

Abstract
Enhanced activation of phosphoinositide 3-kinase (PI3K) is a hallmark of many human tumors because it promotes cell proliferation and survival through several mechanisms. One of these mechanisms is the phosphorylation of the serine and threonine kinase Akt at the cytosolic side of the plasma membrane by phosphoinositide-dependent protein kinase 1 (PDK1), which is recruited and activated by binding to the phosphoinositides produced by PI3K. We previously demonstrated increased nuclear accumulation of PDK1 in cells with enhanced PI3K activity. We report that nuclear PDK1 promoted cell proliferation by suppressing FOXO3A-dependent transcription of the gene encoding p27Kip1 (an inhibitor of cell cycle progression), whereas it enhanced cell survival by inhibiting the activation of c-Jun amino-terminal kinase. Cells with nuclear-localized PDK1 showed anchorage-independent growth, and when injected into mice, these cells induced the formation of solid tumors. In human prostate tumors, cytoplasmic localization of PDK1 correlated only with early-stage, low-risk tumors, whereas nuclear PDK1 localization correlated with high-risk tumors. Together, our findings suggest a role for nuclear-translocated PDK1 in oncogenic cellular transformation and tumor progression in mice and humans.
INTRODUCTION
Growth factor signaling activates phosphoinositide 3-kinase (PI3K) signaling, which regulates many cellular and physiological processes, such as metabolism, proliferation, differentiation, and apoptosis (1, 2). Growth factor-independent growth of cells, a hallmark of many human tumors, is often attributed to the enhanced activation of the PI3K pathway. Not surprisingly, there exists a tumor suppressor network consisting of the phosphatase PTEN (phosphatase and tensin homolog deleted from chromosome 10) (3), which antagonizes the PI3K pathway to curb excessive cellular proliferation. Many human cancers, including prostate cancer (4), are characterized by loss of functional PTEN, which results in tumors that are androgen-independent and represents the second leading cause of cancer mortality in men. Hence, many antitumor therapeutic strategies are focused on inhibiting PI3K and its downstream effectors (5).
Activated PI3K triggers a signaling cascade that results in the localization to the plasma membrane of phosphoinositide-dependent protein kinase 1 (PDK1) and the serine and threonine kinase Akt [also known as protein kinase B (PKB)], and it is here that PDK1 phosphorylates and activates Akt (6–8). Akt is a major oncogenic effector in the PI3K pathway, and it is often found to have enhanced activation in tumors. In addition to Akt, PDK1 also phosphorylates and activates other members of the protein kinase A, protein kinase G, and protein kinase C (AGC) family, such as PKCζ (protein kinase C ζ), p70 S6 kinase, and p90 ribosomal S6 kinase, in the PI3K pathway (9). Once activated, many of these PDK1 substrates, including Akt, translocate to the nucleus and regulate nuclear events such as cell cycle, apoptosis, and gene expression (10). In addition to Akt, accumulating evidence suggests the presence of PI3K in the nucleus (10,11), which is activated by the Ras-like guanosine triphosphatase PI3K enhancer (PIKE) (12). Nuclear PI3K signaling protects cells from apoptosis (13, 14) and is involved in oncogenesis. On the other hand, the nucleus also harbors a tumor suppressor network consisting of promyelocytic leukemia (PML) and protein phosphatase 2A (PP2A). PML-localized PP2A dephosphorylates nuclear phosphorylated Akt (pAkt), thereby terminating growth-promoting signaling (15).
We previously showed that the increased nuclear localization of PDK1 in the nucleus of PTEN-deficient (PTEN−/− ) mouse embryonic fibroblasts (MEFs) is dependent on PI3K activity (16). We identified the hydrophobic nuclear export sequence (NES) in PDK1 and showed that mutation of both NES residues (Leu383 and Phe386) results in constitutive nuclear retention of PDK1 (16). Phosphorylation of a residue adjacent to the NES of PDK1 was proposed to mediate the increased nuclear localization of PDK1 in PTEN−/− cells (17); however, the functional relevance of increased nuclear localization of PDK1 in PTEN−/− cells remains unclear. Here, we showed that nuclear localization of PDK1 increased the nuclear accumulation of pAkt and inhibited FOXO3A-dependent transcription of the gene encoding p27Kip1 to accelerate cell proliferation. In addition, nuclear PDK1 signaling inhibited the activation of c-Jun N-terminal kinase (JNK) and protected cells from apoptosis. Furthermore, transplantation of cells containing nuclear-localized PDK1, but not cells containing wild-type PDK1, to athymic nude mice resulted in robust tumor growth. Finally, we detected a close correlation between extent of nuclear translocation of PDK1 and late-stage human prostate cancer Together, these data suggest a link between the subcellular localization of PDK1 and its tumorigenic potential.
RESULTS
Nuclear-localized PDK1 increases nuclear pAkt abundance, resulting in increased cell growth and proliferation
We previously identified PDK1 as a nucleocytoplasmic shuttling kinase, whose nuclear localization is stimulated by insulin (16) and insulin-like growth factor 1 (16, 17) in a time-dependent manner (fig. S1A). To understand the biological consequences of the nuclear localization of PDK1, we reconstituted PDK1 knockout (PDK1−/−) MEFs with either myc-tagged wild-type PDK1 (designated as W-PDK1−/− cells) or myc-tagged mutant PDK1 with mutations in the NES (16), which renders it constitutively localized to the nucleus (designated as N-PDK1−/− cells). As expected, in the immunofluorescence analysis of these cell lines, W-PDK1−/− cells showed typical cytoplasmic localization of PDK1 (Fig. 1A). In contrast, PDK1 was predominantly confined to the nucleus in N-PDK1−/− cells. Consistent with our previous findings (16), we observed no substantial differences in the extent of phosphorylation of wild-type and mutant PDK1 (fig. S1B), nor did we observe any difference in the in vitro kinase activities when myc-tagged PDK1 proteins were purified from W-PDK1−/− or N-PDK1−/− cells (fig. S1C). In addition, the abundances of several components of the PI3K pathway were comparable in W-PDK1−/− and N-PDK1−/− cells (fig. S1D). PDK1 isolated from N-PDK1−/− cells migrated as a slower band when analyzed by SDS–polyacrylamide gel electrophoresis (SDS-PAGE) than did PDK1 isolated from W-PDK1−/− cells (fig. S1, B to D), possibly as a result of one or more posttranslational modifications, which are currently under investigation.
Fig. 1.
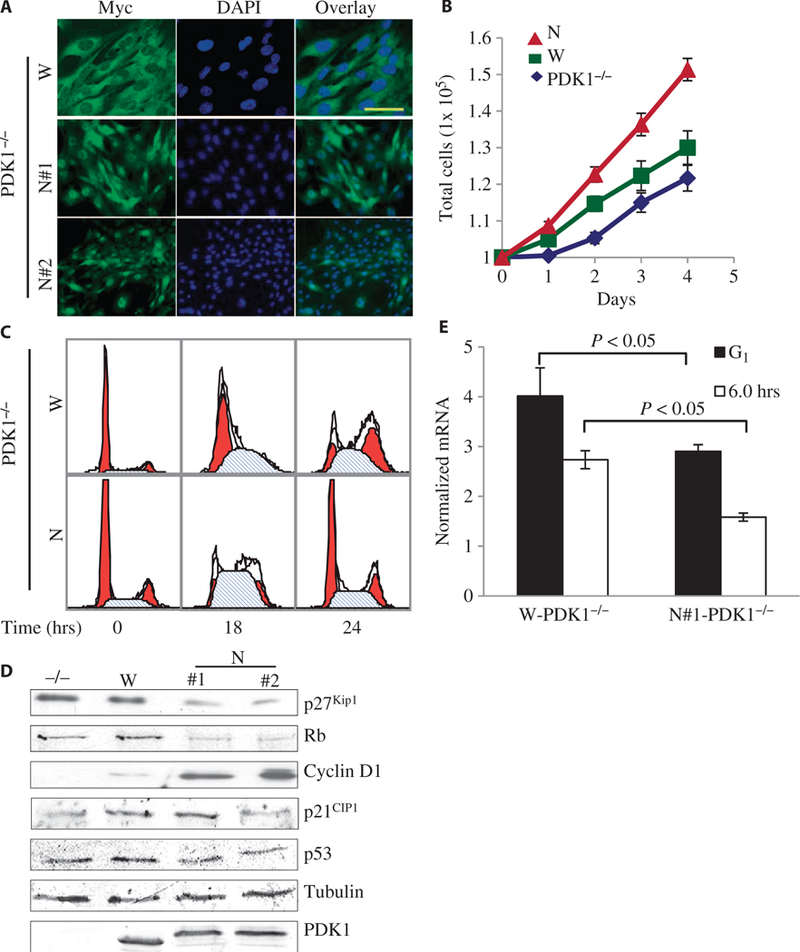
Increased proliferation and accelerated cell cycle in cells with nuclear-localized PDK1. (A) Representative confocal immunofluorescence images of three independent experiments showing the subcellular localization of myc-tagged W-PDK1−/− (W) and nuclear-localized mutant PDK1 (N) in clones of MEFs. PDK1 proteins were detected with an anti-myc antibody (green), and the nuclei were detected with 4’,6’-diamidino-2-phenylindole (DAPI; blue). Scale bar, 50 μm. (B) PDK1−/−, W-PDK1−/−, and N-PDK1−/− cells (1 × 105) were seeded on 60-mm dishes, and cell numbers were counted everyday for 4 days. Values represent means ± SD (n = 3 experiments). (C) Representative flow cytometric analysis data showing the increased G1-to-S progression of N-PDK1−/− cells. MEFs of the indicated genotypes were synchronized and released from G1 phase arrest as described in Materials and Methods. Samples were collected every 6 hours, fixed, and their DNA content was measured. (D) Western blots from asynchronous MEFs of the indicated genotypes showing the amounts of important cell cycle regulatory proteins. For quantification of the three independent experiments, see fig. S3A (n = 3 experiments). (E) Graphical representation of the abundance of p27Kip1 mRNA, which was determined by quantitative polymerase chain reaction (PCR) and normalized to that of mouse TATA box-binding protein (TBP) mRNA. Values represent means ± SD (n = 3 experiments).
Using these cell lines, we first compared the growth rates of the parental wild-type (PDK1+/+) MEFs, as well as those of PDK1 knockout (PDK1−/−), W-PDK1−/−, and N-PDK1−/− MEFs. Consistent with a previous finding (18), PDK1−/− cells had a substantial defect in their rate of proliferation compared with that of parental PDK1+/+ MEFs. This proliferation defect was rescued by reconstitution with myc-WT-PDK1 in W-PDK1−/− MEF to a level comparable with that of parental PDK1+/+ cells (fig. S2A). In contrast, independent clones of MEFs expressing the constitutively nuclear-localized PDK1 (N-PDK1−/−, N#1 and N#2) showed a substantial increase in total cell number (fig. S2A) and in the rate of cell proliferation (Fig. 1B) compared with those of the W-PDK1−/− cells. These results suggest the involvement of nuclear-localized PDK1 in the regulation of cellular proliferation pathways.
To determine the molecular mechanism responsible for the increased proliferation of N-PDK1−/− cells, we compared the cell cycle progressions of W-PDK1−/− and N-PDK1−/− cells by flow cytometric analysis after G1-to-S synchronization and release. The N-PDK1−/− cells showed an accelerated progression through the S phase of the cell cycle (~16 to 18 hours after release) compared to that of the W-PDK1−/− cells (~24 hours after release) (Fig. 1C), indicating possible acceleration through the G1-to-S phase transition in N-PDK1−/− MEFs. Transition from the G1 to the S phase of the cell cycle is inhibited by p27Kip1, which inhibits the cyclin E/CDK2 (cyclin-dependent kinase 2) complex. Cyclin E/CDK2, as well as cyclin D/CDK4, phosphorylates the retinoblastoma (Rb) protein and promotes its degradation (19). Loss of Rb initiates DNA synthesis and the transition into the S phase (20). Because entry into the S phase was accelerated in N-PDK1−/− cells compared to that in W-PDK1−/− cells, we asked whether the functions of p27Kip1 and Rb were compromised in N-PDK1−/− cells. We first examined the abundance of p27Kip1 protein and other signaling molecules that regulate the G1-to-S transition in asynchronous cell cultures under normal growth conditions. The cellular amounts of p27Kip1 and Rb, but not those of p21CIP1 and p53, were substantially reduced in independent clones of N-PDK1−/− cells compared to those in W-PDK1−/− and PDK1−/− cells (Fig. 1D and fig. S3A). Consistent with the reduction in the amount of Rb, we observed a substantial increase in the amounts of cyclin D1 in independent clones of N-PDK1−/− cells compared to those in W-PDK1−/− or PDK1−/− cells (Fig. 1D and fig. S3A).
To determine whether the decreased amounts of p27Kip1 protein and the concomitant accumulation of cyclin E played a role in the accelerated entry of N-PDK1−/− cells into the S phase, we synchronized the cells at the G1-S boundary and released them by the addition of serum. We found that N-PDK1−/− cells showed a marked increase in cyclin E abundance as early as 6 hours after release from synchronization, which was concurrent with their loss of p27Kip1 protein (fig. S2B). On the other hand, the W-PDK1−/− cells displayed a more gradual increase in cyclin E abundance (fig. S2B). To determine whether the decreased amounts of p27Kip1 protein in the N-PDK1−/− cells were the result of suppressed transcription of its gene, we comparedp27Kip1 mRNA abundance in W-PDK1−/− and N-PDK1−/− cells before and after release from the G1-S synchronization. The abundance of p27Kip1 mRNA was substantially reduced in N-PDK1−/− cells compared to that in W-PDK1−/− cells, even at G1 synchronization (Fig. 1E), suggesting that the decreased expression ofp27Kip1, coupled with the increased amount of cyclin E at G1 synchronization (fig. S2B), resulted in the accelerated G1-to-S phase transition in N-PDK1−/− cells and in their overall rate of proliferation.
The inhibition of p27Kip1 expression is often found in tumors with enhanced PI3K and Akt signaling (21). Therefore, we examined the status of pAkt and its downstream targets in asynchronized parental PDK1−/− MEFs, W-PDK1−/− MEFs, and N-PDK1−/− MEFs. We noticed that the extent of phosphorylation of Akt at Thr308, which is mediated by PDK1, was substantially higher in independent clones of N-PDK1−/− cells compared with that in W-PDK1−/− cells (Fig. 2A and fig. S3B). Consistent with an increased activity of Akt, the phosphorylation at Thr32 of the forkhead transcription factor FOXO3A, a downstream target of Akt, was also substantially increased in independent clones of N-PDK1−/− cells (Fig. 2A and fig. S3B) in comparison with W-PDK1−/− cells. These results suggest the possible involvement of nuclear-localized PDK1 in either the activation of nuclear Akt or the retention of pAkt in the nucleus. To identify the sub-cellular source of pAkt, we performed cell fractionation experiments to compare the amounts of Akt phosphorylated at Thr308 in the cytosolic and nuclear fractions of W-PDK1−/− MEFs and N-PDK1−/− MEFs (Fig. 2B and fig. S3C). We found that N-PDK1−/− cells showed substantially more total as well as pAkt at Thr308 in the nuclear fraction than did W-PDK1−/− cells. In addition, immunofluorescence microscopic analysis of W-PDK1−/− MEFs and N-PDK1−/− MEFs revealed an increased nuclear staining of pAkt-Thr308 in N-PDK1−/− cells compared to that in W-PDK1−/− cells (Fig. 2, C and D). Because the activation of Akt results in the phosphorylation and cytoplasmic translocation of FOXO transcription factors, we also compared the cellular localization of FOXO3A in W-PDK1−/− MEFs and N-PDK1−/− MEFs for endogenous FOXO3A either by subcellular fractionation (Fig. 2B and fig. S3C) or by fluorescence microscopic analysis of cells transiently transfected with plasmid encoding green fluorescent protein (GFP)-FOXO3A (Fig. 2, E and F). We found that N-PDK1−/− cells contained substantially more cytoplasmic endogenous (Fig. 2B and fig. S3C) and GFP-tagged (Fig. 2, E and F) FOXO3A than did W-PDK1−/− cells, confirming the suppression of FOXO3A signaling by nuclear-localized PDK1. Because expression of the gene encoding p27Kip1 is dependent on nuclear FOXO3A (22), our results (Figs. 1 and 2) suggest that the nuclear localization of PDK1 increases the accumulation of pAkt in the nucleus, which inhibits FOXO3A and subsequent p27Kip1 expression, resulting in accelerated cell proliferation.
Fig. 2.
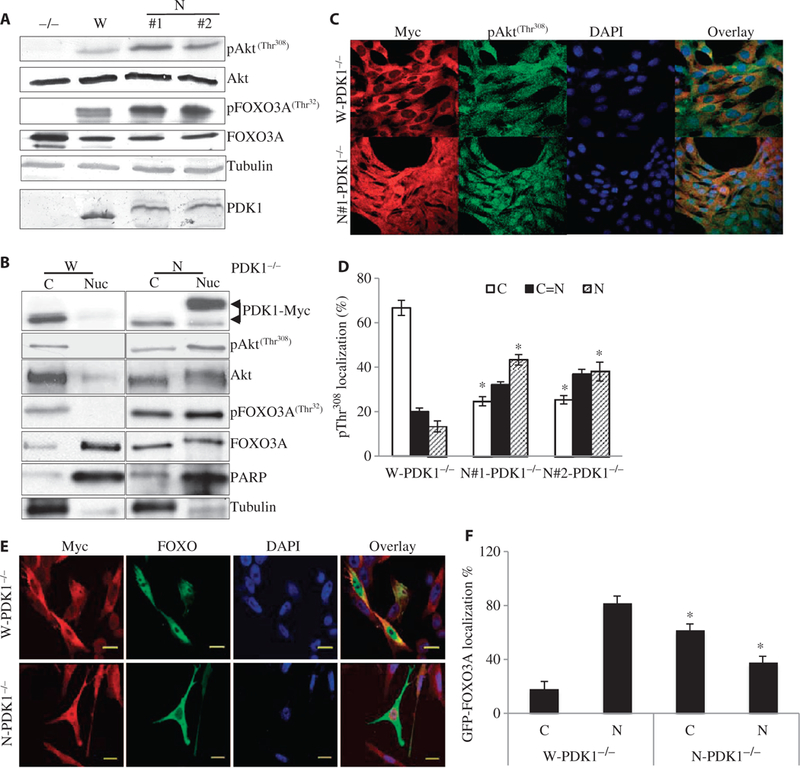
Cells with nuclear-localized PDK1 show a marked increase in nuclear Akt signaling and inhibition of the nuclear localization of FOXO. (A) Asynchronized MEFs of the indicated genotypes were lysed under normal growing conditions. Western blotting analysis was performed with antibodies against the indicated proteins. For quantification of the three independent experiments, see fig. S3B. (B) Cytoplasmic or nuclear fractions prepared from W-PDK1−/− or N-PDK1−/− cells (as described in Materials and Methods) were analyzed by Western blotting with antibodies specific for the indicated proteins. For quantification of these results, see fig. S3C (n = 3 experiments). (C) Comparison of the subcellular localization of Akt phosphorylated atThr308 in W-PDK1−/− and N-PDK1−/− cells under normal growth conditions as determined by confocal microscopy. Cell fixation and immunofluorescence microscopy with antibody against pAkt-Thr308 (second antibody conjugated with Alexa Fluor 488) or with anti-myc antibody (second antibody conjugated with Alexa Fluor 568) were performed as described in Fig. 1A. (D) Quantification of pThr308 staining in W-PDK1−/− and N-PDK1−/− cells from four independent experiments performed in (C). For quantification, 50 cells were counted and scored as cytoplasmic only (C), equal distribution between cytoplasm and nucleus (C = N), or nuclear only (N). The raw numbers were converted into percentages, averaged, and then analyzed for statistical significance. *P < 0.05 versus W-PDK1−/−. (E) GFP-FOXO3A localization in W-PDK1−/− and N-PDK1−/− cells. W-PDK1−/− and N-PDK1−/− cells were transfected with plasmid encoding GFP-FOXO3A. PDK1 localization was determined by confocal immunofluorescence microscopy with an anti-myc antibody and an Alexa Fluor 568-conjugated secondary antibody. Scale bars, 20 μm. (F) Quantification of the subcellular localization of GFP-FOXO3A from the experiment described in (E). n = 3 experiments. *P < 0.05 versus W-PDK1−/− cells.
Nuclear localization of PDK1 promotes cell survival by inhibiting JNK1-mediated proapoptotic signaling
Activation of the nuclear PI3K-Akt signaling pathway protects cells from apoptosis (14); however, the mechanism involved remains unknown. To determine whether nuclear localization of PDK1 had any effect on cell survival, we studied activation of apoptosis by the inflammatory cytokine tumor necrosis factor-α (TNF-α), which is well known to stimulate apoptosis in various cell types. We treated PDK1−/−, W-PDK1−/−, and N-PDK1−/− cells with TNF-α for 6 hours in the presence of actinomycin D, an inhibitor of transcription that is required for TNF-α to stimulate apoptosis (23). We found that TNF-α resulted in the increased detachment of PDK1−/− cells, which was partially rescued in W-PDK1−/− cells (fig. S4A). On the other hand, the independent clones of N-PDK1−/− cells were completely protected from TNF-α-stimulated apoptosis (fig. S4A). TNF-α also markedly increased the cleavage of poly(adenosine 5′-diphosphate–ribose) polymerase (PARP) and caspase-3 (fig. S4B) and enhanced the extent of DNA fragmentation (fig. S4C) in W-PDK1−/− MEFs, but it had little effect on N-PDK1−/− MEFs. We also tested the ability of nuclear PDK1 to protect cells from other proapoptotic stimuli, such as doxorubicin, which induces DNA damage, and ultraviolet (UV) irradiation. N-PDK1−/− cells were protected from apoptosis stimulated by doxorubicin (fig. S4D). In contrast, N-PDK1−/− cells were as sensitive to apoptosis in response to UV irradiation as were W-PDK1−/− cells. UV radiation stimulates apoptosis through multiple pathways including, but not limited to, nuclear and mitochondrial DNA damage, protein oxidation, and cross-linking (fig. S4D). These data suggest that nuclear PDK1 did not inhibit the general machinery that mediates cellular apoptosis, such as caspase-3, but instead suppressed a specific signaling pathway that leads to the onset of the apoptotic response.
When nuclear factor κB-dependent gene expression is suppressed, TNF-α-stimulated JNK activation induces apoptosis (24). To determine the potential effect of nuclear PDK1 on TNF-α–stimulated JNK1 activation, we examined the extent of JNK phosphorylation at Thr183 and Tyr185, which are indicators of TNF-α–stimulated activation of the kinase, in W-PDK1−/− and N-PDK1−/− cell lines. We found that whereas TNF-α robustly stimulated phosphorylation of JNK1 in PDK1−/− and W-PDK1−/− cells, N-PDK1−/− cells completely resisted JNK1 phosphorylation in response to TNF-α (Fig. 3A and fig. S5A). Because TNF-α-induced JNK phosphorylation and activation are biphasic, with only the latter phase being responsible for the proapoptotic function of JNK (24), we performed a time-course study to examine JNK activation in W-PDK1−/− MEFs and N-PDK1−/− MEFs (fig. S5, B and C). Although the activation of JNK in N-PDK1−/− cells was comparable to that in W-PDK1−/− cells at an early time point after exposure to TNF-α, prolonged TNF-α–stimulated JNK activation, which is associated with the induction of apoptosis (24), was markedly inhibited in N-PDK1−/− cells (fig. S5, B and C). We did not detect any substantial differences in the kinetics of phosphorylation of p38 mitogen-activated protein kinase between W-PDK1−/− and N-PDK1−/− cells in response to TNF-α, suggesting that nuclear-localized PDK1 specifically inhibited the activation of JNK. Consistent with the reduced activity of JNK in N-PDK1−/− MEFs, the phosphorylation of the JNK downstream target c-Jun was also markedly inhibited in these cells (fig. S6).
Fig. 3.

Nuclear localization of PDK1 provides protection from apoptosis by inhibiting the JNK pathway. (A) MEFs of the indicated genotypes were treated with TNF-α (5 ng/ml) and actinomycin D (100 ng/ml) or were left untreated for 6 hours. JNK activation was determined by Western blotting analysis with a pJNK-specific antibody, and apoptosis was determined by detection of PARP cleavage. Quantification of three independent experiments is shown in fig. S5A (n = 3 experiments). (B) Transient expression of nuclear PDK1 inhibits JNK1 activation in W-PDK1−/− cells. W-PDK1−/− cells were transiently transfected with plasmid encoding YFP- tagged, nuclear PDK1. Twenty-four hours after transfection, cells were stimulated with TNF-α and actinomycin D for 4 hours. At the end of the treatment, cells were fixed, and the phosphorylation and subcellular localization of c-Jun were determined with an antibody against pSer63 of c-Jun followed by confocal immunofluorescence microscopy. Scale bars, 20 μm. Images are representative of four similar experiments. (C) W-PDK1−/− cells were pretreated with 50 μM SP600125 [a JNK inhibitor (JNKi)] or dimethyl sulfoxide (DMSO) for 1 hour before being treated with TNF-α and actinomycin D as described in (A). The extent of apoptosis was determined by Western blotting analysis with antibodies specific for the indicated proteins. n =3 experiments. (D) N-PDK1−/− cells were pretreated for 4 hours with 10 μM 124005 (an Akt inhibitor, Akti) or DMSO before being treated with TNF-α and actinomycin D as described in (A). The extent of apoptosis was determined by Western blotting with antibodies against the indicated proteins. n = 3 experiments.
To ensure that the suppression of c-Jun phosphorylation in N-PDK1−/− cells was not a result of clonal selection, we transiently expressed yellow fluorescent protein (YFP)–tagged nuclear PDK1 in W-PDK1−/− cells and examined TNF-α-stimulated phosphorylation of c-Jun (Fig. 3B). Only the W-PDK1−/− cells that expressed YFP-tagged nuclear PDK1 were able to block the phosphorylation and nuclear translocation of c-Jun (Fig. 3B). This result suggests that signaling resulting from transiently expressed nuclear-localized PDK1 is sufficient to inhibit JNK activation and c-Jun phosphorylation in cells that otherwise can activate JNK normally.
Next, we asked whether inhibition of JNK was necessary and sufficient for the suppression of TNF-α-stimulated apoptosis in MEFs. We treated W-PDK1−/− cells (which showed normal activation of JNK and apoptosis) with the JNK inhibitor SP600125 before treatment with TNF-α and actinomycin D. The inhibition of JNK by SP600125 effectively protected W-PDK1−/− cells from TNF-α–induced apoptosis (Fig. 3C). On the basis of these findings, we concluded that suppression of JNK activation and phosphorylation is one of the mechanisms by which nuclear PDK1 provided antiapoptotic functions in response to TNF-α. Finally, to determine the mechanism of inhibition of JNK by nuclear PDK1, we examined whether the increased Akt phosphorylation (Figs. 2 and 3A) observed in N-PDK1−/− cells contributed to JNK inhibition and the suppression of apoptosis. We found that inhibition of Akt with the inhibitor 124005, as demonstrated by loss of enhanced FOXO3A phosphorylation, resulted in substantial JNK reactivation and apoptosis in N-PDK1−/− cells (Fig. 3D). Together, our results suggest that the nuclear PDK1-Akt signaling antagonizes TNF-α-stimulated JNK activation and apoptosis.
Constitutive nuclear localization of PDK1 results in oncogenic transformation of MEFs
During our studies, we found that in addition to accelerated cell proliferation and resistance to apoptosis, the N-PDK1−/− cells formed clonal foci when allowed to form confluent monolayers. These observations led us to investigate the possible involvement of nuclear PDK1 in cellular transformation. We first compared the ability of W-PDK1−/− MEFs and N-PDK1−/− MEFs to support anchorage-independent growth in soft agar. We found that the N-PDK1−/− cells formed markedly more colonies on soft agar than did the W-PDK1−/− cells, indicating cellular transformation (Fig. 4, A and B). Furthermore, transfer of N-PDK1−/− cells into athymic nude mice resulted in robust tumor growth at the sites of injection (10 neoplasms per 10 injections) compared with the transfer of W-PDK1−/− cells (0 to 1 neoplasm per 10 injections) (Fig. 4, C and D). Tumors derived from N-PDK1−/− cells grew throughout the duration of the study (Fig. 4E). Hematoxylin and eosin (H&E) staining of the tumors isolated from mice implanted with N-PDK1−/− cells showed extensive nuclear morphology reminiscent of that in solid tumors, such as abnormal nuclear size and fragmentation pattern (Fig. 4F). Immunohistochemistry and Western blotting analysis of isolated tumors or of the tumor-derived cell lines revealed the presence of myc-tagged nuclear PDK1 (Fig. 4, G and H), confirming the presence of cells expressing nuclear-localized PDK1 in the tumors.
Fig. 4.
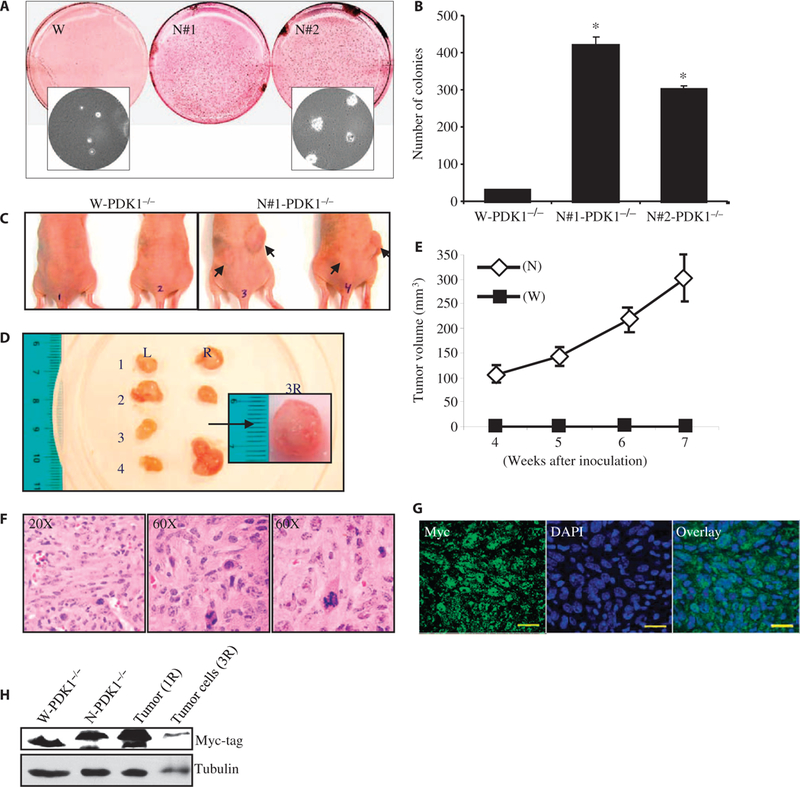
Nuclear localization of PDK1 causes oncogenic transformation of MEFs. (A) Cells (5 × 103) were seeded on 0.4% SeaPlaque agarose plates in triplicate. Three weeks after incubation, the colonies were stained and scanned on a flatbed scanner. The inset picture was taken at ×40 magnification before the colonies were stained. (B) Quantification of soft agar colonies formed from W-PDK1−/− and N-PDK1−/− cells after 3 weeks of growth. Data are means ± SD of three independent experiments. *P < 0.005 versus W-PDK1−/−. (C) In vivo tumorigenesis by N-PDK1−/− cells. Six-week-old athymic nude mice were injected with W-PDK1−/− cells or N-PDK1−/− cells. The arrows indicate tumor growth at the injection site 7 weeks after injection. (D) Size and morphological representation of tumors isolated from mice injected with N-PDK1−/− cells. L, left flank; R, right flank. The numbers correspond to individual mice. Inset picture shows the tumor isolated from the right flank of mouse 3 (tumor 3R) for the purpose of preparing cell lines. Scale bar, 1 cm. (E) Tumor growth curve during the 7 weeks of the study. The data represent the weekly average growth of tumors measured on the right flank of five mice injected with PDK1 cells. Data are means ± SEM. (F) Representative H&E staining image of tumor 4R. (G) Immunohistochemistry (IHC) of tumor 4R with an anti-myc antibody. (H) Representative Western blotting analysis from three separate experiments in which the presence of myc-tagged nuclear PDK1 was confirmed in homogenates derived from cells (tumor ID, 3R) and tumor (tumor ID, 1R). W-PDK1−/− and N-PDK1−/− MEF lanes are controls for the tumor-derived samples.
Late-stage human prostate tumors show an absence of cytoplasmically localized PDK1 with substantially higher amounts of nuclear-localized PDK1−/−
Loss of PTEN is a frequent occurrence in human prostate cancers, and hence, signaling through the PI3K pathway is often enhanced in these tumors. Because increased PI3K pathway activity stimulates the nuclear translocation of PDK1 (fig. S1A) (16), we compared the amounts of nuclear-localized PDK1 in DU145, LnCAP, and PC-3 cells, which are human prostate cancer cell lines. LnCAP and PC-3 cells lack endogenous PTEN and hence have higher basal PI3K pathway activity compared to that of DU145 cells, which express functional PTEN (25). LnCAP and PC-3, but not DU145, cells showed increased nuclear localization of endogenous PDK1 under all of the conditions tested (Fig. 5A). PDK1 derived from LnCAP cells showed a gel mobility shift similar to that of PDK1 isolated from N-PDK1−/− cells (Fig. 5B). We next compared the responses of DU145 and LnCAP cells to TNF-α, doxorubicin, or UV irradiation. Consistent with our observations of N-PDK1−/− cells, LnCAP cells showed substantially greater resistance to apoptosis stimulated by TNF-α or doxorubicin than did DU145 cells (Fig. 5C). Similar to N-PDK1−/− cells, UV radiation was as effective in inducing apoptosis in LnCAP cells as in DU145 cells (Fig. 5C). In addition, transient retroviral expression of nuclear-localized PDK1 in DU145 cells resulted in suppression of TNF-α-stimulated JNK activation and apoptosis (Fig. 5D). These results indicate a cell type-independent role for nuclear PDK1 in providing protection from apoptosis.
Fig. 5.

Nuclear-localized PDK1 protects human prostate tissue-derived cell lines from apoptosis. (A) Representative confocal images showing subcellular localization of PDK1 in LnCAP, PC-3, and DU145 cancer cells. LnCAP, PC-3, and DU145 cells growing on coverslips were fixed and stained with an anti-PDK1 antibody (green). The nuclei were visualized by staining with DAPI (blue). n = 4 experiments. (B) Comparison of PI3K pathway activity between DU145 and LnCAP cells lines. Note the slower mobility of endogenous PDK1 (marked with an asterisk) in LnCAP cells in which most PDK1 were found in the nucleus [see (A)]. (C) Responses of DU145 and LnCAP cells to apoptotic stimuli. Cells were treated for 6 hours with TNF-α (5 ng/ml) and actinomycin D (100 ng/ml), UV-C (30 mJ/m2), or doxorubicin (1 μg/ml) and lysed. Apoptosis was determined by Western blotting analysis with antibodies against the indicated proteins. (D) Retrovirus-mediated expression of nuclear PDK1 protects DU145 cells from TNF-α- stimulated JNK1 activation and apoptosis. Retroviral expression of empty vector or vector expressing nuclear PDK1 and subsequent treatment with TNF-α were performed as described in Materials and Methods. TNF-α and JNK activation and apoptosis were measured in cell lysates as in (C).
To determine whether the increased nuclear localization of PDK1 was associated with human cancers, we studied tissue samples from 88 independent human prostate tumors and examined by immunohistochemistry whether the extent of PDK1 nuclear localization correlated with tumor grade or risk factor. Of the 88 tissue cores examined, 14 showed PDK1 localization only in the cytosol (Fig. 6, A and B). All of these 14 samples were associated with lower tumor risk, lower Gleason score (≤7), and an early stage (I + II) of tumor development (Fig. 6B and table S1). All the cases with high risk, poor differentiation and at an advanced stage showed either nuclear-only localization of PDK1 or both nuclear and cytoplasmic localization. These results suggest the possible involvement of nuclear-localized PDK1 in tumor progression. Combining the data from our results with MEFs, human prostate tumor cell lines, as well as human prostate tissues, we conclude that nuclear translocation of PDK1 is involved in oncogenesis and in tumor progression.
Fig. 6.

Nuclear, but not cytoplasmic, PDK1 is present in high-grade, late-stage human prostate tumor tissues. (A) Representative IHC analysis of sections of human prostate tumor tissues. C, exclusive cytoplasmic localization; C = N, roughly equal distribution between the cytoplasmic and nuclear compartments; N, exclusive nuclear localization. (B)Graphical representation of the subcellular localization of PDK1 as determined by immunohistochemistical analysis of prostate tumor samples. The data represent the percentages of tissue cores showing cytoplasmic-only (C), cytoplasmic and nuclear (C = N), and exclusively nuclear (N) localization of PDK1 for each tumor grade type. No tumors with high risk, with high Gleason score, or at late clinical stage contained cytosolic-only PDK1. See Materials and Methods for an explanation of the data analysis. Statistical significance is indicated as the P value on each panel.
DISCUSSION
We described an oncogenic function for the nuclear translocation of PDK1. We showed that the nuclear localization of PDK1 resulted in the suppression of cellular apoptosis while promoting proliferation. This was achieved without an increase in PDK1 activity, suggesting that subcellular localization was a key determinant of the oncogenic potential of PDK1. Although the details of the mechanism by which enhanced nuclear PDK1 signaling culminates in tumor formation remain to be established, the increased nuclear accumulation of pAkt, as observed in the nucleus of N-PDK1−/− cells, could certainly play a role. In support of this view, increased amounts of nuclear Akt are linked to tumorigenesis (15). Inhibiting the kinase activity of Akt in N-PDK1−/− cells resulted in FOXO3A dephosphorylation, JNK1 reactivation, and apoptosis (Fig. 3D).
PDK1 is mostly cytoplasmic under normal cell culture conditions, which is a result of the presence of a strong NES, which we characterized previously. We and others have shown that growth factors, such as epidermal growth factor or insulin, or loss of PTEN results in increased nuclear localization of PDK1 (16, 17). The molecular mechanism that increases PDK1 nuclear localization remains unclear. Because PDK1 can function in a kinase-independent manner through protein-protein interactions (26) and its nuclear localization is independent of kinase activity (16), it is possible that protein-protein interactions with either nuclear import factors (such as importins) or other proteins may be involved in the nuclear import of PDK1. Uncovering of the nuclear import mechanism for PDK1 could be therapeutically exploited because the cytoplasmic localization of PDK1 was associated only with lower tumor risk in the prostate tumor samples that we examined.
Together with the findings of others (15, 27, 28), our results provide evidence of the functional importance of nuclear PI3K-PDK1-Akt signaling in oncogenesis. On the basis of these results, we propose a model of nuclear PDK1-induced tumorigenesis (fig. S7). In our model, PDK1 shuttles between the cytosol and the nucleus under normal conditions and is localized predominantly in the cytosol because of the presence of a strong NES sequence. Transient nuclear localization of PDK1 may contribute to the phosphorylation and activation of Akt and other potential PDK1 substrate kinases in the nucleus, and this may be essential for normal cell function. Under conditions of enhanced activation of the PI3K signaling pathway, such as after the loss of PTEN, the extent of nuclear localization of PDK1 is enhanced, leading to a sustained and prolonged activation of the nuclear PI3K signaling pathway and downstream events, such as enhanced proliferation and resistance to apoptosis, which lead to cellular transformation. Our results suggest that the chemotherapeutic prevention of the nuclear translocation of PDK1 may provide an effective strategy for the treatment of tumors with enhanced activation of the PI3K pathway.
MATERIALS AND METHODS
Cell lines and plasmids
PDK1+/+ and PDK1−/− MEFs were generated as described previously (29). PDK1−/− MEFs were maintained in Dulbecco’s modified Eagle’s medium (DMEM; Gibco) supplemented with 10% fetal bovine serum (FBS; Gibco) and 1% penicillin and streptomycin (CellGro). The LnCAP subline and DU145 cells were a gift from G. Piazza and were maintained in RPMI 1640 medium (CellGro) supplemented-with 10% FBS and 1% penicillin and streptomycin. To generate W-PDK1−/− and N-PDK1−/− cell lines, wild-type PDK1 and NES mutant form or PDK1 were first subcloned into pMSCV-Puror vector (BD Biosciences) as follows. Wild-type PDK1 was amplified from pcDNA/PDK1/myc (30) by PCR with the following primers: forward, 5′-CCGCTCGAGACTTGGGGCT-CATGGCCAG; reverse, 3′-CATCACCATCACCATTGAATTCC-5′, with the myc tag in frame. To generate pMSCV/nPDK1/myc, Leu383 and Phe386 in the NES of mouse PDK1 (16) were mutated to Ala383 and Asp386 by site-directed mutagenesis. Complementary DNAs (cDNAs) encoding wild-type PDK1 or PDK1L383A/F386D were then subcloned into the pMSCV-Puror vector. Wild-type, W-PDK1−/−, and N-PDK1−/− cells were generated by transfection of cells with the pMSCV/myc-wild-type PDK1 or pMSCV/myc-nPDK1 plasmids. The puromycin-resistant clones were selected by growing the cells in cell culture medium containing puromycin (5 μg/ml).
Cell synchronization and flow cytometric analysis
For cell synchronization, cells were plated at 5 × 105 cells per 100-mm dish containing DMEM supplemented with serum and antibiotics. After 24 hours, the medium was replaced with medium containing 0.1% serum with aphidicolin (2 μg/ml) for 12 hours, and then with serum-free medium with aphidicolin (2 μg/ml) for 8 hours to synchronize the cells in the G1 phase. Cells were released from synchronization by adding DMEM containing 10% FBS. For the experiment involving flow cytometric analysis, cells were collected every 6 hours after release from synchronization by trypsin detachment, washed with 1× phosphate-buffered saline, and fixed in 70% ethanol. DNA was labeled with propidium iodide (Invitrogen) and cleared with ribonuclease H (Invitrogen) before flow cytometric analysis.
Western blotting, immunofluorescence microscopy, immunohistochemistry, and quantification
Tissue and cell lysates were prepared as described previously (31). Protein concentration was determined by Bradford assay. For Western blotting analysis, proteins (25 mg per lane) were separated by SDS-PAGE and detected with antibodies against the following targets: p27Kip1 (Sigma), Rb (Santa Cruz Biotechnology), Akt (BioSource), tubulin (Sigma), JNK1 (Santa Cruz Biotechnology), cyclin E (Santa Cruz Biotechnology), cyclin D1 (Santa Cruz Biotechnology), PTEN (Cell Signaling Technology) PDK1 [Santa Cruz Biotechnology, PKB kinase antibody (E-3)], p21CIP1 (Sigma), caspase-3 (Cell Signaling Technology), and PARP (Cell Signaling Technology). All phospho-specific antibodies were from Cell Signaling Technology. The JNK inhibitor SP600125 and the Akt inhibitor 124005 were from Calbiochem. Western blots were quantified with Scion Image software. For immunofluorescence microscopy, cells were fixed and processed as described previously (16). The secondary antibodies used for immunofluorescence were from Invitrogen [Alexa Fluor 488, catalog nos. A-11001 (anti-mouse) and A-11008 (anti-rabbit), and Alexa Fluor 568, catalog nos. A-11031 (anti-mouse) and A-11011 (antirabbit)]. The specimens were mounted with ProLong Antifad Kit containing DAPI (Invitrogen).
Subcellular fractionation
For the experiment described in Fig. 2B, the cells were grown in five 15-cm dishes for each cell line type in DMEM containing 10% FBS overnight. Subcellular fractionations were performed essentially from these cells as described previously (16).
Quantitative reverse transcription PCR
RNA was isolated from cells after synchronization and release with the RNeasy kit from Qiagen. cDNAs were produced from 1 μg of total RNA with the SuperScript III system with oligo(dT) primers according to the manufacturer’s protocol (Invitrogen). For amplification of the sequence encoding p27Kip1, primers (CDKN1B; forward, 5′ -TCAAACGTGAGAGT- GTCTAACG-3′; reverse, 5′ -CCGGGCCGAAGAGATTTCTG-3′) were used, and quantitative PCR was performed with a Roche LightCycler 480 with the SYBR Green detection method. The normalization was performed with mouse TBP primers (forward, 5′AGAACAATCCAGACTAGCAGCA-3′; reverse, 5′-GGGAACTTCACATCACAGCTC-3′). The experiment was performed three times, with each experiment performed in triplicate.
Soft agar transformation assay and in vivo tumor mice model
Soft agar assays were performed in duplicate according to a protocol previously described (32). For in vivo tumorogenicity experiments, 5 × 106 W-PDK1−/− or N-PDK1−/− cells suspended in Matrigel (BD Biosciences) were injected into the right and left flanks of 6-week-old athymic nude mice. Tumor size was determined weekly with calipers, and tumor volume was determined with the following formula: 0.5(L × W2), where L is the longest diameter and W is the shortest diameter. Tumors isolated from mice after 7 weeks were fixed in 10% formalin, homogenized to produce tumor lysates, or processed to generate cell lines. The formalin-fixed tumors were embedded in paraffin according to a standard procedure, which was followed by deparaffinization and antigen retrieval of the sections for H&E staining to determine tissue morphology or immunofluorescence staining to determine the presence of myc-PDK1 and its localization in tumor tissues.
Retrovirus production and infection
Phoenix Ampho 293T cell lines were transfected with pMSCV-Puror vector or with pMSCV-PDK1 (L383AF386D)–myc–Puror with Lipofect-amine 2000 reagent. Twenty-four hours after transfection, culture media containing either vector alone or nPDK1 virus were filtered through a 0.45-mm filter and used to infect 1 × 106 DU145 cells with polybrene (4 μg/ml). Virus-containing media were removed from the cells after 12 hours and replaced with DMEM containing 10% FBS and 1% penicillin and streptomycin. Thirty-six hours after the initial infection, cells were stimulated with TNF-α and actinomycin D for 6 hours or left untreated. Cell lysates were prepared and analyzed for JNK activation with an antibody specific for JNK pThr183/Tyr185, and apoptosis was determined with an antibody against PARP.
32P Metabolic in vivo labeling and in vitro kinase assays for PDK1 activity
Metabolic 32P in vivo labeling for PDK1 was performed as previously described (33). For in vitro kinase assays, human embryonic kidney 293T cells in fifteen 10-cm dishes were transfected with a total of 100 μg of plasmid encoding hemagglutinin (HA)–tagged kinase-inactive mutant Akt. Eighteen hours after transfection, the medium was changed to DMEM without serum containing 5 μM LY2940025 and 5 μM wortmannin for 12 hours. Four hours before harvesting, the concentrations of LY2940025 and wortmannin were increased to 25 μM in the same medium. The cells were lysed in the cell lysis buffer described earlier, and HA-tagged Akt was immunoprecipitated from cell lysates with anti-HA Sepharose beads (Sigma). After 4 hours, immunoprecipitated samples were washed three times with lysis buffer followed by lysis buffer containing 500 mM NaCl. For HA-Akt elution from the beads, the immunoprecipitates were washed three times with HA-peptide elusion buffer (18 mM Hepes, 5 mM KCl). Elution was performed with 100 mg of HA peptide. The HA peptides were removed by centrifugation of the eluent through the desalting spin column (Pierce). For kinase reactions, PDK1 was immunoprecipitated from the cell lysates of PDK1−/−, W-PDK1−/−, and N-PDK1−/− cells that were maintained under normal logarithmic growth conditions with anti-myc-conjugated Sepharose beads. Immunoprecipitates were washed three times (similar to the HA-Akt samples) with low-salt (150 mM NaCl) or high-salt (500 mM NaCl) lysis buffer followed by three washes with PDK1 kinase buffer (33). The kinase reaction was performed by adding 250 ng of purified HA-Akt to each PDK1 reaction tube containing kinase buffer together with 300 mM nonradioactive adenosine 5′-triphosphate. The kinase reaction was stopped by adding SDS-PAGE loading buffer. The phosphorylation of Akt was measured by Western blotting analysis with an antibody specific for pAkt-Thr308.
Tumor immunohistochemistry
This study was approved by the institutional review board of Shanghai Renji Hospital, affiliated with Shanghai Jiaotong University. A total of 88 cases of primary prostate cancer were retrieved from the Department of Pathology, Renji Hospital. Specimens were formalin-fixed and paraffin-embedded (FFPE) and made into four tissue microarrays. All cases were independently reviewed and graded (according to the World Health Organization consensus Gleason score) by two pathologists. A Gleason score of 2 to 4 is defined as low-grade (well differentiated), 5 to 7 as medium-grade (moderately differentiated), and 8 to 10 as high-grade (poorly differentiated). Clinical stages of these tumors were classified by the tumor-node-metastasis classification either into an early-stage category characterized by stage I to II tumors confined to prostate only or into a late-stage category characterized by stage III to IV tumors beyond the prostate or by metastasis to lymph nodes or other organs. Tumors at an early clinical stage (I + II) and with a Gleason score <7 were categorized into low-risk, whereas other tumors were categorized as high-risk. FFPE tissues sectioned at 4 mm onto slides were dewaxed and rehydrated. Antigen retrieval was performed in a pressure cooker at 110°C for 5 min in retrieval buffer (pH 9.0) (Dako). Endogenous peroxidase activity was blocked with 3% hydrogen peroxide (Dako). Sections were then incubated for 1 hour with antibody against total PDK1 at a dilution of 1:50. Immunocomplexes were detected by incubation with anti-rabbit peroxidase-labeled polymer (Dako) and visualized with diaminobenzidine for 10 min (Dako). Normal rabbit immunoglobulin G diluted to match the concentration of the primary antibody was used as the negative control. All stained slides were scored with a light microscope by a pathologist. A subjective reporting procedure was implemented, recording location (C, cytoplasm only; N, nucleus only; and C + N, both cytoplasm and nucleus) and intensity (mild, +; moderate, ++; and strong, +++) for PDK1.
Supplementary Material
Acknowledgments:
We thank G. Piazza for the LnCAP and DU145 prostate cell lines, D. Hu for the excellent technical support in tissue culture, A. Bandyopadhyay and L. Wang for the help with mice studies, and I.-H. Yeh and the University of Texas Health Science Center at San Antonio Pathology Core for the help with histological and pathological analysis related with mouse tumor samples.
Funding: This work was supported by grants from the NIH (AG26243 to F.L., DK69930 to L.Q.D., and CA079683 to L.-Z.S.), the American Diabetes Association (research award to F.L.), and the American Heart Association Texas Affiliate (Grant-in-Aid no. 0355076Y to L.Q.D.).
Footnotes
Competing interests: The authors declare that they have no competing financial interests.
REFERENCES AND NOTES
- 1.Fruman DA, Meyers RE, Cantley LC, Phosphoinositide kinases. Annu. Rev. Biochem. 67, 481–507 (1998). [DOI] [PubMed] [Google Scholar]
- 2.Zhao L, Vogt PK, Class I PI3K in oncogenic cellular transformation. Oncogene 27, 5486–5496 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Myers MP, Tonks NK, PTEN: Sometimes taking it off can be better than putting it on. Am. J. Hum, Genet. 61, 1234–1238 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cairns P, Okami K, Halachmi S, Halachmi N, Esteller M, Herman JG, Jen J, Isaacs WB, Bova GS, Sidransky D, Frequent inactivation of PTEN/MMAC1 in primary prostate cancer. Cancer Res. 57, 4997–5000 (1997). [PubMed] [Google Scholar]
- 5.Samuels Y, Ericson K, Oncogenic PI3K and its role in cancer. Curr. Opin. Oncol. 18, 77–82 (2006). [DOI] [PubMed] [Google Scholar]
- 6.Vanhaesebroeck B, Alessi DR, The PI3K-PDK1 connection: More than just a road to PKB. Biochem. J. 346, 561–576 (2000). [PMC free article] [PubMed] [Google Scholar]
- 7.Alessi DR, Discovery of PDK1, one of the missing links in insulin signal transduction. Colworth Medal Lecture. Biochem. Soc. Trans. 29, 1–14 (2001). [DOI] [PubMed] [Google Scholar]
- 8.Alessi DR, Deak M, Casamayor A, Caudwell FB, Morrice N, Norman DG, Gaffney P, Reese CB, MacDougall CN, Harbison D, Ashworth A, Bownes M, 3-Phosphoinositide-dependent protein kinase-1 (PDK1): Structural and functional homology with the Drosophila DSTPK61 kinase. Curr. Biol. 7, 776–789 (1997). [DOI] [PubMed] [Google Scholar]
- 9.Mora A, Komander D, van Aalten DM, Alessi DR, PDK1, the master regulator of AGC kinase signal transduction. Semin. Cell Dev. Biol. 15, 161–170 (2004). [DOI] [PubMed] [Google Scholar]
- 10.Neri LM, Borgatti P, Capitani S, Martelli AM, The nuclear phosphoinositide 3-kinase/AKT pathway: A new second messenger system. Biochim. Biophys. Acta 1584, 73–80 (2002). [DOI] [PubMed] [Google Scholar]
- 11.Martelli AM, Faenza I, Billi AM, Manzoli L, Evangelisti C, Falà F, Cocco L, Intranuclear 3′-phosphoinositide metabolism and Akt signaling: New mechanisms for tumorigenesis and protection against apoptosis? Cell. Signal. 18, 1101–1107 (2006). [DOI] [PubMed] [Google Scholar]
- 12.Ye K, Hurt KJ, Wu FY, Fang M, Luo HR, Hong JJ, Blackshaw S, Ferris CD, Snyder SH, Pike. A nuclear GTPase that enhances PI3kinase activity and is regulated by protein 4.1N. Cell 103, 919–930 (2000). [DOI] [PubMed] [Google Scholar]
- 13.Martelli AM, Evangelist C, Billi AM, Manzoli L, Papa V, Cocco L, Intranuclear 3′-phosphoinositide metabolism and apoptosis protection in PC12 cells. Acta Biomed. 78 (Suppl. 1), 113–119 (2007). [PubMed] [Google Scholar]
- 14.Ye K, PIKE/nuclear PI 3-kinase signaling in preventing programmed cell death. J. Cell. Biochem. 96, 463–472 (2005). [DOI] [PubMed] [Google Scholar]
- 15.Trotman LC, Alimonti A, Scaglioni PP, Koutcher JA, Cordon-Cardo C, Pandolfi PP, Identification of a tumour suppressor network opposing nuclear Akt function. Nature 441, 523–527 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lim MA, Kikani CK, Wick MJ, Dong LQ, Nuclear translocation of 3′-phosphoinositide- dependent protein kinase 1 (PDK-1): A potential regulatory mechanism for PDK-1 function. Proc. Natl. Acad. Sci. U.S.A. 100, 14006–14011 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Scheid MP, Parsons M, Woodgett JR, Phosphoinositide-dependent phosphorylation of PDK1 regulates nuclear translocation. Mol. Cell. Biol. 25, 2347–2363 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Williams MR, Arthur JS, Balendran A, van der Kaay J, Poli V, Cohen P, Alessi DR, The role of 3-phosphoinositide-dependent protein kinase 1 in activating AGC kinases defined in embryonic stem cells. Curr. Biol. 10, 439–448 (2000). [DOI] [PubMed] [Google Scholar]
- 19.Coller HA, What’s taking so long? S-phase entry from quiescence versus proliferation. Nat. Rev. Mol. Cell Biol. 8, 667–670 (2007). [DOI] [PubMed] [Google Scholar]
- 20.Slingerland J, Pagano M, Regulation of the Cdk inhibitor p27 and its deregulation in cancer. J. Cell. Physiol. 183, 10–17 (2000). [DOI] [PubMed] [Google Scholar]
- 21.Di Cristofano A, De Acetis M, Koff A, Cordon-Cardo C, Pandolfi PP, Pten and p27KIP1 cooperate in prostate cancer tumor suppression in the mouse. Nat. Genet.222–224 (2001). [DOI] [PubMed] [Google Scholar]
- 22.Stahl M, Dijkers PF, Kops GJ, Lens SM, Coffer PJ, Burgering BM, Medema RH, The forkhead transcription factor FoxO regulates transcription of p27Kip1 and Bim in response to IL-2. J. Immunol. 168, 5024–5031 (2002). [DOI] [PubMed] [Google Scholar]
- 23.Deng Y, Ren X, Yang L, Lin Y, Wu X, A JNK-dependent pathway is required for TNFα-induced apoptosis. Cell 115, 61–70 (2003). [DOI] [PubMed] [Google Scholar]
- 24.Ventura JJ, Hübner A, Zhang C, Flavell RA, Shokat KM, Davis RJ, Chemical genetic analysis of the time course of signal transduction by JNK. Mol. Cell 21, 701–710 (2006). [DOI] [PubMed] [Google Scholar]
- 25.Huang H, Cheville JC, Pan Y, Roche PC, Schmidt LJ, Tindall DJ, PTEN induces chemosensitivity in PTEN-mutated prostate cancer cells by suppression of Bcl-2 expression. J. Biol. Chem. 276, 38830–38836 (2001). [DOI] [PubMed] [Google Scholar]
- 26.Tian X, Rusanescu G, Hou W, Schaffhausen B, Feig LA, PDK1 mediates growth factor-induced Ral-GEF activation by a kinase-independent mechanism. EMBO J. 21, 1327–1338 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Trotman LC, Wang X, Alimonti A, Chen Z, Teruya-Feldstein J, Yang H, Pavletich NP, Carver S, Cordon-Cardo C, Erdjument-Bromage H, Tempst P, Chi SG, Kim HJ, Misteli T, Jiang X, Pandolfi PP, Ubiquitination regulates PTEN nuclear import and tumor suppression. Cell 128, 141–156 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Luo JL, Tan W, Ricono JM, Korchynskyi O, Zhang M, Gonias SL, Cheresh DA, Karin M, Nuclear cytokine-activated IKKα controls prostate cancer metastasis by repressing Maspin. Nature 446, 690–694 (2007). [DOI] [PubMed] [Google Scholar]
- 29.Wang C, Liu M, Riojas RA, Xin X, Gao Z, Zeng R, Wu J, Dong LQ, Liu F, Protein kinase C θ (PKC6)-dependent phosphorylation of PDK1 at Ser504 and Ser532 contributes to palmitate-induced insulin resistance. J. Biol. Chem. 284, 2038–2044 (2009). [DOI] [PubMed] [Google Scholar]
- 30.Wick MJ, Wick KR, Chen H, He H, Dong LQ, Quon MJ, Liu F, Substitution of the autophosphorylation site Thr516 with a negatively charged residue confers constitutive activity to mouse 3-phosphoinositide-dependent protein kinase-1 in cells. J. Biol. Chem. 277, 16632–16638 (2002). [DOI] [PubMed] [Google Scholar]
- 31.Wang L, Balas B, Christ-Roberts CY, Kim RY, Ramos FJ, Kikani CK, Li C, Deng S Reyna N Musi LQ Dong R DeFronzo A, Liu F, Peripheral disruption of the Grb10 gene enhances insulin signaling and sensitivity in vivo. Moi. Cell. Biol. 27, 6497–6505 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Skeen JE, Bhaskar PT, Chen CC, Chen WS, Peng XD, Nogueira V, Hahn-Windgassen A, Kiyokawa H, Hay N, Akt deficiency impairs normal cell proliferation and suppresses oncogenesis in a p53-independent and mTORC1- dependent manner. Cancer Cell 10, 269–280 (2006). [DOI] [PubMed] [Google Scholar]
- 33.Riojas RA, Kikani CK, Wang C, Mao X, Zhou L, Langlais PR, Hu D, Roberts JL, Dong LQ, Liu F, Fine tuning PDK1 activity by phosphorylation at Ser163. J. Biol. Chem. 281, 21588–21593 (2006). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.