

Abstract
This article is one of ten reviews selected from the Annual Update in Intensive Care and Emergency Medicine 2019. Other selected articles can be found online at https://www.biomedcentral.com/collections/annualupdate2019. Further information about the Annual Update in Intensive Care and Emergency Medicine is available from http://www.springer.com/series/8901.
Introduction
Since the first consensus definition of sepsis almost three decades ago [1], our understanding of the clinical characteristics that prognosticate the outcome of this complex syndrome has improved [2], resulting in a simpler classification scheme [3]. The existing definitions, however, remain imprecise and the clinical diagnosis of sepsis corresponds poorly with post hoc presence of infection [4]. Furthermore, the outcome of sepsis depends on factors beyond patient signs and symptoms [5], including age [6], the infection source [7], and the timing and appropriateness of therapeutic interventions [8] (Fig. 1). There is currently a promising shift from predicting outcome to a pathobiology-driven understanding of the heterogeneity in the host response to sepsis, utilizing novel translational high throughput tools and analytic methods to define distinct host response subgroups. It is now well recognized that biological markers improve the classification of sepsis and can facilitate identification of distinct patient subclasses, or endotypes.
Fig. 1.
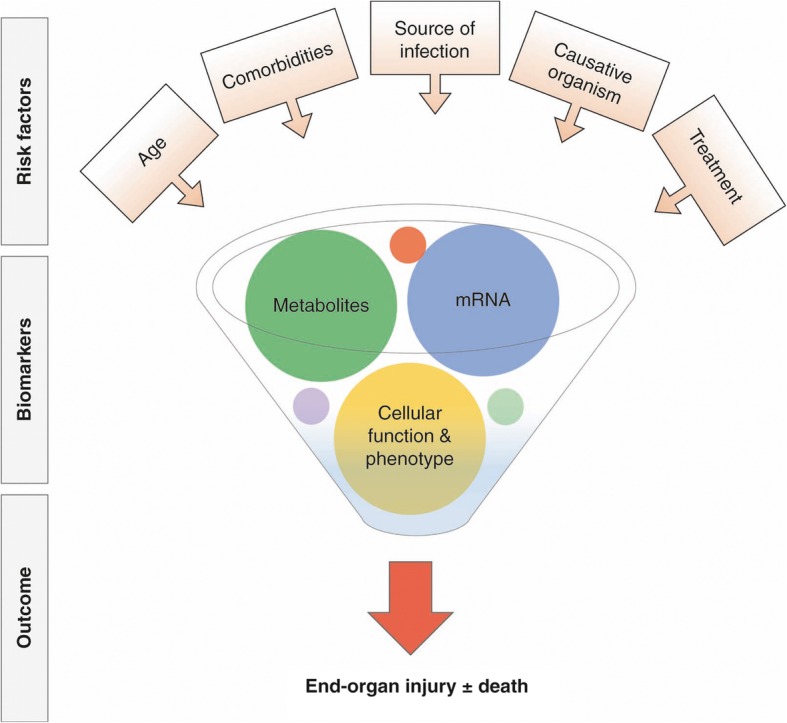
Heterogeneity in critically ill patients with severe infection
Both excessive immune activation [9] and immunosuppression [10] are central to the pathophysiology of sepsis [11]. A clinical measure of immunosuppression is the acquisition of nosocomial infection. However, the development of intensive care unit (ICU)-acquired infection is not unique to patients with sepsis and, in fact, its incidence is comparable to patients admitted to the ICU without infectious conditions [12]. This finding suggests that mechanisms other than the immune response to infection are also contributory, with the key target pathways being (1) endothelial activation, (2) coagulopathy and (3) altered glucose and protein metabolism. Endotypes with derangements in all of these pathways have been identified, with divergent outcomes and differential response to therapies.
In this chapter, we highlight some of the most relevant recent advances in translational biology that assist in deconvoluting heterogeneity in patient response to sepsis. We focus on the use of molecular and metabolomic signatures as well as novel cellular function studies to identify distinct sepsis endotypes (Table 1). Identification of unique biological signatures in patients with sepsis could enable rational enrollment into clinical trials and, more importantly, enhance our approach to the diagnosis, prognosis, as well as individualized treatment to modulate the response to sepsis.
Table 1.
Cohort studies of pathobiology-driven patient phenotypic classification
| Endotypes | Cohort type (n) | Key biomarkers |
|---|---|---|
| Transcriptomic profiling | ||
| Subclass A–C [13] | Pediatric septic shock (98) | 44-gene classifier |
| SRS1 and SRS2 [14] | CAP (265) | 7-gene classifier: IDYRK2, DDNB1IP1, TDRD9, ZAP70, ARL14EP, MDC1 ADGRE3 |
| SRS1_FP and SRS2_FP [15] | Fecal peritonitis (117) | 6-gene classifier: CD163, CDHHC19, MME, FAM89A, ZBP1, B3GNT2 |
| Mars1–4 [16] | All-cause sepsis (306) | 2-gene ratios: Mars1: BPGM:TAP2, Mars2: GADD45A:PCGF5, Mars3: AHNAK:PDCD10, Mars4: IFIT5:GLTSCR2 |
| Inflammopathic, adaptive, coagulopathic [17] | Bacterial sepsis (700) | 33-gene classifier |
| Neutrophil pathways [18] | Sepsis with (29) and without ARDS (28) | OLFM4, LCN2 2, CD24, BPI |
| Metabolomics | ||
| Energy metabolism [27] | Septic shock (39), ICU controls (20) | ↑Glucose, 3-hydroxybutyrate, O-acetylcarnitine, succinate, creatine, creatine phosphate ↓Branched-chain amino acids and arginine |
| Lipid homeostasis and tryptophan catabolism [28] | Septic shock (20) | ↓Unsaturated long-chain PC and LPC, kynurenine |
| Defect in fatty acid-β-oxidation [29] | Community-acquired sepsis survivors (119) and nonsurvivors (31) | Acylcarnitine esters, amino/nucleic acid catabolites, glycolysis/citric acid cycle components |
| Lipid metabolism [30] | SIRS (29), Sepsis (30), sepsis-induced ARDS (31) | γ-glutamylphenylalanine, γ-glutamyltyrosine, 1-arachidonoyl-GPC (20:4), taurochenodeoxycholate, 3-(4-hydroxyphenyl) lactate, sucrose, kynurenine |
| Lipid metabolism [31] | SIRS (42), CAP (67), IAI (60), UTI (73) BSI (26) | SM C22:3 and, lysoPCaC24:0, lysoPCaC26:1, putrescine, lysoPCaC18:0, SM C16:1 |
| Cellular function | ||
| Immunophenotype [34] | Sepsis (148) | Neutrophil, monocyte, and Treg phenotypes |
| Immunophenotype [35] | Sepsis (22) | Lymphocyte PD-1/PD-L1 expression |
| Immunophenotype [36] | Sepsis (505) | Monocyte and CD3, CD4, CD8 Tcell phenotypes |
| Endothelial permeability [37] | Sepsis (35) | Supernatants from whole blood treated with LPS |
| Endothelial permeability [38] | Primary (12) and secondary (6) sepsis | Neutrophils treated with fMLP |
CAP community-acquired pneumonia, PC phosphatidylcholines, LPC lysophosphatidylcholines, IAI intraabdominal infection, UTI urinary tract infection, BSI bloodstream infection, PD programmed death, LPS lipopolysaccharide, fMLP formyl, methionyl-leucyl-phenylalanine, SIRS systemic inflammatory response syndrome
Transcriptomic profiling
Interindividual transcriptome variation in sepsis has been recently evaluated in several large cohorts, with a dysfunctional immune response phenotype being a common theme. Clinical samples for these studies include peripheral blood leukocytes obtained within 24 h of ICU admission from patients with definite or probable infection. Unsupervised hierarchical clustering of approximately 25,000 genome-wide transcriptomic (gene expression microarray and RNA sequencing) profiles is then applied to identify distinct subgroups of patients in a derivation cohort and subsequently the findings are verified in at least one validation cohort. These sophisticated data-driven methods have identified patterns among expressed genes that define molecular subgroups representing different disease states without reference to clinical outcomes, but which could be associated to them. This approach could also define clusters indicative of the individual’s premorbid state (age, comorbidities), illness stage and severity, mortality and genetic predisposition. However, subgroup membership cannot be distinguished on the basis of these clinical characteristics, as was the case for each of the studies summarized later.
One of the first studies to use unsupervised hierarchical clustering to study sepsis subgroups within the ICU population was done in a cohort of 98 children admitted to pediatric ICUs in the United States with septic shock [13]. Three subclasses were identified by differential genome-wide expression patterns: endotype A (29%), endotype B (46%), and endotype C (26%). These three classes differed significantly in clinical phenotypes including ICU mortality (highest in endotype A at 36% relative to 11 and 12% in endotypes B and C, respectively), illness severity and degree of organ failure (both highest in endotype A), and age (youngest in endotype A). The subclasses were also biologically plausible, with the most deranged signaling pathways in endotype A involving repression of genes key to the adaptive immune system, glucocorticoid receptor signaling, as well as to zinc homeostasis.
Similar methodology has recently been applied to the adult critically ill population. Among 265 patients admitted to 29 ICUs in the UK with sepsis due to community-acquired pneumonia (CAP) as part of the Genomic Advances in Sepsis (GAinS) study, transcriptomic profiles defined two sepsis response signatures (SRS1, 41% and SRS2, 59%) [14]. Relative to SRS2 patients, patients in the SRS1 group had a higher 14-day mortality (22% vs. 10%). SRS1 assignment was associated with relative immunosuppression, endotoxin-tolerance, T-cell exhaustion, human leukocyte antigen (HLA) class II downregulation, and metabolic derangements (switch from oxidative phosphorylation to glycolysis). Of the over 3000 differentially expressed genes, seven genes reliably predicted SRS membership. In future studies, patients prospectively assigned into SRS1 may benefit from therapies that boost their immune system and prevent nosocomial infection.
This analytical approach was subsequently used by the same investigators to study the gene expression patterns in 117 patients with fecal peritonitis (FP) [15]. Two distinct groups were again identified (SRS1_FP, 46% and SRS2_FP, 54%), with patients in the SRS1_FP group also having a higher 14-day mortality (19% vs. 4%). The findings were strongly correlated with the SRS groups identified in the CAP study [14], again showing enrichment of endotoxin tolerance and T-cell activation but also in cell death, apoptosis and necrosis. Of the over 1000 differentially expressed genes, a simpler six-gene set was derived that predicted group membership. Of note, when gene expression patterns that distinguished SRS groups were tested in the pediatric cohort described above [13], the same enrichment was not observed (i.e., SRS1_FP [15] and endotype A [13]).
In a group of 306 patients admitted to two ICUs in the Netherlands as part of the Molecular Diagnosis and Risk Stratification of Sepsis (MARS) project, four molecular endotypes (Mars1–4) were identified [16]. Mortality at 28 days differed among the subgroups and was highest in the Mars1 group at 39%, compared to 22% in Mars2, 23% in Mars3 and 33% in Mars4. The Mars1 poor-prognosis endotype had a decrease in expression of genes involved in innate and adaptive immune functions (Toll-like receptor, nuclear factor-κB signaling [NF-κB], antigen presentation, and T-cell receptor signaling) and an increase in expression of cellular metabolic pathways (heme biosynthesis), processes which are both analogous to immune exhaustion. Mars2 and 4 endotype had upregulation of pattern recognition and cytokine pathways (interleukin [IL]-6, NF-κB, interferon signaling, inducible nitric oxide synthase), representing a hyperinflammatory state. Finally, Mars3 was a lower-risk endotype with increased expression of adaptive immune pathways (T-helper cells, natural killer cells, IL-4 signaling, B-cell development), which was highly correlated with the low-risk SRS2 endotype [14]. A two-gene expression ratio was derived to enable classification of each endotype at the time of ICU admission.
In the most recent and comprehensive attempt to identify sepsis subtypes, data from 14 transcriptomic datasets consisting of 700 patients revealed three robust host response clusters across the sepsis spectrum [17]. These were termed: (1) inflammopathic (increased innate and reduced adaptive immune signal marked by increased expression of IL-1 receptor, pattern recognition receptor activity, complement activation); (2) adaptive (reduced innate and high adaptive immune signal with lower mortality, marked by interferon signaling); and (3) coagulopathic (irregularities in the coagulation and complement systems, including platelet degranulation and glycosaminoglycan binding). Similar to the previous analyses, the three groups differed in 30-day mortality, with the highest mortality in the inflammopathic group at 30%, compared to 8% in the adaptive and 25% in the coagulopathic groups. A simplified 33-gene classifier was derived to facilitate cluster assignment. The assignment into the high-mortality inflammopathic cluster corresponded to SRS1 and the low-mortality adaptive cluster to SRS2 [14].
Early transcriptional changes may also have identified patients at risk of sepsis-associated complications at the time of ICU admission. One study from our research group that investigated 57 patients with sepsis found that the differential expression of key mediators of the initial neutrophil response to infection identified patients with acute respiratory distress syndrome (ARDS, n = 29) compared to those with sepsis who did not have ARDS (n = 28), a finding that could not be attributed to the neutrophil count [18].
To date, genome-wide expression studies in sepsis used whole leukocyte populations. However, distinct gene expression patterns are present among subsets of granulocytes and lymphocytes which represent the specialized function of each of these immune cells [19]. Since the transcriptome profile depends on the inflammatory cell type, it is possible that gene expression patterns that distinguish subclasses reflect different leukocyte populations instead of within-cell differences in gene expression. These findings also require validation in large cohorts spanning different countries as variation in ethnic background is a strong determinant of gene expression [20].
Nevertheless, these studies provide evidence of distinct categories of the host response to sepsis and potential novel therapeutic targets based on differentially expressed molecular pathways that distinguish patient endotypes. Furthermore, each study proposed potential ‘downsizing’ of the high-dimensional data into manageable predictive signals that could be incorporated into a simpler point-of-care test, assisting in translating the findings to the bedside.
mRNA and protein signatures
A more established and more feasible method of biological subclassification of patients with sepsis is plasma protein quantification and a vast number of studies have classified sepsis using this approach [21–23], which is beyond the scope of this review. A noteworthy method that could offer a novel way to derive sepsis subclasses is combining molecular and protein biomarkers to predict outcome in patients with septic shock. This approach was used to risk stratify pediatric septic shock using a previously validated risk score consisting of five plasma protein biomarkers (PERSEVERE decision tree) [23] and combining these with four top mortality assessment genes [24]. An improvement was noted in the performance of the risk score estimating the risk of 28-day mortality (PERSEVERE-XP, area under the receiver operating characteristic [AUROC] curve increase from 0.78 to 0.91). The plasma biomarkers were associated with dysfunctional inflammation and cellular injury whereas the genes were related to the tumor protein 53 (TP53, p53), a transcriptional factor functioning as a tumor suppressor, preventing the generation and persistence of cells with genomic damage. Taken together, this approach offers a plausible hypothesis regarding biological pathways that result in a poor outcome due to septic shock.
Metabolomics
Metabolomics is an expanding and less familiar method to decipher heterogeneity in sepsis. It refers to the global assessment of small metabolites in any biological sample, representing a composite ‘snapshot’ of gene expression, enzyme activity, and the physiological landscape [25]. More than 5000 metabolites can be detected in cells, tissues, or biofluids (blood components, urine) using nuclear magnetic resonance (NMR) spectroscopy or mass spectrometry, the latter of which is more sensitive and can detect low-abundant metabolites [26]. The metabolites can include both endogenous (lipids, carbohydrates, amino acids, nucleic acids) and exogenous (microbial components and byproducts) compounds. Alteration in endogenous metabolite concentration can be linked to biological pathways and the magnitude of change relates to the stage of illness, significantly magnifying transcriptome and proteome-level shifts. Studies so far included retrospective specimen collection with small sample sizes.
1H NMR spectroscopy was used to analyze and compare serum samples derived from adults with septic shock and from ICU controls [27]. Sixty metabolites were recognized, 31 of which could distinguish between septic shock and ICU control patients, proposing a composite biomarker pattern that could differentiate between these patient groups. The metabolites were involved in energy metabolism and included glucose, 3-hydroxybutyrate, O-acetylcarnitine, succinate, creatine, creatine phosphate as well a decreased level of branched-chain amino acids and arginine. These results suggest that in early sepsis, metabolites involved in energy metabolism have a role in the pathophysiology of sepsis.
Alteration in metabolites involved in energy metabolism has also been recognized as key in distinguishing sepsis survivors from non-survivors using mass spectrometry to characterize lower concentration metabolites in plasma. In a substudy of the ALBIOS (Albumin Italian Outcome Sepsis) trial which enrolled 1818 patients with severe sepsis or septic shock, day 1 and 7 plasma samples were studied in 20 patients, 45% of whom died by day 28 [28]. The study identified 137 metabolites, many of which were significantly different between survivors and non-survivors. The most notable group of metabolites included a decrease in phosphatidylcholines and lysophosphatidylcholines as well as an increase in kynurenine. This decline in lipid species, particularly long-chain polyunsaturated fatty acids, may lead to increased T-cell activation and an excessive immune response.
The plasma metabolome was similarly studied using mass spectrometry in a subset of 150 patients who were among 1152 individuals with suspected sepsis enrolled in the Community Acquired Pneumonia and Sepsis Outcome Diagnostics (CAPSOD) study [29]. Of the 439 metabolites analyzed, 214 were detected at both day 0 (t0) and at 24 h (t24). Metabolites did not differ at either time point among infectious etiologies (Streptococcus pneumoniae, Staphylococcus aureus or Escherichia coli). There were 76 metabolites at t0 and 128 metabolites at t24 that differed between sepsis survivors and non-survivors at 28 days. Acylcarnitine esters of all fatty acid length (medium- or short-chain) and branched-chain amino acid biochemical group differences were most pronounced between the two patient groups, suggesting that a defect in fatty acid-β-oxidation may occur at the level of the carnitine shuttle. Metabolites comprising fatty acid transport, gluconeogenesis and the citric acid cycle were also differentially deranged.
Another large cohort that used mass spectrometry for metabolomic profiling to study metabolite biomarkers in 60 ICU survivors and 30 ICU non-survivors found that of the 187 metabolites tested, 57 were associated with 28-day mortality [30] and 31 of them were replicated in the CAPSOD validation cohort [29]. These metabolites included diverse lipid, carbohydrate, amino acid and nucleotide products. Higher levels of tyrosine and phenylalanine catabolism products and lower levels of lipid metabolites were associated with mortality. Although this study used the same metabolomic datasets as the previously described study [29], a completely different network of metabolites was identified, which was also predictive of 28-day mortality, implying it is premature to focus on a single metabolite biomarker as several could be implicated in the pathobiology of different stages of sepsis.
The largest study to investigate metabolites using mass spectrometry in patients with sepsis included 406 patients, 268 of whom were included in a discovery cohort (42 patients with systemic inflammatory response syndrome [SIRS], 67 with CAP, 60 with intraabdominal infection, 73 with urinary tract infection and 26 with bloodstream infection) [31]. Again, acylcarnitines and lipids were altered in sepsis relative to SIRS. A sphingolipid SM C22:3 and glycerophospholipid lysoPCaC24:0 model discriminated between these two entities with an AUROC of 0.9. The analysis also indicated great heterogeneity in metabolite patterns depending on the anatomic source of infection and thus a one metabolite model was proposed to prognosticate unfavorable outcome for each infection type.
Cellular function
While molecular, protein and metabolomic biomarkers provide associations between sepsis and its outcome, they cannot determine causality and mechanistic studies linking these associations to sepsis pathobiology are required. Two potentially high throughput techniques available to study ex vivo relationships between cell function and sepsis pathobiology include flow cytometry [32] and electric cell-substrate impedance sensing (ECIS) [33].
Immunophenotyping involves the use of flow cytometry and fluorescent-labeled monoclonal antibodies to simultaneously label multiple cell surface markers, such as those associated with immune dysfunction. In a group of 138 ICU patients recruited from four ICUs in the UK, leukocyte dysfunction defined by a combination of reduced neutrophil CD88 and monocyte HLA-DR as well as an elevated proportion of regulatory T cells (CD4+, CD25++, CD127low) was associated with the development of nosocomial infection [34]. Lymphocyte dysfunction is also present in sepsis and can be quantified by the expression levels of programmed death protein 1 (PD1) and its ligand PDL1, which promote apoptosis. When peripheral blood mononuclear cells from 22 patients with sepsis were compared to healthy controls, both PD1 and PDL1 were higher on all lymphocyte subsets (CD4 T cells and B cells) in patients with sepsis [35]. Similar to transcriptome and metabolome analyses in sepsis, the early innate and adaptive immune status also vary according to infection type [36]. Therefore, the identification of patients with a prespecified source of infection who have an immunophenotype amendable to immunomodulatory therapy could allow for precision medicine-guided therapy.
The endothelium is one of the primary targets in sepsis. Measuring endothelial cell function ex vivo is challenging but our research group developed a novel assay (ECIS) that delineates the heterogeneity in endothelial cell response after exposure to different patient-derived samples. This assay is based on movement of current across a monolayer of endothelial cells, with a decrease in resistance indicative of an increase in vascular permeability. When pulmonary endothelial cells were exposed to lipopolysaccharide (LPS)-stimulated leukocyte supernatants derived from 35 ICU patients with sepsis, substantial heterogeneity in pulmonary endothelial permeability was observed [37]. The same method was used to test the ability of neutrophils from septic patients to induce endothelial damage, demonstrating that neutrophils from septic patients with compared to those without ARDS can induce greater endothelial damage [38]. This in vitro model of vascular permeability may be useful for testing therapeutic agents that could mitigate endothelial injury in early sepsis.
Challenges and future directions
Distinguishing consistent biological heterogeneity in sepsis will necessitate overcoming several technical hurdles. Inclusion criteria ought to be uniform across sites to minimize patient selection bias. The timing of sample collection is critical as endotype assignment is a dynamic process and nearly 50% of patients cross over from one endotype to another over the first 5 days of ICU admission, as demonstrated by serial sampling on sequential days [15]. Similarly, the duration of altered gene expression can vary between patients, and tends to normalize quicker in patients who recover faster [39], emphasizing that standardizing sample collection timing is crucial. Data collection will also need to be standardized to include common clinically meaningful outcomes as studies to date use ICU, hospital, 14-, 28-, 30-, or 90-day mortality. These outcomes represent different endpoints which could be measuring different biological processes. Early deaths are more likely to be directly attributed to the initial episode of sepsis whereas late deaths may represent complications of sepsis beyond nosocomial infections [40]. In the analysis phase, standardizing analytical methods will be important to determine whether sepsis can be categorized into two, three, four or potentially more endotypes.
It is plausible that the host response to sepsis may be nonspecific and could be elicited to different organisms, which invade different organs. Although there is evidence that there may be a shared host response at the transcriptome and metabolome level irrespective of the infection type (Gram-positive sepsis and Gram-negative sepsis) [29, 41] or the anatomic source of infection [15], the studies to date investigating these questions have been relatively small and when pooled data are used [42], there is a host gene expression signature that can discriminate sterile inflammation from bacterial or viral infections. For this reason, the largest transcriptomic analysis to date restricted analysis to only bacterial sepsis [17]. Also, when gene expression data for all-cause sepsis are re-analyzed including patients with only pneumonia and peritonitis, the two most common anatomic sources of infection [7], the proportion of patients assigned to an endotype varies depending on the infection source [16]. Metabolites also vary based on infection source, with CAP having a different metabolite pattern relative to other sites of infection [31]. A recent analysis of the plasma metabolome in H1N1 pneumonia successfully differentiated viral from bacterial culture-positive pneumonia and ventilated ICU controls [43]. Therefore, whether sepsis endotypes are truly independent of infection type and anatomical source will require large-scale prospective cohort studies with enough power to address this question.
In the future, it may be possible to treat distinct manifestations of the host response to sepsis based on signatures representing distinct biological pathways (Fig. 2). Assignment into endotypes could facilitate targeting appropriate therapies to the patient group with a pathway derangement endotype that would most benefit. It would also enable appropriate selection into clinical trials investigating pathway-driven therapeutics that to date have been unsuccessful largely due to the inclusion of a heterogenous group of patients with sepsis.
Fig. 2.
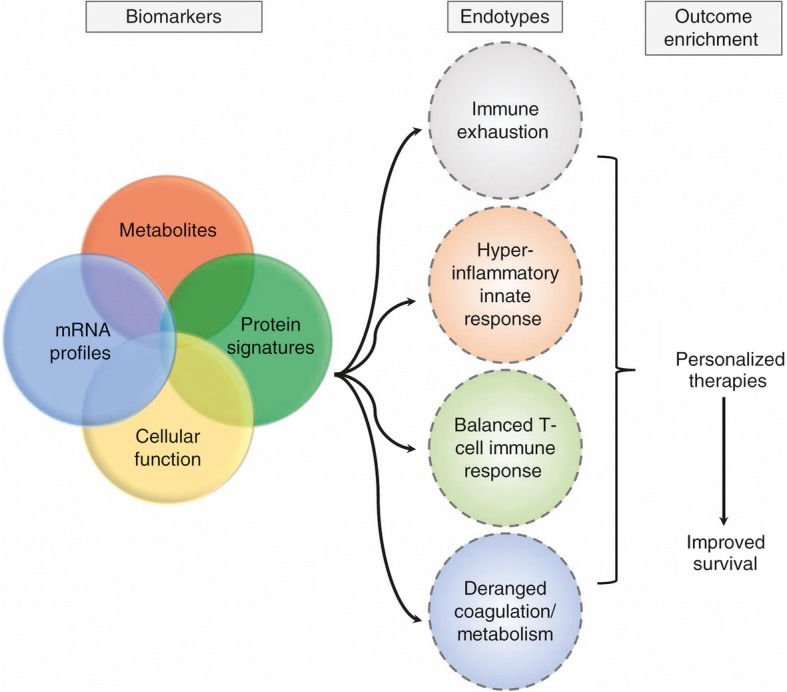
Clustering into pathobiology-driven endotypes
Validation in large, multicenter, and diverse cohorts is needed prior to transitioning from exploration and discovery to testing candidate mRNA, protein, and metabolite models. An overarching future collaborative study aim should include prospective validation in large cohorts to determine whether pathobiology biomarker-driven identification of sepsis endotype at the time of ICU admission can improve clinical outcomes and personalization of treatment.
Conclusion
Applying bioinformatics to integrate systems biology (transcriptomics, proteomics, metabolomics) in combination with functional cellular studies has the potential to identify biological endotypes, which cannot be predicted using clinical covariates alone. Optimizing these novel translational methods will require collaboration, expertise and standardization in patient sample collection, assay performance and data analysis. It is possible that the true nature of the heterogeneity of the host response to sepsis will require a combination of molecular, protein, metabolomic and functional signatures that will lead to an integrated, simple, and clinically useful diagnostic model that could be rapidly used at the time of ICU admission. Hopefully, a parsimonious set of biological markers will be useful to categorize patients into specific sub-groups that would be useful for testing specific new therapies. Ultimately, detection of key biological markers along with clinical indicators at the bedside could improve our approach to precision medicine-guided therapy and outcomes of patients with sepsis.
Acknowledgements
None.
Funding
Publication costs were funded by Dr. Matthay’s NHLBI grant (NHLBI HL134828).
Availability of data and materials
Not applicable.
Authors’ contributions
AL and MAM conceived, prepared, and revised the review. AL and MAM have read and approved the final version of the manuscript.
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Bone RC, Balk RA, Cerra FB, et al. Definitions for sepsis and organ failure and guidelines for the use of innovative therapies in sepsis. Chest. 1992;101:1644–1655. doi: 10.1378/chest.101.6.1644. [DOI] [PubMed] [Google Scholar]
- 2.Gotts JE, Matthay MA. Sepsis: pathophysiology and clinical management. BMJ. 2016;353:i1585. doi: 10.1136/bmj.i1585. [DOI] [PubMed] [Google Scholar]
- 3.Shankar-Hari M, Phillips GS, Levy ML, et al. Developing a new definition and assessing new clinical criteria for septic shock: for the third international consensus definitions for Sepsis and septic shock (Sepsis-3) JAMA. 2016;315:775–787. doi: 10.1001/jama.2016.0289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Klein Klouwenberg PM, Cremer OL, van Vught LA, et al. Likelihood of infection in patients with presumed sepsis at the time of intensive care unit admission: a cohort study. Crit Care. 2015;19:319. doi: 10.1186/s13054-015-1035-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Vincent JL, Opal SM, Marshall JC, Tracey KJ. Sepsis definitions: time for change. Lancet. 2013;381:774–775. doi: 10.1016/S0140-6736(12)61815-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Brakenridge SC, Efron PA, Stortz JA, et al. The impact of age on the innate immune response and outcomes after severe sepsis/septic shock in trauma and surgical intensive care unit patients. J Trauma Acute Care Surg. 2018;85:247–255. doi: 10.1097/TA.0000000000001921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Leligdowicz A, Dodek PM, Norena M, et al. Association between source of infection and hospital mortality in patients who have septic shock. Am J Respir Crit Care Med. 2014;189:1204–1213. doi: 10.1164/rccm.201310-1875OC. [DOI] [PubMed] [Google Scholar]
- 8.Ferrer R, Martinez ML, Goma G, et al. Improved empirical antibiotic treatment of sepsis after an educational intervention: the ABISS-Edusepsis study. Crit Care. 2018;22:167. doi: 10.1186/s13054-018-2091-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Delano MJ, Ward PA. The immune system’s role in sepsis progression, resolution, and long-term outcome. Immunol Rev. 2016;274:330–353. doi: 10.1111/imr.12499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Boomer JS, To K. Chang KC, et al. Immunosuppression in patients who die of sepsis and multiple organ failure. JAMA. 2011;306:2594–2605. doi: 10.1001/jama.2011.1829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hotchkiss RS, Monneret G, Payen D. Sepsis-induced immunosuppression: from cellular dysfunctions to immunotherapy. Nat Rev Immunol. 2013;13:862–874. doi: 10.1038/nri3552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.van Vught LA, Klein Klouwenberg PM, Spitoni C, et al. Incidence, risk factors, and attributable mortality of secondary infections in the intensive care unit after admission for sepsis. JAMA. 2016;315:1469–1479. doi: 10.1001/jama.2016.2691. [DOI] [PubMed] [Google Scholar]
- 13.Wong HR, Cvijanovich N, Lin R, et al. Identification of pediatric septic shock subclasses based on genome-wide expression profiling. BMC Med. 2009;7:34. doi: 10.1186/1741-7015-7-34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Davenport EE, Burnham KL, Radhakrishnan J, et al. Genomic landscape of the individual host response and outcomes in sepsis: a prospective cohort study. Lancet Respir Med. 2016;4:259–271. doi: 10.1016/S2213-2600(16)00046-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Burnham KL, Davenport EE, Radhakrishnan J, et al. Shared and distinct aspects of the sepsis transcriptomic response to fecal peritonitis and pneumonia. Am J Respir Crit Care Med. 2017;196:328–339. doi: 10.1164/rccm.201608-1685OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Scicluna BP, van Vught LA, Zwinderman AH, et al. Classification of patients with sepsis according to blood genomic endotype: a prospective cohort study. Lancet Respir Med. 2017;5:816–826. doi: 10.1016/S2213-2600(17)30294-1. [DOI] [PubMed] [Google Scholar]
- 17.Sweeney TE, Azad TD, Donato M, et al. Unsupervised analysis of transcriptomics in bacterial sepsis across multiple datasets reveals three robust clusters. Crit Care Med. 2018;46:915–925. doi: 10.1097/CCM.0000000000003084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kangelaris KN, Prakash A, Liu KD, et al. Increased expression of neutrophil-related genes in patients with early sepsis-induced ARDS. Am J Phys Lung Cell Mol Phys. 2015;308:L1102–L1113. doi: 10.1152/ajplung.00380.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Palmer C, Diehn M, Alizadeh AA, Brown PO. Cell-type specific gene expression profiles of leukocytes in human peripheral blood. BMC Genomics. 2006;7:115. doi: 10.1186/1471-2164-7-115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Spielman RS, Bastone LA, Burdick JT, Morley M, Ewens WJ, Cheung VG. Common genetic variants account for differences in gene expression among ethnic groups. Nat Genet. 2007;39:226–231. doi: 10.1038/ng1955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hou PC, Filbin MR, Wang H, et al. Endothelial permeability and hemostasis in septic shock: results from the ProCESS trial. Chest. 2017;152:22–31. doi: 10.1016/j.chest.2017.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.van Vught LA, Wiewel MA, Hoogendijk AJ, et al. The host response in patients with sepsis developing intensive care unit-acquired secondary infections. Am J Respir Crit Care Med. 2017;196:458–470. doi: 10.1164/rccm.201606-1225OC. [DOI] [PubMed] [Google Scholar]
- 23.Wong HR, Salisbury S, Xiao Q, et al. The pediatric sepsis biomarker risk model. Crit Care. 2012;16:R174. doi: 10.1186/cc11652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wong HR, Cvijanovich NZ, Anas N, et al. Improved risk stratification in pediatric septic shock using both protein and mRNA biomarkers. PERSEVERE-XP. Am J Respir Crit Care Med. 2017;196:494–501. doi: 10.1164/rccm.201701-0066OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Serkova NJ, Standiford TJ, Stringer KA. The emerging field of quantitative blood metabolomics for biomarker discovery in critical illnesses. Am J Respir Crit Care Med. 2011;184:647–655. doi: 10.1164/rccm.201103-0474CI. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Eckerle M, Ambroggio L, Puskarich MA, et al. Metabolomics as a driver in advancing precision medicine in sepsis. Pharmacotherapy. 2017;37:1023–1032. doi: 10.1002/phar.1974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mickiewicz B, Duggan GE, Winston BW, et al. Metabolic profiling of serum samples by 1H nuclear magnetic resonance spectroscopy as a potential diagnostic approach for septic shock. Crit Care Med. 2014;42:1140–1149. doi: 10.1097/CCM.0000000000000142. [DOI] [PubMed] [Google Scholar]
- 28.Ferrario M, Cambiaghi A, Brunelli L, et al. Mortality prediction in patients with severe septic shock: a pilot study using a target metabolomics approach. Sci Rep. 2016;6:20391. doi: 10.1038/srep20391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Langley RJ, Tsalik EL, van Velkinburgh JC, et al. An integrated clinico-metabolomic model improves prediction of death in sepsis. Sci Transl Med. 2013;5:195ra195. doi: 10.1126/scitranslmed.3005893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rogers AJ, McGeachie M, Baron RM, et al. Metabolomic derangements are associated with mortality in critically ill adult patients. PLoS One. 2014;9:e87538. doi: 10.1371/journal.pone.0087538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Neugebauer S, Giamarellos-Bourboulis EJ, Pelekanou A, et al. Metabolite profiles in sepsis: developing prognostic tools based on the type of infection. Crit Care Med. 2016;44:1649–1662. doi: 10.1097/CCM.0000000000001740. [DOI] [PubMed] [Google Scholar]
- 32.Venet F, Lepape A, Monneret G. Clinical review: flow cytometry perspectives in the ICU— from diagnosis of infection to monitoring of injury-induced immune dysfunctions. Crit Care. 2011;15:231. doi: 10.1186/cc10333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wegener J, Keese CR, Giaever I. Electric cell-substrate impedance sensing (ECIS) as a non-invasive means to monitor the kinetics of cell spreading to artificial surfaces. Exp Cell Res. 2000;259:158–166. doi: 10.1006/excr.2000.4919. [DOI] [PubMed] [Google Scholar]
- 34.Conway Morris A, Datta D, Shankar-Hari M, et al. Cell-surface signatures of immune dysfunction risk-stratify critically ill patients: INFECT study. Intensive Care Med. 2018;44:627–635. doi: 10.1007/s00134-018-5247-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wilson JK, Zhao Y, Singer M, Spencer J, Shankar-Hari M. Lymphocyte subset expression and serum concentrations of PD-1/PD-L1 in sepsis—pilot study. Crit Care. 2018;22:95. doi: 10.1186/s13054-018-2020-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gogos C, Kotsaki A, Pelekanou A, et al. Early alterations of the innate and adaptive immune statuses in sepsis according to the type of underlying infection. Crit Care. 2010;14:R96. doi: 10.1186/cc9031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Leligdowicz A, Chun LF, Jauregui A, et al. Human pulmonary endothelial cell permeability after exposure to LPS-stimulated leukocyte supernatants derived from patients with early sepsis. Am J Phys Lung Cell Mol Phys. 2018;315:L638–L644. doi: 10.1152/ajplung.00286.2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Fox ED, Heffernan DS, Cioffi WG, Reichner JS. Neutrophils from critically ill septic patients mediate profound loss of endothelial barrier integrity. Crit Care. 2013;17:R226. doi: 10.1186/cc13049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Cazalis MA, Lepape A, Venet F, et al. Early and dynamic changes in gene expression in septic shock patients: a genome-wide approach. Intensive Care Med Exp. 2014;2:20. doi: 10.1186/s40635-014-0020-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Goldenberg NM, Leligdowicz A, Slutsky AS, Friedrich JO, Lee WL. Is nosocomial infection really the major cause of death in sepsis? Crit Care. 2014;18:540. doi: 10.1186/s13054-014-0540-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tang BM, McLean AS, Dawes IW, Huang SJ, Cowley MJ, Lin RC. Gene-expression profiling of gram-positive and gram-negative sepsis in critically ill patients. Crit Care Med. 2008;36:1125–1128. doi: 10.1097/CCM.0b013e3181692c0b. [DOI] [PubMed] [Google Scholar]
- 42.Sweeney TE, Wong HR, Khatri P. Robust classification of bacterial and viral infections via integrated host gene expression diagnostics. Sci Transl Med. 2016;8:346ra391. doi: 10.1126/scitranslmed.aaf7165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Banoei MM, Vogel HJ, Weljie AM, et al. Plasma metabolomics for the diagnosis and prognosis of H1N1 influenza pneumonia. Crit Care. 2017;21:97. doi: 10.1186/s13054-017-1672-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Not applicable.