

Abstract
Cross-seeding of misfolded amyloid proteins is believed to induce cross-species infection of prion diseases. In sporadic Alzheimer’s disease (AD), misfolding of 42-residue β-amyloid (Aβ) is widely considered to trigger amyloid plaque deposition. Despite increasing evidence that misfolded Aβ mimics prions, interactions of misfolded 42-residue Aβ42 with more abundant 40-residue Aβ40 in AD are elusive. This study presents in-vitro evidence that a heterozygous E22G pathogenic (“Arctic”) mutation of Aβ40 can enhance misfolding of Aβ via cross-seeding from wild-type (WT) Aβ42 fibril. Thioflavin T (ThT) fluorescence analysis suggested that misfolding of E22G Aβ40 was enhanced by adding 5% (w/w) WT Aβ42 fibril as “seed”, whereas WT Aβ40 was unaffected by Aβ42 fibril seed. 13C SSNMR analysis revealed that such cross-seeding prompted formation of E22G Aβ40 fibril that structurally mimics the seed Aβ42 fibril, suggesting unexpected cross talk of Aβ isoforms that potentially promotes early onset of AD. The SSNMR approach is likely applicable to elucidate structural details of heterogeneous amyloid fibrils produced in cross-seeding for amyloids linked to neurodegenerative diseases.
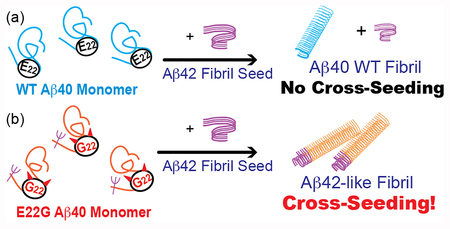
Graphical Abstract
Amyloid diseases, including AD, Parkinson’s and prion diseases, are commonly characterized by misfolded fibrillar aggregates of disease-specific amyloid proteins.1 These amyloid fibrils catalyze self-replication as a “seed” or template by recruiting monomeric proteins into a template-dependent polymerization. Increasing in-vitro and in-vivo evidence suggests that fibril from one amyloid protein can promote misfolding of another amyloid protein via cross-seeding/propagation.2–5 In yeast prions exposure to a misfolded form of a Sup35 prion domain originated from one yeast strain was shown to prompt misfolding of another Sup35 prion domain from a divergent strain through a cross-species barrier.6,7 It is believed that such cross-seeding of prions is essential in cross-species transmission of mammalian prion diseases.3,8,9 However, cross-seeding mechanisms at a molecular level are poorly defined for prions and other amyloid proteins.
AD is an amyloid disease that is linked with misfolding of 39- to 43-residue β-amyloid (Aβ) protein. Aβ is a primary component of the amyloid plaque, a hallmark of AD. Among the Aβ species in AD, 42-residue Aβ (Aβ42) is considered to be the more pathogenic species since it is both more aggregation-prone and toxic than most abundant 40-residue Aβ (Aβ40).1 Furthermore, a lower ratio of Aβ42 to Aβ40 in the plasma is an indicator of AD;10,11 thus, selective misfolding of Aβ42 may trigger initial amyloid plaque deposition. Although early studies reported some degree of cross-seeding on Aβ40 misfolding by Aβ42 fibril seed,12,13 recent studies using improved protocols to prepare fibrils consistently showed negligible or poor enhancement of misfolding of Aβ40 via cross-seeding with Aβ42 fibril seed.14–16 Recent kinetic studies indicated that cross-seeding effects between Aβ42 and Aβ40 may be explained by non-specific surface-catalysis rather than structure-specific template nucleation.15 Thus, unlike prion proteins, cross-seeding effects between the Aβ isoforms have largely been neglected in AD.
In this study, we re-examined the impact of cross-seeding in familial AD (FAD) using an in-vitro model by investigating co-aggregation of WT Aβ species and a pathogenic mutant of Aβ using the E22G Aβ40 associated with “Arctic” FAD. Unlike many pathogenic mutants of Aβ linked with FAD, which generally promote amyloid fibril formation, the E22G mutation was reported to promote the formation of sub-fibrillar diffusible aggregates rather than fibril.17 Here, we present an alternate hypothesis that explains how this E22G mutant and possibly other pathogenic mutants of Aβ can modulate a misfolding pathway of Aβ in FAD via cross-seeding between the mutant and wild-type (WT) Aβ isoforms. Our results suggest that a pathogenic mutation on Aβ can render more abundant Aβ40 prone to aggregation by enhancing interactions between Aβ42 and Aβ40 via cross-seeding. We also introduce SSNMR as a structural tool for investigating cross-seeded amyloids for the first time.
First, we compared misfolding kinetics of both monomeric WT and E22G Aβ40 separately, without any fibril seed, where WT Aβ40 monomer served as a control. Black squares (■) in Fig. 1(a, b) show incubation-time (t) dependence of thioflavin-T (ThT) fluorescence of (a) WT Aβ40 and (b) E22G Aβ40 without fibril seed. ThT fluorescence is a sensitive indicator of amyloid fibril.5,18 E22G Aβ40 and WT Aβ40 (~50 μM) both misfold into fibrils after lag times of 2–7 h. The presence of such a lag time suggests a multi-step misfolding process in which Aβ monomers assemble into an oligomeric intermediate that is undetectable by the ThT assay.19,20 Curve fitting (black lines) using a sigmoidal equation19 produces a quantitative estimate of the lag time. The lag time (τ) of 1.7 h observed for E22G Aβ40 in (b) was considerably shorter than τ of 6.8 h for WT Aβ40 in (a). The lag time of E22G Aβ40 is comparable to that of WT Aβ42 (τ = 3–4 h at ~30 μM Aβ).21 The promoted aggregation of E22G Aβ40 is consistent with previous results.22 Thus, the E22G mutation is likely to promote early onset of AD by enhancing Aβ fibrillization as other pathogenic mutants of Aβ.
Figure 1.

Incubation-time (t) dependence of ThT fluorescence for (a) WT Aβ40 and (b) E22G Aβ40 without seed (■) and with Aβ42 fibril seed at 5% w/w ( ) with fitting curves to kinetic models (solid lines). The WT Aβ40 data showed little difference in the lag time (τ) between the unseeded (6.8 h) and seeded (6.2 h) samples in (a), where τ values were determined by fitting to a sigmoidal equation y(t) = a/{1+exp[−k(t-tC)]} and τ = tC −2/k. Unseeded E22G Aβ40 data in (b) showed intrinsically faster misfolding (τ = 1.7 h) than WT and fit with a sigmoidal curve (black line). The seeded E22G Aβ40 data in (b) showed no lag time, and fit well with the curve based on first-order kinetics equation y(t) =a[1 - exp(−kt)] (red line).
) with fitting curves to kinetic models (solid lines). The WT Aβ40 data showed little difference in the lag time (τ) between the unseeded (6.8 h) and seeded (6.2 h) samples in (a), where τ values were determined by fitting to a sigmoidal equation y(t) = a/{1+exp[−k(t-tC)]} and τ = tC −2/k. Unseeded E22G Aβ40 data in (b) showed intrinsically faster misfolding (τ = 1.7 h) than WT and fit with a sigmoidal curve (black line). The seeded E22G Aβ40 data in (b) showed no lag time, and fit well with the curve based on first-order kinetics equation y(t) =a[1 - exp(−kt)] (red line).
Next, we examined an alternate effect of the mutation by studying the effects of Aβ42 fibril cross-seeding to monomeric E22G Aβ40 and WT Aβ40. The situation mimics the initial phase of plaque deposition in AD after Aβ42 fibril formation. Although E22G Aβ42 may misfold faster than WT Aβ42, we employed here seeding with WT Aβ42 fibril as a benchmark case since its structure and misfolding kinetic properties have been much better characterized.16,23,24 As discussed below, the E22G mutation is likely to stabilize the unique S-shaped triple β structure identified for WT Aβ42 fibril. Red filled circles ( ) in Fig. 1(a, b) show incubationtime dependence of ThT fluorescence of (a) WT Aβ40 and (b) E22G Aβ40 in the presence of seed Aβ42 fibril (5% (w/w)). In (a) nearly no effect of cross-seeding was observed for monomeric WT Aβ40 incubated with Aβ42 fibril seed. The cross-seeding kinetics in (b) shows a marked difference for E22G Aβ40 from the data for WT Aβ40 in (a); the lag time was clearly eliminated by addition of 5% Aβ42 fibril seed. The fitting curve based on the first-order kinetics (see the caption of Fig. 1) better reproduced the experimental data than an optimized sigmoidal curve (Table S1 in SI). These data with additional kinetic data (Fig. S1) convincingly suggest that E22G Aβ40 misfolding was promoted by cross-seeding of Aβ42 fibril despite a common conception that cross-seeding does not easily occur between Aβ40 and Aβ42 isoforms. At the initial phase, monomers of E22G Aβ40 were likely to be converted directly to ThT-detectable fibrils presumably using WT Aβ42 fibril as a template. We observed that WT Aβ40 fibril seed (5% (w/w)) also eliminated a lag time for E22G Aβ40 monomer as well (see Fig. S2). Thus, E22G Aβ40 showed markedly different cross-seeding properties from WT Aβ40.
) in Fig. 1(a, b) show incubationtime dependence of ThT fluorescence of (a) WT Aβ40 and (b) E22G Aβ40 in the presence of seed Aβ42 fibril (5% (w/w)). In (a) nearly no effect of cross-seeding was observed for monomeric WT Aβ40 incubated with Aβ42 fibril seed. The cross-seeding kinetics in (b) shows a marked difference for E22G Aβ40 from the data for WT Aβ40 in (a); the lag time was clearly eliminated by addition of 5% Aβ42 fibril seed. The fitting curve based on the first-order kinetics (see the caption of Fig. 1) better reproduced the experimental data than an optimized sigmoidal curve (Table S1 in SI). These data with additional kinetic data (Fig. S1) convincingly suggest that E22G Aβ40 misfolding was promoted by cross-seeding of Aβ42 fibril despite a common conception that cross-seeding does not easily occur between Aβ40 and Aβ42 isoforms. At the initial phase, monomers of E22G Aβ40 were likely to be converted directly to ThT-detectable fibrils presumably using WT Aβ42 fibril as a template. We observed that WT Aβ40 fibril seed (5% (w/w)) also eliminated a lag time for E22G Aβ40 monomer as well (see Fig. S2). Thus, E22G Aβ40 showed markedly different cross-seeding properties from WT Aβ40.
We next examined whether the cross-seeded Aβ42 fibril influenced the structure of the E22G Aβ40 fibril as a replication template. Transmission electron microscopy (TEM) images (Fig. S3c,d) confirmed formation of E22G Aβ40 fibrils (c) without and (d) with influence of Aβ42 fibril seed. Figure S3e shows that Aβ42 fibrils used as seeds have a different morphology from (d). However, from the TEM data, it is not straightforward to assess structural similarity of E22G Aβ40 and Aβ42 fibrils with site specificity.
Here, we demonstrate that SSNMR analysis solves chemical/structural heterogeneity problems of crossseeded fibrils unlike cryoEM analysis. We prepared E22G Aβ40 fibril samples obtained by incubation of 13C-labeled E22G Aβ40 monomer with 5% (w/w) of unlabeled WT Aβ42 or WT Aβ40 fibril seed and without any seeds. A deliberate use of unlabeled WT Aβ seeds allows us to monitor the structural influence of cross-seeding on the labeled E22G Aβ without chemical heterogeneity issues. In Fig. 2, we compare (a) 2D 13C/13C SSNMR data of 13C-labeled E22G Aβ40 fibril samples obtained by incubation without seed and (b) the corresponding 2D spectra for E22G Aβ40 samples obtained with Aβ42 fibril seed (red) and Aβ40 fibril seed (blue). We also collected (c) control 2D spectra of WT Aβ42 fibril seed (magenta) and WT Aβ40 fibril seed (cyan). The samples were uniformly 13C-,15N-labeled at selected residues (a, b) Gly22, Val24, Ala30, Ile31 and (c) Val24, Ala30, Ile31 (note that WT Aβ lacks Gly22). These residues were selected as their 13C shifts show relatively large differences between the Aβ40 fibril with a U-shaped β-turn-β motif25 and the Aβ42 fibril with a S-shaped triple β-sheet motif.16 The samples for (c) were prepared using the same methods as those for Aβ42 or Aβ40 fibril seeds in (b). Signal assignments are indicated by color-coded lines for different residues (see the inset of a). The SSNMR spectrum for the unseeded E22G Aβ40 fibril in (a) shows more than one set of cross peaks for Ala30 and Ile31 (assignments by solid and dotted lines), clearly demonstrating, at least, two distinct major conformers (i.e. polymorphs). For example, two cross peaks were observed for 13Cα/13Cβ of Ile31 (orange and black arrows) in Fig. 2a. This is consistent with a previous study, which reported up to five structurally distinct polymorphs for E22G Aβ40 fibril.22
Figure 2.

(a) Aliphatic regions of 2D 13C/13C chemical-shift correlation SSNMR spectra of 13C-labeled E22G Aβ40 fibrils prepared without seed Aβ fibril (black). (b) Corresponding 2D spectra of 13C-labeled E22G Aβ40 fibrils prepared with unlabeled seed Aβ42 fibril (red) and seed Aβ40 fibril (blue). (c) Control 2D spectra of 13C-labeled seed Aβ42 fibril (magenta) and seed Aβ40 fibril (cyan) alone, which are equivalent to the seeds used for (b). The isotope-labeled residues for (a, b) are in the inset in (a). The assignments are indicated by color-coded lines (Val24: dark blue, Ala30: magenta, Ile31: green). Except for Gly-22, which does not exist in WT Aβ, the labeled residues for (c) are the same as (a, b). For (a), two major polymorphs are assigned by solid and dotted lines. For (b, c), the spectra in red/magenta and blue/cyan are assigned by solid and dotted lines, respectively. Red/magenta and blue/cyan arrows in (b, c) indicate similarity of spectral positions for Aβ42- and Aβ40-seeded E22G Aβ40 fibrils with those for the seed Aβ42 and Aβ40 fibrils, respectively.
Interestingly, by cross-seeding with Aβ42 fibril, the spectrum of E22G Aβ40 fibril (red) in Fig. 2b was drastically simplified, displaying a single set of strong cross peaks (solid lines), except for a few minor peaks for Ile31. The notable difference between the unseeded and seeded samples clearly demonstrates the influence of the seeding at a molecular level. To our surprise, a 2D 13C spectrum of the 13C-, 15N-labeled Aβ42 seed fibril alone (magenta) with assignments (solid lines) in Fig 2c show very similar spectral features with those for the Aβ42-seeded E22G Aβ40 fibril in Fig. 2b (see Fig. S4a–d also). The peak position of Ile31 13Cβ peak in (c) (magenta arrow) is, for example, consistent with that in (b) (red arrow). In contrast, the E22G Aβ40 fibril sample prepared using Aβ40 fibril seed (blue) in (b) also indicated a single conformer to large extent, but with different spectral features from those prepared with Aβ42 seed (red). The chemical shifts for the blue signals in (b) noted by the dotted lines match up with those for the second species in Fig. 2a but with disagreements for Ala30 and other residues (dotted line; see Table S2 also). Similar trends were observed for the samples labeled at different residues (Val18, Phe19, Ala21, Gly33, Leu34) (Fig. S4e–h and Fig. S5). We confirmed that the spectral pattern for the E22G Aβ40 fibril obtained with WT Aβ40 seed correlates well with the WT Aβ40 fibril used as seed (Fig. 2c cyan), although the patterns are not identical (see Fig. S6 also). For example, peak positions for 13Cβ for Ile31 are very similar (see blue and cyan arrows in Fig. 2b, c). More quantitative analysis using Pearson’s correlation coefficient (Fig. S7) confirmed the results. Thus, E22G Aβ40 is likely to adopt multiple fibril structures similar to Aβ42 fibril with a S-shaped triple β-sheet motif16,26 as well as the Aβ40 fibril with a β-turn-β motif.25 Hence, our spectral fingerprint analyses (Fig. 2 and Fig. S5) revealed that cross-seeding with Aβ42 and Aβ40 fibrils induces misfolding of E22G Aβ40 monomers into a conformer having a similar structure to the seed Aβ42 and Aβ40 fibrils, respectively.
In conclusion, our studies clearly demonstrated that introducing a single site E22G mutation of Aβ40 greatly enhanced misfolding of Aβ40 by cross-seeding with Aβ42 fibril. The present results indicate that the single-site E22G mutation of Aβ40 alters the compatibility between Aβ40 and Aβ42 in cross-seeding while unseeded E22G Aβ40 misfolds into two or more discrete fibril forms, some of which are similar to Aβ42 or Aβ40 fibril. This implies the notion that protein structural plasticity of a mutant Aβ is another critical factor that may drive development of FAD. The results are particularly striking since the heterozygous pathogenic mutation may render more abundant Aβ40 susceptible to plaque formation through cross-seeding from Aβ42 fibril (WT and/or E22G) in AD (additional discussion in SI). Indeed, recent post-mortem studies of Arctic FAD patients suggested association of E22G mutation of Aβ with enhanced deposit of N-terminal truncated Aβ40 in parenchymal plaques, compared with sporadic AD.27 We have also presented the effectiveness of a SSNMR approach to elucidate site-specific structural features for cross-seeded amyloid fibrils for the first time. This approach is likely applicable to examine structural cross talk of various amyloid proteins.
Supplementary Material
Acknowledgements
This work on E22G Aβ40 and WT Aβ42 was supported primarily by the National Institutes of Health (U01 & R01 GM 098033) for YI. Studies on misfolding of Aβ40 were, in part, supported by NIH sub-award (NIH R01AG048793) for YI. We thank Mr. Isamu Matsuda and Takaya Ishiguro for their assistance on kinetic experiments. The authors are also grateful to Drs. Sandra Chimon, Diana Calero, Medhat Shaibat, and Ms. Prakruti Modi for their initial efforts on the E22G Aβ40 system.
Footnotes
Supporting Information Available: Preparation of Aβ fibril samples, additional data and procedures on ThT fluorescence and SSNMR, and TEM analyses are included in the supporting information.
References
- (1).Selkoe DJ Nat. Cell. Biol 2004, 6, 1054. [DOI] [PubMed] [Google Scholar]
- (2).Morales R; Callegari K; Soto C Virus Res. 2015, 207, 106. [DOI] [PubMed] [Google Scholar]
- (3).Jones EM; Surewicz WK Cell 2005, 121, 63. [DOI] [PubMed] [Google Scholar]
- (4).Ross ED; Minton A; Wickner RB Nat. Cell. Biol 2005, 7, 1039. [DOI] [PubMed] [Google Scholar]
- (5).Horvath I; Wittung-Stafshede P Proc. Natl. Acad. Sci. U. S. A 2016, 113, 12473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Tanaka M; Chien P; Yonekura K; Weissman JS Cell 2005, 121, 49. [DOI] [PubMed] [Google Scholar]
- (7).Chernoff YO; Galkin AP; Lewitin E; Chernova TA; Newnam GP; Belenkiy SM Mol. Microbiol 2000, 35, 865. [DOI] [PubMed] [Google Scholar]
- (8).Surewicz WK; Jones EM; Apetri AC Acc. Chem. Res 2006, 39, 654. [DOI] [PubMed] [Google Scholar]
- (9).Prusiner SB Proc. Natl. Acad. Sci. U. S. A 1998, 95, 13363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Graff-Radford NR; Crook JE; Lucas J; Boeve BF; Knopman DS; Ivnik RJ; Smith GE; Younkin LH; Petersen RC; Younkin SG Arch. Neurol 2007, 64, 354. [DOI] [PubMed] [Google Scholar]
- (11).van Oijen M; Hofman A; Soares HD; Koudstaal PJ; Breteler MM B. Lancet Neurol 2006, 5, 655. [DOI] [PubMed] [Google Scholar]
- (12).Hasegawa K; Yamaguchi I; Omata S; Gejyo F; Naiki H Biochemistry 1999, 38, 15514. [DOI] [PubMed] [Google Scholar]
- (13).Ono K; Takahashi R; Ikeda T; Yamada MJ Neurochem. 2012, 122, 883. [DOI] [PubMed] [Google Scholar]
- (14).Pauwels K; Williams TL; Morris KL; Jonckheere W; Vandersteen A; Kelly G; Schymkowitz J; Rousseau F; Pastore A; Serpell LC; Broersen KJ Biol. Chem 2012, 287, 5650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Cukalevski R; Yang XT; Meisl G; Weininger U; Bernfur K; Frohm B; Knowles TPJ; Linse S Chem. Sci 2015, 6, 4215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Xiao Y; Ma B; McElheny D; Parathasarathy S; Hoshi M; Nussinov R; Ishii Y Nat. Struct. Mol. Biol 2015, 22, 499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Nilsberth C; Westlind-Danielsson A; Eckman CB; Condron MM; Axelman K; Forsell C; Stenh C; Luthman J; Teplow DB; Younkin SG; Naslund J; Lannfelt L Nat. Neurosci 2001, 4, 887. [DOI] [PubMed] [Google Scholar]
- (18).Levine HI In Methods in enzymology; Wetzel R, Ed.; Academic Press: San Diego, 1999; Vol. 309, p 274.10507030 [Google Scholar]
- (19).Nielsen L; Khurana R; Coats A; Frokjaer S; Brange J; Vyas S; Uversky VN; Fink AL Biochemistry 2001, 40, 6036. [DOI] [PubMed] [Google Scholar]
- (20).Parthasarathy S; Inoue M; Xiao Y; Matsumura Y; Nabeshima Y.-i.; Hoshi M; Ishii Y J. Am. Chem. Soc 2015, 137, 6480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Xiao Y, Ph. D dissertation. University of Illinois at Chicago, Chicago, IL, 2016. [Google Scholar]
- (22).Norlin N; Hellberg M; Filippov A; Sousa AA; Grobner G; Leapman RD; Almqvist N; Antzutkin ON J. Struct. Biol 2012, 180, 174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Colvin MT; Silvers R; Ni QZ; Can TV; Sergeyev I; Rosay M; Donovan KJ; Michael B; Wall J; Linse S; Griffin RG J. Am. Chem. Soc 2016, 138, 9663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Walti MA; Ravotti F; Arai H; Glabe CG; Wall JS; Bockmann A; Guntert P; Meier BH; Riek R Proc. Natl. Acad. Sci. U. S. A 2016, 113, E4976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Tycko RQ Rev. Biophys 2006, 39, 1. [DOI] [PubMed] [Google Scholar]
- (26).Elkins MR; Wang T; Nick M; Jo H; Lemmin T; Prusiner SB; DeGrado WF; Stohr J; Hong MJ Am. Chem. Soc 2016, 138, 9840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Philipson O; Lord A; Lalowski M; Soliymani R; Baumann M; Thyberg J; Bogdanovic N; Olofsson T; Tjernberg LO; Ingelsson M; Lannfelt L; Kalimo H; Nilsson LNG Neurobiol. Aging 2012, 33 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.