

Abstract
Rosai-Dorfman-Destombes disease is a rare nonmalignant histiocytic condition. Cutaneous disease is common and typically treated with surgical resection, but the optimal medical therapy for inoperable disease has not been determined. We present a 30-year-old man with a large unresectable cutaneous facial mass refractory to corticosteroids and rituximab. He had an excellent response to 13 cycles of lenalidomide 15 mg for 21 out of 28 day cycles and dexamethasone 40 mg/wk with no adverse effects. This is the second case of Rosai-Dorfman-Destombes disease in the literature responsive to lenalidomide-based therapy and confirms the potential efficacy of an immunomodulatory regimen in this disease.
Abbreviation and Acronym: RDD, Rosai-Dorfman-Destombes disease
Rosai-Dorfman-Destombes disease (RDD), also known as sinus histiocytosis with massive lymphadenopathy, is a rare histiocytic condition of the “R” group in the World Health Organization classification of histiocytic disorders.1 This nonmalignant disorder classically presents with bilateral painless cervical lymphadenopathy and is characterized by the accumulation of histiocytes within the affected tissues. More than 40% of cases have extranodal involvement, commonly involving the skin and soft tissues.2 A high clinical suspicion is required to make the diagnosis, which is confirmed by immunohistochemistry, demonstrating an infiltration of the tissues with plasma cells and a large number of histiocytes, the cytoplasm of which expresses S-100 positivity and CD1a negativity, in addition to the histological process of emperipolesis.3 Abundant IgG4-positive plasma cells are often present, and care must be taken to distinguish it from IgG4-related disease.2
Primary cutaneous disease without nodal involvement is rare and tends to have an older age of onset (43.5 years) and a higher predilection for women and Asian descendants,3 whereas in systemic RDD, the age of onset is earlier (20.6 years) and is more commonly seen in men and African descendants.2 Cutaneous lesions are often self-limiting and do not require intervention, and those that are symptomatic or cosmetically unacceptable may be amenable to surgical excision.3 For unresectable or multifocal disease, treatment options include corticosteroids, sirolimus, chemotherapy, targeted therapy, immunomodulatory therapy, and radiation therapy, although these often have suboptimal response rates.2 We present a case of unresectable primary cutaneous RDD with excellent response to lenalidomide and dexamethasone, confirming the efficacy of immunomodulatory therapy in this disease.
Case Description
A 30-year-old First Nations man presented in 2015 with a 2-year history of a progressively enlarging, nonpainful erythematous right-sided facial lesion. Clinical examination revealed a 4×5-cm firm violaceous lesion (Figure 1A).
Figure 1.
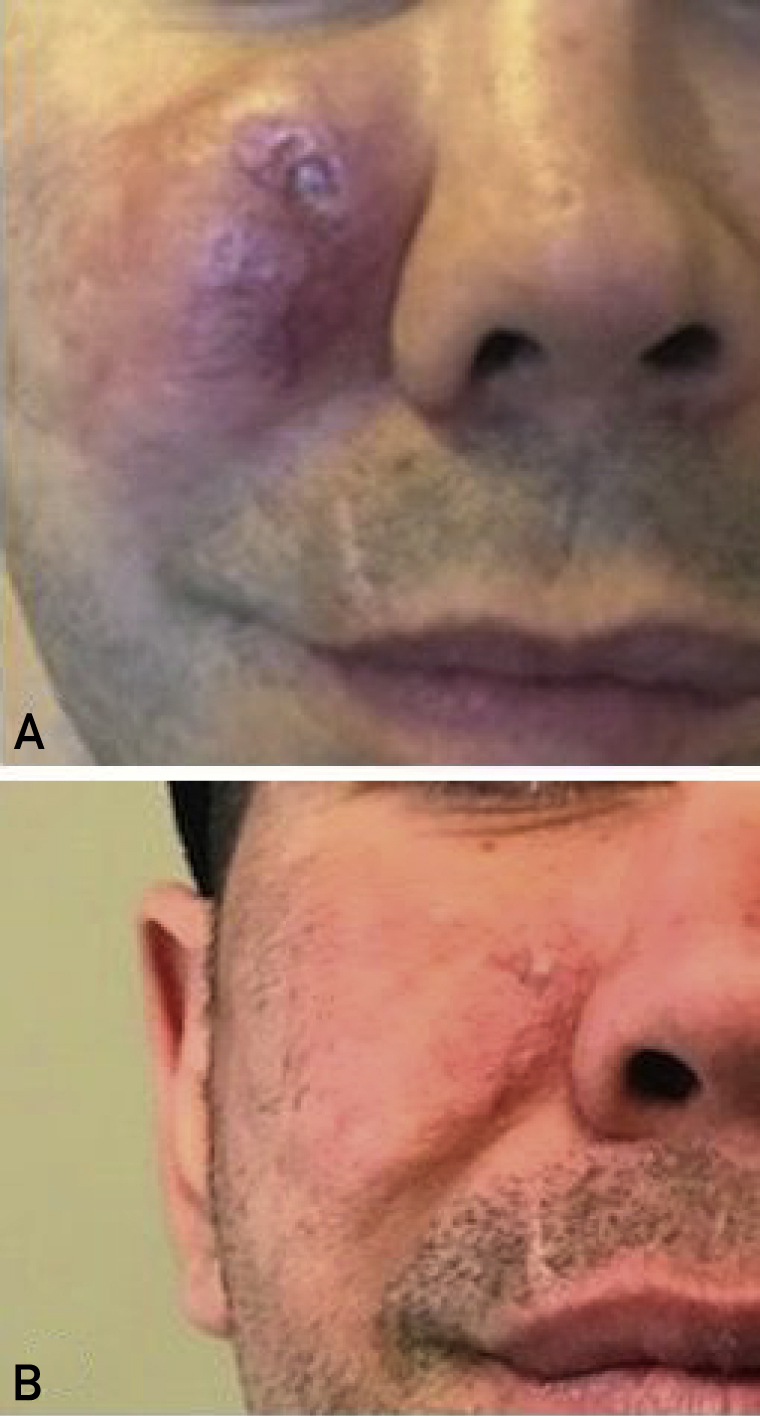
Clinical photographs of the right facial area. A, Large erythematous mass before the commencement of lenalidomide and dexamethasone. B, Resolution of lesion after 12 cycles of lenalidomide and dexamethasone.
Core biopsy revealed histology consistent with RDD (Figure 2). In addition, IgG4 staining revealed only mildly increased numbers of IgG4-positive plasma cells. Complete blood count, differential, creatinine, liver enzyme levels, and serum protein electrophoresis test results were normal. The C-reactive protein level was elevated to 42 mg/L (normal value, <3.1 mg/L); antinuclear antibodies were negative; and the serum IgG4 level was 0.3 g/L (normal value, <1.25 g/L).
Figure 2.
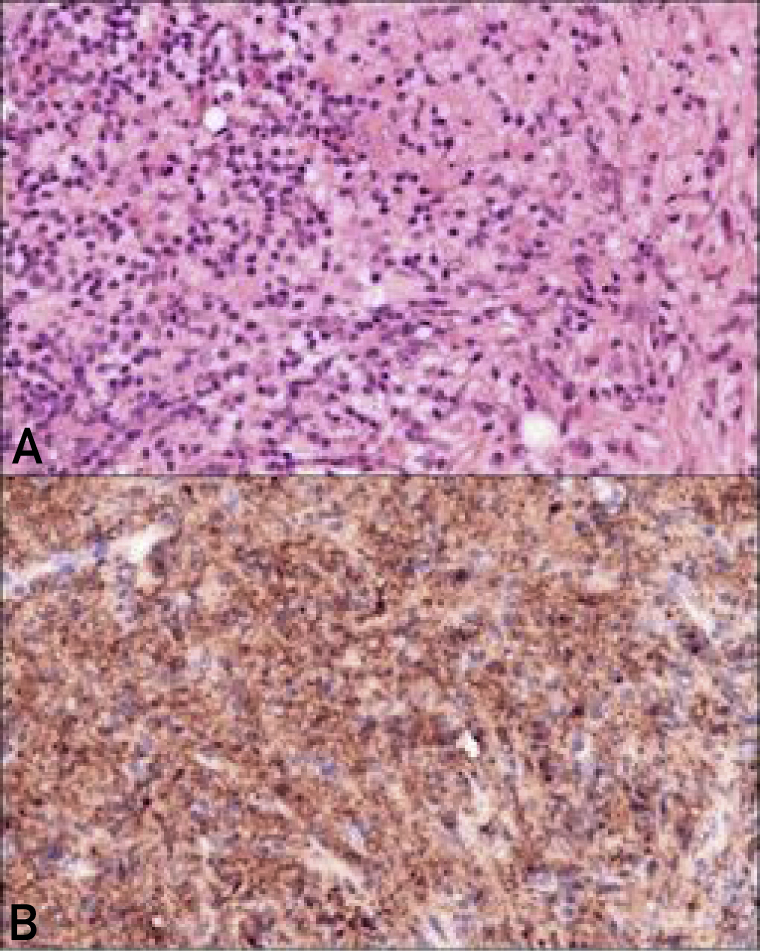
Histology of the facial lesion using S100 staining (magnification 400×). A, Dense proliferation of lymphocytes, plasma cells, and histiocytes with possible emperipolesis. B, Diffuse S100 positivity in the histiocytic component.
The lesion was not amenable to surgery, as resection would result in a large facial defect. He received several intralesional steroid injections with no response. Oral prednisone 50 mg/d every 2 weeks with a slow taper was given with no response. Intravenous rituximab 1 g every 2 weeks for 2 doses was given on September 15, 2016 with no response. On April 10, 2017, he was initiated on a regimen of lenalidomide 15 mg for 21 out of 28 day cycles and dexamethasone 40 mg/wk. At his first follow-up appointment on May 15, 2017, it was felt that the lesion had shrunk in size by 30% and was less firm on palpation. Computed tomography of the head and neck performed on August 30th 2017 had revealed substantial reduction in the size of the right-sided lesion with no bony involvement. At follow-up 1-year after the commencement of treatment, the lesion had almost completely resolved, with some minimal remaining erythema of the skin. (Figure 1B). He had no adverse effects or toxicities with this treatment. Treatment was discontinued on May 12, 2018 after 13 cycles, and the patient remained in remission.
Patients and Methods
Core biopsy was performed to establish a diagnosis, and on the basis of the histopathological findings suggesting dense proliferation of lymphocytes, plasma cells, and pale histiocytes centered within the subcutaneous tissue; emperipolesis; and diffuse S100 positivity in the histiocytic component and negative staining for CD1a, a diagnosis of RDD was made. Additionally, IgG4 staining revealed only mildly increased numbers of IgG4-positive plasma cells. Further imaging confirmed unifocal primary cutaneous RDD.
Response to therapy was based on macroscopic appearance of the lesion and adjunctive imaging.
Discussion
Most primary cutaneous RDD lesions are treated by surgical excision, with an 80% cure rate.3 However, some patients have unresectable, multifocal, or relapsed disease requiring other treatment modalities. There is no uniform approach to the treatment of RDD, and the range of therapeutic options have been summarized in the recent management consensus document.2 Broadly, treatment approaches can be divided into 3 categories: targeted, immunosuppressive, and immunomodulatory. Given the high frequency of B-rapidly accelerated fibrosarcoma proto-oncogene V600 activating sequence variations in Erdheim-Chester disease amenable to treatment with vemurafenib, much effort has been made to find similar molecular targets in RDD. Numerous mutations in kinases including A-rapidly accelerated fibrosarcoma proto-oncogene, mitogen activated protein kinase kinase 1 gene, neuroblastoma-rat sarcoma proto-oncogene, and Kirsten-rat sarcoma proto-oncogene have been reported, but these are rare and tyrosine kinase inhibitors such as imatinib have not been widely used.2 Immunosuppressive agents such as corticosteroids, rituximab, and disease-modifying antirheumatic drugs and chemotherapy are often used, particularly in severe or multifocal disease, with varying degrees of success. Given the putative role of immune dysregulation including increased tumor necrosis factor α and interleukin 6 levels in RDD,4 immunomodulation with thalidomide has been tried in several patients, with responses ranging from partial to complete.2 However, thalidomide is associated with well-known toxicities such as rash and neuropathy and lenalidomide is better tolerated in most patients. To our knowledge, there has been 1 previous report of complete remission of RDD with lenalidomide in a 43-year-old man with multifocal disease who had a partial response to steroids and complete remission to lenalidomide 15 mg/d.4
Conclusion
This case confirms the efficacy and tolerability of lenalidomide and dexamethasone in RDD and, to our knowledge, is the first report of this combination used in unresectable unifocal disease.
Footnotes
Potential Competing Interests: The authors report no competing interests.
References
- 1.Emile J.F., Abla O., Fraitag S., et al. Histiocyte Society Revised classification of histiocytoses and neoplasms of the macrophage-dendritic cell lineages. Blood. 2016;127(22):2672–2681. doi: 10.1182/blood-2016-01-690636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Abla O., Jacobsen E., Picarsic J., et al. Consensus recommendations for the diagnosis and clinical management of Rosai-Dorfman-Destombes disease. Blood. 2018;131(26):2877–2890. doi: 10.1182/blood-2018-03-839753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Al-Khateeb T.H. Cutaneous Rosai-Dorfman disease of the face: a comprehensive literature review and case report. J Oral Maxillofac Surg. 2016;74(3):528–540. doi: 10.1016/j.joms.2015.09.017. [DOI] [PubMed] [Google Scholar]
- 4.Rubinstein M., Assal A., Scherba M., et al. Lenalidomide in the treatment of Rosai-Dorfman disease—a first in use report. Am J Hematol. 2016;19(2):E1. doi: 10.1002/ajh.24225. [DOI] [PubMed] [Google Scholar]