

Graphical Abstract
Keywords: Co-administration, benznidazole, VFV, VNI, Colombiana strain, Trypanosoma cruzi, Chagas disease
Up to now, no vaccines are available for Chagas disease, and the current therapy is largely unsatisfactory. Novel imidazole-based scaffolds of protozoan sterol 14α-demethylase (CYP51) inhibitors have demonstrated potent anti-parasitic activity with no acute toxicity. Presently our aim was to investigate the effectiveness of the experimental 14α-demethylase inhibitor VFV in the mouse models of Trypanosoma cruzi infection using naturally drug-resistant Colombiana strain, under monotherapy and in association with the reference drug, benznidazole (Bz). The treatment with VFV resulted in complete parasitemia suppression and 100 % animal survival when administered orally (given in 10 % DMSO plus 5% Arabic gum) at 25 mg/kg (bid) for 60 days. However, as parasite relapse was found using VFV alone under this treatment scheme, the co-administration of VFV with Bz was assayed giving simultaneously (for 60 days, bid) by oral route, under two different drug vehicles (10 % DMSO plus 5 % Gum Arabic with or without 3% Tween 80). All tested mice groups resulted in >99.9 % of parasitemia decrease and 100 % animal survival. qPCR analysis performed on cyclophosphamide immunosuppressed mice revealed that, although presenting lack of cure, VFV given as monotherapy was 14-fold more active than Bz, and the co-administration of Bz plus VFV (given simultaneously, using 10 % DMSO plus 5 % Gum Arabic as vehicle) resulted in 106-fold lower blood parasitism as compared to the monotherapy of Bz. Another interesting finding was the parasitological cure in 70 % of the animals treated with Bz and VFV when the co-administration was given using the VFV suspension in 10 % DMSO + Arabic gum + Tween 80 (a formulation that we have found to provide a better pharmacokinetics), even after immunosuppression using cyclophosphamide cycles, supporting the promising aspect of the drug co-administration in improving the efficacy of therapeutic arsenal against T. cruzi.
Chagas disease affects more than 6 million people mostly in the poorest areas of Latin America. The available therapy is based on two nitroderivatives, nifurtimox (N) and benznidazole (Bz) that were introduced more than four decades ago and are quite unsatisfactory since both display limited activity (especially in the later chronic stage) and high toxicity. The occurrence of Trypanosoma cruzi strains that are naturally resistant to nitroderivatives [1] represents a special concern calling for the identification of novel trypanocidal candidates and treatment regimens. CYP51 (sterol 14α-demethylase) is the primary target for clinical and agricultural antifungals and has been proven as a relevant target for protozoan infections [2, 3]. Previous studies revealed the high anti-parasitic efficacy of the trypanosomal CYP51 inhibitor VNI, ((R)-N-(1-(2,4-dichlorophenyl)-2-(1H-imidazol-1-yl)ethyl)-4-(5-phenyl-1,3,4-oxadi-azol-2 yl)benzamide) against T. cruzi in vitro and in vivo. However, while being able to cure mice infected with the Bz-susceptible Tulahuen strain (DTU VI) [4], VNI was not able to reach high parasitological cure rates when Bz-resistant T.cruzi strains were employed [5], although in these experiments different drug formulations were used, which might have influenced the cure outcomes. Unfortunately, the two azoles inhibitors of fungal CYP51 (posaconazole and ruvaconazole), although highly effective in pre-clinical studies, presented high levels of therapeutic failure as compared to benznidazole [6]. The lack of translation among these pre-clinical and clinical outcomes has been largely discussed, and some hypothesis raised including the need for more reproducible animal models and readouts [7, 8]. In this sense, highly sensitive in vivo imaging assays and in vitro deeper analysis of sterile cidality [9] claims about the limited ability of posaconazole (and other analogs) to cure T. cruzi experimental infections [10]. However, we can not discard the possibility that the lack of translation between clinical and pre-clinical outcomes could be due to limitations of posaconazole pharmacokinetics [11]. In fact, its maximal concentration in mice plasma does not exceed 5 μM, and the low doses of the drug used in clinical trials for Chagas disease (because of its high cost) resulted in 5 to 10-fold lower concentration in humans than in animal models [12, 11]. In addition to the extremely high cost of posaconazole, another import point is that fungal sterol 14α-demethylases share less than 30 % amino acid sequence identity with the T. cruzi enzyme ortholog [13]. This may also explain at least in part, clinical trial failure and in turn stimulates the analysis of other inhibitors, more closely related to the protozoan enzyme, such as VNI and derivatives (5). Unlike posaconazole, this scaffold does not induce the T. cruzi CYP51 gene expression and does not require an increase in the dosage to sustain its anti-parasitic efficiency over time suggesting that it may have a lower propensity to induce resistance [14]. Also, due to its erratic bioavailability and unpredictable trough plasma concentration, posaconazole has been limited mainly for oropharyngeal or esophageal candidiasis and for prophylaxis in high-risk patients [15], though now, when the intravenous formulation of posaconazole has become available, its clinical use has potential to be extended [16].
Co-administration therapy has been successfully used to treat different pathologies including those triggered by parasitic infections [17, 18]. It has also been largely recommended as promising alternative therapy for CD [19] aiming to improve drug efficacy by allowing (i) to target different cellular elements and metabolic pathways, (ii) to reduce the doses and drug exposure periods thus contributing to the lowering of toxic effects, and (iii) minimizing the risk of drug resistance [20, 21]. In this regard, we investigated the anti-parasitic effect of VNI and VFV [(R)-N-(1-(3,4′-difluorobiphenyl-4-yl)-2-(1H-imidazol-1-yl)ethyl)-4-(5-phenyl-1,3,4oxadiazol-2-yl)benzamide] (the derivative of a broader spectrum of antiprotozoal activity [22]) in mouse models of T.cruzi infection caused by a highly resistant parasite strain (Colombiana) using both monotherapy and in schemes of co-administration with Bz. The in vivo efficacy of VFV was also investigated using different drug formulations such as DMSO with Arabic gum in the presence or not of Tween 80. Our findings demonstrate the benefits of Bz/VFV association, leading to high (>99.9 %) suppression of in vivo infection and reaching considerable rates of parasitological cure (70 %).
Materials and methods
Compounds
Synthesis of VNI and VFV was performed as reported [23]. In this study, the CYP51 inhibitors were diluted using (i) 10 % dimethyl sulfoxide (DMSO) plus 5 % Gum Arabic as we performed in [5] or (ii) 10 % DMSO + 5 % Arabic gum + 3 % Tween 80 – DGAT as we performed in [4, 22]. Benznidazole (Bz) was purchased from Laboratório Farmacêutico do Estado de Pernambuco, LAFEPE, Brazil and dissolved in distilled and sterile water supplemented with 3% Tween 80, which does not cause any detectable effect on mice [24]. Cyclophosphamide (Cy) (Genuxal) was purchased from Baxter Oncology (Frankfurt) and prepared in sterile distilled water [25].
Parasites
Bloodstream trypomastigote (BT) forms of Colombiana (discrete typing unit – DTU I) strain of T. cruzi was obtained from the blood of infected male Swiss mice at the peak of parasitemia [26]. Immediately after the purification step, the parasites were resuspended in RPMI-1640 medium (pH 7.2–7.4) without phenol red (Gibco BRL) supplemented with 10% fetal bovine serum and 2 mM glutamine, as reported previously [27].
In vivo infection
Male Swiss Webster mice were obtained from the Fundação Oswaldo Cruz (FIOCRUZ) animal facilities (ICTB /FIOCRUZ, Rio de Janeiro, Brazil). Mice were housed at a maximum of 05 per cage and kept in a standard room at 20 to 24°C under a 12 h/12 h light/dark cycle. The animals were provided with sterilized water and chow ad libitum. Mice (18 to 23 g) were infected by intraperitoneal (i.p.) route with 5×103 bloodstream trypomastigotes of the Colombiana strain of T. cruzi. Age-matched non-infected mice were maintained under identical conditions [5].
Treatment schemes
The animals were divided into the following groups (5–10 animals per group): uninfected (non-infected and non-treated); untreated (infected but treated only with vehicles); and treated (infected and treated with the compounds). The therapy was performed for 60 days starting at 10 dpi (corresponding to the parasitemia onset in this model of infection), given per oral (po) route, comparing the effectiveness of the monotherapy of each compound (100 mg/kg/day of Bz and 25 mg/kg/bid of VNI and VFV) with the simultaneous administration (60 daily doses given po, 100 mg/kg of Bz and after 15 min, 25 mg/kg of VFV, both only once a day). Only mice with positive parasitemia were used in the infected groups.
Parasitemia and mortality rates
Parasitemia was individually checked by direct microscopic counting of parasites in 5 μL of blood, and mortality rates checked daily until 30 days post-treatment and expressed as the percentage of cumulative mortality (% CM) as described before [25].
Assessment of therapeutic failure
Mice that presented consistent negative parasitemia up to 30 days post-treatment were submitted to three cycles of cyclophosphamide exposure (50 mg/kg/day), each with four consecutive days of administration (ip) and with three days of intervals between each cycle [25]. As reported, assessment of therapeutic failure (or not) was based on the following parasitological methods: blood parasitemia reactivation observed (i) by light microscopy, and (ii) by quantitative real-time polymerase chain reaction (qPCR). Animals presenting negative results for all tests were considered “cured”. For qPCR 500 μL blood was diluted in 1:2 volume of guanidine solution (guanidine-HCl 6M/EDTA 0.2M), and heated for 90 sec. in boiling water. Guanidine-EDTA blood samples were processed using the QIAamp DNA mini kit (QIAGEN), as described by [28]. Quantitative Real-Time PCR Multiplex assays were performed (40 cycles, threshold set at 0.01) for parasite detection targeting the T. cruzi satellite nuclear DNA and the internal amplification control - IAC (pZErO-2 plasmid containing an insert from the A. thaliana aquaporin gene), as described [29]. In uninfected samples, the result of the qPCR was NA (Non-amplified). The standard curves for absolute quantification were constructed with 1/10 serial dilutions of total DNA obtained from a negative GEB sample spiked with 105 parasite equivalents per milliliter of blood (par. eq./mL).
PK studies
Pharmacokinetics and tissue distribution of VNI and VFV were studied using the previously developed protocol that is based on their oxadiazole ring-specific absorption maximum at 291 nm (ε291 36 mM−1 cm−1), which makes this inhibitory scaffold easy to monitor in vivo, with a detection limit of 0.1 μM (4) The compounds were given from two different formulations (3% tween 80, 10% DMSO, and 5 % Arabic gum (DGAT) or 20 % hydroxypropyl-β--cyclodextrin (HPCD)) to compare distinct administration vehicles since our previous studies revealed superior efficacy while using DGAT formulation [4,22] as compared to others vehicles [30, 5]. The compounds were given to mice by oral gavage as a single administration dose of 25 mg/kg. This dose was used in our previous studies (4,5,22,30) and was initially chosen because it is most common for testing azole drugs and drug candidates in mice. To obtain the drug plasma concentration profiles, the blood samples were collected over time (1, 2, 4, 6, 10, 16, and 24 hours after administration, using two mice per each time point), and the tissues were collected four hours after administration as we described previously [22]. For the blood analysis, 30 μl samples of plasma were diluted to 100 μl with PBS, mixed with 100 μl of acetonitrile containing 10 μM ketoconazole (used as an internal standard), the mixture was vortexed, and the drugs were extracted with 300 μl of extraction solution containing 80 % acetonitrile and 20 % water (v/v). After centrifugation at 16,000 g for 10 min the supernatant was transferred to a new tube and dried. For the analysis the samples were re-dissolved in the 500 μl of solvent composed of 50 % acetonitrile and 50 % water and analyzed using reversed-phase high-performance liquid chromatography (HPLC). The HPLC system was equipped with the dual-wavelength UV 2489 detector (Waters) set at 291 and 250 nm and a Symmetry C18 (3.5 μm) 4.6 × 75 mm column. The mobile phase was 55 % 0.01 M ammonium acetate (pH 7.4) and 45 % acetonitrile (v/v) with an isocratic flow rate of 1.0 ml/min. For the tissue distribution analysis, prior to drug extraction approximately 100 mg of each tissue was diluted 5-fold with PBS and homogenized using an IKA Ultra Turrax T8 tissue homogenizer (The Lab World Group, USA), and 100 μl of the homogenate samples was used for the extraction and analysis as described for plasma. VFV, given from the DGAT formulation, was selected for the final set of our experiments because we have found previously that after the second administration its concentration in tissues becomes (and remains) higher than the concentration of VNI [22]. A broader spectrum of antiprotozoal activity [22,23] was another reason for selecting VFV over VNI for further co-administration studies. Finally, because VFV is detectable in plasma for more than 24 hours, it was reasonable to adminster it once a day.
Ethics
The procedures dealing with infected mice and drug treatments were approved by the FIOCRUZ Committee of Ethics for the Use of Animals (CEUA LW16/14) and the procedures dealing with drug administration for the PK experiments were approved by the IACUC of Meharry Medical College.
Results
Given as monotherapy (25 mg/kg/bid, diluted in 10 % dimethyl sulfoxide (DMSO) plus 5 % Gum Arabic), VFV suppressed completely the parasitemia in infected animals, showing the effect similar to that of VNI (25 mg/kg/bid, prepared in 10 % dimethyl sulfoxide (DMSO) plus 5 % Gum Arabic) (Table 1) and Bz (100 mg/kg/day, its optimal dose) (Table 1). All treated groups reached 100 % of mice survival during the whole period of analysis (Table 1), but after the end of monotherapy, a natural parasitemia relapse was observed in all animals exposed to monotherapies of Bz and VFV but only in 3 out of five mice in the VNI group (Table 1). To confirm the therapeutic outcomes, all the animals were further immunosuppressed by 12 cycles of cyclophosphamide (Cy) and then monitored for another four weeks by counting the parasite load in the blood samples using light microscopy. We found that 20 % of the VNI group displayed a sustained negative parasitemia (which was also consistently negative according to the results of qPCR blood analysis), and VFV presented about 14-fold reduction of the blood parasite load measured by qPCR as compared to Bz (Table 2).
Table 1 -.
Swiss male mice infected with bloodstream trypomastigotes of Colombiana strain (n=5) and submitted to mono (VFV, VNI and Bz) and co-administration therapy (Bz and VFV). VNI and VFV were diluted in 10 % DMSO plus 5 % Gum Arabic.
| Parasitemia peak (mean ± SD number parx104/mL) | % Parasitemia suppression | % Animal survival | Natural relapse after the therapy | |
|---|---|---|---|---|
| Bz 60 days | 0 ± 0 | 100 | 100 | 5 out of 5 mice |
| VNI 60 days | 0.2 ± 0.4 | 99.9 | 100 | 3 out of 5 mice |
| VFV 60 days | 0.2 ± 0.4 | 99.9 | 100 | 5 out of 5 mice |
| Bz+VFV (simultaneous)** | 1.4 ± 3.1 | 99.9 | 100 | 3 out of 5 mice |
Simultaneous therapy: 60 days of Bz + VFV (once a day)
Table 2 -.
qPCR analysis of Swiss male mice infected with bloodstream trypomastigotes of Colombiana strain (n=5) and submitted to co-administration therapy (Bz and VFV). VFV was diluted in 10 % DMSO plus 5 % Gum Arabic.
| % Negative blood qPCR* | Blood parasite load determined by qPCR | |
|---|---|---|
| Bz+VFV (simultaneous)** | 0 | 123 ± 53 |
| Bz | 0 | 13038 ± 13612 |
| VNI | 20 | 372 ± 528 |
| VFV | 0 | 905 ± 854 |
Surviving mice
Simultaneous therapy: 60 days of Bz + VFV (once a day)
In the co-administration assays, the simultaneous administration of Bz and VFV (both compounds given once a day, 100 mg/kg and 25 mg/kg, respectively) revealed that besides reaching >99 % parasitemia decline at the peak and 100 % animal survival (Figure 1), two of five mice remained negative after the end of the co-administration (Table 1). After Cy administration, although none remained negative according to the light microscopy analysis, the qPCR demonstrated a substantial reduction in the blood parasite load, reaching more than 100-fold decrease in comparison with the Bz monotherapy (Table 2). In these experiments, as well as in the monotherapy experiments VFV was administered to mice using 10 % DMSO plus 5 % Gum Arabic as a vehicle, which (because of VNI and VFV hydrophobicity) was most likely not the best formulation.
Figure 1:
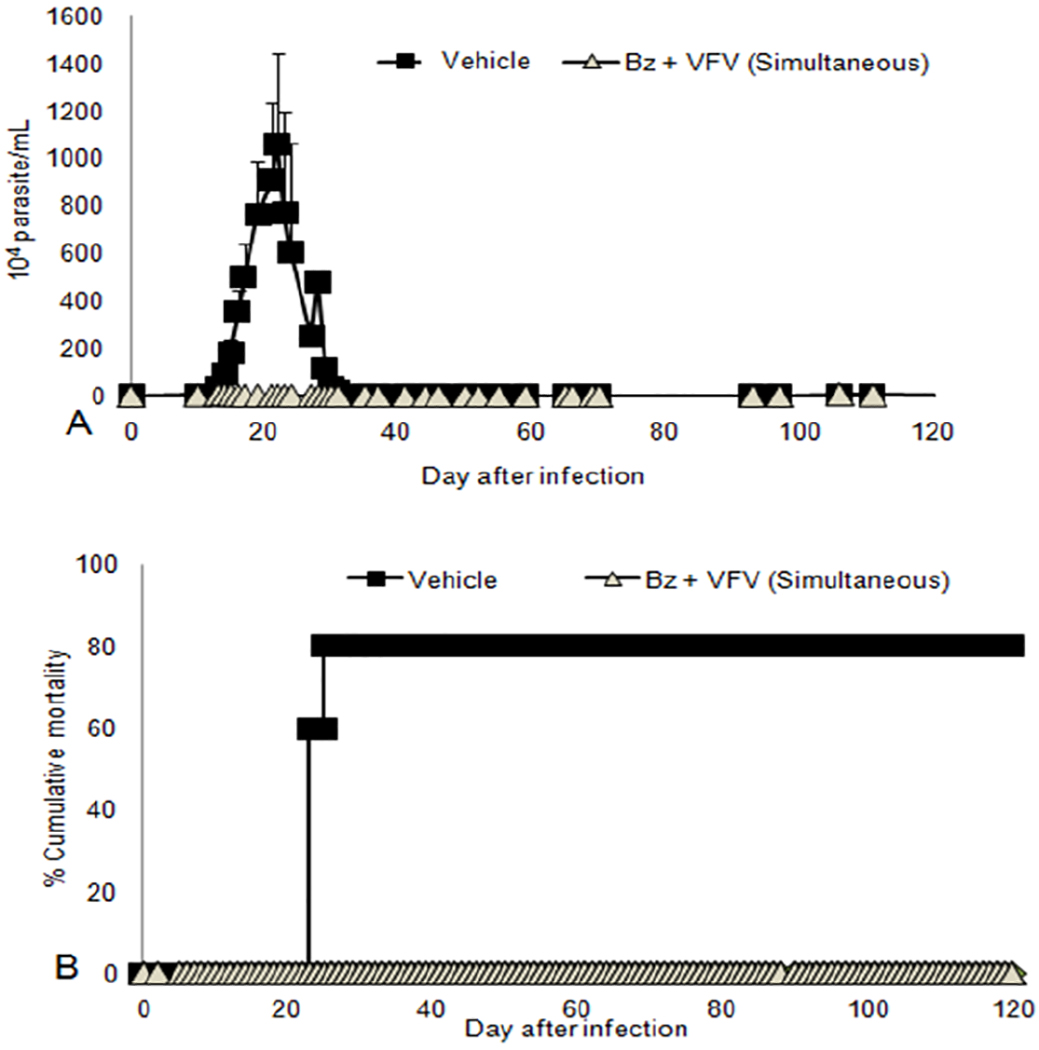
Effect of VFV (25 mg/kg/day, in 10 % dimethyl sulfoxide (DMSO) plus 5 % Gum Arabic administered simultaneously with Benznidazole (Bz) – 100 mg/kg/day) for 60 days in the experimental mouse model of Chagas disease caused by the Colombiana strain of Trypanosoma cruzi (n=5). (A) Parasitemia levels and (B) Percentage of cumulative mortality.
Next, to compare two alternative vehicles that in our previous in vivo studies produced better results, we analyzed the PK of VNI and VFV given from 20% hydroxypropyl-β-cyclodextrin (HPCD) (30) or from Tween 80 (3% Tween 80, 10% DMSO, 5% Arabic gum) (DGAT) (4,22). Because the PK studies indicated a three-fold higher bioavailability when Tween 80 was added to the vehicle (Figure 2, Table 3), further studies were conducted, now using the DGAT formulation (10% DMSO + 5% Arabic gum + 3% Tween 80 (DGAT) as the vehicle. The data corroborated the high efficacy of Bz, VFV, and BZ + VFV in suppressing the parasitemia levels (> 99.8 % at the peak), leading to 100 % animal survival, while in the untreated infected group all animals died (Fig. 3, Table 4). After the drug administration, we found a natural parasitemia relapse in 6, 5 and 2 out of 10 mice for groups Bz, VFV, and the co-administration Bz plus VFV, respectively. After immunosuppression, again, we found that while Bz-treated group reached 30 % of clear blood parasite load, the co-administration resulted in 7 negative samples by qPCR (one out of 8 negative samples was lost during the qPCR procedure), thus reaching 70 % of cure when Bz + VFV were given using DGAT vehicle (Table 4).
Figure 2:

Pharmacokinetics of VNI and VFV in mice after a single oral administration dose of 25 mg/kg depending on formulation: DGAT (3% Tween 80, 10% DMSO, 5% Arabic gum) or HPCD (20% hydroxypropyl-β-cyclodextrin). 16 mice were used for testing of each drug and two mice were used for each of the 8 independent experimental points. A. Plasma concentration profiles. B. Tissue distribution 4 hours after administration. Panel A, p< 0.001 between VNI/DGAT and VNI/HPCD; and p<0.0015 between VFV/DGAT and VFV/HPCD. Panel B, ★ p<0.015 ; ◆p< 0.02 (Two-Way-Anova).
Table 3 -.
PK profile data for VNI and VFV given from HPCD and DGAT formulations.
| Compound | Formulation | Tmax, hours | Cmax, μM [μg/mL] | AUC, μg × h/L | Life time, hoursa |
|---|---|---|---|---|---|
| VNI | DGAT | 2 | 39±2 [20] | 163 | 16 |
| HPCD | 2 | 30±4 [15] | 55 | 10 | |
| VFV | DGAT | 4 | 24±2 [11] | 159 | >26 |
| HPCD | 4 | 14±2 [8] | 64 | <24 |
Life time, hours: The time until compound becomes undetectable given by per oral route (below 0.1 μM, which is the detection limit). References: 4 and 22.
Figure 3:

Effect of VFV (25 mg/kg/day) administered simultaneously with Benznidazole (Bz) - 100mg/kg/day) for 60 days in the experimental mouse model of Chagas disease caused by the Colombiana strain of Trypanosoma cruzi (n=10). (A) Parasitemia levels and (B) Percentage of cumulative mortality. VFV was diluted in 10 % DMSO + 5% Arabic gum + 3 % Tween 80 – DGAT.
Table 4 -.
Swiss male mice infected with bloodstream trypomastigotes of Colombiana strain (n=10) and submitted to mono (VFV and Bz) and co-administration therapy (Bz and VFV). VFV was diluted using 10 % DMSO + 5 % arabic gum + 3 % Tween 80 - (DGAT) and Bz dissolved in water + 3 % Tween 80.
| Parasitemia peak (mean ± SD number parx104/mL) | % Parasitemia suppression | % Animal survival | Natural relapse after the therapy | % Blood parasite clearence (negative qPCR) | |
|---|---|---|---|---|---|
| Bz 60 days | 0 ± 0 | 100 | 100 | 6 out of 10 mice | 30 |
| VFV 60 days | 5.7 ± 5.1 | 99.8 | 100 | 5 out of 10 mice | 0 |
| Bz+VFV | 0 ± 0 | 100 | 100 | 2 out of 10 mice | 70* |
One out of the 8 negative samples was lost during the qPCR procedure
Discussion
There is an urgent need for safer and more efficacious drugs for Chagas disease, a neglected silent life-threatening illness that causes more than 7,000 annual deaths, with about 25 million people at risk of infection in endemic areas of Latin America, affecting mainly rural populations in areas of low resources, where human contact with the vectors is frequent [31]. Despite the fact that vector control and blood transmission regional initiatives have successfully achieved substantial reduction in the number of new acute cases [32], other concerns still remain as challenges, including the disease globalization, the existence of alternative routes of transmission, such as mother-to-child or oral transmission through contaminated food [33, 34], and particularly the high variability in the drug sensitivity across multiple strains that comprise the genetically diverse population of T. cruzi. Therapeutic failures are well documented in Chagas disease under the use of Bz and nifurtimox, which seems to depend on the interplay of the genetic background of the T. cruzi strains and their mammalian hosts, the drug access and accumulation in different environments, and the host immune response [35, 36].
Thus, as the available chemotherapy for CD has substantial limitations, diverse pre-clinical approaches have been used in an attempt to identify new therapeutic alternatives, including the screening of natural and chemical libraries [37], the repositioning of drugs already licensed to other diseases [38], the synthesis and validation of specific inhibitors targeting parasite molecules [39], as well as the use of co-administration of licensed drugs with promising novel candidates [19, 21]. Among the target-directed anti-parasitic approaches, inhibitors of the sterol biosynthesis pathway, in particular, those related to the CYP51 enzyme, have been widely studied on experimental infection with T. cruzi [21, 40] and two of them, the antifungal drug posaconazole and the prodrug of another antifungal drug candidate ravuconazole (E1224) have been recently moved to clinical trials but unfortunately displayed limited treatment success in chronic chagasic patients [41].
Because we found previously that VNI, our experimental inhibitor of Tulahuen T. cruzi CYP51, though suppressing the parasitemia and preventing mortality, was unable to achieve the complete parasitological cure in the mouse models of Colombiana T. cruzi infection (20 day treatment, 25 mg/kg/bid, administered using 10 % DMSO plus 5 % Arabic gum) [5], in this study we mostly concentrated on the CYP51 structure-based VNI derivative VFV, since VFV has a broader spectrum of activity as the protozoan CYP51 inhibitor and was shown to be more potent than VNI against the Y strain T. cruzi [42] and Leishmania [22].
The present studies were conducted employing a longer period of treatment (60 days) using Bz (as reference drug at its optimal dose (100 mg/kg/day) for mouse T.cruzi infection [5, 25]) and the imidazole molecules (VNI and VFV) under monotherapy regimen and also addressing the scheme of drug co-administration (using two different vehicles, 10 % dimethyl sulfoxide (DMSO) plus 5 % Arabic gum [5] and 10% DMSO + 5% Arabic gum + 3% Tween 80 – DGAT. The later drug – formulation was chosen to improve pharmacological status as our PK studies demonstrated higher bioavailability and tissue distribution when Tween was added to the vehicle solution. plus 5 % Arabic gum) was more effective than Bz in reducing the blood parasitism measured by qPCR analysis of blood samples from treated mice submitted to Cy injection. VFV resulted in more than 14-fold reduction in blood parasite DNA that corresponds to lower and subpatent blood parasitism. Another significant finding was the improved efficacy of VFV when administrated in co-administration with Bz resulting in >100-fold less blood parasitism measured by qPCR than Bz alone. Interestingly, exposure to Bz plus VFV induced 70 % blood parasite clearance when the second vehicle was used (10% DMSO + 5% Arabic gum + 3% Tween 80 – DGAT), supporting the PK studies that demonstrated higher plasma levels and better tissue distribution of VFV when Tween 80 is added to the vehicle solution and corroborating the previous findings using acute and chronic T.cruzi mouse infection (Tulahuen strain) that demonstrated high rates of cure when these CYP51 inhibitors were given using DGAT solution [4]. Also, the promising aspect of this co-administration drug scheme reinforces the potential use of CYP51 inhibitors along with Bz as therapeutic arsenal against T. cruzi. Other studies also revealed the successful use of drug co-administration in experimental animal models of T. cruzi infection, e.g., amidine compounds and Bz [25] and also other compound classes including posaconazole and Bz (24,40,41,42). The advantages of VFV include its low cellular (EC25>50 μM, NIH/3T3 [23]) and in vivo toxicity (NOAEL> 200 mg/kg [30]), weak influence on the major human drug-metabolizing cytochromes P450 (e.g., CYP3A4 IC50=3.6 μM vs. IC50=0.14 μM for posaconazole), moderate hepatic clearance, and the ability to penetrate different tissues and organs [23].
To summarize, this study supports the use of co-administration schemes of anti-parasitic drugs as future potential clinical evaluation of Chagas disease. The curative action of Bz/VFV co-administrations was achieved in a well-established acute infection model with the Colombiana strain using optimal doses of compounds that did not exert any signs of animal toxicity until the endpoint (>120 days of following up). These findings accumulate evidence that it is possible to achieve a better therapeutic outcome using Bz in co-administration with other drug candidates that target other metabolic pathways of the parasite. Several other pathogen-induced diseases such as tuberculosis, leprosy, HAT and HIV infection reached better efficacy with co-administration of drugs with different mechanisms of action. The co-administration drug treatment not only can boost the effect of the different therapeutic compounds but may also aid in avoiding the development of parasite chemotherapeutic resistance [19]. Indeed, more than one new drug is needed for each so that co-administration can be employed to avoid drug resistance and to provide back-up drugs when resistance emerges [34]. The presented results encourage further investigations aiming to achieve therapeutic and affordable orally administrated drugs, as alternatives for those millions of chagasic patients.
Acknowledgments
The authors thank the Program for Technological Development in Tools for Health (PDTIS-Fiocruz) for the facilities on the Real-Time PCR RPT09A platform and RPT11G.
Financial support
The present study was supported by grants from Fundação Carlos Chagas Filho de Amparo a Pesquisa do Estado do Rio de Janeiro (FAPERJ), Conselho Nacional Desenvolvimento científico e Tecnológico (CNPq), Fundação Oswaldo Cruz, PDTIS, PAPI/CNPq/Fiocruz, CAPES. MNCS and CB are research fellows of CNPq. MNCS and CB are CNE researchers. G.I.L was supported by National Institutes of Health grant GM067871 through the NIGMS.
References
- 1.Zingales B, Miles MA, Moraes CB, Luquetti A, Guhl F, Schijman AG, Ribeiro I. 2014. Drug discovery for Chagas disease should consider Trypanosoma cruzi strain diversity. Mem Inst Oswaldo Cruz. 22: 1–2. DOI: 10.1590/0074-0276140156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lockhart SR, Fothergill AW, Iqbal N, Bolden CB, Grossman NT, Garvey EP, Brand SR, Hoekstra WJ, Schotzinger RJ, Ottinger E, Patterson TF,Wiederhold NP. 2016. The Investigational Fungal Cyp51 Inhibitor VT-1129 Demonstrates Potent In Vitro Activity against Cryptococcus neoformans and Cryptococcus gattii. Antimicrob Agents Chemother. 60 (4): 2528–31. DOI: 10.1128/AAC.02770-15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lepesheva GI, Waterman MR. 2011. Sterol 14alpha-demethylase (CYP51) as a therapeutic target for human trypanosomiasis and leishmaniasis. Curr Top Med Chem.11: 2060–71. DOI : 10.2174/156802611796575902 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Villalta F, Dobish MC, Nde PN, Kleshchenko YY, Hargrove TY, Johnson CA, Waterman MR, Johnston JN, Lepesheva GI. 2013. VNI cures acute and chronic experimental Chagas disease. J Infect Dis 208:504–511. DOI: 10.1093/infdis/jit042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Soeiro MNC, de Souza EM, da Silva CF, Batista DDGJ, Batista MM, Pavao BP, Araújo JS, Aiub CA, da Silva PB, Britto C, Kim K, Sulikowski G, Hargrove TY, Waterman MR, Lepesheva GI. 2013. In vitro and in vivo studies of the antiparasitic activity of sterol 14-demethylase (CYP51) inhibitor VNI against drug-resistant strains of Trypanosoma cruzi. Antimicrob Agents Chemother 57:4151–4163. DOI: 10.1128/AAC.00070-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Molina I, Salvador F, Sánchez-Montalvá A. 2014. Posaconazole versus benznidazole for chronic Chagas’ disease. N Engl J Med. 371 (10):966 DOI: 10.1056/NEJMc1407914 [DOI] [PubMed] [Google Scholar]
- 7.Moraes CB, Giardini MA, Kim H, Franco CH, Araujo-Junior AM, Schenkman S, Chatelain E, Freitas-Junior LH. 2014. Nitroheterocyclic compounds are more efficacious than CYP51 inhibitors against Trypanosoma cruzi: implications for Chagas disease drug discovery and development.16, (4): 4703 DOI: 10.1038/srep04703 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chatelain E, Konar N. 2015. Translational challenges of animal models in Chagas disease drug development: a review. Drug Des Devel Ther. 19, (9)4807–23. DOI: 10.2147/DDDT.S90208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cal M, Ioset JR, Fügi MA, Mäser P, Kaiser M. 2016. Assessing anti-T. cruzi candidates in vitro for sterile cidality. Int J Parasitol Drugs Drug Resist. 6 (3):165–170. DOI: 10.1016/j.ijpddr.2016.08.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Francisco AF, Lewis MD, Jayawardhana S, Taylor MC, Chatelain E, Kelly JM. 2015. Limited Ability of Posaconazole To Cure both Acute and Chronic Trypanosoma cruzi Infections Revealed by Highly Sensitive In Vivo Imaging. Antimicrob Agents Chemother. 59 (8):4653–61. DOI: 10.1128/AAC.00520-15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lepesheva GI, Friggeri L, Waterman MR. 2018. CYP51 as drug targets for fungi and protozoan parasites: past, present and future. Parasitology. 12: 1 – 17. DOI: 10.1017/S0031182018000562 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Molina I, Salvador F, Sánchez-Montalvá A. 2015. The use of posaconazole against Chagas disease. Curr Opin Infect Dis. 28 (5): 397 – 407.DOI: 10.1097/QCO.0000000000000192 [DOI] [PubMed] [Google Scholar]
- 13.Lepesheva GI, Waterman MR. 2011. Sterol 14alpha-demethylase (CYP51) as a therapeutic target for human trypanosomiasis and leishmaniasis. Curr Top Med Chem. 11 (16):2060–71. DOI: 10.2174/156802611796575902 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lepesheva GI, Hargrove TY, Anderson S, Kleshchenko Y, Furtak V, Wawrzak Z, Villalta F, Waterman MR. 2010. Structural Insights into Inhibition of Sterol 14 alpha-Demethylase in the Human Pathogen Trypanosoma cruzi. J Biol Chem. 285 (33) : 25582–25590. DOI: 10.1074/jbc.M110.133215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pappas PG, Kauffman CA, Andes DR, Clancy CJ, Marr KA, Ostrosky-Zeichner L, Reboli AC, Schuster MG, Vazquez JA, Walsh TJ, Zaoutis TE, Sobel JD. 2016. Clinical practice guideline for the management of candidiasis: 2016 Update by the Infectious Diseases Society of America. Clin. Infect. Dis 62, e1–e50 17. DOI: 10.1093/cid/civ933 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jeong W, Haywood P, Shanmuganathan N, Lindsay J, Urbancic K, Ananda-Rajah MR, Chen SCA, Bajel A, Ritchie D, Grigg A, Seymour JF, Peleg AY, Kong DCM, and Slavin MA 2016. Safety, clinical effectiveness and trough plasma concentrations of intravenous posaconazole in patients with haematological malignancies and/or undergoing allogeneic haematopoietic stem cell transplantation: off-trial experience. J. Antimicrob. Chemother 71, 3540–3547. DOI: 10.1093/jac/dkw322 [DOI] [PubMed] [Google Scholar]
- 17.Stickles AM, Smilkstein MJ, Morrisey JM, Li Y, Forquer IP, Kelly JX, Pou S, Winter RW, Nilsen A, Vaidya AB, Riscoe MK. 2016. Atovaquone and ELQ-300 combination therapy: A novel dual-site cytochrome bc1 inhibition strategy for malaria. Antimicrob Agents Chemother. 00791–16. DOI: 10.1128/AAC.00791-16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chekwube AI, Onyema EI, Ikenna UE, Ezeokonkwo RC. 2014. Effect of diminazene aceturate, levamisole and vitamin C combination therapy in rats experimentally infected with Trypanosoma brucei brucei. Asian Pac J Trop Med. 7(6):438–45. DOI: 10.1016/S1995-7645(14)60071-7 [DOI] [PubMed] [Google Scholar]
- 19.Coura JR Present situation and new strategies for Chagas disease chemotherapy a proposal. 2009. Mem Inst Oswaldo Cruz. 104(4): 549–554. DOI: org/10.1590/S0074-02762009000400002 [DOI] [PubMed] [Google Scholar]
- 20.McKerrow JH, Doyle PS, Engel JC, Podust LM, Robertson SA, Ferreira R, Saxton T, Arkin M, Kerr ID, Brinen LS, Craik CS. 2009. Two approaches to discovering and developing new drugs for Chagas disease. 104:263–9. DOI: org/10.1590/S0074-02762009000900034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bahia MT, Diniz Lde F, Mosqueira VC. 2014. Therapeutical approaches under investigation for treatment of Chagas disease. 9:1225–37. DOI: org/10.1517/13543784.2014.922952 [DOI] [PubMed] [Google Scholar]
- 22.Lepesheva GI, Hargrove TY, Rachakonda G, Wawrzak Z, Pomel S, Cojean S, Nde PN, Nes WD, Locuson CW, Calcutt MW, Waterman MR, Daniels JS, Loiseau PM, Villalta F. 2015. VFV as a New Effective CYP51 Structure-Derived Drug Candidate for Chagas Disease and Visceral Leishmaniasis. J Infect Dis 212: 1439–1448. DOI: 10.1093/infdis/jiv228 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hargrove TY, Kim K, Soeiro MNC, da Silva CF, Batista DD, Batista MM, Yazlovitskaya MS Waterman MR, Sulikowski GA, Lepesheva GI. 2012. CYP51 structures and structure-based development of novel, pathogen-specific inhibitory scaffolds. Int J Parasitol Drugs Drug Resist. 2: 178–186. DOI: 10.1128/AAC.01294-15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Da Silva CF; Lima AJ; Romanha MM; Policarpo AJ; Stephens ASJ; Som PCE; Boykin DW and Soeiro MNC 2011. In vitro trypanocidal activity of DB745B and other novel arylimidamides against Trypanosoma cruzi. J Antimicrob Agents Chemother. 66: 1295 –1297. DOI: 10.1128/AAC.01236-07 [DOI] [PubMed] [Google Scholar]
- 25.Guedes-da-Silva FH, Batista DGJ, da Silva CF, Meuser MB, Simões-Silva MR, de Araújo JS, Ferreira CG, Moreira OC, Britto C, Lepesheva GI, Soeiro MNC. 2015. Different therapeutic outcomes of benznidazole and VNI treatments in different genders in mouse experimental models of Trypanosoma cruzi infection. Antimicrob Agents Chemother. 59:7564 –7570. DOI: 10.1128/AAC.01294-15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Batista DG, Batista MM, de Oliveira GM, do Amaral PB, Lannes-Vieira J, Britto CC, Junqueira A, Lima MM, Romanha AJ, Sales Junior PA, Stephens CE, Boykin DW, Soeiro MNC. 2010. Arylimidamide DB766, a potential chemotherapeutic candidate for Chagas’ disease treatment. Antimicrob Agents Chemother 54:2940–2952. DOI: 10.1128/AAC.01617-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Timm BL, da Silva PB, Batista MM, da Silva FHG, da Silva CF, Tidwell RR, Patrick DA, Jones SK, Bakunov SA, Bakunova SM, Soeiro MNC. 2014. In vitro and in vivo biological effects of novel arylimidamide derivatives against trypanosoma cruzi. Antimicrob. Agents Chemother. 58:3720–3726. DOI: 10.1128/AAC.02353-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Moreira OC, Ramírez JD, Velázquez E, Melo MF, Lima-Ferreira C, Guhl F, Sosa-Estani S, Marin-Neto JA, Morillo CA, Britto C. 2013. Towards the establishment of a consensus real-time qPCR to monitor Trypanosoma cruzi parasitemia in patients with chronic Chagas disease cardiomyopathy: a substudy from the BENEFIT trial. Acta Trop. 125 (1): 23–31. DOI: 10.1016/j.actatropica.2012.08.020 [DOI] [PubMed] [Google Scholar]
- 29.Duffy T, Cura CI, Ramirez JC, Abate T, Cayo NM, Parrado R, Bello ZD, Velazquez E, Muñoz-Calderon A, Juiz NA, Basile J, Garcia L, Riarte A, Nasser JR, Ocampo SB, Yadon ZE, Torrico F, de Noya BA, Ribeiro I, Schijman AG. 2013. Analytical performance of a multiplex Real-Time PCR assay using TaqMan probes for quantification of Trypanosoma cruzi satellite DNA in blood samples. PLoS Negl Trop Dis. 7(1). DOI: 10.1371/journal.pntd.0002000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Guedes-da-Silva FH, Batista DG, Da Silva CF, De Araújo JS, Pavão BP, Simões-Silva MR, Batista MM, Demarque KC, Moreira OC, Britto C, Lepesheva GI, Soeiro MN. 2017. Antitrypanosomal Activity of Sterol 14α-Demethylase (CYP51) Inhibitors VNI and VFV in the Swiss Mouse Models of Chagas Disease Induced by the Trypanosoma cruzi Y Strain. Antimicrob Agents Chemother. 24. 61: (4). DOI: 10.1128/AAC.02098-16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.WHO. 2016. World Health Organization; http://www.who.int/topics/chagas_disease/en/, acessed in 04/29/16. [Google Scholar]
- 32.Dias JC. 2009. Emilination of Chagas disease transmission: perspectives. Mem Inst Oswaldo Cruz. 104 1: 41–5. DOI: org/10.1590/S0074-02762009000900007 [DOI] [PubMed] [Google Scholar]
- 33.Gascón J, Bern C, Pinazo MJ. 2010. Chagas disease in Spain, the United States and other non-endemic countries. Acta Trop. 115(1–2):22–7. [DOI] [PubMed] [Google Scholar]
- 34.WHO. 2015. The World Health Report. World Health Organization, Geneva. [Google Scholar]
- 35.Romanha AJ, Castro SL, Soeiro Mde N, Lannes-Vieira J, Ribeiro I, Talvani A, Bourdin B, Blum B, Olivieri B, Zani C, Spadafora C, Chiari E, Chatelain E, Chaves G, Calzada JE, Bustamante JM, Freitas-Junior LH, Romero LI, Bahia MT, Lotrowska M, Soares M, Andrade SG, Armstrong T, Degrave W, Andrade Zde A. 2010. In vitro and in vivo experimental models for drug screening and development for Chagas disease. Mem Inst Oswaldo Cruz. 105(2):233–8. DOI: org/10.1590/S0074-02762010000200022 [DOI] [PubMed] [Google Scholar]
- 36.Coura JR, de Castro SL. 2002. A critical review on Chagas disease chemotherapy. Mem Inst Oswaldo Cruz . 97(1):3–24. DOI: org/10.1590/S0074-02762002000100001 [DOI] [PubMed] [Google Scholar]
- 37.Soeiro Mde N, de Castro SL. 2011. Screening of Potential anti-Trypanosoma cruzi Candidates: In Vitro and In Vivo Studies. Open Med Chem J. 5:21–30. DOI: 10.2174/1874104501105010021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kaiser M, Mäser P, Tadoori LP, Ioset JR, Brun R. 2015. Antiprotozoal Activity Profiling of Approved Drugs: A Starting Point toward Drug Repositioning. PLoS One. 10(8). DOI: org/10.1371/journal.pone.0135556 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tagoe DNA, Kalejaiye TD, de Koning HP. 2015. The ever unfolding story of cAMP signaling in trypanosomatids: vive la difference! Front. Pharmacol 6:185 DOI: 10.3389/fphar.2015.00185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hoekstra WJ, Hargrove TY, Wawrzak Z, da Gama Jaen Batista D, da Silva CF, Nefertiti AS, Rachakonda G, Schotzinger RJ, Villalta F, Soeiro Mde N,Lepesheva GI. 2015. Clinical Candidate VT-1161’s Antiparasitic Effect In Vitro, Activity in a Murine Model of Chagas Disease, and Structural Characterization in Complex with the Target Enzyme CYP51 from Trypanosoma cruzi. Antimicrob Agents Chemother. 60(2):1058–66. DOI: 10.1128/AAC.02287-15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Molina I, Salvador F, Sánchez-Montalvá A. 2015. The use of posaconazole against Chagas disease. Cur Opin Infect Dis 5: 397–407. DOI: 10.1097/QCO.0000000000000192 [DOI] [PubMed] [Google Scholar]
- 42.Cherkesova TS, Hargrove TY, Vanrell MC, Ges I, Usanov SA, Romano PS, Lepesheva GI (2014) Sequence variation in CYP51A from the Y strain of Trypanosoma cruzi alters its sensitivity to inhibition. FEBS Lett 588: 3878–3885. DOI: 10.1016/j.febslet.2014.08.030 [DOI] [PMC free article] [PubMed] [Google Scholar]