency of 30. Cells were spun at 13,000 rpm for 10 mins at 4 °C. The bottom layer containing the lipid fraction was collected and dried under vacuum. For rat cortical neuron and human samples the protocol was adjusted as follows. Media was removed from wells (rat cortical neurons: ×3 wells of a 24 well plate; human iPS-derived neurons: ×1 well of a 6 well plate). 1 ml of cold sterile 0.9% NaCl (made with LCMS grade water) was added to wells and cells were scraped. Cell suspensions were added to Eppendorf tubes and the protocol for chloroform/methanol extraction was followed as above. Dried lipid samples were dissolved in 50 μl 65:30:5 (v/v/v) acetonitrile:isopropanol:water. 5μl of dissolved sample was injected into the LCMS using separate injections for positive and negative ionization modes. Identification of lipids was made on the basis of column retention time and chemical formula. Details of LCMS protocol utilized were as per (Smulan et al., 2016). Samples were injected onto the LC/MS twice – once for positive and once for negative ionization – and the data were analyzed separately. A software package, LipidSearch, that identifies lipids based on exact mass and fragmentation pattern, aligns peaks among multiple LC/MS runs and performs peak integration in an automated fashion was used for data analysis (although we did not use the function of aligning between runs as all samples compared in any heatmap were run at the same time). A peak quality metric in the software contributes to validation and quality control metrics. To enhance the rigorous nature of the QC analysis, a “pool” sample consisting of a mixture of several uL from each biological samples was run. This created a representative sample that is run multiple times to get a measure of technical reproducibility for each metabolite. A CV (standard deviation / average) was calculated for technical replicates and peaks with CV>0.4 were rejected. 0.3-fold and 0.1-fold dilutions of this pooled sample were also run to confirm samples were in the linear range of detection for each lipid, or whether the detector is close to saturation or nearing the lower limit of detection. Peaks with R<0.9 for this dilution series were rejected. Finally, there was an additional “reject” flag in the software and all lipids with this flag were rejected. A total signal value for each sample was calculated as a measure of total lipid material per sample. The same lipid class analysis on the raw peak areas and on the peak areas normalized to this total signal was performed. Individual lipid data were analyzed in the same way. These values are used to calculate averages and fold changes. We incorporated an internal standard (TG45 – 15:15:15) in to yeast preparations as an additional test to confirm similar results were achieved normalizing to internal standard versus total lipid signal. To maintain consistency between models and because all analysis was a relative measure rather than a quantitative analysis, normalizing to total lipid signal was maintained throughout. All samples to be compared were run on the same run and controls incorporated to check there was no peak drift within a run. The running order of samples was randomized (to minimize potential contribution due to technical variation). Lipids: DG-diglyceride;TG-triglyceride;CL-cardiolipin;LPC-lysophosphatidylcholine;LPE-lysophosphotidyethanolamine;LPI-lysophosphatidylinositol;LPS-lysophosphatidylserine;PC-phosphatidylcholine;PE-phosphatidylethanolamine;PG-phosphatidylglycerol;PI-phosphatidylinositol;PS-phosphatidylserine; AcCa- acylcarnitine; Cer- ceramide; CerG1/G2-glucosylceramide; CerP- ceramide phosphate; ChE- cholesterol; MG- monoglyceride; DG-diglyceride; SM- sphingomyelin; SO- sphigosine; CL-cardiolipin; LPG- lysophosphatidylglycerol;
Heatmap construction
To construct heatmaps log2 values of normalized lipid counts were calculated for each lipid and each lipid was median centered across samples. These values were averaged for each of the 3 replicates to make the figures where only the averaged values are shown. Abundance levels were calculated by median-summarizing control samples, followed by log2-transformation and median centering of all the lipids. All heatmaps were visualized and images exported with Java TreeView. Bars indicating saturation levels were constructed using a custom R script. All heatmaps and colored bar labels were compiled in Adobe Illustrator. Yeast: Baseline abundance (Abd) of each lipid species is indicated by a red/blue bar on the left of the heat map (relative scale from −3 to 3, see key). Baseline abundance was calculated on relative amount of each lipid species in the vector strain with 0 nM inducer. Yellow/Blue heatmap coloring is a representation of a given lipid species relative to the median of the logs across all samples for that lipid species; data for 0, 2, 5 and 10 nM of inducer are shown (relative log scale from −3 to 3, see key). Status of saturation (presence of double bonds [DB]) of each lipid species is indicated by grey (>1 DB) or black (0 DB) bar on the right of the heat map. Rat Cortical Neurons: Baseline abundance (Abd) of each lipid species is indicated by a red/blue bar on the left of the heat map (relative scale from -3 to 3, see key). Baseline abundance was calculated on the relative amount of each lipid species at MOI1 for the vector control at day 14 and day 20. Bar shown represents Abd for day 20 (both time points were similar and can be compared in Fig S3A. Yellow/Blue heatmap coloring is a representation of a given lipid species relative to the median of the logs across all samples for that lipid species. The relative log scale (−3 to 3) for yellow/blue representation is indicated by a smaller yellow/blue bar on the left of the heatmap. Status of saturation (presence of double bonds [DB]) of each lipid species is indicated by grey (>1 DB) or black (0 DB) bar on the right of the heat map. Human Neurons: Baseline abundance (Abd) of each lipid species is indicated by a red/blue bar on the left of the heat map (relative scale from −3 to 3, see key). Baseline abundance was calculated on the basis of the relative amount of each lipid species in the vector control. Degree of baseline abundance (Baseline Abd Scale) is indicated by a smaller red/blue bar on the left of the heatmap. Yellow/Blue heatmap coloring is a representation of a given lipid species relative to the median of the logs across all samples for that lipid species. The relative log scale (−3 to 3) for yellow/blue representation is indicated by a smaller yellow/blue bar on the left of the heatmap. Status of saturation (presence of double bonds [DB]) of each lipid species is indicated by grey (>1 DB) or black (0 DB) bar on the right of the heat map.
Cell viability flow cytometry assay
10 μg/mL propidium iodide (Sigma P4864) was added to 180 μl of diluted uninduced and induced yeast cultures (in duplicate) at the 12hr time point. A MACSQuant VYB cytometer with a 96-well plate platform (Miltenyi Biotech) was used to measure samples. 10,000 events were collected per sample. Using the FlowJo software, dead cells were gated in the Y3 fluorescence channel (661/20 filter).
Microscopy
Yeast cells in logarithmic phase were induced with 10nm estradiol or uninduced (control) for 12 hours. Cells were centrifuged, media was removed, and cells were washed once in PBS. Cells were stained with 1ug/ml BODIPY (Life Technologies, D3922) for 10 mins and washed twice in PBS before microscopy. ImageJ (integrated density) was used to quantify differences between uniduced and induced. An n of 22 yeast cells per condition was used to generate a t-test p value.
Rat cortical neurons and human iPS-derived neurons were cultured in 8 well glass microscopy chambers and transduced with αS Lentivirus at different MOI as indicated. Media was removed from live cells and cells were washed once in PBS. Cells were fixed in freshly prepared 4% formaldehyde solution for 20 mins and then washed once with PBS. Cells were stained with HA antibody to detect αS expression and with HOECHST (Invitrogen, H3570) for nuclear staining and with 1 μg/ml BODIPY (Life Technologies, D3922). Single images were taken using a Nikon Eclipse Ti microscope. ImageJ (integrated density) was used to quantify differences in LD. An n of 16 cells per condition was used in rat cortical neurons to generate t-test p values. An n of 7 cells per condition was used in human cortical neurons to generate t-test p values.
Western blots
Protein analysis was performed by extracting cellular extracts from yeast, rat cortical neuron and human iPS-derived neuron samples. In general, samples were boiled with 4X NuPAGE LDS Sample Buffer (Invitrogen, NP0007), centrifuged and run on NuPAGE 4–12% Bis-Tris Midi gels with NuPAGE MES buffer (Novex, Life Technologies, NP0002). Western blots for trafficking of CPY were performed using protein samples generated from the same cells as were lipid profiled. ImageJ was used to quantify ER accumulation of CPY and values analyzed as appropriate by t-test at n=3 for each treatment type. Samples were prepared as usual but run on NuPAGE 8% Bis-Tris Midi gels in NuPAGE MOPS SDS running buffer (Novex, Life Technologies, NP0001). Proteins were transferred to PVDF membranes using the iBLOT2 system (Invitrogen, IB24001). Membranes were blocked in 5% milk PBS-T or Rocklands blocking buffer (MB-070) and blotted where appropriate with the following antibodies:
| Protein | Secondary | Company | Ref Code |
|---|---|---|---|
| αSynuclein | Mouse | BD | BDB610786 |
| CPY | Mouse | Invitrogen | A6428 |
| PGK1 | Rabbit | Antibodies Online | ABIN568371 |
| GFP | Mouse | Roche | 11814460001 |
| HA | Rat | Roche | Clone 3F10, 12013819001 |
| Tubulin | Mouse | Sigma | Clone B-5-1-2, T5168 |
| SCD1 | Mouse | Abcam | ab19862 |
| Actin | Rabbit | Abcam | ab8227 |
| Transferrin | Rabbit | Abcam | ab84036 |
| DJ-1 | Rabbit | - | (Baulac et al., 2004) |
| Phosphorylated αS | Rabbit | Abcam | ab168381 |
FA and choline treatments
Yeast and neuron FA treatment (fatty acids have been shown to be taken up in multiple cell types: (Alexander et al., 1998; Black and DiRusso, 2003, 2007; Ebert et al., 2003; Edmond et al., 1987; Melton et al., 2011; Taib et al., 2013; Zou et al., 2002)) was performed in CSM.Gal and neurobasal medium, respectively. FAs [OA (Sigma O1383), palmitic acid (Sigma P5585), palmitOA (Sigma 76169), stearic acid (Sigma 85679)] were diluted in FA free bovine serum albumin (Sigma A8806) and supplemented in to media. Choline (choline chloride Sigma C7527) was diluted in water and supplemented in to CSM.gal media for yeast experiments.
αS toxicity models in rat neurons and human neurons
Rat embryonic cortical neurons and NGN2-induced human neurons expressing human αS were used as relevant neuronal models for αS-mediated perturbation of lipid homeostasis. Isolated and enriched neurons were maintained in adherent monolayer culture. These neurons were transduced with Lentiviral constructs harboring neuron-specific human synapsin promoter and human αS transgene or a control gene (i.e., GFP in the same vector backbone).
Preparation and maintenance of rat cortical neurons
Rat embryos from anesthetized pregnant Sprague-Dawley rats (Charles-River Laboratories) at embryonic day 18 were harvested by cesarean microdissection under a stereoscope. Dissected cortices were collected in HBSS on ice. Cells were dissociated with Accumax (Innovative Cell Technologies) and DNase (40 U/μl) treatment at 37 °C for 25 min followed by gentle triutration with sterile Pasteur pipette in complete neurobasal medium (Life Technologies) supplemented with B27 (Life Technologies), glutamine (0.5 mM), β–mercaptoethanol (25 μM), penicillin (100 IU/ml) and streptomycin (100 μg/ml). Cell suspension was filtered through a 70 μM cell strainer to remove tissue debris and clumps. For seeding the cells, flat-bottom polystyrene plates [96-well, 24-well, 6-well] were coated with poly-ornithine and laminin and the 8-well glass chambers (Lab-Tek; 70378–81) were coated with poly-D-Lysine. The isolated cell suspension was seeded at a density of 40,000 cells (per well of 96-well plate), 200,000 cells (per well of 24-well plate), 1,000,000 cells (per well of 6-well plate) or 88,000 cells (per well of 8-well glass chamber) in neurobasal medium with previously mentioned supplements. The spent medium was exchanged with fresh complete neurobasal media (without β–mercaptoethanol) after the first 4 days of incubation. Neuronal morphology was observed under an inverted phase contrast microscope. The neurons exhibit progressively robust neurite extension that forms a network with neighboring neurons and have minimal non-neuronal-like cells after 4 DIV.
NGN2 induced human neuron differentiation protocol
The induced human neurons (cell line is called CORR-1) were generated as described previously (Soldner et al., 2011) and maintained as feeder free cells in defined, serum-free media (mTeSR, Stem Cell Technologies). To generate the NGN2-inducible iPSC line, virus was produced as described previously (Pang et al., 2011) with FUW-TetO-Ngn2-P2A-Puromycin (Addgene plasmid #52047) and FUW-M2rtTA (Addgene plasmid #20342). The iPSC line was transduced with each virus at an MOI of 30 and expanded as feeder free cells in mTesr. Neural induction was achieved with minor modifications to previous protocols (Zhang et al., 2013); briefly on day zero of the differentiation the NGN2-iPSC line was dissociated with accutase (Stem Cell Technologies) and plated in mTesr media (supplemented with 10 μM ROCK inhibitor Y-27632 and 2 μg/mL Doxycycline) at 750,000 cells/well on a matrigel coated well of a 6-well plate. On day one of the differentiation, the culture media was changed to DMEM/F12 supplemented with N2 (Gibco, Cat No. 17502–048), B27 (Gibco, Cat No. 17504–044), non-essential amino acids, GlutaMAX, 2 μg/mL puromycin and 2 μg/mL doxycycline. For days 3–7 the cells were maintained in DMEM/F12 supplemented with N2, B27, NEAA, GlutaMAX and 2 μg/mL doxycycline. On day 7 the cells were dissociated with accutase and replated in PEI coated 6-well plate in DMEM/F12 supplemented with N2, B27, 10 ng/mL BDNF, 10 ng/mL GDNF, 2 mM cAMP, 0.4 μM ascorbic acid, 2 μg/mL laminin, 10 μM Y-27632 and 0.5 μM AraC. The neurons were maintained in BrainPhys media (Stem Cell Technologies) supplemented with N2, B27, 10 ng/mL BDNF, 10 ng/mL GDNF, 2 mM cAMP, 0.4 μM ascorbic acid and 2 μg/mL laminin with half media changes every 2–3 days for 7 days before viral transduction.
Lentivirus constructs and virus preparation
pLV-hSyn Lentiviral expression vector under the synapsin (hSyn) promoter was obtained from Addgene (Addgene #22909) (Nathanson et al., 2009). For Lentiviral constructs, expression vectors (pLV-hSyn-hSNC or pLV-hSyn-mGFP) were generated with the human synapsin promoter upstream of the human αS(hSNC) or a monomeric GFP (mGFP) cDNA. To prepare the Lentivirus, psPAX2 packaging vector and pMD2.G envelope vector along with the pLV-hSyn expression vector were transfected in 90% confluent monolayer of adherent 293T cells in 10 cm plates using Lipofectamine2000 (Invitrogen) transfection reagent following the manufacture’s instruction. Lentivirus was harvested from the supernatant of the transfected 293T cells at 2, 3 and 4 days post-transfection. Virus from the collected supernatant was purified using Lenti-X Maxi Purification Kit (Clontech) according to the protocol provided by the manufacturer. The purified virus was concentrated with Lenti X concentrator (Clontech) according to the instructions. Viral pellet was resuspended in Neurobasal media (Life Technologies). Viral titer was determined using Lentivirus-Associated p24 ELISA Kit (Cell BioLabs) according to the manufacturer’s protocol.
Viral transduction of rat primary cortical cultures and human neural cells
Rat cortical cultures were transduced with various multiplicities of infection (MOI) of Lentivirus at day in vitro (DIV) 4. Differentiated human neurons were transduced with the Lentiviral preparation 7 days post differentiation.
DsiRNA treatment of rat cortical cultures and human neurons
Predesigned Dicer-Substrate siRNA (DsiRNA) for target genes were ordered from IDT and preparations were transfected into the neuronal cultures using Lipofectamine RNAiMAX Reagent (ThermoFisher, 13778075) using manufacturer’s protocol. All experiments involving DsiRNAs and fatty acid treatments were performed with viral titer of MOI5. In many cases, multiple DsiRNA were used for ATP measurements in rat cortical neurons and graphs presented are representative of DsiRNA trends. Neurons transduced with Lentiviral preparations and/or transfected with DsiRNAs were processed for neuronal toxicity assay after indicated time points. As a readout of neuronal toxicity, cell viability was measured by quantifying cellular ATP content using ViaLight Plus Cytotoxicity BioAssay Kit (Lonza). Cell Titer Blue Cell Viability Assay (Promega) was employed as an additional viability readout. Adenylate Kinase assays were performed using the ToxiLight™ bioassay kit (Lonza).
RTPCR to determine gene depletion in rat cortical neurons following DsiRNA treatment
Having unsuccessfully trialed many antibodies for our protein knockdown targets in rat cortical neurons, we transitioned to RTPCR to get the most accurate readout for degree of expression knockdown. RNA was extracted from cells using the Ambion Cells-to-CT kit (ThermoFisher Scientific A25603) according to the manufacturers protocol. RTPCR was performed using the Ambion Cells-to-CT kit coupled with pre-mixed primers specific for rat genes that were targeted for depletion (Taqman Gene Expression Assays, ThermoFisher Scientific).
Rat cortical neuron treatment with SCD inhibitors
SCD inhibitors (HY19762 and HY15700, MedChemexpress) and (ab142089, Abcam) were diluted in DMSO and added to rat cortical neurons at concentrations indicated in figures.
Immunoblotting
Neuronal cultures were harvested to detect various proteins by lysing the cells in RIPA buffer (25 mM HEPES, pH 7.5, 150 mM NaCl, 0.25% Deoxychloate, 10% Glycerol, 25 mM NaF, 10 mM MgCl2, 1 mM EDTA, 1% TritonX-100, 0.5 mM PMSF, protease inhibitor cocktail from Roche). Protein samples from total cell lysate were processed under denaturing conditions by adding 1X LDS sample buffer with 100 mM DTT. Processed cell lysate was separated in 4–12% NuPage Bis-Tris polyacrylamide gel (Life Technologies) and transferred onto PVDF membranes using iBlot protein transfer apparatus (Life Technologies). Membranes were blocked in 5% non-fat dry milk in PBST for 1 hour at room temperature followed by incubation with primary antibodies in 5% non-fat dry milk in PBST at 4°C overnight with gentle rocking. The blots were then washed with PBST for 3 X 15 minutes and incubated with secondary antibodies conjugated to IRDye 680 or 800 (1:10,000, Rockland) or to HRP (1:10,000, Sigma) in 5% non-fat dry milk in PBST for 1 hour at room temperature. After three 15 min washes with PBST, blots were developed with SuperSignal West Femto maximum sensitivity chemiluminescent substrate (ThermoFisher Scientific) by ChemiDoc MP Imaging System and analyzed by Image Lab software (Bio-Rad).
C. elegans Model for Dopaminergic Neuron Degeneration
UA44 animals bearing an a-syn::GFP transgene under the control of the dat-1 DAergic neuron specific promoter simultaneously express αS and GFP in the eight C. elegans dopaminergic (DA-ergic) neurons (4 CEP, 2 ADE and 2 PDE). UA44 animals were crossed in fat-7 mutants, which were raised on fat-6 RNAi for the duration of the experiments. The extent to which DA neurons degenerated in wild-type and fat-6;fat-7 animals was measured over a 10-day period beginning with the first day of adulthood. Degeneration of dopaminergic neurons was measured as described (Tucci et al., 2011). The extent to which DAergic neurons degenerated in wild-type animals expressing αS (UA44) and fat-6;fat-7 animals in the UA44 background was measured over a 10-day period beginning with the first day of adulthood. N = 55 animals, UA44; 65 animals, UA44;fat-6;fat-7.
Patient αS Triplication and Genetically Corrected Lines Neuronal Differentiation
Patient triplication and genetically corrected lines were obtained from EBiSC [https://cells.ebisc.org/EDi001-A] [https://cells.ebisc.org/EDi001-A-4] (Devine et al., 2011). Neurogenin 2 Induced Neuron (Ngn2-iN) Differentiation iPSCs were transduced with 3 lentiviruses on day 0 (Maherali et al., 2008; Vierbuchen et al., 2010; Zhang et al., 2013): Ngn2 (pTet-O-Ngn2-puro), rtTA (FUdeltaGW-rtTA) and GFP (Tet-O-FUW-EGFP). FUdeltaGW-rtTA was a gift from Konrad Hochedlinger (Addgene plasmid #19780). Tet-O-FUW-EGFP was a gift from Marius Wernig (Addgene plasmid #30130). pTet-O-Ngn2-puro was a gift from Marius Wernig (Addgene plasmid #52047). Neural induction was essentially as per (Zhang et al., 2013) with some minor modifications. Briefly, on day 0, lines were plated in mTesr media (supplemented with 10 M ROCK inhibitor Y-27632 and 2 μg/mL Doxycycline) on a matrigel coated 6-well plates. On day 1, media was changed to KSR media containing DOX (2 μg/ml). On day 2, media was changed to KSR:N2B (1:1) with DOX (2 μg/ml) and puromycin (5 μg/ml). On day 3 media was changed to N2B supplemented with B27 with DOX and puromycin. On day 4, media was changed to NBM media with B27, DOX (2 μg/ml), puromycin, growth factors (BDNF/CNTF/GDNF) and ROCK inhibitor. From day 5 onwards cells were maintained in NBM media containing B27, DOX, purpomycin and growth factors. Cells were profiled on day 23 (as per protocols above). Data were normalized to total positive lipid ion signal and to total protein and both gave similar results.
Human embryonic stem cell (hESC) culture and neuronal differentiation
Maintenance and neuronal differentiation of the hESC line BGO1 (NIH code: BG01; BresaGen, Inc., Athens, GA) and the isogeneic genetically engineered BGO1-SNCAE46K line carrying the E46K mutation in SNCA have been described in detail before (Soldner et al., 2011; Soldner et al., 2016). In brief, hESCs were maintained on mitomycin C-inactivated mouse embryonic fibroblast feeder layers in hESC medium (DMEM/F12 supplemented with 15% FBS (Hyclone), 5% KnockOut Serum Replacement, 1 mM glutamine, 1% nonessential amino acids, 0.1 mM β-mercaptoethanol (Sigma) and 4 ng ml−1 FGF2 (R&D systems). Cultures were passaged every 5–7 days either by trituration or enzymatically with collagenase type IV (Invitrogen; 1.5 mg ml−1). To induce neuronal differentiation, hESCs were harvested using 1.5 mg ml−1 collagenase type IV (Invitrogen), separated from the MEF feeder cells by gravity, gently triturated and cultured for 8 days in non-adherent suspension culture dishes (Corning) in EB medium (DMEM (Invitrogen) supplemented with 20% KnockOut Serum Replacement (Invitrogen), 0.5 mM glutamine (Invitrogen), 1% nonessential amino acids (Invitrogen), 0.1 mM β-mercaptoethanol (Sigma)) supplemented with 50 ng ml−1 human recombinant Noggin (Peprotech) and 1,000 nM dorsomorphin (Stemgent). Subsequently human EBs were plated onto poly-L-ornithine (15 μg ml−1, Sigma), laminin (1 μg ml−1Sigma), fibronectin (2 μg ml−1 Sigma) coated tissue culture dishes in N2 medium (Kim et al., 2003) supplemented with 50 ng ml−1 human recombinant Noggin (Peprotech), 1,000 nM dorsomorphin (Stemgent) and FGF2 (20 ng ml−1, R&D systems). After 8 days, neural rosette-bearing EBs were cut out by microdissection, dissociated using 0.05% trypsin/EDTA solution (Invitrogen) and subsequently expanded on poly-L-ornithine, laminin and fibronectin coated cell culture dishes a density of 5 × 105 cells per cm2 in N2 medium supplemented with FGF2 (20 ng ml−1, R&D systems). Proliferating NPCs were passaged 5 times before induction of terminal differentiation into neurons by growth factor withdrawal in N2 medium supplemented with ascorbic acid (Sigma). Differentiated neurons were passaged using Accutase (Stem cell technology) 12 days after withdrawal of growth factors and harvested for analysis at day 38 of terminal differentiation. An n of 6 was analyzed for BGO1 and BGO1-SNCAE46K lines.
Mouse Experiments
All animal procedures were approved by the Institutional Animal Care and Use Committee at BWH (IACUC protocol #05022). B6-Tg (SNCA*WT) mice were generated by micro-injection into C57BL/6J one-cell embryosB6N.Cg-Tg(SNCA*E46K)3Elan/J mice (E46K) were purchased from Jackson Laboratories. We chose this mouse because 1) it is genomically humanized and thus closer to the human fPD genetic state and 2) it has aS tg expression in striatal and cortical regions. An initial characterization that includes evaluating DAergic integrity is found online (https://www.michaeljfox.org/files/MJFF_SfN_aSyn_Poster.pdf). This mouse had no significant changes in striatal dopamine and/or SNpc TH+ cell counts at 4, 8 or 12 mos vs. Bl6 controls. Nonetheless, compared to all other published aS mice, this BAC-E46K showed the most promising trend toward reduced TH counts at 12 mos, so we chose it for the requested comparison.
Western blot analyses
For western blotting, 10 μg total protein of RIPA extracts of dissected mouse brain regions were electroblotted onto nitrocellulose membranes (Millipore, Bedford, MA). For improved immunodetection of αS (monomers of which are prone to washing off filters), the membranes were fixed in 0.4% paraformaldehyde (PFA) for 20 min. After washing in phosphate-buffered saline (PBS), membranes were blocked for 1 h at RT in PBST (phosphate-buffered saline with 0.2% Tween-20) containing 5% bovine serum albumin (BSA). Blots were then incubated with human-specific αS antibody (15G7, Enzo; 1:500). After washing with PBST, membranes were probed with appropriate secondary antibodies (1:5000, American Qualex, CA), visualized with enhanced chemiluminescence (ECL, PerkinElmer, Boston, MA). Proteins were normalized to b-actin (A5441, Sigma; 1:3000) used as a loading control. Quantification of signal intensities was performed as described (Nuber et al., 2008).
Pole test
Mice were placed on top of a 50 cm vertical pole (all-thread metal rod) with a diameter of 1 cm and tested for their ability to descend from a round (‘assistant’) platform (2.5 cm diameter; head-down guidance). The test consisted of 3 consecutive trials with inter-trial pause of 5 min. Average times were calculated for each mouse. N=6 for each mouse type.
Wire test
Animals were placed on the hanging wire. The test consisted of 3 consecutive trials for each mouse with a 10 min interval between each repetition. Maximal time was 90 sec. Average times to endure on the wire were calculated for each mouse. WT N=6, E46K N=5.
Mouse brain biochemical analyses
Cortical Unsaturated fatty acids (UFA), DG and TG were measured in total cortical lipid extracts (Lipid Extraction Kit; ab211044; Abcam) using a colorimetric enzymatic assay (UFA: Cell Biolabs, Inc. STA-613; DG: Cell Biolabs, Inc. MET-5028; TG: abcam, ab65336). Cortical proteins were extracted in RIPA buffer (TBS+, 1% NP-40, 0.5% sodium deoxycholate, 0.1% sodium dodecyl sulphate) and the extraction step followed by ultracentrifugation for 30 min at 120,000g.
Cell lines and cell culture for inclusion assays
Stable M17D/αS-3K cell pools (Dettmer et al., 2015a) and the doxycycline-inducible cell line M17DTR/αS-3K::YFP//RFP (Dettmer et al., 2017) were generated as described previously from human neuroblastoma cells (BE (2)-M17, called M17D; ATCC number CRL-2267). Cells were cultured at 37 °C, 5% CO2 in ‘DMEM complete’, i.e. in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% FBS, 50 units per mL penicillin, 50 μg per mL streptomycin and 2 mM L-glutamine.
αS inclusion formation assay
Expression of αS 3K::YFP in M17D-TR/αS-3K::YFP//RFP cells (RFP expression is constitutive) was induced by adding 1 μg/mL dox and inclusion formation was followed over 24 h using the IncuCyte Zoom 2000 platform (Essen Biosciences). Images (red, green, bright field) were taken every 2 h. To measure inclusion formation, we created the processing definition ‘Inclusions’ (see (Dettmer et al., 2017) for details) and inclusion signals were normalized to the constitutive RFP signal.
OA loading of αS-inclusion-forming neuroblastoma cells
Dox-inducible M17D-TR/αS-3K::YFP//RFP cells were plated in 384-well plates in DMEM complete without FBS. The day after, BSA/OA complexes were mixed with fresh DMEM complete medium just before application to the cultures (Sharon et al., Neuron, 2003). The complexes were prepared by mixing BSA with OA at a molar ratio of 1:5 (Cayman Chemical, 90260) in binding buffer (10 mM Tris HCl [pH 8.0], 150 mM NaCl) followed by incubation at 37 °C for 30 min. Control wells were incubated in parallel with BSA alone. After 6 h incubation, cells were induced by adding 1μg/ml dox. Inclusion formation was followed over 24 h via Incucyte Zoom 2000 and quantified as described previously.
SCD1 knockdown in αS-inclusion-forming neuroblastoma cells
M17D-TR/αS-3K::YFP//RFP cells were plated in 96-well plates (25% confluent). The day after plating, 30μl OptiMEM + 2.5μl Trifecta RNAi (either control or SCD1) (IDT TriFECTa DsiRNA Kit - SCD1 human) and 25ul OptiMEM + 1.5μl Lipofectamine RNAiMAX Transfection Reagent (Life Technologies, 13778150) were prepared and mixed together. 10μl of this mix was added to the 100μl volume in the 96-well plate for 48 h. Cells were dox-induced and inclusion formation was monitored for 24 h via Incucyte. Cells were lysed and knockdown efficiency was monitored by immunoblotting.
Scd1 inhibition in αS-inclusion-forming neuroblastoma cells
M17D-TR/αS-3K::YFP//RFP cells were treated with Scd1 inhibitor 24 h after plating on 384-well plates, at 1 or 10μM (HY19762 or HY15700, MedChemexpress) and immediately induced via dox, followed by Incucyte-based analysis.
Crosslinking and sequential extraction of treated neuroblastoma cells
M17D/αS-3K cells were plated in 10cm dishes. For OA treatment, the BSA/OA mix was added as described above for 30 h (to mimic the 6h incubation time then the 24 h induction in the other experiment). For the SCD1 inhibitor experiment, cells were treated for 24 h at a final concentration of 10μM of the drug. After that, cells were collected to perform intact cell crosslinking and sequential extraction as described before (Dettmer, Nat.com. 2015). Briefly, the 10 cm dishes were splitted into 4 tubes: 3 different tubes for the crosslinking experiment with different amount of DSG and a fourth one for the sequential extraction. BCA assay was performed on the crosslinked samples to match samples that have similar protein concentration as an indication of protein-to-crosslinker ratio. The quality of the crosslinking was evaluated via dimer:monomer ratio on DJ-1 control blots. Sequential extraction efficiency was evaluated by distribution of cytosolic (marker: DJ-1) and the membrane fraction (marker Tfr). BCA assay and matching of samples that have similar protein con-centration as an indication of similar protein-to-crosslinker ratios. The most important criterion for data inclusion was equal crosslinking observed for the DJ-1 control blots (no apparent differences in DJ-1 dimer:monomer ratios was a pre-established criterion).
Immunoblotting
Protein concentrations were determined by BCA assay (Thermo Scientific) following the manufacturer’s directions. Samples were prepared for electrophoresis by the addition of NuPAGE LDS sample buffer and boiling for 10 min. 20μg of total protein were loaded per lane. Samples were electrophoresed on NuPAGE 4–12% Bis-Tris gels with NuPAGE MES-SDS running buffer and the SeeBlue Plus2 MW marker. After electrophoresis, gels were electroblotted onto Immobilon-Psq 0.2 mm PVDF membrane (Millipore) for 90 min at 400 mA constant current at 4 °C in 25 mM Tris, 192 mM glycine, 20% methanol transfer buffer. After transfer, membranes were incubated in 0.4% paraformaldehyde, PBS for 30 min at RT, rinsed twice with PBS, stained with 0.1% Ponceau S in 5% acetic acid, rinsed with water and blocked in 0.2% IBlock solution (PBS containing 0.1% (v/v) Tween 20 (PBS-T) and 0.2% (w/v) IBlock) for either 30 min at RT or overnight at 4 °C. After blocking, membranes were incubated in primary antibody in 0.2% IBlock with 0.02% sodium azide for either 1 h at RT or overnight at 4 °C. Membranes were washed 3 × 10 min in PBS-T at RT and incubated (45 min at RT) in horseradish peroxidase-conjugated secondary antibody (GE Healthcare) diluted 1:10,000 in 0.2% IBlock solution. Membranes were then washed 3 × 10 min in PBS-T and developed with SuperSignal West Dura (Thermo Scientific).
Supplementary Material
Highlights.
αS impacts lipid homeostasis, triggering excess oleic acid (OA) & diglycerides (DG)
Triglycerides & lipid droplets protect against toxicity by sequestering OA and DG
Stearoyl-CoA desaturase (SCD) inhibition rescues αS toxicity & neuron degeneration
SCD inhibition decreases αS inclusions, increases αS multimerization and solubility
Acknowledgements
We thank Linda Clayton, Luke Whitesell, Gerry Fink, Maeve Bonner, Charles Serhan, Maja Radulovic, Ian Cheeseman, Tim Bartels, Can Kayatekin, Joe Negri, Meichen Liao and Andrew Newman for valuable discussions and input; Melissa Duquette and Molly Rajsombath for technical assistance; Hana El Samad and her lab for estradiol constructs; and Lisa Freinkman, Bena Chan, Caroline Lewis, Tenzin Kunchok at the Metabolomics Core, Whitehead Institute for Biomedical Research. We thank Christina Muratore at the iPSC Neurohub at the Ann Romney Center for Neurologic Diseases for providing cell lines, reagents and assistance. We are grateful to Nicole Boucher for her administrative support. Audrey Madden, Robert Burger, Brooke Bevis provided lab support. SDK was supported by the Austrian Science Fund FWF, DK Molecular Enzymology Project W901 and NAWI Graz. SF and SL were supported by the JPB Foundation. SL was an HHMI Investigator. Supported by a fellowship from the Jane Coffin Childs Memorial Fund for medical research (MB), NIH grants NS065743 (GPH), GM102155 (MAW), NS103123 (SN), NS099328 (UD), NS083845 (DS). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript. Much of this work was performed in the laboratory of and under the supervision of Dr. Susan Lindquist, who sadly passed away before completion of this manuscript. We recognize her significant involvement, contribution and commitment to this work.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of Interests
DS is a director and consultant to Prothena Biosciences.
References
- Adibhatla RM, and Hatcher JF (2007). Role of Lipids in Brain Injury and Diseases. Future Lipidol 2, 403–422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander JJ, Snyder A, and Tonsgard JH (1998). Omega-oxidation of monocarboxylic acids in rat brain. Neurochem Res 23, 227–233. [DOI] [PubMed] [Google Scholar]
- Anderson JP, Walker DE, Goldstein JM, de Laat R, Banducci K, Caccavello RJ, Barbour R, Huang J, Kling K, Lee M, et al. (2006). Phosphorylation of Ser-129 is the dominant pathological modification of alpha-synuclein in familial and sporadic Lewy body disease. J Biol Chem 281, 29739–29752. [DOI] [PubMed] [Google Scholar]
- Aranda-Diaz A, Mace K, Zuleta I, Harrigan P, and El-Samad H (2017). Robust Synthetic Circuits for Two-Dimensional Control of Gene Expression in Yeast. ACS Synth Biol 6, 545–554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartels T, Choi JG, and Selkoe DJ (2011). alpha-Synuclein occurs physiologically as a helically folded tetramer that resists aggregation. Nature 477, 107–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baulac S, LaVoie MJ, Strahle J, Schlossmacher MG, and Xia W (2004). Dimerization of Parkinson’s disease-causing DJ-1 and formation of high molecular weight complexes in human brain. Mol Cell Neurosci 27, 236–246. [DOI] [PubMed] [Google Scholar]
- Black PN, and DiRusso CC (2003). Transmembrane movement of exogenous long-chain fatty acids: proteins, enzymes, and vectorial esterification. Microbiol Mol Biol Rev 67, 454–472, table of contents. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Black PN, and DiRusso CC (2007). Yeast acyl-CoA synthetases at the crossroads of fatty acid metabolism and regulation. Biochim Biophys Acta 1771, 286–298. [DOI] [PubMed] [Google Scholar]
- Bodner CR, Dobson CM, and Bax A (2009). Multiple tight phospholipid-binding modes of alpha-synuclein revealed by solution NMR spectroscopy. J Mol Biol 390, 775–790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boutet E, El Mourabit H, Prot M, Nemani M, Khallouf E, Colard O, Maurice M, Durand-Schneider AM, Chretien Y, Gres S, et al. (2009). Seipin deficiency alters fatty acid Delta9 desaturation and lipid droplet formation in Berardinelli-Seip congenital lipodystrophy. Biochimie 91, 796–803. [DOI] [PubMed] [Google Scholar]
- Braak H, Rub U, Schultz C, and Del Tredici K (2006). Vulnerability of cortical neurons to Alzheimer’s and Parkinson’s diseases. J Alzheimers Dis 9, 35–44. [DOI] [PubMed] [Google Scholar]
- Burre J, Sharma M, and Sudhof TC (2014). alpha-Synuclein assembles into higher-order multimers upon membrane binding to promote SNARE complex formation. Proc Natl Acad Sci U S A 111, E4274–4283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cartwright BR, Binns DD, Hilton CL, Han S, Gao Q, and Goodman JM (2015). Seipin performs dissectible functions in promoting lipid droplet biogenesis and regulating droplet morphology. Mol Biol Cell 26, 726–739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chandra S, Gallardo G, Fernandez-Chacon R, Schluter OM, and Sudhof TC (2005). Alpha-synuclein cooperates with CSPalpha in preventing neurodegeneration. Cell 123, 383–396. [DOI] [PubMed] [Google Scholar]
- Chang D, Nalls MA, Hallgrimsdottir IB, Hunkapiller J, van der Brug M, Cai F, International Parkinson’s Disease Genomics, C., and Me Research, T., Kerchner GA, Ayalon G, et al. (2017). A meta-analysis of genome-wide association studies identifies 17 new Parkinson’s disease risk loci. Nat Genet 49, 1511–1516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X, and Goodman JM (2017). The collaborative work of droplet assembly. Biochim Biophys Acta 1862, 1205–1211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen YP, Song W, Huang R, Chen K, Zhao B, Li J, Yang Y, and Shang HF (2013). GAK rs1564282 and DGKQ rs11248060 increase the risk for Parkinson’s disease in a Chinese population. J Clin Neurosci 20, 880–883. [DOI] [PubMed] [Google Scholar]
- Choi W, Zibaee S, Jakes R, Serpell LC, Davletov B, Crowther RA, and Goedert M (2004). Mutation E46K increases phospholipid binding and assembly into filaments of human alpha-synuclein. FEBS Lett 576, 363–368. [DOI] [PubMed] [Google Scholar]
- Collier TJ, Srivastava KR, Justman C, Grammatopoulous T, Hutter-Paier B, Prokesch M, Havas D, Rochet JC, Liu F, Jock K, et al. (2017). Nortriptyline inhibits aggregation and neurotoxicity of alpha-synuclein by enhancing reconfiguration of the monomeric form. Neurobiol Dis 106, 191–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Covino R, Ballweg S, Stordeur C, Michaelis JB, Puth K, Wernig F, Bahrami A, Ernst AM, Hummer G, and Ernst R (2016). A Eukaryotic Sensor for Membrane Lipid Saturation. Mol Cell 63, 49–59. [DOI] [PubMed] [Google Scholar]
- Davidson WS, Jonas A, Clayton DF, and George JM (1998). Stabilization of alpha-synuclein secondary structure upon binding to synthetic membranes. J Biol Chem 273, 9443–9449. [DOI] [PubMed] [Google Scholar]
- De Smet CH, Vittone E, Scherer M, Houweling M, Liebisch G, Brouwers JF, and de Kroon AI (2012). The yeast acyltransferase Sct1p regulates fatty acid desaturation by competing with the desaturase Ole1p. Mol Biol Cell 23, 1146–1156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dettmer U, Newman AJ, Luth ES, Bartels T, and Selkoe D (2013). In vivo cross-linking reveals principally oligomeric forms of alpha-synuclein and beta-synuclein in neurons and non-neural cells. J Biol Chem 288, 6371–6385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dettmer U, Newman AJ, Soldner F, Luth ES, Kim NC, von Saucken VE, Sanderson JB, Jaenisch R, Bartels T, and Selkoe D (2015a). Parkinson-causing alpha-synuclein missense mutations shift native tetramers to monomers as a mechanism for disease initiation. Nat Commun 6, 7314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dettmer U, Newman AJ, von Saucken VE, Bartels T, and Selkoe D (2015b). KTKEGV repeat motifs are key mediators of normal alpha-synuclein tetramerization: Their mutation causes excess monomers and neurotoxicity. Proc Natl Acad Sci U S A 112, 9596–9601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dettmer U, Ramalingam N, von Saucken VE, Kim TE, Newman AJ, Terry-Kantor E, Nuber S, Ericsson M, Fanning S, Bartels T, et al. (2017). Loss of native alpha-synuclein multimerization by strategically mutating its amphipathic helix causes abnormal vesicle interactions in neuronal cells. Hum Mol Genet 26, 3466–3481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devine MJ, Ryten M, Vodicka P, Thomson AJ, Burdon T, Houlden H, Cavaleri F, Nagano M, Drummond NJ, Taanman JW, et al. (2011). Parkinson’s disease induced pluripotent stem cells with triplication of the alpha-synuclein locus. Nat Commun 2, 440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dickson DW (2012). Parkinson’s disease and parkinsonism: neuropathology. Cold Spring Harb Perspect Med 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ebert D, Haller RG, and Walton ME (2003). Energy contribution of octanoate to intact rat brain metabolism measured by 13C nuclear magnetic resonance spectroscopy. J Neurosci 23, 5928–5935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edmond J, Robbins RA, Bergstrom JD, Cole RA, and de Vellis J (1987). Capacity for substrate utilization in oxidative metabolism by neurons, astrocytes, and oligodendrocytes from developing brain in primary culture. J Neurosci Res 18, 551–561. [DOI] [PubMed] [Google Scholar]
- Epand RM, So V, Jennings W, Khadka B, Gupta RS, and Lemaire M (2016). Diacylglycerol Kinase-epsilon: Properties and Biological Roles. Front Cell Dev Biol 4, 112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fei W, Li H, Shui G, Kapterian TS, Bielby C, Du X, Brown AJ, Li P, Wenk MR, Liu P, et al. (2011). Molecular characterization of seipin and its mutants: implications for seipin in triacylglycerol synthesis. J Lipid Res 52, 2136–2147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foley P (2010). Lipids in Alzheimer’s disease: A century-old story. Biochim Biophys Acta 1801, 750–753. [DOI] [PubMed] [Google Scholar]
- Galvagnion C (2017). The Role of Lipids Interacting with alpha-Synuclein in the Pathogenesis of Parkinson’s Disease. J Parkinsons Dis 7, 433–450. [DOI] [PubMed] [Google Scholar]
- Gould N, Mor DE, Lightfoot R, Malkus K, Giasson B, and Ischiropoulos H (2014). Evidence of native alpha-synuclein conformers in the human brain. J Biol Chem 289, 7929–7934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grippa A, Buxo L, Mora G, Funaya C, Idrissi FZ, Mancuso F, Gomez R, Muntanya J, Sabido E, and Carvalho P (2015). The seipin complex Fld1/Ldb16 stabilizes ER-lipid droplet contact sites. J Cell Biol 211, 829–844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gurry T, Ullman O, Fisher CK, Perovic I, Pochapsky T, and Stultz CM (2013). The dynamic structure of alpha-synuclein multimers. J Am Chem Soc 135, 3865–3872. [DOI] [PubMed] [Google Scholar]
- Hofbauer HF, Schopf FH, Schleifer H, Knittelfelder OL, Pieber B, Rechberger GN, Wolinski H, Gaspar ML, Kappe CO, Stadlmann J, et al. (2014). Regulation of gene expression through a transcriptional repressor that senses acyl-chain length in membrane phospholipids. Dev Cell 29, 729–739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jo E, Fuller N, Rand RP, St George-Hyslop P, and Fraser PE (2002). Defective membrane interactions of familial Parkinson’s disease mutant A30P alpha-synuclein. J Mol Biol 315, 799–807. [DOI] [PubMed] [Google Scholar]
- Kim JH, Panchision D, Kittappa R, and McKay R (2003). Generating CNS neurons from embryonic, fetal, and adult stem cells. Methods Enzymol 365, 303–327. [DOI] [PubMed] [Google Scholar]
- Klemann C, Martens GJM, Sharma M, Martens MB, Isacson O, Gasser T, Visser JE, and Poelmans G (2017). Integrated molecular landscape of Parkinson’s disease. NPJ Parkinsons Dis 3, 14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurat CF, Natter K, Petschnigg J, Wolinski H, Scheuringer K, Scholz H, Zimmermann R, Leber R, Zechner R, and Kohlwein SD (2006). Obese yeast: triglyceride lipolysis is functionally conserved from mammals to yeast. J Biol Chem 281, 491–500. [DOI] [PubMed] [Google Scholar]
- Lesage S, Anheim M, Letournel F, Bousset L, Honore A, Rozas N, Pieri L, Madiona K, Durr A, Melki R, et al. (2013). G51D alpha-synuclein mutation causes a novel parkinsonian-pyramidal syndrome. Ann Neurol 73, 459–471. [DOI] [PubMed] [Google Scholar]
- Licker V, Turck N, Kovari E, Burkhardt K, Cote M, Surini-Demiri M, Lobrinus JA, Sanchez JC, and Burkhard PR (2014). Proteomic analysis of human substantia nigra identifies novel candidates involved in Parkinson’s disease pathogenesis. Proteomics 14, 784–794. [DOI] [PubMed] [Google Scholar]
- Listenberger LL, Han X, Lewis SE, Cases S, Farese RV Jr., Ory DS, and Schaffer JE (2003). Triglyceride accumulation protects against fatty acid-induced lipotoxicity. Proc Natl Acad Sci U S A 100, 3077–3082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lockshon D, Olsen CP, Brett CL, Chertov A, Merz AJ, Lorenz DA, Van Gilst MR, and Kennedy BK (2012). Rho signaling participates in membrane fluidity homeostasis. PLoS One 7, e45049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lucke C, Gantz DL, Klimtchuk E, and Hamilton JA (2006). Interactions between fatty acids and alpha-synuclein. J Lipid Res 47, 1714–1724. [DOI] [PubMed] [Google Scholar]
- McMaster CR (2017). From yeast to humans: Roles of the Kennedy pathway for phosphatidylcholine synthesis. FEBS Lett. [DOI] [PubMed] [Google Scholar]
- Melton EM, Cerny RL, Watkins PA, DiRusso CC, and Black PN (2011). Human fatty acid transport protein 2a/very long chain acyl-CoA synthetase 1 (FATP2a/Acsvl1) has a preference in mediating the channeling of exogenous n-3 fatty acids into phosphatidylinositol. J Biol Chem 286, 30670–30679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nathanson JL, Yanagawa Y, Obata K, and Callaway EM (2009). Preferential labeling of inhibitory and excitatory cortical neurons by endogenous tropism of adeno-associated virus and lentivirus vectors. Neuroscience 161, 441–450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nielsen J (2009). Systems biology of lipid metabolism: from yeast to human. FEBS Lett 583, 3905–3913. [DOI] [PubMed] [Google Scholar]
- Nuber S, Petrasch-Parwez E, Winner B, Winkler J, von Horsten S, Schmidt T, Boy J, Kuhn M, Nguyen HP, Teismann P, et al. (2008). Neurodegeneration and motor dysfunction in a conditional model of Parkinson’s disease. J Neurosci 28, 2471–2484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Obi K, Akiyama H, Kondo H, Shimomura Y, Hasegawa M, Iwatsubo T, Mizuno Y, and Mochizuki H (2008). Relationship of phosphorylated alpha-synuclein and tau accumulation to Abeta deposition in the cerebral cortex of dementia with Lewy bodies. Exp Neurol 210, 409–420. [DOI] [PubMed] [Google Scholar]
- Outeiro TF, and Lindquist S (2003). Yeast cells provide insight into alpha-synuclein biology and pathobiology. Science 302, 1772–1775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pagac M, Cooper DE, Qi Y, Lukmantara IE, Mak HY, Wu Z, Tian Y, Liu Z, Lei M, Du X, et al. (2016). SEIPIN Regulates Lipid Droplet Expansion and Adipocyte Development by Modulating the Activity of Glycerol-3-phosphate Acyltransferase. Cell Rep 17, 1546–1559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pang ZP, Yang N, Vierbuchen T, Ostermeier A, Fuentes DR, Yang TQ, Citri A, Sebastiano V, Marro S, and Südhof TC (2011). Induction of human neuronal cells by defined transcription factors. Nature 476, 220–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perrin RJ, Payton JE, Barnett DH, Wraight CL, Woods WS, Ye L, and George JM (2003). Epitope mapping and specificity of the anti-alpha-synuclein monoclonal antibody Syn-1 in mouse brain and cultured cell lines. Neurosci Lett 349, 133–135. [DOI] [PubMed] [Google Scholar]
- Petschnigg J, Wolinski H, Kolb D, Zellnig G, Kurat CF, Natter K, and Kohlwein SD (2009). Good fat, essential cellular requirements for triacylglycerol synthesis to maintain membrane homeostasis in yeast. J Biol Chem 284, 30981–30993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pranke IM, Morello V, Bigay J, Gibson K, Verbavatz JM, Antonny B, and Jackson CL (2011). alpha-Synuclein and ALPS motifs are membrane curvature sensors whose contrasting chemistry mediates selective vesicle binding. J Cell Biol 194, 89–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qi Y, Sun L, and Yang H (2017). Lipid droplet growth and adipocyte development: mechanistically distinct processes connected by phospholipids. Biochim Biophys Acta 1862, 1273–1283. [DOI] [PubMed] [Google Scholar]
- Rochet JC, Outeiro TF, Conway KA, Ding TT, Volles MJ, Lashuel HA, Bieganski RM, Lindquist SL, and Lansbury PT (2004). Interactions among alpha-synuclein, dopamine, and biomembranes: some clues for understanding neurodegeneration in Parkinson’s disease. J Mol Neurosci 23, 23–34. [DOI] [PubMed] [Google Scholar]
- Sastry PS (1985). Lipids of nervous tissue: composition and metabolism. Prog Lipid Res 24, 69–176. [DOI] [PubMed] [Google Scholar]
- Schell H, Hasegawa T, Neumann M, and Kahle PJ (2009). Nuclear and neuritic distribution of serine-129 phosphorylated alpha-synuclein in transgenic mice. Neuroscience 160, 796–804. [DOI] [PubMed] [Google Scholar]
- Schmitt F, Hussain G, Dupuis L, Loeffler JP, and Henriques A (2014). A plural role for lipids in motor neuron diseases: energy, signaling and structure. Front Cell Neurosci 8, 25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharon R, Bar-Joseph I, Frosch MP, Walsh DM, Hamilton JA, and Selkoe DJ (2003). The formation of highly soluble oligomers of alpha-synuclein is regulated by fatty acids and enhanced in Parkinson’s disease. Neuron 37, 583–595. [DOI] [PubMed] [Google Scholar]
- Sharon R, Goldberg MS, Bar-Josef I, Betensky RA, Shen J, and Selkoe DJ (2001). alpha-Synuclein occurs in lipid-rich high molecular weight complexes, binds fatty acids, and shows homology to the fatty acid-binding proteins. Proc Natl Acad Sci U S A 98, 9110–9115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singleton AB, Farrer M, Johnson J, Singleton A, Hague S, Kachergus J, Hulihan M, Peuralinna T, Dutra A, Nussbaum R, et al. (2003). alpha-Synuclein locus triplication causes Parkinson’s disease. Science 302, 841. [DOI] [PubMed] [Google Scholar]
- Smulan LJ, Ding W, Freinkman E, Gujja S, Edwards YJ, and Walker AK (2016). Cholesterol-Independent SREBP-1 Maturation Is Linked to ARF1 Inactivation. Cell Rep 16, 9–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snead D, and Eliezer D (2014). Alpha-synuclein function and dysfunction on cellular membranes. Exp Neurobiol 23, 292–313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soldner F, Laganière J, Cheng AW, Hockemeyer D, Gao Q, Alagappan R, Khurana V, Golbe LI, Myers RH, and Lindquist S (2011). Generation of isogenic pluripotent stem cells differing exclusively at two early onset Parkinson point mutations. Cell 146, 318–331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soldner F, Stelzer Y, Shivalila CS, Abraham BJ, Latourelle JC, Barrasa MI, Goldmann J, Myers RH, Young RA, and Jaenisch R (2016). Parkinson-associated risk variant in distal enhancer of alpha-synuclein modulates target gene expression. Nature 533, 95–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soper JH, Roy S, Stieber A, Lee E, Wilson RB, Trojanowski JQ, Burd CG, and Lee VM (2008). Alpha-synuclein-induced aggregation of cytoplasmic vesicles in Saccharomyces cerevisiae. Mol Biol Cell 19, 1093–1103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stockl M, Fischer P, Wanker E, and Herrmann A (2008). Alpha-synuclein selectively binds to anionic phospholipids embedded in liquid-disordered domains. J Mol Biol 375, 1394–1404. [DOI] [PubMed] [Google Scholar]
- Szymanski KM, Binns D, Bartz R, Grishin NV, Li WP, Agarwal AK, Garg A, Anderson RG, and Goodman JM (2007). The lipodystrophy protein seipin is found at endoplasmic reticulum lipid droplet junctions and is important for droplet morphology. Proc Natl Acad Sci U S A 104, 20890–20895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taib B, Bouyakdan K, Hryhorczuk C, Rodaros D, Fulton S, and Alquier T (2013). Glucose regulates hypothalamic long-chain fatty acid metabolism via AMP-activated kinase (AMPK) in neurons and astrocytes. J Biol Chem 288, 37216–37229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tardiff DF, Jui NT, Khurana V, Tambe MA, Thompson ML, Chung CY, Kamadurai HB, Kim HT, Lancaster AK, Caldwell KA, et al. (2013). Yeast reveal a “druggable” Rsp5/Nedd4 network that ameliorates alpha-synuclein toxicity in neurons. Science 342, 979–983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trimbuch T, Beed P, Vogt J, Schuchmann S, Maier N, Kintscher M, Breustedt J, Schuelke M, Streu N, Kieselmann O, et al. (2009). Synaptic PRG-1 modulates excitatory transmission via lipid phosphate-mediated signaling. Cell 138, 1222–1235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tucci ML, Harrington AJ, Caldwell GA, and Caldwell KA (2011). Modeling dopamine neuron degeneration in Caenorhabditis elegans. Methods Mol Biol 793, 129–148. [DOI] [PubMed] [Google Scholar]
- van Dijk KD, Berendse HW, Drukarch B, Fratantoni SA, Pham TV, Piersma SR, Huisman E, Breve JJ, Groenewegen HJ, Jimenez CR, et al. (2012). The proteome of the locus ceruleus in Parkinson’s disease: relevance to pathogenesis. Brain Pathol 22, 485–498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volles MJ, and Lansbury PT Jr. (2007). Relationships between the sequence of alpha-synuclein and its membrane affinity, fibrillization propensity, and yeast toxicity. J Mol Biol 366, 1510–1522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner A, and Daum G (2005). Formation and mobilization of neutral lipids in the yeast Saccharomyces cerevisiae. Biochem Soc Trans 33, 1174–1177. [DOI] [PubMed] [Google Scholar]
- Wakamatsu M, Ishii A, Ukai Y, Sakagami J, Iwata S, Ono M, Matsumoto K, Nakamura A, Tada N, Kobayashi K, et al. (2007). Accumulation of phosphorylated alpha-synuclein in dopaminergic neurons of transgenic mice that express human alpha-synuclein. J Neurosci Res 85, 1819–1825. [DOI] [PubMed] [Google Scholar]
- Walker DG, Lue LF, Adler CH, Shill HA, Caviness JN, Sabbagh MN, Akiyama H, Serrano GE, Sue LI, Beach TG, et al. (2013). Changes in properties of serine 129 phosphorylated alpha-synuclein with progression of Lewy-type histopathology in human brains. Exp Neurol 240, 190–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walther TC, Chung J, and Farese RV Jr. (2017). Lipid Droplet Biogenesis. Annu Rev Cell Dev Biol. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang CW, Miao YH, and Chang YS (2014a). Control of lipid droplet size in budding yeast requires the collaboration between Fld1 and Ldb16. J Cell Sci 127, 1214–1228. [DOI] [PubMed] [Google Scholar]
- Wang H, Becuwe M, Housden BE, Chitraju C, Porras AJ, Graham MM, Liu XN, Thiam AR, Savage DB, Agarwal AK, et al. (2016). Seipin is required for converting nascent to mature lipid droplets. Elife 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L, Das U, Scott DA, Tang Y, McLean PJ, and Roy S (2014b). alpha-synuclein multimers cluster synaptic vesicles and attenuate recycling. Curr Biol 24, 2319–2326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang W, Perovic I, Chittuluru J, Kaganovich A, Nguyen LT, Liao J, Auclair JR, Johnson D, Landeru A, Simorellis AK, et al. (2011). A soluble alpha-synuclein construct forms a dynamic tetramer. Proc Natl Acad Sci U S A 108, 17797–17802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westphal CH, and Chandra SS (2013). Monomeric synucleins generate membrane curvature. J Biol Chem 288, 1829–1840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang P, and Reue K (2017). Lipin proteins and glycerolipid metabolism: Roles at the ER membrane and beyond. Biochim Biophys Acta 1859, 1583–1595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Pak C, Han Y, Ahlenius H, Zhang Z, Chanda S, Marro S, Patzke C, Acuna C, and Covy J (2013). Rapid single-step induction of functional neurons from human pluripotent stem cells. Neuron 78, 785–798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou Z, DiRusso CC, Ctrnacta V, and Black PN (2002). Fatty acid transport in Saccharomyces cerevisiae. Directed mutagenesis of FAT1 distinguishes the biochemical activities associated with Fat1p. J Biol Chem 277, 31062–31071. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.