

Abstract
The somatic mutations in ATP binding cleft of the tyrosine kinase binding domain of EGFR are known to occur in 15–40% of non-small cell lung cancer (NSCLC) patients. Although first and second generation anti-EGFR inhibitors are widely used to treat these patients, their therapeutic efficacy is modest and often results in adverse effects or drug resistance. Therefore, there is a need to develop novel as well as safe anti-EGFR drugs. The rapid emergence of computational drug designing provided a great opportunity to both discover and predict the efficacy of novel EGFR inhibitors from plant sources. In the present study, we designed several chemical analogues of edible curcumin (CUCM) compound and assessed their drug likeliness, ADME and toxicity properties using a diverse range of advanced computational methods. We also have examined the structural plasticity and binding characteristics of EGFR wild-type and mutant forms (S769L and K846R) against ligand molecules like Gefitinib, native CUCM, and different CUCM analogues. Through multidimensional experimental approaches, we conclude that CUCM-36 ((1E,4Z,6E)-1-(3,4-Diphenoxyphenyl)-5-hydroxy-7-(4-hydroxy-3-phenoxyphenyl)-1,4,6-heptatrien-3-one) is the best anti-EGFR compound with high drug-likeness, ADME properties, and low toxicity properties. CUCM-36 compound has demonstrated better affinity towards both wild-type (ΔG is −8.5 kcal/Mol) and mutant forms (V769L & K846R; ΔG for both is >−9.20 kcal/Mol) compared to natural CUCM and Gefitinib inhibitor. This study advises the future laboratory assays to develop CUCM-36 as a novel drug compound for treating EGFR positive non-small cell lung cancer patients.
Keywords: Curcumin analogue, EGFR genetic, Molecular docking, Novel compound, Mutations
1. Introduction
The lung carcinoma (LC) is a leading form of lung disease causing huge morbidity and mortality worldwide. This disease starts in the lung as a primary metastatic growth and then spreads to other parts of the body. The two main forms of LC are small cell lung cancer (SCLC) and non-small cell lung carcinoma (NSCLC) (Collins et al., 2007). The classical symptoms of LC are losing weight, breathing difficulties, cough (often with blood) and pain in the chest (Wang et al., 2016). LC has become the 4th leading reason for the hospitalization of respiratory disease patients (Salim et al., 2011). The leading cause of LC associated mortality (up to 85% of LC) is due to non-small cell lung cancer (NS-CLC). LC develops because of genetic as well as epigenetic changes of the cellular genome. The comprehensive molecular dissection of NS-CLC has laid the foundation to develop novel small drug molecules targeting mutations in EGFR, ALK, K-Ras, B-Raf, c-MET, NKX2-1, LKB1 genes, which are critical to the disease progression (Stella et al., 2013). Out of these LC genes, approximately 10–40% NS-CLC patients demonstrate activating mutations in EGFR gene.
The EGFR gene encodes a transmembrane epidermal growth factor receptor protein that once activated (by ligand binding), transduces the signals that are important for cellular proliferation, differentiation, migration, and survival (Stewart et al., 2015). Therefore, targeting ATP binding cleft of the tyrosine kinase binding domain of EGFR by potential inhibitors (like Gefitinib and Erlotinib) has become an attractive treatment strategy for treating patients suffering from NS-CLC (Zhang et al., 2012). Interestingly, these EGFR inhibitors show strong binding affinity with mutant forms of EGFR compared to the native form, and they were initially seen to be giving encouraging results for treating NS-CLC patients. However, the emergence of acquired drug resistance in patients limits its usage in clinical settings (Stella et al., 2012). The acquired drug resistance of EGFR is attributed to the threonine to methionine substitution at residue position 790 (Zhang et al., 2012). The underlying molecular cause of this drug resistance is assumed to be due to the mutation led steric interferences in the EGFR and inhibitor binding characteristics. Although some irreversible inhibitors like CL 387–785 and HK Inh-272 are developed to counter the acquired resistance of EGFR molecule, they are found to modify the covalent bonds in EGFR protein structure, thus limiting their practical application (Sato et al., 2012). Therefore, there is a need to search and develop novel as well as safe treatment regimes (for treating NS-CLC patients) which can easily counteract the drug resistance induced by EGFR mutations.
The traditional compounds obtained from nature are proven to be a potential source of several anti-cancer lead molecules (Banaganapalli et al., 2013b). Most of the successful anti-cancer drugs currently being used are derived from natural products or their analogues (Mondal et al., 2012). In this context, Curcumin (CUCM) (diferuloylmethane), a plant polyphenol (extracted from turmeric plants) is well known for its potential low toxic anti-cancer activity (see Fig. 1). The effectiveness of CUCM in treating lung, colon, breast and prostate cancers is already well reported (Starok et al., 2015). The CUCM compound is known to act against several molecular targets like EGFR, PKB/Akt, NF-κB, and MAPK inside the cancer cells (Kasi et al.,2016). In breast cancer cell lines, CUCM is demonstrated to inhibit the expression of EGFR and also induces the apoptosis (Sun et al., 2012). The chemically synthesized CUCM is being intensively studied to enhance its properties. However, whether CUCM or its analogues show similar effects to shut down EGFR expression (both in wild and mutant forms) in lung cancer cells is not yet investigated.
The classical laboratory investigations demand expensive drug compound (analogues) synthesis by series of chemical methods and laboratory investigations involving cellular systems and animal models. In contrary, the rapid development of bioinformatics discipline has provided a great opportunity for designing the anti-EGFR inhibitor compounds with desired specificity and sensitivity. Computational approaches have proven to be highly reliable in predicting the mutation induced drug resistance and also to design resistance evading drugs. The computational approaches built on machine learning and pattern classification methods (decision trees, support vector machine, and neural networks) can potentially classify the pathogenic mutations, create the three-dimensional protein structures, assist in designing therapeutic inhibitors and also predict the resistance of target proteins towards these inhibitor molecules. Owing to the lack of substantial amount of data in this direction, we sought to design novel CUCM analogues which can competitively inhibit the ATP binding cleft of tyrosine kinase domain in both native and mutated forms of EGFR molecules.
Fig. 1.
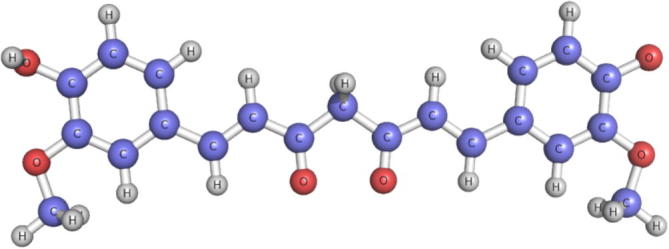
Molecular Structure of native CUCM compound.
2. Methods
2.1. Designing the curcumin analogues library
CUCM consists of two aromatic phenolic groups connected by unsaturated carbonyl moieties. We used two aromatic phenolic moieties (consists of four functional groups -R1, -R2, -R3 and -R4) to design different chemical derivatives of CUCM. The linkers (functional chemical groups), electron donors (—OH, —CH2—CH3, —CH3) and electron acceptors (—NO2) were manually replaced or modified at four functional group of the two aryl rings. Through an exhaustive combination of the two aryl rings and four chemical substituents, we generated a unique library of 50 CUCM analogues. Table 1 reveals the aryl group substitutions of the CUCM compound we generated in the presented study.
Table 1.
List of Linkers (electron donor and electron acceptor) used at R1, R2, R3, R4 sites of curcumin compound used in generating multiple curcumin analogues.
| Compounds | R1 | R2 | R3 | R4 |
|---|---|---|---|---|
| CURCUMIN | —OH | —OCH3 | —OCH3 | —OH |
| CUCM-1 | —OCH3 | —OH | —OH | —OCH3 |
| CUCM-2 | —OCH3 | —OCH3 | —OH | —OCH3 |
| CUCM-3 | —OCH3 | —OCH3 | —OCH3 | —OCH3 |
| CUCM-4 | —OCH3 | —ONH2 | —ONH2 | —OCH3 |
| CUCM-5 | —OH | —ONH2 | —ONH2 | —OH |
| CUCM-6 | —ONH2 | —OH | ONH(CH2CH3) | —ONH2 |
| CUCM-7 | —OH | —OH | —O(OH) | —OH |
| CUCM-8 | —OH | —ONH2 | —OCH3 | —OH |
| CUCM-9 | —OH | —OH | —OH | —OH |
| CUCM-10 | —OCH2CH2CH3 | —OCH2CH2CH3 | —H2CH2CH CH | —OCH2CH2CH3 |
| CUCM-11 | —OCH2CH2C C | —OH | —OCH2CH2C C | —OH |
| CUCM-12 | —OCH2CH CHCH3 | —OCH2CH2C C | —OCH2CH2C C | —OH |
| CUCM-13 | —OH | —OCH2CH2C C | —OCH2CH2C C | —OH |
| CUCM-14 | —ONH2 | —OH | —OCH2CH2C C | —OH |
| CUCM-15 | —OH | —OH | —OH | —ONH2 |
| CUCM-16 | —OCH2CH2C C | —OH | —OH | ONH(OH) |
| CUCM-17 | —OCH CH | —ONH2 | —OCH CH | —OH |
| CUCM-18 | —OH | —OCH CH | —OH | —OCH2(OH) |
| CUCM-19 | —OCH CH | —OCH CH | —OCH CH | —OCH2(OH) |
| CUCM-20 | —OCH CH | —OCH CH | —OCH2NH2 | —OH |
| CUCM-21 | —OCH2NH | —OH | —OH | —OH |
| CUCM-22 | —OCH2NH2 | —OCH2NH2 | —OCH2NH2 | —OCH2NH2(OH) |
| CUCM-23 | —OH | —OH | —OCH2NH2 | —OCH2(OH) |
| CUCM-24 | —OH | ONH(OH) | —ONH(OH) | —OCH2C6H5 |
| CUCM-25 | —OCH2(OH) | —OH | —OH | —OH |
| CUCM-26 | —OCH2(OH) | ONH(OH) | —OH | —OH |
| CUCM-27 | —OH | ONH(OH) | —ONH(OH) | —O(CH2(OH) |
| CUCM-28 | —OH | —OH | —ONH(OH) | —OH |
| CUCM-29 | —OCH2(OH) | —OCH2(OH) | —OCH2(OH) | —OCOCH3 |
| CUCM-30 | —OCH2NH3 | —OCH2(OH) | —OH | —OCOCH3 |
| CUCM-31 | —OCH2NH3 | —OH | —OCH2(OH) | —OH |
| CUCM-32 | —OCH2NH2(OH) | —OH | —OCH2(OH) | —OCOCH3 |
| CUCM-33 | —OCH2NH2(OH) | —OCH2NH2(OH) | —OCH2(OH) | —O(OCH3) |
| CUCM-34 | —OCH2(OH) | —OCH2(OH) | —OCH2NH2(OH) | —OH |
| CUCM-35 | —OCH2C6H5 | —OCH2C6H5 | —OCH2C6H5 | —OH |
| CUCM-36 | —OH | —OH | —OCH2C6H5 | —OH |
| CUCM-37 | —OCH2C6H5 | —OH | —OCH2C6H5 | —OCH2(OH) |
| CUCM-38 | —O(CH2(OH) | —O(CH2(OH) | —O(CH2(OH) | —OCH2(OH) |
| CUCM-39 | —O(CH2(OH) | —OH | —O(CH2(OH) | —OH |
| CUCM-40 | —OCOCH3 | —OCOCH3 | —OH | —OH |
| CUCM-41 | —OH | —OCOCH3 | —OCOCH3 | —OCH2(OH) |
| CUCM-42 | —OH | —OH | —OCOCH3 | —OCH2NH3 |
| CUCM-43 | —OCOCH3 | —OCOCH3 | —OCOCH3 | —OCH2NH3 |
| CUCM-44 | —O(OCH3) | —O(OCH3) | —OH | —OCH2NH2(OH) |
| CUCM-45 | —OH | —OH | —O(OCH3) | —OCH2NH2(OH) |
| CUCM-46 | —O(OCH3) | —OH | —O(OCH3) | —OCH2(OH) |
| CUCM-47 | —O(OCH3) | —OH | —OH | —OCH2C6H5 |
| CUCM-48 | —ONH(OCH3) | —ONH(OCH3) | —OH | —OH |
| CUCM-49 | —ONH(OCH3) | —OH | —ONH(OCH3) | —OH |
| CUCM-50 | —ONH(OCH3) | —ONH(OCH3) | —ONH(OCH3) | —OCOCH3 |
2.2. Determination of drug likeliness properties
The 3D topology of energy minimized coordinates of all CUCM analogues was described with the help of PRODRG tool (Schuttelkopf and van Aalten, 2004). Generally, natural bioactive molecules comprise functional chemical groups which possess certain properties that are similar to known drugs. Therefore, calculating the molecular properties of these bioactive compounds is significant drug discovery and development. Herein, we have used Molinspiration program (accessible at http://www.molinspiration.com/) to estimate the drug-likeness of CUCM analogues. This web server has several options to design any chemical compound either manually, or by intaking the query compounds in the format of canonical SMILES for calculating their molecular properties as well as bioactivity scores. Molinspiration web server calculates milogP (partition coefficient), TPSA (topological polar surface area), mass (molecular size), natoms (range of atoms), range of O or N (number of hydrogen bond acceptor), range of OH (number of hydrogen bond donor), nrotb (range of rotatable bonds) and molecular volume (volume of drug distribution) characteristics for any bioactive compound. The Molinspiration server is also used to estimate the drug likeliness of designed analogues by predicting the bioavailability scores of G-protein-coupled receptors (GPCR) ligands, ion channel modulator, enzymes and nuclear receptors (Lipinski 2004). Overall different molecular properties are taken into consideration while screening the potential lead CUCM analogue from a large pool of query molecules we designed in this study.
2.3. Determination of ADME-Tox properties
The ADME (absorption, digestion, metabolism, and excretion) and toxicity (mutagenic, tumorigenic, irritant) properties are directly related to the biological effect of drugs and their metabolic fate in an organism. Therefore, we determined ADME properties of potential CUCM analogues and their possible effects on health using Variable Nearest Neighbor ADMET (vNN-ADMET) web server (https://vnnadmet.bhsai.org/vnnadmet/home.xhtml) (Schyman et al., 2017). This web server can process both prebuilt or customized ADMET models by accepting one or more query molecules in canonical SMILES format as an input. This web server calculates the structural distance between molecules to construct ADMET models of potential CUCM analogues. These ADMET models quickly assess some of the important properties like cytotoxicity, mutagenicity, cardiotoxicity, drug-drug interactions, microsomal stability and the likelihood of causing liver injury of any potential drug candidates.
2.4. Constructing Gefitinib sensitive and resistance mutations in 3-dimensional structure of EGFR molecule and domain mapping
In this study, we studied the differential interaction of both Gefitinib (N-(3-Chloro-4-fluorophenyl)-7-methoxy-6-(3-morpholinopropoxy)quinazolin-4-amine) (a known EGFR inhibitor) sensitive and resistant mutations of EGFR molecules. For this purpose, we have initially downloaded the EGFR wild-type protein structure from Protein Databank (PDB ID: 5XWD, chain A (Matsuda et al., 2018) and 3GOP, chain A (Red Brewer et al., 2009)). This protein structure served as a template for constructing EGFR mutant forms. For two mutant models, we used homology-based computer modeling tool like Modeller9v11(Webb abd Sali, 2016). The full-length amino acid sequence of EGFR (in FASTA format) extracted from KEGG gene database (entry number KE1956) was used to incorporate mutated against wild-type amino acid residues for providing input to modeler program. The Modeller is an easily accessible web interface, which depends on protein NMR information to satisfy spatial restraints in creating probability density function for determining atomic locations in the protein models. This method aligns the input amino acid sequences and template protein structures. The built protein models, whether native or mutant forms were energy minimized, by using Gromacs program (Pronk et al., 2013). The structure quality of energy-minimized protein models is assessed with the help of validation tools like Procheck (Laskowski et al., 1996) and ProSA (Wiederstein and Sippl, 2007). PyMOL (http://pymol.sourceforge.net) program was used in the visualization and analysis of all the protein structures built. The NCBI conserved domain search (https://www.ncbi.nlm.nih.gov/Structure/cdd/wrpsb.cgi) web server is used in identifying and mapping the gefitinib-sensitive and resistant mutations in the EGFR molecule.
2.5. Analysis of structural drifts in mutated EGFR
To estimate the structural drifts in mutated EGFR molecules, we have superposed the Cα traces and backbone atoms of 3D structures using Yasasra (Krieger and Vriend, 2014). The exact structural fit (in term of Root Mean Square Deviation-RMSD values) between two amino acid residues or whole polypeptide chains of EGFR is measured. RMSD value is a quantitative metric of structural resemblance between two atomic coordinates when superimposed on each other (Banaganapalli et al., 2016).
2.6. Protein-drug interaction
In this study, we used AutoDock 4.0 (Morris et al., 2008) to execute a docking simulation of Gefitinib and CUCM analogues against EGFR protein using a Lamarckian Genetic Algorithm (LGA). Throughout the procedure, the ligand molecule was maintained in the flexible form and protein in its rigid form. The equal distribution of polar hydrogens and Gasteiger charges is ensured for both protein and ligand molecules, before initiating the molecular docking procedure. The histidine amino acids (at delta-HD1 or epsilon-HE2 positions) on a protein molecule were neutralized using edit Histidine hydrogens option in Autodock MGL tool. The grid parameter file (calculate the grid map of protein-ligand) was prepared using the default parameters of a grid of 60 × 60 × 60 points in x, y, and z directions and center spacing of the grid is 0.367 Å (approximately 1/4 of the length of c–c covalent bond). Finally, a docking file with different set parameters was prepared with AutoDock tool. The corresponding LGA parameters were set to default settings, which includes 150 runs, 150 conformational possibilities, 50 populations and 2,50,0000 energy evaluations. For docking procedure, translation parameters were set at 0.2 Å; the quaternion to 5.0 Å; the torsion angle to 0.5 Å, and the RMS cluster tolerance level to 0.75 Å (Banaganapalli et al., 2013a). At the end of docking step, ligand molecules which showed the maximum binding energy in the protein-ligand docking complex were selected. The resultant complex structures were explored using Pymol program (Yuan et al., 2016).
3. Results
3.1. The physicochemical screening of Curcumin derivatives
The physiochemical and pharmaceutical properties such as miLogP value, molecular weight, number of hydrogen bond acceptors, number of hydrogen bond donors, and number of rotatable bonds for each CUCM analogue were analyzed. These properties were evaluated against Lipinski's rule of five that predicts drug-likeness of the potential drug compound. Lipinski's rule of five states that most of the molecules with good membrane permeability will have LogP ≤5, molecular weight ≤500, the number of hydrogen bond acceptors ≤10, and the number of hydrogen bond donors ≤5. Hence, all the 50 chemical analogues of CUCM were evaluated for various parameters that would help to adjudge the particular substance to be a probable drug. Accordingly, we observed that 17 CUCM analogues are found to be compliant to the Lipinski's requirement for a potential drug compound (Table 2). The biophysical scores of all those 17 CUCM analogues were as follows, miLogPvalue is <4.63, MW is <423.42 kDa, TPSA is <140 Å, nON is <9, noHNH is <5, RB is <10 and molecular volume is >374.99 g/mol. Rotatable bonds are important for conformational changes of molecules and also determines the binding characteristics between receptors and their ligand molecules. It has been reported that the number of rotatable bonds should be ≤10 for passing oral bioavailability criteria (Priya et al., 2015). The CUCM analogues under investigation had low to high number of rotatable bonds (0–8 in general).
Table 2.
Drug-likeness physico-chemical properties of curcumin analogues by Molinspiration.
| Compound | miLogP | TPSA (Å) | MW (kDa) | nON | nOHNH | nrotb | Volume (g/mol) |
|---|---|---|---|---|---|---|---|
| CUCM-1 | 3.05 | 96.22 | 368.38 | 6 | 3 | 7 | 331.83 |
| CUCM-2 | 3.35 | 85.23 | 382.41 | 6 | 2 | 8 | 349.36 |
| CUCM-3 | 3.66 | 74.23 | 396.44 | 6 | 1 | 9 | 366.89 |
| CUCM-4 | 2.83 | 126.28 | 398.42 | 8 | 5 | 9 | 356.34 |
| CUCM-7 | 2.62 | 127.44 | 356.33 | 7 | 5 | 6 | 305.76 |
| CUCM-8 | 2.63 | 122.25 | 369.37 | 7 | 5 | 7 | 326.56 |
| CUCM-9 | 2.43 | 118.21 | 340.33 | 6 | 5 | 5 | 296.77 |
| CUCM-14 | 3.06 | 122.25 | 407.42 | 7 | 5 | 9 | 365.86 |
| CUCM-16 | 3.12 | 128.48 | 423.42 | 8 | 5 | 10 | 374.99 |
| CUCM-17 | 3.54 | 111.25 | 407.42 | 7 | 4 | 10 | 366.43 |
| CUCM-18 | 2.71 | 116.45 | 396.39 | 7 | 4 | 9 | 351.26 |
| CUCM-25 | 2.10 | 127.44 | 370.36 | 7 | 5 | 7 | 322.56 |
| CUCM-26 | 2.36 | 128.48 | 399.40 | 8 | 5 | 9 | 352.49 |
| CUCM-36 | 4.63 | 107.22 | 416.43 | 6 | 4 | 7 | 369.15 |
| CUCM-39 | 1.76 | 136.68 | 400.38 | 8 | 5 | 9 | 348.35 |
| CUCM-42 | 2.42 | 131.48 | 413.43 | 8 | 5 | 10 | 369.15 |
| CUCM-45 | 2.66 | 137.71 | 415.40 | 9 | 5 | 10 | 361.48 |
| Curcumin | 3.05 | 96.22 | 368.38 | 6 | 3 | 7 | 331.83 |
MiLogP = molinspiration Octonal/water partition coefficient; nON = number of H-bond acceptor; nOHNH = number of H-bond donors; nrotb = number of rotatable bonds; MW = molecular weight; TPSA = total polar surface area and molecular volume.
3.2. Molecular properties of Curcumin analogues obtained from Molinspiration
Fig. 2 reveals the drug-likeliness model scores generated by molsoft program, where blue color refers to drug-like behavioral properties, and green color refers to non-drug-like properties. Drug-like compounds show prediction values in a positive value, and non-drug-like compounds show zero or negative values. The drug-likeness prediction scores for CUCM compound (with a score of −0.66) and most (11/17; 64.70%) of its chemical analogues were negative. Only CUCM-7 (with a score of 0.37), CUCM-14 (with a score of 0.22), CUCM-26 (with a score of 0.21), CUCM-36 (with a score of 0.22), CUCM-42 (with a score of 0.27) and CUCM-45 (with a score of 0.26) analogues showed positive values. The drug-likeliness scores of these CUCM analogues are comparable to the standard anti-EFGR drug Gefitinib (1.26). Therefore, it is assumed that CUCM-7, CUCM-14, CUCM-26, CUCM-36, CUCM-42, CUCM-45, are good bioactive molecules which can potentially act as inhibitors of EGFR.
Fig. 2.

Drug-likeness model score of newly designed CUCM analogues, native CUCM compound and Gefitinib, a standard anti-EGFR drug. Positive score for any query compound indicates its drug potential.
3.3. ADMET predictions
The pharmacokinetics and safety profile, simply known as ADME-Tox of the CUCM analogues was predicted by the variable nearest neighbor computational method. ADME influences the drug levels and kinetics of drug exposure to the tissues. Of the six drug like CUCM analogues, selected in the previous stage, 5 compounds have shown negative predictions for ADMET endpoints. The compounds CUCM-7 and CUCM-26 showed negative predictions for three endpoints like Human Liver Microsomal (HLM) Stability test, acts as an inhibitor for Cyp2C9 and Cyp2C19 enzymes. However, in addition to HLM, Cyp2C9 and Cyp2C19 enzymes, native CUCM and its CUCM-14, CUCM-42 and CUCM-45 analogues act as inhibitors for matrix metalloproteinases (MMP). Only CUCM-36 ((1E,4Z,6E)-1-(3,4-Diphenoxyphenyl)-5-hydroxy-7-(4-hydroxy-3-phenoxyphenyl)-1,4,6-heptatrien-3-one) compound was predicted to be a potential drug compound as it showed negative predictions for all 15 ADMET endpoints of vNN method. The maximum recommended therapeutic dose of this CUCM-36 compound is 421 mg per day (Table 3).
Table 3.
ADME-TOX predictions of curcumin analogues by variable nearest neighbor (vNN) method.
| Compound | Cyto-toxicity | HLM | Cyp1A2 inhibitor | Cyp3A4 inhibitor | Cyp2D6 inhibitor | Cyp2C9 inhibitor | Cyp2C19 inhibitor | BBB | P-gp inhibitor & substrate | MMP | AMES | MRTD (mg/day) |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| CUCM-7 | No | Yes | No | No | No | Yes | Yes | No | No | No | No | 226 |
| CUCM-14 | No | Yes | No | No | No | Yes | Yes | No | No | Yes | No | 242 |
| CUCM-26 | No | Yes | No | No | No | Yes | Yes | No | No | No | No | 218 |
| CUCM-36 | No | No | No | No | No | No | No | No | No | No | No | 421 |
| CUCM-42 | No | Yes | No | No | No | Yes | Yes | No | No | Yes | No | 249 |
| CUCM-45 | No | No | Yes | No | No | No | No | No | No | No | Yes | 406 |
| Curcumin | No | Yes | No | No | No | Yes | Yes | No | No | Yes | No | 428 |
*HLM = Human Liver Microsomal Stability, Cyp1A2 = Cytochrome p450 1A2, Cyp3A4 = Cytochrome p450 3A4, Cyp2D6 = Cytochrome p450 2D6, Cyp2C9 = Cytochrome p450 2C9, Cyp2C19 = Cytochrome p450 2C19, BBB = blood brain barrier, P-gp = glycoprotein, MMP = metallo matrix protein, MRTD = maximum recommended therapeutic dose.
3.4. EGFR molecular modeling and determination of structural divergence
The identification of mutant protein structure is essential to understand how the amino acid substitution can change structural features of proteins. Therefore, we constructed two mutant forms of EGFR protein models (S769L & K846R) by comparative modeling methods using MODELLER sever, which provided approximately 100 models as an output. We selected the best predicted model based on the RMSD and template modeling (TM) scores, which were generated when native and mutant EGFR proteins were aligned against each other. All the EGFR mutant models showed an RMSD value of <2.0 and a TM value in between 0.5 and 1, confirming their good topology. The stereochemical and geometrical parameters of the built EGFR models was assessed through PROCHECK and PROSA servers. The PROCHECK analysis revealed that for both mutated models, 95% their amino acids are located in the allowed region. The Ramachandran plot constructed for EGFR models showed that 99.1% amino acids were present in the allowed regions (Fig. 3), and only 0.9% amino acids in disallowed regions confirming the reliability of built EGFR models. The G-factor value representing the dihedral angles in side chains of EGFR mutated model is found to be 1.0. This value is well within the permitted range to confirm the normality of the protein structure. The ProSA-web analysis of EGFR muted models (S769L and K846R) showed overall model quality (Z-score) values −10.02 and −10.23, respectively. These values are within the range characteristic of native proteins indicating good quality of the built model. The overall quality or Z score of the EGFR and EGFR mutated models reveals a similar correlation of the energy pattern between X-ray structures. The Z-score measures the deviation of total energy of the structure concerning an energy distribution derived from random conformations.
Fig. 3.

The stereochemical quality analysis of EGFR wildtype and mutant protein models by Procheck [Ramachandran plot – favored region is red in color; allowed region is yellow in color; disallowed region is white in color] and ProSa [overall model quality (black dots indicate the match between experimentally solved protein structures distinguished by dark blue (X-ray) and light blue (NMR); and residue model quality graphs (amino acid energies, +ve values regions are error part of the structure, whereas −ve value region considered to high structural quality)].
3.5. Superimposition analysis
The biophysical orientation of a three-dimensional protein structure could determine its stability, ligand binding efficiency, and other associated functional properties. In this study, we analyzed the EGFR structural drifts (in terms of RMSD scores) among Gefitinib resistant (V769L) and sensitive (K846R) mutations of EGFR molecule (Fig. 4). The amino acid sequence similarity among both forms of EGFR molecules is 99.33%. The structurally similar amino acid residues or whole polypeptide chain levels show the RMSD values in between 0 and <2.0 Å. The larger the RMSD value between two query structures indicates their dissimilarity, and zero means they are identical in structure. The RMSD values of EFGR mutants at whole 3-dimensional structures and at amino acid residue levels is found to be 0.38 Å and 2.675 Å for V769L and 0.23 Å and 2.835 Å for K846R, respectively. These findings highlight the subtle structural changes caused by substituted amino acid residues in the structure of native EGFR protein molecule.
Fig. 4.

The superimposition of EGFR wildtype and mutant V769L& K846R proteins in PyMOL software (circle zoom view).
3.6. Domain analysis
The NCBI domain scan has predicted six functional domains in EGFR protein molecule, of which domain 1, Receptor L domain is localized in between 57th and168th amino acids. This single-stranded right-hand beta-helix domain creates the bilobal ligand binding site in EGFR protein. The second one, Furin-like cysteine rich domain localized in between 185th and 355th amino acids functions like protease domain. The third one, Receptor L3 domain performs similar functions like domain 1 of EGFR. The fourth one, growth factor receptor domain IV located in between 505 and 637 amino acids, plays an important role in interacting with furin-like domain of EGFR. The fifth domain is transmembrane domain located in between 634 and 677 amino acids. The sixth one, catalytic domain of the protein tyrosine kinase located in between 704th and 1016th amino acids catalyzes the transfer of the gamma-phosphoryl group from ATP to tyrosine (Tyr) residues in protein substrates. The V769L (gefitinib sensitizing) and K846R (gefitinib resistance) mutations we analyzed in this study are localized in protein tyrosine kinase (6th domain) of EGFR molecule.
3.7. Molecular docking
The Gefitinib, CUCM and shortlisted CUCM-36 compound were initially energy minimized by adding partial surface charges using PRODRG web server. Then, these energy minimized structures were used in molecular docking against both native and mutant forms of EGFR to assess their inhibitory properties (Fig. 5). Molecular docking results showed that gefitinib and CUCM- 36 compounds interact with ATP binding cleft of EGFR via non-covalent interactions (hydrogen bonding). The gefitinib was determined to release the binding energy (ΔG) of −7.5 kcal/Mol and forms 3 hydrogen bonds with the LYS745, Gly768 and Glu863 of the EGFR ATP binding pocket. The binding energy of Gefitinib with mutated EGFR (K846R) is −8.1 kcal/Mol and forms 3 hydrogen bonds with Lys745, Arg748 and Glu 762 amino acids. Whereas, with V769L mutant form of EGFR, Gefitinib forms only one hydrogen bond with Leu862 amino acid and releases the binding energy of −6.8 kcal/Mol. The parent CUCM compound has shown a better binding affinity than Gefitinib towards EGFR molecule (Table 4). With native EGFR molecule, CUCM forms hydrogen bonds with Arg23, Ser246, Lys261 and Asn604 and releases the binding energy (ΔG is −7.8 kcal/Mol). The binding affinity of mutated EGFR is seen to be higher for both K846R (ΔG is −8.0 kcal/Mol) and V769L (ΔG is −8.1 kcal/Mol) forms. Interestingly, the CUCM-36 compound shows higher affinity while binding to V769L (ΔG is −9.60 kcal/Mol) compared to the K846R-mutant (ΔG is −9.2 kcal/Mol) and native EGFR (ΔG is −8.5 kcal/Mol) molecules. These results show that the CUCM-36 performs better than CUCM and Gefitinib compounds in effectively inhibiting EGFR molecule both in native, and mutant (gefitinib-sensitive and resistant) forms.
Fig. 5.

The visualization of molecular docking analysis of wild type and mutant forms of EGFR molecule against CUCM-36, native CUCM compound and Gefitinib drugs.
Table 4.
Molecular Docking Analysis Results of Curcumin, CUCM-36 and Gefitinib Compounds.
| Drug | Protein | Binding energy* (kcal/Mol) | No of H bonds (drug-enzyme) | Interacting amino acids |
|---|---|---|---|---|
| Gefitinib | EGFR | −7.5 | 3 | Lys745, Gly768 and Glu863 |
| EGFR(V769L) | −6.8 | 1 | Leu862 | |
| EGFR(K846R) | −8.2 | 3 | Lys745, Arg748 and Glu762 | |
| Curcumin | EGFR | −7.8 | 5 | Arg23, Ser246, Lys261 and Asn604 |
| EGFR(V769L) | −8.0 | 7 | Arg23, Ser246, Arg244, Lys261 and Asn604 | |
| EGFR(K846R) | −8.1 | 3 | Gly696, Arg429 and Arg705 | |
| CUCM-36 | EGFR | −8.5 | 3 | Lys745, Lys754, Arg748 |
| EGFR(V769L) | −9.60 | 4 | Lys745, Arg748, Gly863, Ala864 | |
| EGFR(K846R) | −9.2 | 4 | Lys745, Arg748, Val765, Gly863 | |
The change in binding free energy is related to the inhibition constant as per the following the equation: ΔG = RT in Ki, where R is the gas constant 1.987 cal K−1 Mol−1, and T is the absolute temperature assumed to be 298.15 K.
4. Discussion
Targeting EGFR molecule with drugs like Gefitinib and Imatinib remains as a first line therapy for lung cancer treatment (Gridelli et al., 2011). Gefitinib is a small molecule drug that competitively inhibits the binding of ATP at the active site and modulates tyrosine kinase activity of EGFR. The EGFR sequencing of 10% of gefitinib responders of NSCLC revealed the evidence of somatic gain-of-function mutations in its tyrosine kinase domain. The Gefitinib responders’ rate is up to 40% among the patients belonging to Asian ethnic group, non-smokers and those presenting adenocarcinoma histology (Chan and Hughes, 2015). Approximately 77% of these clinical responders to Gefitinib revealed mutations in EGFR gene as compared to the 7% of NSCLC who are refractory to gefitinib (Sharma et al., 2007). Most of the EGFR mutations are found to be clustered around ATP-binding pocket within the tyrosine kinase domain (exons 18–24). However, deletion mutations at exon 19 and point mutation at exon 21 (L858R) represent the most common EGFR mutations (Shigematsu et al., 2005) and their presence indicates sensitivity to gefitinib. Gefitinib treatment sometimes leads to the phenomenon of acquired resistance in around 45–60% of NSCLC tumors, through the accumulation of T90M mutation in exon 20 of EGFR molecule. This EGFR mutation leads to constitutive activation of downstream signaling pathways and promotes cellular growth and proliferation. The high cost, numerous side effects and secondary resistance caused by the acquisition of new mutations due to Gefitinib therapy pose a big challenge for using them in lung cancer treatment (Hong et al., 2016).
Owing to the adverse effects caused by chemotherapeutic agents, there has been an increasing interest in using multitargeted, inexpensive, innocuous and readily available phytomedicine or nutraceuticals for treating diseases like lung cancer (Hosseini and Ghorbani, 2015). Especially, if these herbal medicine compounds are derivatives of ethnic food agents, then it makes them more acceptable from perspective of safety and effectiveness. The availability of EGFR-tyrosine kinase structure has provided an opportunity to virtually screen potential active anti-EGFR compounds (Choowongkomon et al., 2010). CUCM, a phenolic compound [1, 7-bis (4-hydroxy-3-methoxyphenyl)-1, 6-heptadien-3, 5-Dione] derived from plant Curcuma longa is traditionally used as an edible agent (in the form of turmeric power) to fight inflammation and microbial infections due to its versatile pharmacological properties (Gupta et al., 2013). CUCM molecule acts against a diverse range of therapeutically important molecular targets of cancer signaling pathways such as EGFR, Ras, p53, AKT, Wnt-β catenin, PI3K, and mTOR, etc. (Kasi et al., 2016). The current literature suggests that CUCM can block proliferation, transformation, and invasion of lung cancer cells both in vitro and in vivo (Aggarwal and Harikumar, 2009). CUCM shows various effects on cancer cells like G1/S arrest and apoptosis induction (Karunagaran et al., 2005).
Phase 1 and II clinical trials have shown that CUCM compound is orally well tolerated and have no dose-limiting toxicity (Gupta et al., 2013). However, the relative poor bioavailability due to intestinal absorption, rapid metabolism, and systemic elimination limits the clinical usage of CUCM (Hatcher et al., 2008). Therefore, numerous efforts have been made to enhance the metabolic stability and anti-proliferative activity of CUCM by designing CUCM derivatives, which resemble native compound but possess modified chemical side chains on functional moieties (Vyas et al., 2013). In recent decades, some studies have successfully synthesized new CUCM analogues [symmetrical 1, 5-diarylpentadienone molecules with extra alkoxy substitutions] and demonstrated them to possess 30 times additional growth-suppressive activity compared to their native counterpart (Park et al., 2013). Furthermore, these analogues have down-regulated the expression of beta-catenin, k-ras, cyclin D1, c-myc, at a 1/8th concentration at which normal CUCM shows its effect (Ohori et al., 2006).
In this report, we have designed 50 different chemical derivatives of CUCM compound with the aim of identifying the differential affinity of CUCM derivatives against Gefitinib sensitive and resistant forms of EGRF molecule. The presence of a —OH group at the C4, C15 positions and —OCH3 group at C5, C16 positions in C21H20O6 [(1E, 6E)-1, 7–bis (4-hydroxy-3-methoxyphenyl) hepta1,6-diene-3-5-dione] did not favor the bioavailability of CUCM. Hence, the introduction of polar groups, such as hydroxy or methoxy, around the aryl moiety of CUCM is likely to enhance the bioactivity. In the CUCM-36 ((1E, 4Z, 6E)-1-(3,4-Diphenoxyphenyl)-5-hydroxy-7-(4-hydroxy-3-phenoxyphenyl)-1,4,6-heptatrien-3-one) analogue, an additional reactive —OH groups at C4, C5, C15 positions, and an aromatic hydrocarbon ring (—CH2C6H5) at the C16 position were added to the native CUCM (C21H20O6) molecule. This chemical modification might have contributed to increased bioavailability of the modified CUCM analogue compared with native CUCM compound. Additionally, the presence of additional—OH groups at C4, C5 and C16 positions in CUCM-36 appears to have favored the anti-EGFR activity. In the other CUCM analogues, less polar side chains are present at R1 (—OH), R2 (—OCH3), R3 (—OCH3) and R4 (—OH) positions which could have affected their overall molecular activity and bioavailability. In this study, we designed CUCM-36 which can effectively inhibit both Gefitinib sensitive (K846R located in exon 21) and resistant (V769L located in exon 20) mutations of EGFR molecule with higher affinity.
In conclusion, this report describes the atomic scale modification of edible CUCM to design CUCM-36 analogue as a probable drug for targeting EGFR mutations. Computational testing showed that this CUCM analogue is the best probable anti-EGFR drug, due to its drug-likeness, ADME properties, and low toxicity properties. When compared to the known inhibitors like native CUCM or Gefitinib, CUCM-36 showed better efficacy in binding ATP binding cleft of EGFR in both native and mutant forms. Our multidimensional drug screening approaches demonstrate the utility of computational tools in designing and rapid preliminary screening of potential anti-EGFR drug compounds from natural compounds. This study confirms that computational protocols are highly efficient in discovering potential anti-EGFR drug compounds with both minimal resources and less technical expertise. However, our prediction approaches cannot fully elucidate the complex drug metabolism reactions taking place inside the human body. Therefore, we recommend future studies to synthesize CUCM-36 compound chemically, and test its EFGR inhibitory action as well as drug metabolism in cell lines and animal models.
Acknowledgments
This project was funded by the Deanship of Scientific Research (DSR) at King Abdulaziz University, under Grant no. G-482-140-38. The authors, therefore, acknowledge the DSR for technical and financial support.
Footnotes
Peer review under responsibility of King Saud University.
References
- Aggarwal B.B., Harikumar K.B. Potential therapeutic effects of curcumin, the anti-inflammatory agent, against neurodegenerative, cardiovascular, pulmonary, metabolic, autoimmune and neoplastic diseases. Int. J. Biochem. Cell Biol. 2009;41:40–59. doi: 10.1016/j.biocel.2008.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banaganapalli B., Mohammed K., Khan I.A., Al-Aama J.Y., Elango R., Shaik N.A. A computational protein phenotype prediction approach to analyze the deleterious mutations of human MED12 gene. J. Cell Biochem. 2016;117:2023–2035. doi: 10.1002/jcb.25499. [DOI] [PubMed] [Google Scholar]
- Banaganapalli B., Mulakayala C., Gowsia D., Mulakayala N., Pulaganti M., Shaik N.A., Anuradha C.M., Rao R.M., Al-Aama J.Y., Chitta S.K. Synthesis and biological activity of new resveratrol derivative and molecular docking: dynamics studies on NFkB. Appl. Biochem. Biotechnol. 2013;171:1639–1657. doi: 10.1007/s12010-013-0448-z. [DOI] [PubMed] [Google Scholar]
- Banaganapalli B., Mulakayala C., Pulaganti M., Mulakayala N., Anuradha C.M., Suresh Kumar C., Shaik N.A., Yousuf Al-Aama J., Gudla D. Experimental and computational studies on newly synthesized resveratrol derivative: a new method for cancer chemoprevention and therapeutics? OMICS. 2013;17:568–583. doi: 10.1089/omi.2013.0014. [DOI] [PubMed] [Google Scholar]
- Chan B.A., Hughes B.G. Targeted therapy for non-small cell lung cancer: current standards and the promise of the future. Transl. Lung Cancer Res. 2015;4:36–54. doi: 10.3978/j.issn.2218-6751.2014.05.01. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choowongkomon K., Sawatdichaikul O., Songtawee N., Limtrakul J. Receptor-based virtual screening of EGFR kinase inhibitors from the NCI diversity database. Molecules. 2010;15:4041–4054. doi: 10.3390/molecules15064041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins L.G., Haines C., Perkel R., Enck R.E. Lung cancer: diagnosis and management. Am. Fam. Phys. 2007;75:56–63. [PubMed] [Google Scholar]
- Gridelli C., De Marinis F., Di Maio M., Cortinovis D., Cappuzzo F., Mok T. Gefitinib as first-line treatment for patients with advanced non-small-cell lung cancer with activating epidermal growth factor receptor mutation: review of the evidence. Lung Cancer. 2011;71:249–257. doi: 10.1016/j.lungcan.2010.12.008. [DOI] [PubMed] [Google Scholar]
- Gupta S.C., Patchva S., Aggarwal B.B. Therapeutic roles of curcumin: lessons learned from clinical trials. AAPS J. 2013;15:195–218. doi: 10.1208/s12248-012-9432-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hatcher H., Planalp R., Cho J., Torti F.M., Torti S.V. Curcumin: from ancient medicine to current clinical trials. Cell. Mol. Life Sci. 2008;65:1631–1652. doi: 10.1007/s00018-008-7452-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong D., Zhang G., Zhang X., Lian X. Pulmonary toxicities of gefitinib in patients with advanced non-small-cell lung cancer: a meta-analysis of randomized controlled trials. Medicine (Baltimore) 2016;95:e3008. doi: 10.1097/MD.0000000000003008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hosseini A., Ghorbani A. Cancer therapy with phytochemicals: evidence from clinical studies. Avicenna J. Phytomed. 2015;5:84–97. [PMC free article] [PubMed] [Google Scholar]
- Karunagaran D., Rashmi R., Kumar T.R. Induction of apoptosis by curcumin and its implications for cancer therapy. Curr. Cancer Drug Targets. 2005;5:117–129. doi: 10.2174/1568009053202081. [DOI] [PubMed] [Google Scholar]
- Kasi P.D., Tamilselvam R., Skalicka-Wozniak K., Nabavi S.F., Daglia M., Bishayee A., Pazoki-Toroudi H., Nabavi S.M. Molecular targets of curcumin for cancer therapy: an updated review. Tumour Biol. 2016;37:13017–13028. doi: 10.1007/s13277-016-5183-y. [DOI] [PubMed] [Google Scholar]
- Krieger E., Vriend G. YASARA view – molecular graphics for all devices – from smartphones to workstations. Bioinformatics. 2014;30:2981–2982. doi: 10.1093/bioinformatics/btu426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laskowski R.A., Rullmannn J.A., MacArthur M.W., Kaptein R., Thornton J.M. AQUA and PROCHECK-NMR: programs for checking the quality of protein structures solved by NMR. J. Biomol. NMR. 1996;8:477–486. doi: 10.1007/BF00228148. [DOI] [PubMed] [Google Scholar]
- Lipinski C.A. Lead- and drug-like compounds: the rule-of-five revolution. Drug Discov. Today Technol. 2004;1:337–341. doi: 10.1016/j.ddtec.2004.11.007. [DOI] [PubMed] [Google Scholar]
- Matsuda T., Ito T., Takemoto C., Katsura K., Ikeda M., Wakiyama M., Kukimoto-Niino M., Yokoyama S., Kurosawa Y., Shirouzu M. Cell-free synthesis of functional antibody fragments to provide a structural basis for antibody-antigen interaction. PLoS One. 2018;13:e0193158. doi: 10.1371/journal.pone.0193158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mondal S., Bandyopadhyay S., Ghosh M.K., Mukhopadhyay S., Roy S., Mandal C. Natural products: promising resources for cancer drug discovery. Anticancer Agents Med. Chem. 2012;12:49–75. doi: 10.2174/187152012798764697. [DOI] [PubMed] [Google Scholar]
- Morris, G.M., Huey, R., Olson, A.J. 2008. Using AutoDock for ligand-receptor docking. Curr. Protoc. Bioinformatics. Unit 8 14 (Chapter 8). [DOI] [PubMed]
- Ohori H., Yamakoshi H., Tomizawa M., Shibuya M., Kakudo Y., Takahashi A., Takahashi S., Kato S., Suzuki T., Ishioka C., Iwabuchi Y., Shibata H. Synthesis and biological analysis of new curcumin analogues bearing an enhanced potential for the medicinal treatment of cancer. Mol. Cancer Ther. 2006;5:2563–2571. doi: 10.1158/1535-7163.MCT-06-0174. [DOI] [PubMed] [Google Scholar]
- Park W., Amin A.R., Chen Z.G., Shin D.M. New perspectives of curcumin in cancer prevention. Cancer Prev. Res. (Phila) 2013;6:387–400. doi: 10.1158/1940-6207.CAPR-12-0410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Priya R., Sumitha R., Doss C.G., Rajasekaran C., Babu S., Seenivasan R., Siva R. Molecular docking and molecular dynamics to identify a novel human immunodeficiency virus inhibitor from alkaloids of Toddalia asiatica. Pharmacogn. Mag. 2015;11:S414–422. doi: 10.4103/0973-1296.168947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pronk S., Pall S., Schulz R., Larsson P., Bjelkmar P., Apostolov R., Shirts M.R., Smith J.C., Kasson P.M., van der Spoel D., Hess B., Lindahl E. GROMACS 4.5: a high-throughput and highly parallel open source molecular simulation toolkit. Bioinformatics. 2013;29:845–854. doi: 10.1093/bioinformatics/btt055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Red Brewer M., Choi S.H., Alvarado D., Moravcevic K., Pozzi A., Lemmon M.A., Carpenter G. The juxtamembrane region of the EGF receptor functions as an activation domain. Mol. Cell. 2009;34:641–651. doi: 10.1016/j.molcel.2009.04.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salim E.I., Jazieh A.R., Moore M.A. Lung cancer incidence in the arab league countries: risk factors and control. Asian Pac. J. Cancer Prev. 2011;12:17–34. [PubMed] [Google Scholar]
- Sato T., Watanabe H., Tsuganezawa K., Yuki H., Mikuni J., Yoshikawa S., Kukimoto-Niino M., Fujimoto T., Terazawa Y., Wakiyama M., Kojima H., Okabe T., Nagano T., Shirouzu M., Yokoyama S., Tanaka A., Honma T. Identification of novel drug-resistant EGFR mutant inhibitors by in silico screening using comprehensive assessments of protein structures. Bioorg. Med. Chem. 2012;20:3756–3767. doi: 10.1016/j.bmc.2012.04.042. [DOI] [PubMed] [Google Scholar]
- Schuttelkopf A.W., van Aalten D.M. PRODRG: a tool for high-throughput crystallography of protein-ligand complexes. Acta Crystallogr. D Biol. Crystallogr. 2004;60:1355–1363. doi: 10.1107/S0907444904011679. [DOI] [PubMed] [Google Scholar]
- Schyman P., Liu R., Desai V., Wallqvist A. vNN Web Server for ADMET Predictions. Front. Pharmacol. 2017;8:889. doi: 10.3389/fphar.2017.00889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma S.V., Bell D.W., Settleman J., Haber D.A. Epidermal growth factor receptor mutations in lung cancer. Nat. Rev. Cancer. 2007;7:169–181. doi: 10.1038/nrc2088. [DOI] [PubMed] [Google Scholar]
- Shigematsu H., Lin L., Takahashi T., Nomura M., Suzuki M., Wistuba I.I., Fong K.M., Lee H., Toyooka S., Shimizu N., Fujisawa T., Feng Z., Roth J.A., Herz J., Minna J.D., Gazdar A.F. Clinical and biological features associated with epidermal growth factor receptor gene mutations in lung cancers. J. Natl. Cancer Inst. 2005;97:339–346. doi: 10.1093/jnci/dji055. [DOI] [PubMed] [Google Scholar]
- Starok M., Preira P., Vayssade M., Haupt K., Salome L., Rossi C. EGFR inhibition by curcumin in cancer cells: a dual mode of action. Biomacromolecules. 2015;16:1634–1642. doi: 10.1021/acs.biomac.5b00229. [DOI] [PubMed] [Google Scholar]
- Stella G.M., Luisetti M., Inghilleri S., Cemmi F., Scabini R., Zorzetto M., Pozzi E. Targeting EGFR in non-small-cell lung cancer: lessons, experiences, strategies. Respir. Med. 2012;106:173–183. doi: 10.1016/j.rmed.2011.10.015. [DOI] [PubMed] [Google Scholar]
- Stella G.M., Scabini R., Inghilleri S., Cemmi F., Corso S., Pozzi E., Morbini P., Valentini A., Dore R., Ferrari S., Luisetti M., Zorzetto M. EGFR and KRAS mutational profiling in fresh non-small cell lung cancer (NSCLC) cells. J. Cancer Res. Clin. Oncol. 2013;139:1327–1335. doi: 10.1007/s00432-013-1444-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stewart E.L., Tan S.Z., Liu G., Tsao M.S. Known and putative mechanisms of resistance to EGFR targeted therapies in NSCLC patients with EGFR mutations-a review. Transl. Lung Cancer Res. 2015;4:67–81. doi: 10.3978/j.issn.2218-6751.2014.11.06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun X.D., Liu X.E., Huang D.S. Curcumin induces apoptosis of triple-negative breast cancer cells by inhibition of EGFR expression. Mol. Med. Rep. 2012;6:1267–1270. doi: 10.3892/mmr.2012.1103. [DOI] [PubMed] [Google Scholar]
- Vyas A., Dandawate P., Padhye S., Ahmad A., Sarkar F. Perspectives on new synthetic curcumin analogues and their potential anticancer properties. Curr. Pharm. Des. 2013;19:2047–2069. [PMC free article] [PubMed] [Google Scholar]
- Wang H., Liu X., Rice S.J., Belani C.P. Pulmonary rehabilitation in lung cancer. PM R. 2016;8:990–996. doi: 10.1016/j.pmrj.2016.03.010. [DOI] [PubMed] [Google Scholar]
- Webb, B., Sali, A. 2016. Comparative protein structure modeling using modeller. Curr. Protoc. Bioinformatics 54, 5 6 1–5 6 37. [DOI] [PMC free article] [PubMed]
- Wiederstein M., Sippl M.J. ProSA-web: interactive web service for the recognition of errors in three-dimensional structures of proteins. Nucleic Acids Res. 2007;35:W407–410. doi: 10.1093/nar/gkm290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan S., Chan H.C.S., Filipek S., Vogel H. PyMOL and Inkscape bridge the data and the data visualization. Structure. 2016;24:2041–2042. doi: 10.1016/j.str.2016.11.012. [DOI] [PubMed] [Google Scholar]
- Zhang Z., Lee J.C., Lin L., Olivas V., Au V., LaFramboise T., Abdel-Rahman M., Wang X., Levine A.D., Rho J.K., Choi Y.J., Choi C.M., Kim S.W., Jang S.J., Park Y.S., Kim W.S., Lee D.H., Lee J.S., Miller V.A., Arcila M., Ladanyi M., Moonsamy P., Sawyers C., Boggon T.J., Ma P.C., Costa C., Taron M., Rosell R., Halmos B., Bivona T.G. Activation of the AXL kinase causes resistance to EGFR-targeted therapy in lung cancer. Nat. Genet. 2012;44:852–860. doi: 10.1038/ng.2330. [DOI] [PMC free article] [PubMed] [Google Scholar]