

Abstract
Epidemiologic studies, including prospective birth cohort investigations, have implicated maternal immune activation in the etiology of neuropsychiatric disorders. Maternal infectious pathogens and inflammation are plausible risk factors for these outcomes and have been associated with schizophrenia, autism spectrum disorders, and bipolar disorder. Concurrent with epidemiologic work are animal models of prenatal immune activation, which have documented behavioral, neurochemical, neuroanatomic, and neurophysiologic disruptions that mirror phenotypes observed in these neuropsychiatric disorders. Epidemiologic studies of maternal immune activation offer the advantage of directly evaluating human populations, but are limited with respect to the inability to uncover pathogenic mechanisms. Animal models, on the other hand, are limited with regard to their generalizability to psychiatric disorders, but have made significant strides toward discovering causal relationships and biological pathways between maternal immune activation and neuropsychiatric phenotypes. Incorporating these risk factors in “reverse translational” animal models of maternal immune activation has yielded a wealth of data supporting the predictive potential of the epidemiologic studies. To further enhance the translatability between epidemiology and basic science, we propose a complementary approach that includes deconstructing neuropsychiatric outcomes of maternal immune activation into key pathophysiologically defined phenotypes that are identifiable in humans and animals, and that evaluates the inter-species concordance regarding interactions between maternal immune activation and genetic as well as epigenetic factors, including processes involving intergenerational disease transmission.
INTRODUCTION
Epidemiologic studies over the past decades have repeatedly implicated prenatal environmental factors, including maternal immune activation (MIA), in the etiology of neuropsychiatric illnesses(1, 2). Maternal infectious pathogens and inflammation are plausible risk factors for these outcomes and have been associated with schizophrenia(1-3), autism spectrum disorders(4-11) and bipolar disorder(12, 13). Concurrent with the epidemiologic work are animal models of MIA that have documented behavioral, neurochemical, neuroanatomic, and neurophysiologic disruptions in the offspring(14-17). The use of animal models in translational research aims at complementing this work by establishing causal relationships, identifying cellular and molecular mechanisms, and exploring potential therapeutic interventions(14-17). Despite progress on addressing these questions, there remain challenges regarding how to best approach the bidirectional “translation” between the findings of the epidemiologic and basic neuroscience studies of schizophrenia and other disorders.
The Epidemiological Perspective
A proliferation of epidemiologic studies have implicated maternal infection as a risk factor for neuropsychiatric illness(1-13). While early epidemiologic studies, which made use of ecologic data, suggested associations between influenza epidemics and schizophrenia among those exposed in utero, these findings were inconsistent. Such studies have been supplanted in more recent years with birth cohort studies that utilize prospectively acquired serologic biomarkers of infection and/or inflammation in individual pregnancies. As discussed in the next section, an increasing number of publications suggest associations between maternal infectious or inflammatory biomarkers and schizophrenia, autism spectrum disorders, and bipolar disorder(1, 2, 6, 14-16, 22). Other studies, which have utilized prospective data on maternal infection acquired from records on clinically diagnosed infections, have also yielded evidence in support of these exposures as risk factors(1, 6).
The Basic Science Perspective
Motivated by epidemiologic findings, a plethora of animal models of MIA have been established over the last two decades. As extensively reviewed elsewhere(14-17), one class of animal models is based on prenatal exposure to live pathogens, such as influenza virus(23-26) or Toxoplasma gondii (T. gondii). These models are particularly useful for the verification of causal relationships in epidemiological studies that assess the role of specific infectious pathogens. Another class of animal models makes use of immune-activating agents that primarily stimulate the innate immune system, such as the bacterial endotoxin, lipopolysaccharide (LPS), or the synthetic double-stranded RNA analog, polyriboinosinic-polyribocytidilic acid (poly(I:C)(14, 15, 17, 25, 27-29). These were developed initially to test whether imbalances in maternal and/or fetal cytokines may be critical for the association between prenatal infection and postnatal brain pathology(30, 31). An important refinement of this second class of models was the application of individual cytokines as immune-activating agents(9, 32, 33). This approach aims at addressing whether specific cytokines, or cytokine networks, mediate the association between MIA and neuropsychiatric illnesses. A third class of models is based on specific immunopathological processes that have been implicated in the etiology of neuropsychiatric illnesses. Two prominent examples of this class are animal models of maternal exposure to autism-related maternal autoantibodies(34-37) and allergic disorders/asthma(38-40).
The majority of current MIA models are based on maternal exposure to non-virulent, immune-activating agents such as the viral mimetic poly(I:C)(14-17, 53). While this experimental approach offers some clear advantages(14-17, 53), it does not reproduce the full spectrum of immune responses normally induced by infectious pathogens. For example, this method falls short in stimulating pathogen-specific humoral and cellular immune reactions, which may be part of the mechanism mediating the negative effects of maternal infection on the offspring. As discussed in detail elsewhere(53), one reason why the field shifted from MIA models that are based on exposure to infectious pathogens is that they require stringent biosafety levels, which in turn cannot be easily realized in many research laboratories. Another reason for the increasing popularity of non-virulent immune activating agents such as poly(I:C) in MIA models is that they allow basic scientists to tightly control the intensity and duration of the (innate) maternal immune response. This, in turn, allows researchers to identify sub- and supra-threshold effects of MIA on brain and behavioral functions in the offspring(28, 31), and whether these are influenced by the precise gestational timing of MIA(41, 42, 54-56). Finally, MIA models using non-virulent immune activating agents were initially developed with the aim of exploring whether the association between prenatal infection and neurodevelopmental abnormalities is mediated by specific infectious processes, or alternatively, by components of immune responses that are commonly triggered by various infections, including cytokines(30, 31, 57). Although the latter seem to be crucial contributing factors to many associations(2, 15), it is unlikely that distinct pathogens mediate the negative effects of maternal infection on the offspring through the same immune responses and pathophysiological mechanisms(58). To fully appreciate and approach this complexity, we believe that the field would benefit from a revival and extension of experimental approaches that make use of prenatal exposure to distinct virulent and non-virulent agents.
Another limitation of MIA models is that they typically exclude “real-life” influences in humans other than the primary exposure of interest, which may mediate or modify the effects of the exposures, and play contributory roles in disease outcomes. This limitation may also be one of the reasons why the findings from animal models of MIA appear more robust in terms of their effects on adult brain functions, as compared to epidemiological studies that explore the association between markers of infection and risk of mental disorders (see next section). In keeping with their “artificial” nature, the findings from animal models of MIA may also be associated with the potential of providing data that are not borne out by human studies. The recent discovery that maternal segmented filamentous bacteria (SFB) mediate the association between prenatal poly(I:C)-induced immune activation and autism-related brain abnormalities in mouse offspring(59) is an illustrative example. SFB are a family of autochthonous, apathogenic bacteria occurring in the ileum of rodents and other vertebrate species and have been shown to potently influence immune development and functions in mice(60). Contrary to mice, however, it remains controversial whether, and to what extent, SFB modulate the same immune parameters in humans(61-63).
The use of cross-species approaches in MIA models is one possible solution to minimize the potential of overinterpreting or oversimplifying the findings obtained in a certain animal species or strain (64-71). While the majority of these models have been developed in rodent species, most notably rats and mice, some have recently been extended to species that are evolutionarily and ethologically close to humans, including rhesus monkeys(8, 64, 69, 70). Whereas rodents are separated evolutionarily from humans by more than 70 million years, rhesus monkeys diverged from human evolution approximately 25 million years ago and thus exhibit greater similarity to humans in terms of genetics, immunology, neurobiology, and behavior (71,72). Compared with rodents, rhesus monkeys are also more comparable to humans regarding placental physiology, gestational timelines, pre- and postnatal brain development, and cortical architecture(71, 72). Thus, the inclusion of species that are more similar genetically to humans (e.g., rhesus monkeys) can aid in interpreting the outputs of rodent MIA models in terms of what they might mean for pathological symptoms in humans(65-68), thereby enhancing the cross-species transfer of information and translatability to the clinical condition in humans(71, 72)
REVIEW OF THE FINDINGS OF MIA STUDIES OF NEUROPSYCHIATRIC OUTCOMES AND RELATEDNESS OF FINDINGS BETWEEN EPIDEMIOLOGY AND BASIC SCIENCE
A key question is how the findings from these two disciplines can complement and inform one another with regard to furthering our understanding of the role of MIA in neuropsychiatric outcomes. In particular, we consider “reverse translational” approaches to this question, that is, whether human findings on maternal infection can “predict’ parallel findings in experimental model systems. We first consider the parallels between the findings in epidemiologic and animal studies for schizophrenia, bipolar disorder, and autism spectrum disorders. Since a full review of the findings of MIA and neuropsychiatric outcomes is beyond the scope of this article, we highlight some key results and refer the reader to several comprehensive reviews(1, 2, 6, 14-17, 22). Below, we focus on the potential areas of concordance between epidemiologic and basic science studies for each of these disorders.
Schizophrenia
To date, MIA and offspring psychiatric outcomes have been most commonly investigated for schizophrenia. We focus here on select findings that are based on biomarkers of infection. Though not all findings have been replicated, key epidemiologic results include associations between maternal infectious pathogens (influenza virus, herpes simplex virus (HSV)), T. gondii, rubella, and bacterial pathogens) and inflammatory biomarkers (cytokines, C-reactive protein) and schizophrenia(1, 19). Maternal exposure to influenza during early to mid-gestation, as quantified by antibody in maternal sera, has been associated with a threefold increased risk of schizophrenia in the Child Health and Development Study (CHDS), based on a large birth cohort in northern California(3). Elevated T. gondii IgG has been related to a twofold elevation in schizophrenia risk in this same birth cohort(73). Maternal genital/reproductive infections have also been related to schizophrenia in this cohort(74). Maternal exposure to HSV-2 has been associated with non-affective and affective psychoses in the National Collaborative Perinatal Project(75) though not in the birth cohorts of the CHDS or the Finnish Prenatal Studies (FiPS), which is based on a large national birth cohort in Finland (76). Neonatal antibodies to T. gondii and cytomegalovirus have been associated with non-affective psychosis in adulthood(77). In our study of maternal cytokines in the CHDS, we observed that increased interleukin-8 (IL-8) was related to schizophrenia(78). In the FiPS, we found that maternal C-reactive protein (CRP), a non-specific biomarker of inflammation, was associated with an increased risk of schizophrenia(19). Since it is unlikely that associations between biomarkers of inflammation are accounted for by one or a small group of infections, these findings may point to a common pathogenic pathway by which different infections give rise to schizophrenia.
Since their initial establishment, animal models of MIA have repeatedly documented structural and functional phenotypes that are implicated in schizophrenia and related psychotic disorders(2, 14-17). On the basis of early epidemiologic findings on maternal influenza and schizophrenia(1), Fatemi et al. pioneered an experimental mouse model of prenatal exposure to human influenza virus in mice(23-26, 31). As reviewed elsewhere(14, 15), maternal influenza infection in mice led to a variety of behavioral, neurochemical, morphological, and transcriptional changes in the offspring, many of which are implicated in schizophrenia and related disorders. These findings are thus strongly related to, and provide experimental support for, the association between maternal influenza infection and risk of schizophrenia(1, 3, 76). Since then, many additional investigations based on “reverse translational” animal models of MIA have yielded a wealth of new data supporting the predictive potential of the epidemiologic studies. For example, deficits in sensorimotor gating, impairments in selective or sustained attention, deficiencies in working memory, and hyper-responsiveness to psychotomimetic drugs have been found in various rodent models of MIA, including prenatal exposure to influenza virus, the viral mimetic poly(I:C), the bacterial endotoxin LPS, and selected inflammatory cytokines(14-17). Some of these deficits show a maturational delay in their appearance and can be mitigated by symptomatic or preventive treatments with antipsychotic medications(14-17).
Notably, the fact that prenatal exposure to various immune-activating agents can elicit similar phenotypes is consistent with epidemiological findings suggesting that the association between MIA and schizophrenia is not limited to a single infectious or inflammatory condition(1, 15). Despite the similarities between MIA models, however, there are also some notable differences between the models with respect to the nature of brain and behavioral changes. For example, whereas prenatal poly(I:C) exposure in rats and mice has been shown to induce cellular, neurochemical and behavioral phenotypes that are characteristic of a hyperdopaminergic state(27, 79, 80), prenatal LPS exposure may rather induce a hypodopaminergic state in adult rodent offspring(81). Prenatal LPS exposure in the rhesus monkey was also found to cause a significant increase in global white matter volume(64), whereas an opposite pattern (i.e., decreased white matter volume) was observed in rhesus monkey offspring born to influenza-infected mothers(69). Besides the notable influence of prenatal timing and the genetic background discussed above, such differences may arise because different immunogens can induce a distinct set of neuroimmune abnormalities across brain development, and consequently, may lead to differing long-term deficits in brain structure and function. This notion would also be consistent with epidemiological findings that appear to suggest that not all infectious pathogens have the same potential to increase neuropsychiatric disease risk(1, 13). As discussed more extensively below, a closer examination of the commonalities and differences between the mediating factors and outcomes of distinct MIA models should help to further address this important issue.
Another question is whether animal models can also “predict” certain epidemiological associations. While comparatively little work has been conducted, our recent findings support this assertion. We developed an environmental “two-hit” model in mice, in which prenatal exposure to mild but physiologically relevant MIA served as the “first hit”, and subchronic exposure to unpredictable, psychological stressors in pubescence as the “second hit”(50). Hence, this multifactorial model incorporates two environmental risk factors that have each been associated with increased risk of psychiatric disorders such as schizophrenia. We showed that combined exposure to the two environmental adversities acted in synergy to induce psychosis-related neural and behavioral abnormalities in adult mice(50). These results provided the first evidence suggesting that prenatal immune adversities can function as a neurodevelopmental disease primer, which in turn can increase the offspring’s vulnerability to the detrimental neuropathological effects of subsequent stress exposure during pubescence(50). These basic-science findings have recently been translated to a large population-based epidemiological study, which comprised nearly 1 million Danish persons born between 1980 and 1998(82). In that study, Danish nationwide registers were linked to estimate the independent and joint effects of exposure to prenatal infection and peripubertal psychological trauma on the risk of schizophrenia(82). Prenatal exposure to infection was defined based on hospital admissions with an infection during pregnancy, whereas exposure to traumatizing experiences during the period of peripuberty (from age 8 to 14 years) was defined according to Danish standards and included parental deaths, maltreatment or physical and/or sexual abuse, and maternal and paternal histories of crime and occupational situations(82). Confirming the hypothesis initially put forward by the environmental “two-hit” model in mice(50), the Danish population-based epidemiological study demonstrated that exposure to prenatal infection and peripubertal psychological trauma was associated with a significantly higher risk of developing schizophrenia (in males) compared to exposure to either insult alone, and the interaction between infection and trauma attained statistical significance(82). These findings suggest that the cross-fertilization between basic research in animals and risk factor epidemiology may offer the potential of predicting yet undiscovered associations between MIA and neuropsychiatric illnesses.
Bipolar disorder
Thus far, only a few epidemiologic studies have evaluated MIA in relation to bipolar disorder in offspring. Our group has demonstrated that maternal influenza, documented by antibodies in prenatal sera(19) and physician diagnoses(83), has been associated with a fivefold increased risk of bipolar disorder. While most other studies suggest no association between maternal infectious pathogens and bipolar disorder(13), one study found that maternal exposure to the type I strain of T. gondii was related to an increased risk of affective psychoses in offspring, which includes bipolar disorder(84).
Even though animal models of MIA have not specifically explored their validity for bipolar disorder, some of the experimentally induced phenotypes may be relevant for this neuropsychiatric illness as well. For example, deficits in sensorimotor gating, as seen in various rodent MIA models(2, 14-17), are also present in acutely manic(85) and remitted bipolar disorder patients(86). Moreover, several animal studies have reported the emergence of depression-like behaviors in offspring exposed to MIA(87, 88). The latter phenotypes may not only be relevant for unipolar depression, but also for depressive episodes in bipolar disorder. The investigation of other core behavioral symptoms of bipolar disorder, such as poor decision-making, altered risk-taking behavior, impulsivity, and loss of inhibitory control remain unexplored in MIA models. Additional work is also necessary to evaluate whether MIA-induced deficits can be mitigated by pharmacological treatments used in bipolar disorder, including the mood stabilizer lithium, and anticonvulsants such as valproate and lamotrigine(89).
Autism
In recent years, maternal infection and inflammation have been investigated in relation to autism spectrum disorders. Although findings are mixed, and more work is necessary, evidence has emerged linking maternal inflammation to risk of autism spectrum disorder in offspring. In the FiPS birth cohort, our group demonstrated that elevated maternal levels of CRP, a nonspecific biomarker of inflammation, in early to mid-gestation, was related to an increased risk of autism spectrum disorders in offspring(4). However, in the Early Markers of Autism (EMA) study in California, maternal mid-pregnancy CRP levels were related to a decreased risk of autism spectrum disorders(90). In studies of cytokines and chemokines in archived maternal serum samples in the EMA study, significantly increased levels of these analytes were related to autism spectrum disorders(5, 11). In amniotic fluid samples from a Danish study, several cytokines including tumor necrosis factor-alpha (TNFα) and several inflammatory interleukins were related to autism spectrum disorders in offspring(7). Moreover, maternal fever has been associated with autism. Although replication of these findings is necessary, they suggest that MIA may also be related to autism spectrum disorders. Consistent with this interpretation, other maternal immune factors, including maternal autoantibodies targeting fetal proteins, have been associated with increased autism spectrum disorder risk in the offspring (for review see (91)). These findings include significant associations between paired maternal antibody reactivity to fetal brain proteins with the 37 and 73kDa molecular weight bands and diagnosis of ASD in children(92). Within proteins corresponding to the 37-, 39-, and 73 kDa bands, maternal autoantibodies recognized seven developmentally regulated proteins in the fetal brain, including lactate dehydrogenase A and B, stress-induced phosphoprotein 1, and collapsin response mediator proteins 1 and 2(91, 92). Several of these proteins are critical for normal brain development, including neuronal migration and neural network formation.
Animal models further support the hypothesis that MIA is an environmental risk factor for autism spectrum disorders. For example, prenatal exposure to the viral mimetic poly(I:C), the bacterial endotoxin LPS, or allergies/asthma, can all induce behavioral abnormalities that are reminiscent of core symptoms of autism spectrum disorders, including deficits in social interaction and communication as well as high levels of repetitive behaviors(9, 15, 38, 93). These manipulations also cause brain morphological and cellular abnormalities implicated in autism spectrum disorders, including abnormal cerebellar development, impaired expression of the extracellular matrix protein reelin, and altered synapse density and neural connectivity(2, 15, 25). Importantly, some of these rodent findings have been extended to rhesus monkeys, both at the behavioral and brain morphological levels(8, 64, 70, 94).
FUTURE RESEARCH
Broadening the Concepts to the Study of MIA and Pathophysiologically Defined Phenotypes
One key unanswered question is whether there are particular factors that account for differences in psychiatric outcomes following MIA. There do not seem to be clear differences between these disorders with regard to gestational timing of MIA, though larger sample sizes are needed given the reduction in statistical power that results from stratification of analyses by periods of pregnancy. Similarly, there are inadequate data to permit comparisons between disorders on the effects of individual maternal infections, and a lack of information on the intensity of the immune response. Although the study of interaction of MIA with parental or offspring genes that cluster with particular disorders is a promising research direction, only a few studies of MIA, discussed further below, have evaluated possible gene × environment (G×E) interactions, and none to our knowledge have compared them between different disorders.
While there is clear merit in conducting such comparisons, our view is that this question might be addressed more effectively by asking how the field might move from the study of MIA and specific psychiatric outcomes to improving our understanding of the connections of this risk factor to key elements of these disorders. One approach is to deconstruct the psychiatric outcomes into their essential psycho- and neuropathological components. This approach is conceptually closer to research of MIA in animal models and thus appears more likely to maximize the extent to which findings in animal models can be translated to humans, and back-translated to animals (Figure 1), and advance the field by opening new avenues for the discovery of novel etiopathogenic factors and pathways. In this regard, animal models of MIA will be key to identifying neurobiological pathways leading to discrete pathological outcomes that may (or may not) cross current diagnostic boundaries.
Figure 1.
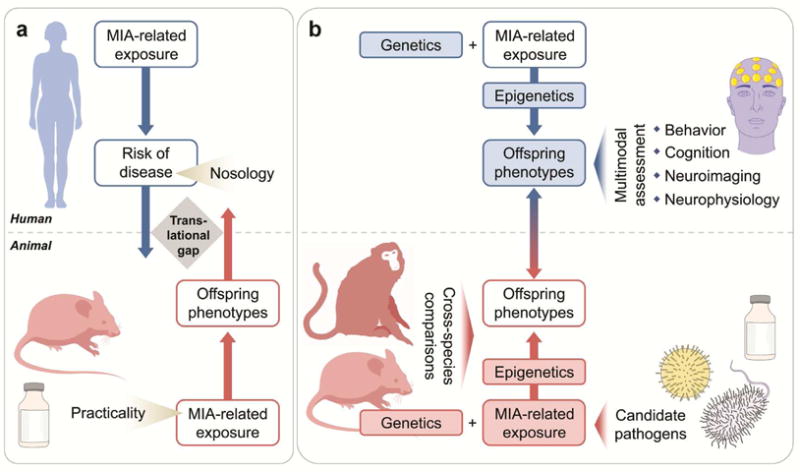
Contribution of epidemiologic and basic science studies to translational research of maternal immune activation in neuropsychiatric disorders. (a) Schematic illustration of the prevailing research approaches in both disciplines. With a few exceptions, epidemiologic studies of MIA generally aim at establishing associations between infectious, inflammatory, or other immune exposures and risk of certain neuropsychiatric disorders, the latter of which are defined by the current nosologic system. On the other hand, most animal models of MIA are single-factor models in which the isolated effects of MIA-related exposures are investigated with respect to behavioral, cognitive, neuroimaging, and neurophysiologic phenotypes in the offspring. For practical reasons, these models are often implemented in rodent species and are based on artificial immune-activating agents (e.g., synthetic doublestranded RNA) that do not require stringent biosafety precautions. The outcome of these epidemiological and basic science approaches is often a lack of analogy, resulting in a translational gap that can undermine their translational validity. (b) Schematic illustration of epidemiological and basic science approaches that can maximize the bi-directional translational validity through the modification of research concepts and the addition of supplementary research modules. In these alternate approaches, the objective of assessing MIA exposure-disease risk associations in epidemiologic designs is complemented with or even replaced by: 1) attempts to explore the effects of MIA on specific behavioral, cognitive, neuroimaging, and neurophysiologic phenotypes, which are free of nosologic constraints; 2) the concomitant study of genetic and epigenetic factors; 3) the establishment of multifactorial animal models that incorporate genetic/epigenetic risk factors and MIArelated exposures that involve epidemiologically established infectious pathogens and other immune factors such as inflammatory mediators. In addition, cross-species comparisons involving animal species with advanced cortical development will further enhance the bi-directional translatability between epidemiologic and basic science studies.
Here, we illustrate this using the example of schizophrenia. Cognitive and neuroimaging anomalies are key components of the phenotype of schizophrenia(95), and are also amenable to modeling in animals. Thus far, however, only a few studies have attempted to determine whether exposure to MIA confers vulnerability to these and other phenotypic characteristics of schizophrenia. In the CHDS birth cohort, we demonstrated that maternal exposure to infection (influenza, toxoplasmosis) was associated with impairments in executive functions, including set-shifting abnormalities in the Wisconsin Card Sorting Test and disruptions in performance on the Trail Making Test(96). Elevated maternal IL-8 levels, which were associated with risk of schizophrenia in this cohort, were also related to ventricular enlargement(97). Notably, similar phenotypes, including impairments in executive functions and ventricular enlargement, are also observed in adult rats and mice exposed to MIA(2, 14, 15, 52, 98-102) suggesting that there is bi-directional translational validity between the outcomes in the animal model and the epidemiologic/clinical condition.
The application of more systematic methods of classification of outcomes that are based on biologically relevant phenotypes(103) to MIA models may offer further promise: the studies reviewed above suggest that this exposure alters neurobiological and behavioral functions that cannot be simply mapped onto a particular diagnostic phenotype. Parenthetically, prenatal exposure to infectious or inflammatory adversity may be viewed as a general vulnerability factor for developmental disturbances rather than a disease-specific risk factor. This view is compatible with the Research Domain Criteria (RDoC) system, which capitalizes on biological determinism to explain the pathogenesis of distinct psychiatric symptoms and focuses on endophenotypes rather than nosologic entities(103). Shifting the research focus on classification of neurobiological outcomes rather than nosologic entities likely minimizes strict disease-to-model correspondence, a major challenge of translational studies (Figure 1).
The potential for realizing these research aims exists. There are several large, existing population-based birth cohorts with biospecimens and data on early development that have been linked to national databases on psychiatric outcomes(19, 104, 105). Subjects from these cohorts have the potential to be located through national registries and followed up for neurobiological outcomes, and biomarkers of maternal infection and inflammation have already been assessed in mothers of cases and matched controls from these cohorts. Given their translational value, the findings from animal models of MIA could facilitate the selection of biobehavioral outcomes to be investigated in corresponding human epidemiologic studies.
Conducting such research in prospective birth cohort studies, however, requires either long intervals of follow-up or identifying a sample of cases who belonged to a birth cohort. Given the relatively rare outcome, the limitations inherent to such research include loss to follow-up with consequent bias and small sample sizes. One potential solution to this problem is exemplified by a recent study of maternal/childhood micronutrient supplementation(106). In this study, pregnant mothers and their neonates were supplemented with phosphatidylcholine (aimed to deliver choline to the offspring’s brain), and received neurophysiologic testing of sensorimotor gating during infancy and of neurocognition during childhood. Compared to unsupplemented offspring, children assigned to phosphatidylcholine supplementation had an increased likelihood of normal inhibition of the P50 auditory evoked response, a biomarker of improved sensorimotor gating, and fewer attention problems and social withdrawal in early childhood(106). The P50 auditory evoked response is mediated by a specific cholinergic receptor, the α7-nAChR, which is encoded by the CHRNA7 gene. These findings were also recapitulated in a mouse model, in which choline supplementation of wild-type mice led to improvement in sensory inhibition of this auditory evoked response, whereas there were no beneficial effects in mice that were heterozygous or mutant for the CHRNA7 gene(107).
Transdisciplinary Approaches for Advancing the Understanding of the Role of Genetics and Epigenetics in the Context of MIA
Several genes identified from genome wide association studies, including those within the major histocompatibility complex (MHC) locus(108) and complement C4(109), encode proteins that play important roles in immune functioning and in neurodevelopment. With the exception of rare copy number variants(110, 111), mutations in these and other individual genes are generally associated with relatively small increases in odds of psychiatric outcomes, such as schizophrenia(112) and autism spectrum disorders(113), but are hypothesized to confer larger disease susceptibility by interacting with environmental exposures such as MIA. Notably, the assessment of environmental exposures is sometimes required to detect genetic effects, as demonstrated, for example, by the findings of a genome-wide study of association and interaction with maternal CMV infection(114) and a study which demonstrated an interaction between maternal pyelonephritis and family history of psychosis(115). Hence, environmental factors such as MIA may unmask the (statistical and biological) significance of certain genetic variations.
Recently, Mendelian randomization (MR) approaches, combined with genome wide association studies have been used to interrogate the genetic architecture of biomarkers of inflammation and infection. In one study, summary association results from large consortia of candidate gene or genome-wide association studies were included in concert with MR methods to evaluate associations between soluble interleukin-6 receptor, C-reactive protein levels and schizophrenia(116). The findings revealed a protective effect of CRP and a risk-increasing effect of sIL-6R on schizophrenia risk, possibly accounted for by early life infection. While this study was conducted in (mostly) non-pregnant adults, it provides proof of concept for the use of measures of the genetic architecture of response to infectious agents in the mother as a proxy for risk to offspring after maternal infection. Although MR studies are not without limitations, this work has the potential to investigate MIA in neuropsychiatric disorders on birth cohorts in which only information on candidate genes, but not maternal infection, are available. This approach could be broadly applicable to many cohorts since prospective data on infections during pregnancy are not widely ascertained, while genetic markers can be assessed using biospecimens that are more readily obtainable. Studies that use MR methods coupled with GWAS to explore a role of infection in triggering autoimmune disorders and inflammation, combined with evaluation of shared genetic variance for autoimmune disorders in separate cohorts, offer further promise.
To date, however, few epidemiologic studies have conducted a thorough evaluation of risk genes in relation to MIA in studies of neuropsychiatric outcomes(114). A major reason is that such studies require availability of DNA and a measure of maternal infection or inflammation during pregnancy, follow-up of the offspring for the neuropsychiatric outcomes, as well as large sample sizes given that studies of G×E interaction generally require greater statistical power. The integration and evaluation of putative risk genes in animal models of MIA may offer a complementary approach to the investigation of G×E interactions in human studies (Figure 1). This has been a fruitful strategy to unravel interactions between MIA and selected risk genes of neuropsychiatric illnesses(117), including CHRNA7(49), disrupted-in-schizophrenia 1 (DISC1)(46, 47), neuregulin 1 (NRG1)(48), and Nurr1 (NR4A2)(118). Besides providing evidence for additive effects on brain and behavioral abnormalities resembling aspects of major mental illnesses, these basic science studies show that prenatal immune activation can interact with selected risk genes to produce novel neurobehavioral phenotypes that are not apparent in animals harboring the genetic variant alone(117). For example, while mutations in the dopamine-related transcription factor Nurr1 and poly(I:C)-induced MIA in mice exert additive effects on locomotor hyperactivity and sensorimotor gating deficits, the combination of the two is required to impair attentional shifting and sustained attention(118). Similarly, the combination of mild MIA (induced by subthreshold doses of poly(I:C)) and mutations in DISC1 is necessary to impair social interaction in adult mouse offspring(46, 47).
Transdisciplinary approaches involving epidemiology and basic science may also advance our understanding of the role of epigenetics in the context of MIA. Epigenetic factors, defined as non-heritable alterations in the genome, are becoming increasingly recognized in the etiology of neuropsychiatric disorders(20, 21, 40). It has already been demonstrated that prenatal environmental factors such as smoking(119) are linked to epigenetic alterations and to psychiatric disorders. In addition, advanced paternal age has been related to an increased rate of de novo mutations(120) and to autism(121) and schizophrenia(122), and may have transgenerational effects(121). While MIA has not been examined to date in relation to epigenetic effects and de novo mutations in human populations, recent research in animal models suggests that epigenetic modifications may be a critical molecular mechanism by which MIA can mediate changes in brain development and function(20, 40, 43, 123-126). Lasting epigenetic changes in response to MIA have been identified in various brain areas, including cortical and subcortical regions(40, 123-125), and in specific cell types such as microglia(40). Furthermore, using the maternal poly(I:C) administration model in mice, we have recently provided the first piece of evidence showing that MIA-induced behavioral abnormalities and whole-genome transcriptional changes can be transmitted across generations without additional immune exposures(126). These transgenerational effects were mediated via the paternal but not maternal lineage and were present for at least three generations, pointing towards epigenetic inheritance via male gametes. Further studies on the identification of epigenetic and transgenerational effects in MIA-induced neurodevelopmental disorders may help identify complex patterns of transgenerational disease transmission beyond genetic inheritance. Conceivably, the consideration of ancestral histories of infection may be a useful approach for developing new preventive treatment strategies against infection-mediated neurodevelopmental disorders.
Implications of research in MIA for health policy and prevention
This work has the potential for significant impact on future health policy and prevention. Many infectious agents are preventable with relatively straightforward public health measures (127). In work from our group, we estimated that the population attributable risk for schizophrenia following exposure to 3 maternal infections (influenza, T. gondii, and genital reproductive infections), was 30% in the CHDS birth cohort, suggesting that the number of cases in this population that are preventable by elimination of these infections from the population can be reduced by as much as one third(1, 127). T. gondii can be prevented by the use of simple hygienic measures, such as the use of gloves when gardening or changing cat litter boxes, adequate cooking of meat before consumption, and washing kitchen knives after cutting meats, fruits, and vegetables(128). The occurrence of many sexually transmitted infections can be reduced by the use of barrier contraceptives, antimicrobials, educational programs to promote safe sex, delaying first sexual contacts, and partner notification(129, 130). Maternal immunization during pregnancy is another health policy option with potential ramifications for prevention of neuropsychiatric disorders. Though not always offering complete protection, influenza vaccination is a mainstay for the prevention of this virus(131). These measures are scalable to large populations given their relatively low cost and practicality. Most influenza vaccines appear safe in terms of maternal, fetal or neonatal complications, even if given to pregnant women(132-135) and several health organizations recommend prophylactic influenza vaccination for this population (132).
CONCLUSIONS
Accumulating evidence suggests that maternal exposure to infectious and inflammatory insults is related to the etiology of major neuropsychiatric illnesses. The bi-directional translation of epidemiologic to basic neuroscience studies offers the promise of developing a more complete and nuanced understanding of MIA in relation to biological mechanisms. We suggest future studies aimed at capitalizing on the integration of these disciplines, particularly in relation to pathophysiologic entities that transcend diagnostic boundaries, and assessing multi-factorial models including genetic and epigenetic factors, in MIA-induced psychopathology.
Acknowledgments
Grant Sponsor: Alan S. Brown, M.D., M.P.H.: National Institute of Environmental Health Sciences (grant number: 1R01ES019004); Urs Meyer, Ph.D.: Swiss National Science Foundation (grant number: 310030_169544).
Footnotes
Conflict of Interest: The authors report no competing interests.
References
- 1.Brown AS, Derkits EJ. Prenatal infection and schizophrenia: A review of epidemiologic and translational studies. Am J Psychiatry. 2010;167:261–280. doi: 10.1176/appi.ajp.2009.09030361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Estes ML, McAllister AK. Maternal immune activation: Implications for neuropsychiatric disorders. Science. 2016;353:772–777. doi: 10.1126/science.aag3194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Brown AS, Begg MD, Gravenstein S, Schaefer CA, Wyatt RJ, Bresnahan M, Babulas VP, Susser ES. Serologic evidence of prenatal influenza in the etiology of schizophrenia. Arch Gen Psychiatry. 2004;61:774–780. doi: 10.1001/archpsyc.61.8.774. [DOI] [PubMed] [Google Scholar]
- 4.Brown AS, Sourander A, Hinkka-Yli-Salomäki S, McKeague IW, Sundvall J, Surcel HM. Elevated maternal C-reactive protein and autism in a national birth cohort. Mol Psychiatry. 2014;19:259–264. doi: 10.1038/mp.2012.197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jones KL, Croen LA, Yoshida CK, Heuer L, Hansen R, Zerbo O, DeLorenze GN, Kharrazi M, Yolken R, Ashwood P, Van de Water J. Autism with intellectual disability is associated with increased levels of maternal cytokines and chemokines during gestation. Mol Psychiatry. 2017;22:273–279. doi: 10.1038/mp.2016.77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jiang HY, Xu LL, Shao L, Xia RM, Yu ZH, Ling ZX, Yang F, Deng M, Ruan B. Maternal infection during pregnancy and risk of autism spectrum disorders: A systematic review and meta-analysis. Brain Behav Immun. 2016;58:165–172. doi: 10.1016/j.bbi.2016.06.005. [DOI] [PubMed] [Google Scholar]
- 7.Abdallah MW, Larsen N, Grove J, Norgaard-Pedersen B, Thorsen P, Mortensen EL, Hougaard DM. Amniotic fluid chemokines and autism spectrum disorder: An exploratory study utilizing a Danish historic birth cohort. Brain Behav Immun. 2012;26:170–176. doi: 10.1016/j.bbi.2011.09.003. [DOI] [PubMed] [Google Scholar]
- 8.Bauman MD, Iosif AM, Ashwood P, Braunschweig D, Lee A, Schumann CM, Van de Water J, Amaral DG. Maternal antibodies from mothers of children with autism alter brain growth and social behavior development in the rhesus monkey. Transl Psychiatry. 2013;3:e278. doi: 10.1038/tp.2013.47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Choi GB, Yim YS, Wong H, Kim S, Kim H, Kim SV, Hoeffer CA, Littman DR, Huh JR. The maternal interleukin-17a pathway in mice promotes autism-like phenotypes in offspring. Science. 2016;351:933–939. doi: 10.1126/science.aad0314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zerbo O, Qian Y, Yoshida C, Fireman BH, Klein NP, Croen LA. Association between influenza infection and vaccination during pregnancy and risk of autism spectrum disorder. JAMA Pediatr. 2017;171:e163609. doi: 10.1001/jamapediatrics.2016.3609. [DOI] [PubMed] [Google Scholar]
- 11.Goines PE, Croen LA, Braunschweig D, Yoshida CK, Grether JK, Hansen R, Kharrazi M, Ashwood P, van de Water J. Increased midgestational IFN-γ, IL-4 and IL-5 in women bearing a child with autism. A case-control study. Mol Autism. 2011;2:13. doi: 10.1186/2040-2392-2-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Canetta SE, Bao Y, Co MD, Ennis FA, Cruz J, Terajiima M, Shen L, Kellendonk C, Schaefer CA, Brown AS. Serological documentation of maternal influenza exposure and bipolar disorder in adult offspring. Am J Psychiatry. 2014;171:557–563. doi: 10.1176/appi.ajp.2013.13070943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Brown A. The Kraepelinian dichotomy from the perspective of prenatal infectious and immunologic insults. Schizophr Bull. 2015;41:786–791. doi: 10.1093/schbul/sbv063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Meyer U. Prenatal poly(I:C) exposure and other developmental immune activation models in rodent systems. Biol Psychiatry. 2014;75:307–315. doi: 10.1016/j.biopsych.2013.07.011. [DOI] [PubMed] [Google Scholar]
- 15.Meyer U, Feldon J, Fatemi SH. In-vivo rodent models for the experimental investigation of prenatal immune activation effects in neurodevelopmental brain disorders. Neurosci Biobehav Rev. 2009;33:1061–1079. doi: 10.1016/j.neubiorev.2009.05.001. [DOI] [PubMed] [Google Scholar]
- 16.Harvey L, Boksa P. Prenatal and postnatal animal models of immune activation: Relevance to a range of neurodevelopmental disorders. Dev Neurobiol. 2012;72:1335–1348. doi: 10.1002/dneu.22043. [DOI] [PubMed] [Google Scholar]
- 17.Meyer U, Feldon J. Epidemiology-driven neurodevelopmental animal models of schizophrenia. Prog Neurobiol. 2010;90:285–326. doi: 10.1016/j.pneurobio.2009.10.018. [DOI] [PubMed] [Google Scholar]
- 18.Rothman KJ, Greenland S. Modern Epidemiology Philadelphia. Lippincott Williams & Wilkins; 1998. [Google Scholar]
- 19.Canetta S, Sourander A, Surcel H-M, Hinkka-Yli-Salomäki S, Leiviskä J, Kellendonk C, McKeague IW, Brown AS. Elevated maternal C-reactive protein and increased risk of schizophrenia in a national birth cohort. Am J Psychiatry. 2014;171:960–968. doi: 10.1176/appi.ajp.2014.13121579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Weber-Stadlbauer U. Epigenetic and transgenerational mechanisms in infection-mediated neurodevelopmental disorders. Transl Psychiatry. 2017;7:e1113. doi: 10.1038/tp.2017.78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bohacek J, Mansuy IM. Molecular insights into transgenerational non-genetic inheritance of acquired behaviours. Nat Rev Genet. 2015;16:641–652. doi: 10.1038/nrg3964. [DOI] [PubMed] [Google Scholar]
- 22.Oliveira J, Oliveira-Maia AJ, Tamouza R, Brown AS, Leboyer M. Infectious and immunogenetic factors in bipolar disorder. Acta Psychiatr Scand. 2017 doi: 10.1111/acps.12791. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fatemi SH, Emamian ES, Kist D, Sidwell RW, Nakajima K, Akhter P, Shier A, Sheikh S, Bailey K. Defective corticogenesis and reduction in reelin immunoreactivity in cortex and hippocampus of prenatally infected neonatal mice. Mol Psychiatry. 1999;4:145–154. doi: 10.1038/sj.mp.4000520. [DOI] [PubMed] [Google Scholar]
- 24.Fatemi SH, Reutiman TJ, Folsom TD, Huang H, Oishi K, Mori S, Smee DF, Pearce DA, Winter C, Sohr R, Juckel G. Maternal infection leads to abnormal gene regulation and brain atrophy in mouse offspring: Implications for genesis of neurodevelopmental disorders. Schizophr Res. 2008;99:56–70. doi: 10.1016/j.schres.2007.11.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Shi L, Smith SE, Malkova N, Tse D, Su Y, Patterson PH. Activation of maternal immune system alters cerebellar development in the offspring. Brain Behav Immun. 2009;23:116–123. doi: 10.1016/j.bbi.2008.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Fatemi SH, Folsom TD, Rooney RJ, Mori S, Kornfield TE, Reutiman TJ, Kneeland RE, Liesch SB, Hua K, Hsu J, Patel DH. The viral theory of schizophrenia revisited: Abnormal placental gene expression and structural changes with lack of evidence for H1N1 viral presence in placentae of infected mice or brains of exposed offspring. Neuropharmacology. 2012;62:1290–1298. doi: 10.1016/j.neuropharm.2011.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zuckerman L, Rehavi M, Nachman R, Weiner I. Immune activation during pregnancy in rats leads to a postpubertal emergence of disrupted latent inhibition, dopaminergic hyperfunction, and altered limbic morphology in the offspring: A novel neurodevelopmental model of schizophrenia. Neuropsychopharmacology. 2003;28:1778–1789. doi: 10.1038/sj.npp.1300248. [DOI] [PubMed] [Google Scholar]
- 28.Meyer U, Feldon J, Schedlowski M, Yee BK. Towards an immuno-precipitated neurodevelopmental animal model of schizophrenia. Neurosci Biobehav Rev. 2005;29:913–947. doi: 10.1016/j.neubiorev.2004.10.012. [DOI] [PubMed] [Google Scholar]
- 29.Romero E, Guaza C, Castellano B, Borrell J. Ontogeny of sensorimotor gating and immune impairment induced by prenatal immune challenge in rats: implications for the etiopathology of schizophrenia. Mol Psychiatry. 2010;15:372–383. doi: 10.1038/mp.2008.44. [DOI] [PubMed] [Google Scholar]
- 30.Gilmore JH, Jarskog LF. Exposure to infection and brain development: Cytokines in the pathogenesis of schizophrenia. Schizophr Res. 1997;24:365–367. doi: 10.1016/s0920-9964(96)00123-5. [DOI] [PubMed] [Google Scholar]
- 31.Shi L, Fatemi SH, Sidwell RW, Patterson PH. Maternal influenza infection causes marked behavioral and pharmacological changes in the offspring. J Neurosci. 2003;23:297–302. doi: 10.1523/JNEUROSCI.23-01-00297.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Smith SE, Li J, Garbett K, Mirnics K, Patterson PH. Maternal immune activation alters fetal brain development through interleukin-6. J Neurosci. 2007;27:10695–10702. doi: 10.1523/JNEUROSCI.2178-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Girard S, Tremblay L, Lepage M, Sebire G. IL-1 receptor antagonist protects against placental and neurodevelopmental defects induced by maternal inflammation. J Immunol (Baltimore, Md: 1950) 2010;184:3997–4005. doi: 10.4049/jimmunol.0903349. [DOI] [PubMed] [Google Scholar]
- 34.Martinez-Cerdeno V, Camacho J, Fox E, Miller E, Ariza J, Kienzle D, Plank K, Noctor SC, Van de Water J. Prenatal exposure to autism-specific maternal autoantibodies alters proliferation of cortical neural precursor cells, enlarges brain, and increases neuronal size in adult animals. Cereb Cortex. 2016;26:374–383. doi: 10.1093/cercor/bhu291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Braunschweig D, Duncanson P, Boyce R, Hansen R, Ashwood P, Pessah IN, Hertz-Picciotto I, Van de Water J. Behavioral correlates of maternal antibody status among children with autism. J Autism Dev Disord. 2012;42:1435–1445. doi: 10.1007/s10803-011-1378-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Braunschweig D, Golub MS, Koenig CM, Qi L, Pessah IN, Van de Water J, Berman RF. Maternal autism-associated IgG antibodies delay development and produce anxiety in a mouse gestational transfer model. J Neuroimmunol. 2012;252:56–65. doi: 10.1016/j.jneuroim.2012.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Martin LA, Ashwood P, Braunschweig D, Cabanlit M, van de Water J, Amaral DG. Stereotypies and hyperactivity in rhesus monkeys exposed to IgG from mothers of children with autism. Brain Behav Immun. 2008;22:806–816. doi: 10.1016/j.bbi.2007.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Schwartzer JJ, Careaga M, Coburn MA, Rose DR, Hughes HK, Ashwood P. Behavioral impact of maternal allergic-asthma in two genetically distinct mouse strains. Brain Behav Immun. 2017;63:99–107. doi: 10.1016/j.bbi.2016.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Schwartzer JJ, Careaga M, Chang C, Onore CE, Ashwood P. Allergic fetal priming leads to developmental, behavioral and neurobiological changes in mice. Transl Psychiatry. 2015;5:e543. doi: 10.1038/tp.2015.40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Vogel Ciernia A, Careaga M, LaSalle JM, Ashwood P. Microglia from offspring of dams with allergic asthma exhibit epigenomic alterations in genes dysregulated in autism. Glia. 2018;66:505–521. doi: 10.1002/glia.23261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Meyer U, Nyffeler M, Yee BK, Knuesel I, Feldon J. Adult brain and behavioral pathological markers of prenatal immune challenge during early/middle and late fetal development in mice. Brain Behav Immun. 2008;22:469–86. doi: 10.1016/j.bbi.2007.09.012. [DOI] [PubMed] [Google Scholar]
- 42.Meyer U, Nyffeler M, Engler A, Urwyler A, Schedlowski M, Knuesel I, Yee BK, Feldon J. The time of prenatal immune challenge determines the specificity of inflammation-mediated brain and behavioral pathology. J Neurosci. 2006;26:4752–4762. doi: 10.1523/JNEUROSCI.0099-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Richetto J, Massart R, Weber-Stadlbauer U, Szyf M, Riva MA, Meyer U. Genome-wide DNA methylation changes in a mouse model of infection-mediated neurodevelopmental disorders. Biol Psychiatry. 2017;81:265–276. doi: 10.1016/j.biopsych.2016.08.010. [DOI] [PubMed] [Google Scholar]
- 44.Borrell J, Vela JM, Arevalo-Martin A, Molina-Holgado E, Guaza C. Prenatal immune challenge disrupts sensorimotor gating in adult rats. Implications for the etiopathogenesis of schizophrenia. Neuropsychopharmacology. 2002;26:204–215. doi: 10.1016/S0893-133X(01)00360-8. [DOI] [PubMed] [Google Scholar]
- 45.Missault S, Van den Eynde K, Vanden Berghe W, Fransen E, Weeren A, Timmermans JP, Kumar-Singh S, Dedeurwaerdere S. The risk for behavioural deficits is determined by the maternal immune response to prenatal immune challenge in a neurodevelopmental model. Brain Behav Immun. 2014;42:138–146. doi: 10.1016/j.bbi.2014.06.013. [DOI] [PubMed] [Google Scholar]
- 46.Abazyan B, Nomura J, Kannan G, Ishizuka K, Tamashiro KL, Nucifora F, Pogorelov V, Ladenheim B, Yang C, Krasnova IN, Cadet JL, Pardo C, Mori S, Kamiya A, Vogel MW, Sawa A, Ross CA, Pletnikov MV. Prenatal interaction of mutant DISC1 and immune activation produces adult psychopathology. Biol Psychiatry. 2010;68:1172–1181. doi: 10.1016/j.biopsych.2010.09.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lipina TV, Zai C, Hlousek D, Roder JC, Wong AH. Maternal immune activation during gestation interacts with DISC1 point mutation to exacerbate schizophrenia-related behaviors in mice. J Neurosci. 2013;33:7654–7666. doi: 10.1523/JNEUROSCI.0091-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.O’Leary C, Desbonnet L, Clarke N, Petit E, Tighe O, Lai D, Harvey R, Waddington JL, O’Tuathaigh C. Phenotypic effects of maternal immune activation and early postnatal milieu in mice mutant for the schizophrenia risk gene neuregulin-1. Neuroscience. 2014;277:294–305. doi: 10.1016/j.neuroscience.2014.06.028. [DOI] [PubMed] [Google Scholar]
- 49.Wu WL, Adams CE, Stevens KE, Chow KH, Freedman R, Patterson PH. The interaction between maternal immune activation and alpha 7 nicotinic acetylcholine receptor in regulating behaviors in the offspring. Brain Behav Immun. 2015;46:192–202. doi: 10.1016/j.bbi.2015.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Giovanoli S, Engler H, Engler A, Richetto J, Voget M, Willi R, Winter C, Riva MA, Mortensen PB, Feldon J, Schedlowski M, Meyer U. Stress in puberty unmasks latent neuropathological consequences of prenatal immune activation in mice. Science. 2013;339:1095–1099. doi: 10.1126/science.1228261. [DOI] [PubMed] [Google Scholar]
- 51.Deslauriers J, Larouche A, Sarret P, Grignon S. Combination of prenatal immune challenge and restraint stress affects prepulse inhibition and dopaminergic/GABAergic markers. Prog Neuropsychopharmacol Biol Psychiatry. 2013;45:156–164. doi: 10.1016/j.pnpbp.2013.05.006. [DOI] [PubMed] [Google Scholar]
- 52.Piontkewitz Y, Arad M, Weiner I. Abnormal trajectories of neurodevelopment and behavior following in utero insult in the rat. Biol Psychiatry. 2011;70:842–851. doi: 10.1016/j.biopsych.2011.06.007. [DOI] [PubMed] [Google Scholar]
- 53.Meyer U, Feldon J. To poly(I:C) or not to poly(I:C): Advancing preclinical schizophrenia research through the use of prenatal immune activation models. Neuropharmacology. 2012;62:1308–1321. doi: 10.1016/j.neuropharm.2011.01.009. [DOI] [PubMed] [Google Scholar]
- 54.Fortier ME, Luheshi GN, Boksa P. Effects of prenatal infection on prepulse inhibition in the rat depend on the nature of the infectious agent and the stage of pregnancy. Behav Brain Res. 2007;181:270–277. doi: 10.1016/j.bbr.2007.04.016. [DOI] [PubMed] [Google Scholar]
- 55.Aguilar-Valles A, Luheshi GN. Alterations in cognitive function and behavioral response to amphetamine induced by prenatal inflammation are dependent on the stage of pregnancy. Psychoneuroendocrinology. 2011;36:634–648. doi: 10.1016/j.psyneuen.2010.09.006. [DOI] [PubMed] [Google Scholar]
- 56.Meehan C, Harms L, Frost JD, Barreto R, Todd J, Schall U, Shannon Weickert C, Zavitsanou K, Michie PT, Hodgson DM. Effects of immune activation during early or late gestation on schizophrenia-related behaviour in adult rat offspring. Brain Behav Immun. 2017;63:8–20. doi: 10.1016/j.bbi.2016.07.144. [DOI] [PubMed] [Google Scholar]
- 57.Meyer U, Feldon J, Yee BK. A review of the fetal brain cytokine imbalance hypothesis of schizophrenia. Schizophr Bull. 2009;35:959–972. doi: 10.1093/schbul/sbn022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Labouesse MA, Langhans W, Meyer U. Long-term pathological consequences of prenatal infection: beyond brain disorders. Am J Physio Regul Integrative Comp Physiol. 2015;309:R1–R12. doi: 10.1152/ajpregu.00087.2015. [DOI] [PubMed] [Google Scholar]
- 59.Kim S, Kim H, Yim YS, Ha S, Atarashi K, Tan TG, Longman RS, Honda K, Littman DR, Choi GB, Huh JR. Maternal gut bacteria promote neurodevelopmental abnormalities in mouse offspring. Nature. 2017;549:528–532. doi: 10.1038/nature23910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ivanov II, Littman DR. Segmented filamentous bacteria take the stage. Mucosal Immunol. 2010;3:209–212. doi: 10.1038/mi.2010.3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Yin Y, Wang Y, Zhu L, Liu W, Liao N, Jiang M, Zhu B, Yu HD, Xiang C, Wang X. Comparative analysis of the distribution of segmented filamentous bacteria in humans, mice and chickens. ISME J. 2013;7:615–621. doi: 10.1038/ismej.2012.128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Caselli M, Cassol F, Gentili V, Di Luca D. Genome sequences of segmented filamentous bacteria in animals: Implications for human research. Gut Microbes. 2012;3:401–405. doi: 10.4161/gmic.20736. [DOI] [PubMed] [Google Scholar]
- 63.Jönsson BA, Rylander L, Lindh C, Rignell-Hydbom A, Giwercman A, Toft G, Pedersen HS, Ludwicki JK, Góralczyk K, Zvyezday V, Spanó M, Bizzaro D, Bonefeld-Jorgensen EC, Manicardi GC, Bonde JP, Hagmar L. Inuendo. Inter-population variations in concentrations, determinants of and correlations between 2,2′,4,4′,5,5′-hexachlorobiphenyl (CB-153) and 1,1-dichloro-2,2-bis (p-chlorophenyl)-ethylene (p,p′-DDE): A cross-sectional study of 3161 men and women from Inuit and European populations. Environ Health. 2005;4:27. doi: 10.1186/1476-069X-4-27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Willette AA, Lubach GR, Knickmeyer RC, Short SJ, Styner M, Gilmore JH, Coe CL. Brain enlargement and increased behavioral and cytokine reactivity in infant monkeys following acute prenatal endotoxemia. Behav Brain Res. 2011;219:108–115. doi: 10.1016/j.bbr.2010.12.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Cosgrove VE, Kelsoe JR, Suppes T. Toward a valid animal model of bipolar disorder: How the research domain criteria help bridge the clinical-basic science divide. Biol Psychiatry. 2016;79:62–70. doi: 10.1016/j.biopsych.2015.09.002. [DOI] [PubMed] [Google Scholar]
- 66.Peleg-Raibstein D, Feldon J, Meyer U. Behavioral animal models of antipsychotic drug actions. Handb Exp Pharmacol. 2012:361–406. doi: 10.1007/978-3-642-25761-2_14. [DOI] [PubMed] [Google Scholar]
- 67.Silverman JL, Yang M, Lord C, Crawley JN. Behavioural phenotyping assays for mouse models of autism. Nat Rev Neurosci. 2010;11:490–502. doi: 10.1038/nrn2851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Arguello PA, Gogos JA. Modeling madness in mice: One piece at a time. Neuron. 2006;52:179–196. doi: 10.1016/j.neuron.2006.09.023. [DOI] [PubMed] [Google Scholar]
- 69.Short SJ, Lubach GR, Karasin AI, Olsen CW, Styner M, Knickmeyer RC, Gilmore JH, Coe CL. Maternal influenza infection during pregnancy impacts postnatal brain development in the rhesus monkey. Biol Psychiatry. 2010;67:965–973. doi: 10.1016/j.biopsych.2009.11.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Machado CJ, Whitaker AM, Smith SE, Patterson PH, Bauman MD. Maternal immune activation in nonhuman primates alters social attention in juvenile offspring. Biol Psychiatry. 2015;77:823–832. doi: 10.1016/j.biopsych.2014.07.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Bauman MD, Schumann CM. Advances in nonhuman primate models of autism: Integrating neuroscience and behavior. Exp Neuorol. 2018;299:252–265. doi: 10.1016/j.expneurol.2017.07.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Phillips KA, Bales KL, Capitanio JP, Conley A, Czoty PW, t Hart BA, Hopkins WD, Hu SL, Miller LA, Nader MA, Nathanielsz PW, Rogers J, Shively CA, Voytko ML. Why primate models matter. Am J Primatol. 2014;76:801–827. doi: 10.1002/ajp.22281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Brown AS, Schaefer CA, Quesenberry CP, Jr, Liu L, Babulas VP, Susser ES. Maternal exposure to toxoplasmosis and risk of schizophrenia in adult offspring. Am J Psychiatry. 2005;162:767–773. doi: 10.1176/appi.ajp.162.4.767. [DOI] [PubMed] [Google Scholar]
- 74.Babulas V, Factor-Litvak P, Goetz R, Schaefer CA, Brown AS. Prenatal exposure to maternal genital and reproductive infections and adult schizophrenia. Am J Psychiatry. 2006;163:927–929. doi: 10.1176/ajp.2006.163.5.927. [DOI] [PubMed] [Google Scholar]
- 75.Buka SL, Cannon TD, Torrey EF, Yolken RH. Maternal exposure to herpes simplex virus and risk of psychosis among adult offspring. Biol Psychiatry. 2008;63:809–815. doi: 10.1016/j.biopsych.2007.09.022. [DOI] [PubMed] [Google Scholar]
- 76.Cheslack-Postava K, et al. Maternal exposure to sexually transmitted infections and schizophrenia among offspring. Schizophr Res. 2015;166:255–260. doi: 10.1016/j.schres.2015.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Blomstrom A, Karlsson H, Gardner R, Jorgensen L, Magnusson C, Dalman C. Associations between maternal infection during pregnancy, childhood infections, and the risk of subsequent psychotic disorder—A Swedish cohort study of nearly 2 million individuals. Schizophr Bull. 2016;42:125–133. doi: 10.1093/schbul/sbv112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Brown AS, Hooton J, Schaefer CA, Zhang H, Petkova E, Babulas V, Perrin M, Gorman JM, Susser ES. Elevated maternal interleukin-8 levels and risk of schizophrenia in adult offspring. Am J Psychiatry. 2004;161:889–895. doi: 10.1176/appi.ajp.161.5.889. [DOI] [PubMed] [Google Scholar]
- 79.Vuillermot S, Weber L, Feldon J, Meyer U. A longitudinal examination of the neurodevelopmental impact of prenatal immune activation in mice reveals primary defects in dopaminergic development relevant to schizophrenia. J Neurosci. 2010;30:1270–1287. doi: 10.1523/JNEUROSCI.5408-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Ozawa K, Hashimoto K, Kishimoto T, Shimizu E, Ishikura H, Iyo M. Immune activation during pregnancy in mice leads to dopaminergic hyperfunction and cognitive impairment in the offspring: A neurodevelopmental animal model of schizophrenia. Biol Psychiatry. 2006;59:546–554. doi: 10.1016/j.biopsych.2005.07.031. [DOI] [PubMed] [Google Scholar]
- 81.Carvey PM, Chang Q, Lipton JW, Ling Z. Prenatal exposure to the bacteriotoxin lipopolysaccharide leads to long-term losses of dopamine neurons in offspring: A potential, new model of Parkinson’s disease. Front Biosci. 2003;8:s826–837. doi: 10.2741/1158. [DOI] [PubMed] [Google Scholar]
- 82.Debost JP, Larsen JT, Munk-Olsen T, Mortensen PB, Meyer U, Petersen L. Joint effects of exposure to prenatal infection and peripubertal psychological trauma in schizophrenia. Schizophr Bull. 2017;43:171–179. doi: 10.1093/schbul/sbw083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Parboosing R, Bao Y, Shen L, Schaefer CA, Brown AS. Gestational influenza and bipolar disorder in adult offspring. JAMA Psychiatry. 2013;70:677–685. doi: 10.1001/jamapsychiatry.2013.896. [DOI] [PubMed] [Google Scholar]
- 84.Xiao J, Buka SL, Cannon TD, Suzuki Y, Viscidi RP, Torrey EF, Yolken RH. Serological pattern consistent with infection with type I Toxoplasma gondii in mothers and risk of psychosis among adult offspring. Microbes Infect. 2009;11:1011–1018. doi: 10.1016/j.micinf.2009.07.007. [DOI] [PubMed] [Google Scholar]
- 85.Perry W, Minassian A, Feifel D, Braff DL. Sensorimotor gating deficits in bipolar disorder patients with acute psychotic mania. Biol Psychiatry. 2001;50:418–424. doi: 10.1016/s0006-3223(01)01184-2. [DOI] [PubMed] [Google Scholar]
- 86.Giakoumaki SG, Roussos P, Rogdaki M, Karli C, Bitsios P, Frangou S. Evidence of disrupted prepulse inhibition in unaffected siblings of bipolar disorder patients. Biol Psychiatry. 2007;62:1418–1422. doi: 10.1016/j.biopsych.2006.12.002. [DOI] [PubMed] [Google Scholar]
- 87.Khan D, Fernando P, Cicvaric A, Berger A, Pollak A, Monje FJ, Pollak DD. Long-term effects of maternal immune activation on depression-like behavior in the mouse. Transl Psychiatry. 2014;4:e363. doi: 10.1038/tp.2013.132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Ronovsky M, Berger S, Molz B, Berger A, Pollak DD. Animal models of maternal immune activation in depression research. Curr Neuropharmacol. 2016;14:688–704. doi: 10.2174/1570159X14666151215095359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Grunze H, Vieta E, Goodwin GM, Bowden C, Licht RW, Moller HJ, Kasper S. The World Federation of Societies of Biological Psychiatry (WFSBP) guidelines for the biological treatment of bipolar disorders: update 2009 on the treatment of acute mania. World J Biol Psychiatry. 2009;10:85–116. doi: 10.1080/15622970902823202. [DOI] [PubMed] [Google Scholar]
- 90.Zerbo O, Traglia M, Yoshida C, Heuer LS, Ashwood P, Delorenze GN, Hansen RL, Kharrazi M, Van de Water J, Yolken RH, Weiss LA, Croen LA. Maternal mid-pregnancy C-reactive protein and risk of autism spectrum disorders: The early markers for autism study. Transl Psychiatry. 2016;6:e783. doi: 10.1038/tp.2016.46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Edmiston E, Ashwood P, Van de Water J. Autoimmunity, autoantibodies, and autism spectrum disorder. Biol Psychiatry. 2017;81:383–390. doi: 10.1016/j.biopsych.2016.08.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Braunschweig D, Ashwood P, Krakowiak P, Hertz-Picciotto I, Hansen R, Croen LA, Pessah IN, van de Water J. Autism: Maternally derived antibodies specific for fetal brain proteins. Neurotoxicology. 2008;29:226–231. doi: 10.1016/j.neuro.2007.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Malkova NV, Yu CZ, Hsiao EY, Moore MJ, Patterson PH. Maternal immune activation yields offspring displaying mouse versions of the three core symptoms of autism. Brain Behav Immun. 2012;26:607–616. doi: 10.1016/j.bbi.2012.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Careaga M, Murai T, Bauman MD. Maternal immune activation and autism spectrum disorder: from rodents to nonhuman and human primates. Biol Psychiatry. 2017;81:391–401. doi: 10.1016/j.biopsych.2016.10.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Mesholam-Gately RI, Giuliano AJ, Goff KP, Faraone SV, Seidman LJ. Neurocognition in first-episode schizophrenia: a meta-analytic review. Neuropsychology. 2009;23:315–336. doi: 10.1037/a0014708. [DOI] [PubMed] [Google Scholar]
- 96.Brown AS, Vinogradov S, Kremen WS, Poole JH, Deicken RF, Penner JD, McKeague IW, Kochetkova A, Kern D, Schaefer CA. Prenatal exposure to maternal infection and executive dysfunction in adult schizophrenia. Am J Psychiatry. 2009;166:683–690. doi: 10.1176/appi.ajp.2008.08010089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Ellman LM, Deicken RF, Vinogradov S, Kremen WS, Poole JH, Kern DM, Tsai WY, Schaefer CA, Brown AS. Structural brain alterations in schizophrenia following fetal exposure to the inflammatory cytokine interleukin-8. Schizophr Res. 2010;121:46–54. doi: 10.1016/j.schres.2010.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Zuckerman L, Weiner I. Maternal immune activation leads to behavioral and pharmacological changes in the adult offspring. J Psychiatr Res. 2005;39:311–323. doi: 10.1016/j.jpsychires.2004.08.008. [DOI] [PubMed] [Google Scholar]
- 99.Li Q, Cheung C, Wei R, Hui ES, Feldon J, Meyer U, Chung S, Chua SE, Sham PC, Wu EX, McAlonan GM. Prenatal immune challenge is an environmental risk factor for brain and behavior change relevant to schizophrenia: Evidence from MRI in a mouse model. PLoS One. 2009;4:e6354. doi: 10.1371/journal.pone.0006354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Bitanihirwe BK, Weber L, Feldon J, Meyer U. Cognitive impairment following prenatal immune challenge in mice correlates with prefrontal cortical AKT1 deficiency. Int J Neuropsychopharmacol. 2010;13:981–996. doi: 10.1017/S1461145710000192. [DOI] [PubMed] [Google Scholar]
- 101.Richetto J, Calabrese F, Riva MA, Meyer U. Prenatal immune activation induces maturation-dependent alterations in the prefrontal GABAergic transcriptome. Schizophr Bull. 2014;40:351–361. doi: 10.1093/schbul/sbs195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Wallace J, Marston HM, McQuade R, Gartside SE. Evidence that aetiological risk factors for psychiatric disorders cause distinct patterns of cognitive deficits. Eur Neuropsychopharmacol. 2014;24:879–889. doi: 10.1016/j.euroneuro.2013.12.005. [DOI] [PubMed] [Google Scholar]
- 103.Insel T, Cuthbert B, Garvey M, Heinssen R, Pine DS, Quinn K, Sanislow C, Wang P. Research domain criteria (RDoC): Toward a new classification framework for research on mental disorders. Am J Psychiatry. 2010;167:748–751. doi: 10.1176/appi.ajp.2010.09091379. [DOI] [PubMed] [Google Scholar]
- 104.Andersen AM, Olsen J. The Danish National Birth Cohort: selected scientific contributions within perinatal epidemiology and future perspectives. Scand Public Health. 2011;39:115–120. doi: 10.1177/1403494811407674. [DOI] [PubMed] [Google Scholar]
- 105.Magnus P, Irgens LM, Haug K, Nystad W, Skjaerven R, Stoltenberg C. Cohort profile: the Norwegian Mother and Child Cohort Study (MoBa) Int J Epidemiol. 2006;35:1146–1150. doi: 10.1093/ije/dyl170. [DOI] [PubMed] [Google Scholar]
- 106.Ross RG, Hunter SK, Hoffman MC, McCarthy L, Chambers BM, Law AJ, Leonard S, Zerbe GO, Freedman R. Perinatal phosphatidylcholine supplementation and early childhood behavior problems: Evidence for CHRNA7 moderation. Am J Psychiatry. 2016;173:509–516. doi: 10.1176/appi.ajp.2015.15091188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Stevens KE, Choo KS, Stitzel JA, Marks MJ, Adams CE. Long-term improvements in sensory inhibition with gestational choline supplementation linked to alpha7 nicotinic receptors through studies in CHRNA7 null mutation mice. Brain Res. 2014;1552:26–33. doi: 10.1016/j.brainres.2014.01.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Corvin A, Morris DW. Genome-wide association studies: findings at the major histocompatibility complex locus in psychosis. Biol Psychiatry. 2014;75:276–283. doi: 10.1016/j.biopsych.2013.09.018. [DOI] [PubMed] [Google Scholar]
- 109.Sekar A, Bialas AR, de Rivera H, Davis A, Hammond TR, Kamitaki N, Tooley K, Presumey J, Baum M, Van Doren V, Genovese G, Rose SA, Handsaker RE, Daly MJ, Carroll MC, Stevens B, McCarroll SA. Schizophrenia risk from complex variation of complement component 4. Nature. 2016;530:177–183. doi: 10.1038/nature16549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Marshall CR, et al. Contribution of copy number variants to schizophrenia from a genome-wide study of 41,321 subjects. Nature Genet. 2017;49:27–35. doi: 10.1038/ng.3725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Pinto D, et al. Functional impact of global rare copy number variation in autism spectrum disorders. Nature. 2010;466:368–372. doi: 10.1038/nature09146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Schizophrenia Working Group of the Psychiatric Genomics Consortium. Biological insights from 108 schizophrenia-associated genetic loci. Nature. 2014;511:421–427. doi: 10.1038/nature13595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Weiss LA, Arking DE, Daly MJ, Chakravarti A. A genome-wide linkage and association scan reveals novel loci for autism. Nature. 2009;461:802–808. doi: 10.1038/nature08490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Borglum AD, Demontis D, Grove J, Pallesen J, Hollegaard MV, Pedersen CB, Hedemand A, Mattheisen M, Uitterlinden A, Nyegaard M, Orntoft T, Wiuf C, Didriksen M, Nordentoft M, Nothen MM, Rietschel M, Ophoff RA, Cichon S, Yolken RH, Hougaard DM, Mortensen PB, Mors O. Genome-wide study of association and interaction with maternal cytomegalovirus infection suggests new schizophrenia loci. Mol Psychiatry. 2014;19:325–333. doi: 10.1038/mp.2013.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Clarke MC, Tanskanen A, Huttunen M, Whittaker JC, Cannon M. Evidence for an interaction between familial liability and prenatal exposure to infection in the causation of schizophrenia. Am J Psychiatry. 2009;166:1025–1030. doi: 10.1176/appi.ajp.2009.08010031. [DOI] [PubMed] [Google Scholar]
- 116.Hartwig F, Borges M, Horta B, Bowden J, Davey Smith G. Inflammatory biomarkers and risk of schizophrenia: A 2-sample mendelian randomization study. JAMA Psychiatry. 2017;74:1226–1233. doi: 10.1001/jamapsychiatry.2017.3191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Ayhan Y, McFarland R, Pletnikov MV. Animal models of gene-environment interaction in schizophrenia: A dimensional perspective. Prog Neurobiol. 2016;136:1–27. doi: 10.1016/j.pneurobio.2015.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Vuillermot S, Joodmardi E, Perlmann T, Ogren SO, Feldon J, Meyer U. Prenatal immune activation interacts with genetic Nurr1 deficiency in the development of attentional impairments. J Neurosci. 2012;32:436–451. doi: 10.1523/JNEUROSCI.4831-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Niemela S, Sourander A, Surcel HM, Hinkka-Yli-Salomaki S, McKeague IW, Cheslack-Postava K, Brown AS. Prenatal nicotine exposure and risk of schizophrenia among offspring in a national birth cohort. Am J Psychiatry. 2016;173:799–806. doi: 10.1176/appi.ajp.2016.15060800. [DOI] [PubMed] [Google Scholar]
- 120.Kong A, Frigge ML, Masson G, Besenbacher S, Sulem P, Magnusson G, Gudjonsson SA, Sigurdsson A, Jonasdottir A, Wong WS, Sigurdsson G, Walters GB, Steinberg S, Helgason H, Thorleifsson G, Gudbjartsson DF, Helgason A, Magnusson OT, Thorsteindottir U, Stefansson K. Rate of de novo mutations and the importance of father’s age to disease risk. Nature. 2012;488:471–475. doi: 10.1038/nature11396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Reichenberg A, Gross R, Weiser M, Bresnahan M, Silverman J, Harlap S, Rabinowitz J, Shulman C, Malaspina D, Lubin G, Knobler HY, Davidson M, Susser E. Advancing paternal age and autism. Arch Gen Psychiatry. 2006;63:1026–1032. doi: 10.1001/archpsyc.63.9.1026. [DOI] [PubMed] [Google Scholar]
- 122.Malaspina D, Harlap S, Fennig S, Heiman D, Nahon D, Feldman D, Susser ES. Advancing paternal age and the risk of schizophrenia. Arch Gen Psychiatry. 2001;58:361–367. doi: 10.1001/archpsyc.58.4.361. [DOI] [PubMed] [Google Scholar]
- 123.Basil P, Li Q, Dempster EL, Mill J, Sham PC, Wong CC, McAlonan GM. Prenatal maternal immune activation causes epigenetic differences in adolescent mouse brain. Transl Psychiatry. 2014;4:e434. doi: 10.1038/tp.2014.80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Hollins SL, Zavitsanou K, Walker FR, Cairns MJ. Alteration of imprinted Dlk1-Dio3 miRNA cluster expression in the entorhinal cortex induced by maternal immune activation and adolescent cannabinoid exposure. Transl Psychiatry. 2014;4:e452. doi: 10.1038/tp.2014.99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Reisinger SN, Kong E, Khan D, Schulz S, Ronovsky M, Berger S, Horvath O, Cabatic M, Berger A, Pollak DD. Maternal immune activation epigenetically regulates hippocampal serotonin transporter levels. Neurobiol Stress. 2016;4:34–43. doi: 10.1016/j.ynstr.2016.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Weber-Stadlbauer U, Richetto J, Labouesse MA, Bohacek J, Mansuy IM, Meyer U. Transgenerational transmission and modification of pathological traits induced by prenatal immune activation. Mol Psychiatry. 2017;22:102–112. doi: 10.1038/mp.2016.41. [DOI] [PubMed] [Google Scholar]
- 127.Brown AS, Patterson PH. Maternal infection and schizophrenia: Implications for prevention. Schizophr Bull. 2011;37:284–290. doi: 10.1093/schbul/sbq146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Kijlstra A, Jongert E. Control of the risk of human toxoplasmosis transmitted by meat. International journal for parasitology. 2008;38:1359–1370. doi: 10.1016/j.ijpara.2008.06.002. [DOI] [PubMed] [Google Scholar]
- 129.Low N, Broutet N, Adu-Sarkodie Y, Barton P, Hossain M, Hawkes S. Global control of sexually transmitted infections. Lancet. 2006;368:2001–2016. doi: 10.1016/S0140-6736(06)69482-8. [DOI] [PubMed] [Google Scholar]
- 130.Stoner B. Sexually transmitted infections: overview. In: HK H, SR Q, editors. International Encyclopedia of Public Health. San Diego, CA: Academic Press; 2008. [Google Scholar]
- 131.Kilbourne ED. Influenza. New York, NY: Plenum Medical Book Co; 1987. [Google Scholar]
- 132.Seasonal influenza and 2009 H1N1 influenza vaccination coverage among pregnant women–10 states, 2009-10 influenza season. MMWR Morb Mortal Rep. 2010;59:1541–1545. [PubMed] [Google Scholar]
- 133.Black SB, Shinefield HR, France EK, Fireman BH, Platt ST, Shay D. Effectiveness of influenza vaccine during pregnancy in preventing hospitalizations and outpatient visits for respiratory illness in pregnant women and their infants. Am J Perinatol. 2004;21:333–339. doi: 10.1055/s-2004-831888. [DOI] [PubMed] [Google Scholar]
- 134.Heinonen OP, Shapiro S, Monson RR, Hartz SC, Rosenberg L, Slone D. Immunization during pregnancy against poliomyelitis and influenza in relation to childhood malignancy. Int J Epidemiol. 1973;2:229–235. doi: 10.1093/ije/2.3.229. [DOI] [PubMed] [Google Scholar]
- 135.Hviid A, Svanstrom H, Molgaard-Nielsen D, Lambach P. Association between pandemic influenza A(H1N1) vaccination in pregnancy and early childhood morbidity in offspring. JAMA Pediatr. 2017;171:239–248. doi: 10.1001/jamapediatrics.2016.4023. [DOI] [PubMed] [Google Scholar]