

Abstract
To explore the developmental reorganization of the three-dimensional genome of the brain in the context of neuropsychiatric disease, we monitored chromosomal conformations in differentiating neural progenitor cells. Neuronal and glial differentiation was associated with widespread developmental remodeling of the chromosomal contact map and included interactions anchored in common variant sequences that confer heritable risk for schizophrenia. We describe cell type–specific chromosomal connectomes composed of schizophrenia risk variants and their distal targets, which altogether show enrichment for genes that regulate neuronal connectivity and chromatin remodeling, and evidence for coordinated transcriptional regulation and proteomic interaction of the participating genes. Developmentally regulated chromosomal conformation changes at schizophrenia-relevant sequences disproportionally occurred in neurons, highlighting the existence of cell type–specific disease risk vulnerabilities in spatial genome organization.
Graphical Abstract
INTRODUCTION:
Chromosomal conformations, topologically associated chromatin domains (TADs) assembling in nested fashion across hundreds of kilobases, and other “three-dimensional genome” (3DG) structures bypass the linear genome on a kilo- or megabase scale and play an important role in transcriptional regulation. Most of the genetic variants associated with risk for schizophrenia (SZ) are common and could be located in enhancers, repressors, and other regulatory elements that influence gene expression; however, the role of the brain’s 3DG for SZ genetic risk architecture, including developmental and cell type–specific regulation, remains poorly understood.
RATIONALE:
We monitored changes in 3DG after isogenic differentiation of human induced pluripotent stem cell–derived neural progenitor cells (NPCs) into neurons or astrocyte-like glial cells on a genome-wide scale using Hi-C. With this in vitro model of brain development, we mapped cell type–specific chromosomal conformations associated with SZ risk loci and defined a risk-associated expanded genome space.
RESULTS:
Neural differentiation was associated with genome-wide 3DG remodeling, including pruning and de novo formations of chromosomal loopings. The NPC-to-neuron transition was defined by the pruning of loops involving regulators of cell proliferation, morphogenesis, and neurogenesis, which is consistent with a departure from a precursor stage toward postmitotic neuronal identity. Loops lost during NPC-to-glia transition included many genes associated with neuron-specific functions, which is consistent with non-neuronal lineage commitment. However, neurons together with NPCs, as compared with glia, harbored a much larger number of chromosomal interactions anchored in common variant sequences associated with SZ risk. Because spatial 3DG proximity of genes is an indicator for potential coregulation, we tested whether the neural cell type–specific SZ-related “chromosomal connectome” showed evidence of coordinated transcriptional regulation and proteomic interaction of the participating genes.
To this end, we generated lists of genes anchored in cell type–specific SZ risk-associated interactions. Thus, for the NPC-specific interactions, we counted 386 genes, including 146 within the risk loci and another 240 genes positioned elsewhere in the linear genome but connected via intrachromosomal contacts to risk locus sequences. Similarly, for the neuron-specific interactions, we identified 385 genes: 158 within risk loci and 227 outside of risk loci. Last, for glia-specific interactions, we identified 201 genes: 88 within and 113 outside of risk loci. We labeled the genes located outside of schizophrenia risk loci as “risk locus–connect,” which we define as a collection of genes identified only through Hi-C interaction data, expanding—depending on cell type—by 50 to 150% the current network of known genes overlapping risk sequences that is informed only by genome-wide association studies. This disease-related chromosomal connectome was associated with “clusters” of coordinated gene expression and protein interactions, with at least one cluster strongly enriched for regulators of neuronal connectivity and synaptic plasticity and another cluster for chromatin-associated proteins, including transcriptional regulators.
CONCLUSION:
Our study shows that neural differentiation is associated with highly cell type–specific 3DG remodeling. This process is paralleled by an expansion of 3DG space associated with SZ risk. Specifically, developmentally regulated chromosomal conformation changes at SZ-relevant sequences disproportionally occurred in neurons, highlighting the existence of cell type–specific disease risk vulnerabilities in spatial genome organization.
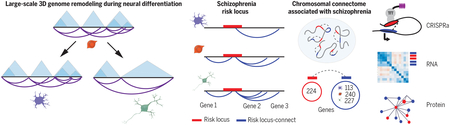
3DG remodeling across neuronal differentiation with parallel expansion of SZ risk space. (Left) Chromatin conformation assays reveal pruning of short-range loops in neurons along with widening of TADs upon differentiation from NPCs. (Right) Cell type–specific chromatin interactions, functionally validated with CRISPR assays, expand the network of known risk-associated genes (blue circle), which show evidence for coregulation at the transcriptomic and proteomic levels.
Spatial genome organization is highly regulated and critically important for normal brain development and function (1). Many of the risk variants contributing to the heritability of complex genetic psychiatric disorders are located in noncoding sequences (2), presumably embedded in “three-dimensional genome” (3DG) structures important for transcriptional regulation, such as chromosomal loop formations that bypass linear genome on a kilobase (or megabase) scale and topologically associated domains (TADs) (3) that assemble in nested fashion across hundreds of kilobases (4–7). By linking noncoding schizophrenia-associated genetic variants with distal gene targets, 3DG mapping with Hi-C (3, 8) and other genome-scale approaches could inform how higher-order chromatin organization affects genetic risk for psychiatric disease. To date, only a very limited number of Hi-C datasets exist for the human brain: two generated from bulk tissue of developing forebrain structures (7) and adult brain (9) and one from neural stem cells (10). Although such datasets have advanced our understanding of the genetic risk architecture of psychiatric disease (7, 11), 3DG mapping from postmortem tissue lacks cell type–specific resolution and may not capture higher-order chromatin structures sensitive to the autolytic process (12). We monitored developmentally regulated changes in chromosomal conformations during the course of isogenic neuronal and glial differentiation, describing large-scale pruning of chromosomal contacts during the transition from neural progenitor cells (NPCs) to neurons. Furthermore, we uncovered an expanded 3DG risk space for schizophrenia—with a functional network of disease-relevant regulators of neuronal connectivity, synaptic signaling, and chromatin remodeling—and demonstrate neural cell type–specific coordination at the level of the chromosomal connectome, transcriptome, and proteome.
Results
Neural progenitor differentiation is associated with dynamic 3DG remodeling
We applied in situ Hi-C to map the 3DG of two male human induced pluripotent stem cell (hiPSC)–derived neural progenitor cells (NPCs) (13), together with isogenic populations of induced excitatory neurons (“neuron”) generated through viral overexpression of the transcription factor NGN2 (14) and differentiations of astrocyte-like glial cells (“glia”) (Fig. 1, A and B, and table S1) (15). Transcriptome RNA sequencing (RNA-seq) comparison with published datasets (16) confirmed that the NPCs, but not glia, from subjects S1 and S2 clustered together with NPCs from independent donors, whereas S1 and S2 NGN2 neurons closely aligned with directed differentiation forebrain neurons (17) and prenatal brain datasets (fig. S1, A and B). As with our transcriptomic datasets, hierarchical clustering of our Hi-C datasets after initial processing (fig. S2A) also showed clear separation by cell type (Fig. 1A and fig. S2B). Genome-scale interaction matrices were enriched for intrachromosomal conformations (fig. S2C), with the exception of the negative control (“No Ligase”) NPC library, in which we omitted the ligase step (Materials and methods) and observed an interaction map with no signal due to the loss of chimeric fragments (fig. S2D). Given the observed correlation between technical replicates of Hi-C assays from the same donor and cell type, and the correlation between cell type–specific Hi-C from the two donors (Pearson correlation of PC1, Rtechnical replicates, range = 0.970 to 0.979; Rsubject1-subject 2 by cell type, range = 0.962 to 0.970), we pooled by cell type for subsequent analyses (fig. S2E).
Fig. 1. Neural differentiation is associated with large-scale remodeling of the 3D genome.

(A) (Top) Derivation scheme of isogenic cell types from two male control cell lines. Pink oval, donor hiPSC; orange, NPC; green, neuron; purple, glia. (Bottom) Hierarchical clustering of intrachromosomal interactions (Materials and methods) from six in situ Hi-C libraries. a and b are technical replicates of the same library; height corresponds to the distance between libraries (Materials and methods) (fig. S2B). (B) Immunofluorescent staining of characteristic cell markers for NPCs (Nestin and SOX2), neurons (TUJ1 and MAP2), and glia (Vimentin and S100β). (C) Venn diagram of loop calls specific to and shared by different subsets of cells, including previously published GM12878 lymphoblastoid Hi-C data. (D) Gene ontology (GO) enrichment (significant terms only) of genes overlapping anchors of loops shared by NPCs, neurons, and glia but absent in GM12878. (E) (Left) Cell-type pooled whole-genome heatmaps at 500-kb resolution (fig. S2C). (Right) “Arc map” showing intrachromosomal interactions at 40-kb resolution of the q-arm of chr17 for isogenic neurons, NPCs, and glia, as indicated, from subject 2. RNA-seq tracks for each cell type shown on top of arc maps. Green, neuron; orange, NPC; purple, glia. (F) FPKM gene expression of CUX2 across three cell types with heatmap zoomed in on CUX2 loop (black arrow) (fig. S3). (G) Number of loops specific to each cell type (not shared with other cell types) with one anchor in an A compartment and another in a B compartment (pink), both in B compartments (red), or both in A compartments (blue). (H) (Left) Box-and-whisker distribution plot of TAD size across four cell types. (Right) Median TAD length for each of the four cell types. (I) Heatmaps at 40-kb resolution for a 3-Mb window at the CDH2 locus on chr18. (Bottom) Nested TAD landscape in glia with multiple subTADs (black arrows) called, which (top) is absent from neuronal Hi-C. RNA-seq tracks: green, neuron; purple, glia (figs. S1 to S5).
We first focused on intrachromosomal loop formations, which are conservatively defined as distinct contacts between two loci in the absence of similar interactions in the surrounding sequences (3). Our comparative analyses included published (3) in situ Hi-C data from the B lymphocyte–derived cell line GM12878 (table S1). When analyzed with the HiCCUPS pipeline (5- and 10-kb loop resolutions combined, sub-sampled to 372 million valid-intrachromosomal read pairs to reflect the library with the fewest reads after filtration) (3), 17,767 distinct loops were called: n = 3118 (17.5%) were shared among all four cell types, whereas n = 5068 (28.5%) were specific to only one of the four cell types (Fig. 1C). Biologically relevant terms such as “central nervous system development,” “forebrain development,” and “neuron differentiation” were among the top gene ontology (GO) enrichments from genes overlapping loops shared between NPCs, glia, and neurons (brain-specific) but not identified in lymphocytes (Fig. 1D and table S2), indicating strong tissue-specific loop signatures that were also confirmed in individual cell types (fig. S3A and tables S3 to S6).
Unexpectedly, there was a reduction (~40 to 50% decrease) in the total number of chromosomal loops in neurons relative to isogenic glia and NPCs (fig. S3, B and C). Reduced densities of chromosomal conformations were also evident in genome browser visualization of chromosomal arms, including chr17q (Fig. 1E). Although both glia and NPCs harbored ~13,000 loop formations, only 7206 were identified in neurons (Fig. 1C; fig. S3, B and C; and table S1), including 442 neuron-specific loop formations. One such neuron-specific loop was at CUX2, a transcription factor whose expression marks a subset of cortical projection neurons (18) and that is highly expressed in our NGN2-induced neurons (Fig. 1F and fig. S3, D and E). Examples of loops lost in neurons include one spanning the Ca2+ channel and dystonia-risk gene, ANO3 (fig. S3F) (19). Furthermore, NPCs, neurons, and glia had similar proportions of loops anchored in solely active (A) compartments, solely inactive (B) compartments, or in both, indicating no preferential loss of either active or inactive loops in neurons (Fig. 1G). However, among the genes overlapping anchors of loops that underwent pruning during the course of the NPC-to-neuron transition, regulators of cell proliferation, morphogenesis, and neurogenesis ranked prominently in the top 25 GO terms with significant enrichment (Benjamini-Hochberg corrected P < 10−6 – 10−12) (fig. S3G and table S4B), which is consistent with a departure from precursor stage toward postmitotic neuronal identity (20). Likewise, loops lost during NPC-to-glia transition were significantly enriched (Benjamini-Hochberg corrected P < 10−3 – 10−6) for neuron-specific functions, including “transmission across chemical synapse,” “γ-aminobutyric acid (GABA) receptor activation,” and “postsynapse” (fig. S3G and table S4C), which is consistent with non-neuronal lineage commitment.
We defined “loop genes” as genes that either have gene body or transcription start site (TSS) overlap with a loop anchor (5- or 10-kb bins forming the points of contact in a chromatin loop). Genes with loop-bound gene bodies (one-tailed Z test, Zrange = 42.1 to 59.2, P < 10−324 for all) or loop-bound TSS (one-tail Z-test, Zrange = 15.2 to 28.8, Prange < 2.32 × 10−52 to 4.40 × 10−182) both showed significantly greater expression [mean log10(FPKM + 1); FPKM, fragments per kilobase of exon per million fragments mapped] than that of background (all genes for all brain cell types) (fig. S4A), suggesting that looping architecture was associated with increased gene expression. Furthermore, 3% of loops shared by NPCs, neurons, and glia (brain-specific loops) interconnected a brain expression quantitative trait locus (eQTL) single-nucleotide polymorphism (SNP) with its destined target gene(s), representing significant enrichment over background as determined with 1000 random distance- and functional annotation–matched loop samplings, (random sampling, one-sided empirical P = 0.012) (Materials and methods) (fig. S4B).
We aimed to confirm that the observed net loss of loop formations during the NPC-to-neuron transition could be replicated across a variety of independent cell culture and in vivo approaches and was not specific to our methodological choice of NGN2-induction. We conducted an additional Hi-C experiment on cells differentiated from hiPSC-NPCs by means of a non-NGN2 protocol that used only differentiation medium and yielded a heterogeneous population of hiPSC-forebrain-neurons in addition to a small subset of glia (17). In addition, we reanalyzed Hi-C datasets generated from a mouse model of neural differentiation, consisting of mouse embryonic stem cell (mESCs), mESC-derived NPCs (mNPC), and cortical neurons (mCN) differentiated from the mNPCs via inhibition of the Sonic Hedgehog (SHH) pathway (21). To examine whether such genome-wide chromosomal loop remodeling also occurred in the developing brain in vivo, we reanalyzed Hi-C data from human fetal cortical plate (CP), mostly composed of young neurons, and forebrain germinal zone (GZ), primarily harboring dividing neural precursor cells in addition to a smaller subset of newly generated neurons (7). Across both the hiPSC-NPC-to-forebrain neuron and mESC-mNPC-mCN differentiation, in vitro neurons showed a 20% decrease in loops compared with their neural progenitors (fig. S4, C and D). Consistent with this, in vivo CP (neuron) compared with GZ (progenitor) showed a 13% decrease in loops genome-wide (fig. S4E). The highly replicative cell types included here, mouse ESCs and human lymphoblastoid GM12878 cells, exhibited loop numbers very similar to their neuronal counterparts (fig. S4, D and E), suggesting that the changes in 3DG architecture from NPC to neurons do not simply reflect a generalized effect explained by mitotic potential.
Along with having fewer total loops, neurons exhibited a greater proportion of longer-range (>100 kb) loops than did NPCs or glia (two-sample two-tailed Kolmogorov-Smirnov test, KSrange = 0.1269 to 0.2317, P < 2.2 × 10−16 for three comparisons: Neu versus NPC/Glia/GM) (fig. S5A). Likewise, in each of the alternative in vitro and in vivo analyses considered above, neurons exhibited a greater proportion of longer-range (>100 kb) loops than did NPCs or glia [two-sample two-tailed Kolmogorov-Smirnov test, KS = 0.0427, P = 1.5 × 10−5 for hiPSC-NPC versus forebrain neuron; KS = 0.0936, P = 1.1 × 10−16 for mESC-NPC versus mCN; KS = 0.0663, P = 2.04 × 10−8 for fetal CP (neuron) compared with GZ (progenitor)] (fig. S5, B, C, D, and E). Therefore, multiple in vitro and in vivo approaches comparing, in human and mouse, neural precursors to young neurons consistently show a reduced number of loops in neuron-enriched cultures and tissues, primarily affecting shorter-range loops.
Consistent with studies in peripheral tissues reporting conservation of the overall loop-independent TAD landscape across developmental stages, tissues, and species (when considering syntenic loci) (10, 22), overall TAD landscapes (3) remained similar between neurons, glia, and NPCs. Nonetheless, TADs also showed a subtle (~10%) increase in average size in neurons compared with isogenic NPCs, independent of the differentiation protocol applied (Wilcoxon-Mann-Whitney test, P < 5.3 × 10−6) (Fig. 1H and fig. S5, F and G), as highlighted here at a 3.4-Mb TAD at the CDH2 cell adhesion gene locus (Fig. 1I). TAD remodeling may therefore reflect restructuring of nested subdomains within larger neuronal TADs (tables S7 and S8). To examine whether such developmental reorganization of the brain’s spatial genomes was associated with a generalized shift in chromatin structure, we applied the assay for transposase accessible chromatin with high-throughput sequencing (ATAC-seq) to map open chromatin sequences before and after NGN2-neuronal induction (table S1). Genome-wide distribution profiles for transposase-accessible chromatin were only minimally different between NPCs and neurons (fig. S5H) and further revealed that both NPCs and neurons showed low to moderate chromatin accessibility [–2.5 < log2(ATAC signal) < 1] for ≥89% of the anchor sequences comprising cell type–specific and shared “brain” loops in our cell culture system (fig. S5I). These findings, taken together, point to widespread 3DG changes during the NPC-to-neuron transition and NPC-to-glia transition in human and mouse brain that are unlikely attributable to global chromatin accessibility differences. This includes highly cell type–specific signatures in gene ontologies of differentiation-induced loop prunings, reflecting neuronal and glial (non-neuronal) lineage commitment (fig. S3, A and G, and table S4, B and C), and a subtle widening of average loop and TAD length in young neurons (Fig. 1H and fig. S5, A to G).
Chromosomal contacts associated with schizophrenia risk sequences
Because many schizophrenia risk variants lie in noncoding regions in proximity to several genes, we predicted that chromosomal contact mapping could resolve putative regulatory elements capable of conferring schizophrenia risk via their physical proximity (bypassing linear genome) to the target gene, as has been demonstrated in tissue in vivo (7, 11). We overlaid our cell type–specific interactions onto the 145 risk loci associated with schizophrenia risk (2, 23). Because only very few loops (defined as distinct pixels with greater contact frequency than neighboring pixels on a contact map) (3) were associated with schizophrenia risk loci (n = 212, 81, and 17 loops in NPC, glia, and neurons, respectively) (table S9), we applied an established alternative approach to more comprehensively explore the 3DG in context of disease-relevant sequences (7). This approach defines interactions as those filtered contacts that stand out over the global background and applies binomial statistics to identify chromosomal contacts anchored at disease-relevant loci (7). To begin, we examined the 40 loci with strongest statistical evidence for colocalization of an adult postmortem brain eQTL and schizophrenia genome-wide association study (GWAS) signal (24). Chromosomal contacts were called for 29 of the 46 eQTLs present in the 40 loci, with 8 of 29 (28%) of the loci showing significant interactions (binomial test, −log q value range = 1.33 to 11.0) between the eQTL-SNPs (eSNPs) in the one contact anchor and the transcription start site of the associated gene(s) in the other anchor (table S10). We conclude that ~30% of risk locus–associated eQTLs with strong evidence for colocalization with GWAS signal bypass the linear genome and are in physical proximity to the proximal promoter and transcription start site of the target gene, resonating with previous findings in fetal brain tissue that used a similar contact mapping strategy (7).
Cell type–specific contact maps with 10-kb-wide bins, queried for the schizophrenia-associated loci, frequently revealed differential chromosomal conformations in NPCs, glia, and neurons. For example, the risk locus upstream of the PROTOCADHERIN cell adhesion molecule gene clusters (chromosome 5), which is critically relevant for neuronal connectivity in developing and adult brain (fig. S6A) (25, 26), showed through both observed/expected interaction matrix (27) and global background-filtered contact mapping (7) a bifurcated bundle of interactions in NPCs, with one bundle emanating to sequences 5′ and the other bundle to sequences 3′ from the locus. In neurons, the 3′ bundle was maintained, but the 5′ bundle was “pruned,” whereas glia showed the opposite pattern; these differences between the three cell types were highly significant (observed/expected Wilcoxon rank sum P < 10−9 to 10−15) (Fig. 2, A to C). Dosage of the noncoding schizophrenia risk-SNP (rs111896713) at the PCDH locus significantly increased the expression of multiple PROTOCADHERIN genes (PCDHA2, PCDHA4, PCDHA7, PCDHA8, PCDHA9, PCDHA10, and PCDHA13) in adult frontal cortex of a large cohort of 579 individuals, including cases with schizophrenia and controls (fig. S6B and table S11) (28). The affected genes were interconnected to the disease-relevant noncoding sequence in neurons and NPCs but not in glia (fig. S6C). Therefore, cell type–specific Hi-C identified chromosomal contacts anchored in schizophrenia-associated risk sequences that affected expression of the target gene(s). On the basis of earlier chromosome conformation capture assays at the site of candidate genes, the underlying mechanisms may include alterations in transcription factor and other nucleoprotein binding at loop-bound cis-regulatory elements (5) or even local disruption of chromosomal conformations (6).
Fig. 2. Cell type–specific chromosomal contact maps at schizophrenia risk loci.

(A) Juicebox observed/expected interaction heatmaps at 10-kb resolution for the risk-associated clustered PCDH locus chr5:140023665−140222664 for NPC, glia, and neurons as indicated. (Far right) Grayscale heatmap depicts areas of highly cell-specific contact enrichments: upstream genes including ANKHD1 (dotted rectangle “A” and arrowhead) and downstream PCDH gene clusters (dotted rectangle “B” and arrows). Clustered PCDH gene expression patterns are available in fig. S6A. (B) Violin plots of observed/expected interaction values in the regions A and B highlighted in (A). (C) Map of contacts identified by binomial statistics. Red box with dashed black line represents the schizophrenia risk locus, dotted boxes regions “A” and “B” in heatmaps. (D) Cell-type resolved contact map of 10-kb bins (bold, black vertical lines) within risk sequenceson chr12 (left), chrX (middle), and chr5 (right); NPC (orange), neuron (green), glia (purple); −log q value, significance of contact between schizophrenia risk locus and each 10-kb bin; gene models (“Genes”) below with SNP-loop target gene highlighted in red. (E) Epigenomic editing (CRISPRa with nuclease-deficient dCas9 in NPCs) for three risk SNP-target gene pairs and their respective control sequences (top), measured with quantitative reverse transcription polymerase chain reaction (RT-PCR) (fold change from baseline) for VP64 (middle) and VPR (bottom) transcriptional activators. (F) Quantitative PCR gene expression changes upon directing catalytically active Cas9 to schizophrenia risk-associated credible SNPs (vertical red dashes with rsIDs) interacting via chromosomal contacts with promoters of ASCL1, EFNB1, and MATR3 in NPCs. Targeting strategy and contact distances depicted above; *P < 0.05, **P < 0.01, ***P < 0.0001 (figs. S6 and S7).
Transcriptional profiles of hiPSC-derived NPCs and neurons most closely resemble those of the human fetus in the first trimester (29); moreover, a portion of the genetic risk architecture of schizophrenia matches to regulatory elements that are highly active during prenatal development (30). We surveyed in our Hi-C datasets seven loci encompassing 36 “credible” (potentially causal) schizophrenia-risk SNPs with known chromosomal interactions in fetal brain to genes important for neuron development and function (7). We found that risk-associated chromosomal contacts were conserved between our hiPSC-NPCs and the published human fetal CP and germinal zone Hi-C datasets (7) for five of the seven loci (71%) tested (CHRNA2, EFNB1, MATR3, PCDH, and SOX2, but not ASCL1 or DRD2) (table S12). To test the regulatory function of these conserved risk sequence-bound conformations, we performed single-guide RNA (sgRNA)–based epigenomic editing experiments on isogenic antibiotic-selected NPCs that stably express nuclease-deficient dCas9-VP64 (31, 32) or dCas9-VPR (33, 34) transactivators (table S13). Previous studies in peripheral cell lines succeeded in inducing gene expression changes by placing dCas9-repressor fusion proteins at the site of chromosomal contacts separated by up to 2 Mb of linear genome from the promoter target (35). We tested ASCL1-, EFNB1-, MATR-3, and SOX2-bound chromosomal contacts separated by 200-to 700-kb interspersed sequences (Fig. 2, D and E; fig. S7A; and table S14). Pools of five individual sgRNAs directed against a risk-associated non-coding sequence bypassing 225 and 355 kb of genome consistently resulted in significantly decreased expression of ASCL1 [one-way analysis of variance (ANOVA), FVP64(2, 15) = 22.20, P < 0.0001; Dunnett’s PVP64 = 0.023] and EFNB1 target genes [one-way ANOVA, FVP64(2, 6) = 14.47, P = 0.0051, Dunnett’s PVP64 = 0.0356; FVPR(2, 6) = 1.46, P = 0.0111, Dunnett’s PVPR = 0.0088], in comparison with positive (promoter-bound) and negative (linear genome) control sgRNAs. Epigenomic editing of risk sequence 500 to 600 kb distant from the SOX2 and MATR3 loci did not alter target gene expression (Fig. 2, D and E, and fig. S7, A and B), which could reflect practical limitations in nonintegrative transfection-based (as opposed to viral) methods, impact of epigenetic landscape, or suboptimal guide RNA positioning (34), further limited by the 10-kb contact map resolution. Because portions of the MATR3-bound risk sequences are embedded in repressive chromatin, we directed five sgRNAs for Cas9 nuclease mutagenesis toward a 138–base pair (bp) sequence within a MATR3 long-range contact that was enriched with trimethyl-histone H3K27me3, commonly associated with Polycomb repressive chromatin remodeling, in order to disrupt it (fig. S7, C to E). This strategy produced a significant increase in MATR3 expression upon ablation of the putative repressor sequence, whereas targeting MATR3 (linear genome) control sequence remained ineffective (fig. S7, D and E). We conducted additional genomic mutagenesis assays, with sgRNAs directly overlapping credible SNPs participating in chromatin contacts with ASCL1, EFNB1, EP300, MATR3, PCDHA7, PCDHA8, and PCDHA10 (table S10). Cas9 nuclease deletion of interacting credible SNPs significantly increased gene expression of ASCL1, EFNB1, and EP300 (Prange = 0.0053 to 0.04, trange = 2.449 to 4.265) (Fig. 2F and fig. S7F). Similar targeting of four credible SNPs upstream of the clustered PCDH locus significantly decreased levels, by ~50 to 60%, of PCDHA8 and PCDHA10 (Prange = 0.0122 to 0.0124, trange = 4.326 to 4.343), two of the genes whose expression increased with dosage of the risk SNP rs111896713 in adult postmortem brain (figs. S6C and S7G). Taken together, our (epi)genomic editing assays (fig. S7H) demonstrate that chromosomal contacts anchored in schizophrenia risk loci potentially affect target gene expression across hundreds of kilobases, which is consistent with predictions from chromosomal conformation maps from hiPSC-derived brain cells described here, and from developing (7, 11) and adult (5) human brain tissue.
Cell type–specific schizophrenia-related chromosomal connectomes are associated with gene co-regulation and protein-protein association networks
Having shown that the chromosomal contact maps anchored in sequences associated with schizophrenia heritability undergo cell type–specific regulation (Fig. 2, A to C), are reproducible in neural cell culture and fetal brain (table S12), frequently harbor risk-associated eQTLs (table S10), and bypass extensive stretches of linear genome to affect target gene expression in genomic and epigenomic editing assays (Fig. 2, D to F, and fig. S7), we investigated chromosomal contacts for all 145 GWAS-defined schizophrenia risk loci together (23) (tables S15 to S17). We refer to the resulting “network” of risk loci and their 3D proximal genes as the “schizophrenia-related chromosomal connectome.”
Earlier studies in adult brain had shown that open chromatin-associated histone modification and other “linear epigenome” mappings strongly link the genetic risk architecture of schizophrenia specifically with neuronal, as opposed to non-neuronal, chromatin (36), which would suggest that similar cell-specific signatures may emerge in the risk-associated 3DG. Neurons and NPCs, but not the isogenic glia, showed a high preponderance of chromosomal contacts with schizophrenia-associated risk loci (Fig. 3A). There were 1203 contacts involving schizophrenia risk sequences that were highly specific to neurons (median distance between risk and target bins = 510 kb), 1100 highly specific for NPCs (median distance between risk and target bins = 520 kb), whereas only 425 highly specific for glia (median distance between risk and target bins = 580 kb) (Fig. 3A; figs. S8, A and B; and tables S15 to S17). There were also unexpectedly robust cell type– and gene ontology–specific signatures, including genes associated with neuronal connectivity and synaptic signaling (Fig. 3B and tables S18 and S19). Separate analysis of the Psychiatric Genomics Consortium “PGC2” 108 risk loci (2) yielded similar results (fig. S9, A and B).
Fig. 3. Expanded GWAS risk connectome is associated with gene coregulation.

(A) Counts of highly cell type–specific contacts associated with schizophrenia risk in each of the three hiPSC-derived cell types. (B) GO enrichment of genes located in schizophrenia risk contacts in NPCs (left), neurons (middle), and glia (right). (C) (Left) Schematic workflow of analyses performed with cell type–specific contact genes, distinguished as “risk locus” and “risk locus–connect” genes. (Middle) Venn diagram of genes located in the 145 loci and those found in cell type–specific contacts, with numbers in blue indicating “risk locus–connect” genes. (Right) Schematic workflow of analyses performed with combined set of “risk locus” and “risk locus–connect” genes. (D) RNA Pearson transcriptomic correlation heatmaps consisting of risk locus and risk locus–connect genes derived from the cell type–specific contacts of NPCs (left), neurons (middle), and glia (right). Organization scores (|r|avg) for each heatmap are reported with P values from sampling analysis. Schematics above heatmaps are representations of each cell type’s particular connectome (blue oval) and frequency distribution of organization scores from permutation analyses of randomly generated heatmaps (red, observed organization score of heatmap being tested). The gray bar corresponds to n genes that have at least 1 count per million in RNA-seq dataset out of the total number of genes and are used to construct the heatmap; red and blue bars indicate how many of the genes in the heatmap are in a risk locus (red) and are risk locus–connect (blue). Fuchsia, neuron connectivity/synaptic function genes; yellow, chromatin remodeling genes as determined from gene ontology analysis in (E). Additional information on coexpression clusters is provided in tables S22 and S23 (figs. S8 and S9).
Because spatial 3DG proximity of genes is an indicator for potential coregulation (37), we tested whether the neural cell type–specific schizophrenia-related chromosomal connectome showed evidence of coordinated transcriptional regulation and proteomic interaction of the participating genes. To this end, we generated lists of genes anchored in the most highly cell type–specific schizophrenia risk–associated contacts (Materials and methods) (Fig. 3C, fig. S8B, and table S18). Thus, for the NPC-specific contacts, we counted 386 genes, including 146 within the risk loci and another 240 genes positioned elsewhere in the linear genome but connected via an intrachromosomal contact to within-risk-locus sequences. Similarly, for the neuron-specific contacts, we identified 385 genes, including 158 within risk loci and 227 outside of risk loci (Fig. 3C). Last, for glia-specific contacts, we identified 201 genes, including 88 within and 113 outside of risk loci. We labeled the intrachromosomal contact genes located outside of schizophrenia risk loci as “risk locus-connect,” which we define as a collection of genes identified only through Hi-C interaction data, expanding—depending on cell type—by 50 to 150% the current network of known genes overlapping risk sequences that is informed only by GWAS (Fig. 3C).
To examine whether such types of disease-associated, cell type–specific chromosomal connectomes were linked to a coordinated program of gene expression, we analyzed a merged transcriptome dataset (comprised of 47 hiPSC-NPC and 47 hiPSC-forebrain-neuron RNA-seq libraries from 22 schizophrenia and control donors not related to the those of our Hi-C datasets) (16). We examined pair-wise correlations of the collective sets of the 386 NPC, 385 neuron, and 201 glia genes representing “risk locus” and “risk locus–connect” genes (cell type–specific “risk connectomes”). The risk connectome for each cell type showed extremely strong pair-wise correlations, with two of the largest clusters visualized on the neuron and NPC correlation matrices involving an admixture of 354 “risk locus” and “risk locus–connect” genes each, and similarly 181 genes from the glia matrix (Fig. 3D and table S20). The averaged gene-by-gene transcript correlation index for each matrix overall, defined here as “organization score” (|r|avg), for the NPCs, neurons, and glia were 0.22 to 0.25. Such levels of organized gene expression were robustly significant for NPC and neurons, after controlling for linear genomic distance (1000 random samplings, |r|avg, P < 0.001 for NPC and for neuron; P = 0.041 for glia) (Fig. 3D, fig. S9E, and table S21). There were four large clusters in the correlation matrices of the neuronal and NPC risk connectome: neuronal connectivity and synaptic signaling proteins (neuron cluster 1 and NPC cluster 2) and epigenetic regulators (neuron cluster 2 and NPC cluster 1). For example, within neuron cluster 1 (Fig. 3D, middle), 62 of 125 genes encoded neural cell adhesion and synaptic molecules, voltage-gated ion channels, and other neuron-specific genes (Fig. 3E and tables S22 and S23). We thus conclude that the chromosomal connectomes associated with schizophrenia risk are cell type–specific, with the neuronal risk connectome particularly enriched for genes pertaining to neuronal connectivity, synaptic signaling, and chromatin remodeling (Fig. 3, D and E). Analyses of the subset of PGC2 risk loci (108 and 145) provided similar results (fig. S9, C to F). Additionally, organization scores for neuron cluster 1 and cluster 2 genes were similar between hiPSC-derived NPCs and forebrain neurons from schizophrenia cases (n = 47) and control (n = 47), suggesting that many risk locus–connect and risk locus genes are coregulated across individuals (fig. S9H).
Numerous proteins encoded by risk locus and risk locus–connect genes were associated with synaptic signaling (table S24). The cell type–specific risk locus–connect and risk locus genes show significant protein-protein interaction network effects for NPCs (P = 0.0004) and neurons (P = 0.009) but not glia (Fig. 4A, figs. S10 to S12, and table S24) when examined by using the STRING database v10.5 (38, 39). We observed many proteomic clusters, including large groups of epigenomic regulators associated with the SWI/SNF (SWItch/Sucrose Non-Fermentable) chromatin remodeling complex and histone lysine methyltransferases and demethylases (Fig. 4A and figs. S10 and S11), many of which were the genes identified in NPC cluster 1 and neuron cluster 2 of the transcriptome analysis (Fig. 3, D and E). The transcriptomic correlation heatmaps for these protein networks (“STRING” genes), when compared with randomly generated subset heatmaps from the overall (“Full”) schizophrenia-related chromosomal connectome (Fig. 3D), had higher organization scores in NPCs and neurons (NPC |r|avg = 0.2963, P = 0.007; neuron |r|avg = 0.2877, P = 0.008, glia |r|avg = 0.2225, P = 0.595, STRING versus full permutation test) (Materials and methods) (Fig. 4B, figs. S13 to S15, and table S21). Because the transcriptomic correlation heatmap for the schizophrenia-related chromosomal connectome was significantly decreased by the removal specifically of the NPC STRING protein network genes (P < 10−3) (table S24), this subset of STRING-interacting proteins may drive the observed orchestrated coregulation. Within these transcriptome- and proteome-based regulatory networks were numerous occasions of coregulated (RNA) and interacting (protein) risk locus and risk locus–connect genes that share the same TAD, including CDC20, which regulates dendrite development (40, 41) and is associated at the protein level with RNF220, an E3 ubiquitin-ligase and β-catenin stabilizer (Fig. 4C) (42).
Fig. 4. Expanded GWAS risk connectome is linked to protein-protein association networks.

(A) Overview and representative examples (zoomed in) of protein-protein association networks in NPCs (left), neurons (middle), and glia (right). Numbers of edges connecting the proteins in each network and STRING-computed P values are reported below. Gray bar indicates the subset of these genes whose proteins are involved in the network out of the total number of genes from cell type–specific interactions; red and blue bars indicate how many of the genes in the network are in a risk locus (red) and are risk locus–connect (blue). (B) Comparison of organization scores between the full RNA transcriptomic correlation heatmaps (brown) (Fig. 3D) and the “STRING” heatmaps (tan) (figs. S13 to S15), consisting of only those genes in protein networks for each cell type. Permutation test, **P < 0.01. (C) Representative neuronal TAD landscape (chr1, ~2 Mb) depicting a schizophrenia risk–associated locus (red) with its risk locus–connect genes (blue), MED8, MPL, CDC20, and RNF220, which are members of the neuronal schizophrenia protein network (green circle). CDC20 and RNF220 interact at the protein level (green circle with gray border). (D) (Left) Liquid chromotography–selected reaction monitoring (LC-SRM) mass spectrometry (MS) was performed on dorsolateral prefrontal cortex (DLPFC) tissue from 43 adult postmortem brains (23 schizophrenia, 20 control). (Middle) 182 neuronal proteins were reliably quantified, and four of them were observed to have associations in the neuron protein network in (A). (Right) GABBR1, GRM3, GRIN2A, and GRIA1 proteins were found to have significantly more correlated expression than expected by random permutation analysis. Additional information on protein-protein interactions is provided in figs. S9 to S15.
To examine whether such coregulation could be representative of the prefrontal cortex proteome of the adult brain, we screened a newly generated mass spectrometry–based dataset of 182 neuronal proteins, the majority of which were synaptic, quantified from prefrontal cortex of n = 23 adult schizophrenia and n = 20 control subjects (table S25) (43). Among the 182 proteins, there were four from the risk-associated neuronal protein network (Fig. 4D): GABAB receptor subunit GABBR1 and ionotropic (GRIA1 and GRIN2A) and metabotropic glutamate receptor subunits (GRM3). Protein-protein correlation scores were significantly higher for these four risk-associated proteins than expected from random permutation analysis from the pool of 182 proteins (P < 0.002) across patients and controls. We conclude that the schizophrenia-related chromosomal connectome, tethering other portions of the genome to the sequences associated with schizophrenia heritability, provides a structural foundation for a functional connectome that reflects coordinated regulation of gene expression and interactions within the proteome.
Discussion
Neural progenitor differentiation into neurons and glia is associated with dynamic remodeling of chromosomal conformations, including loss of many NPC-specific chromosomal contacts, with differentiation-induced loop pruning primarily affecting a subset of genes important for neurogenesis (NPC-to-neuron loss) and neuronal function (NPC-to-glia loss). These findings broadly resonate with a recent report linking neural differentiation to multiple scales of 3DG folding, governed by multiple mechanisms, including CTCF-dependent loop alterations, repressive chromatin remodeling, and cell- and lineage-specific transcription factor networks (21). Our results suggest that developmental 3DG remodeling affects a substantial portion of sequences that confer liability for schizophrenia; furthermore, these genes in 3D physical proximity with schizophrenia-risk variants show a surprisingly strong correlation at the level of the transcriptome and proteome. How might the disease-relevant reorganization of the spatial genome (the “chromosomal connectome”) provide a structural foundation for coordinated regulation of expression? Recent Hi-C studies in mouse brain showed that chromosomal contacts preferentially occurred between loci targeted by the same transcription factors (21), and likewise, multiple schizophrenia risk loci could converge on intra- and interchromosomal hubs sharing a similar regulatory architecture including specific enhancers as well as transcription and splicing factors (44–46). Intriguingly, the three major functional categories associated with the genetic risk architecture of schizophrenia—neuronal connectivity, synaptic signaling, and chromatin remodeling (47, 48)—were heavily represented within the cell type–specific chromosomal connectomes of neurons and NPCs described here (Fig. 3, B and E) and in whole tissue in vivo (7, 11). Cell type–specific 3DG reorganization during the course of neural progenitor differentiation, as shown here, could therefore have profound implications for our understanding of the genetic underpinnings of psychiatric disease. For example, inclusion of the cell type–specific risk (sequence)–associated chromosomal connectome may lead to refinements of cumulative schizophrenia risk allele burden estimates, including “polygenic risk score” (PRS) or “biologically informed multilocus profile scores” (BIMPS), which currently only explain a small portion of disease risk (49). Cell type–specific intersection of 3DG and genetic risk maps are of clinical interest beyond psychiatric disorders; for example, risk variants that confer susceptibility to auto-immune disease were embedded in physically interacting chromosomal loci in lymphoblastoid cells (50). Our 3DG maps from neural progenitors and their isogenic neurons and glia are accessible through the PsychENCODE Knowledge Portal (https://synapse.org) and more than double the number of currently available Hi-C datasets from human brain (7, 9, 10), providing investigators with a resource to chart the expanded genome space associated with cognitive and neuropsychiatric disease in context of cell type–specific remodeling of chromosomal conformations during early development.
Materials and methods
In situ Hi-C from hiPSC-derived cells
In situ Hi-C libraries were generated from 2 million to 5 million cultured hiPSC-derived NPCs, glia, and neurons as described in (3) without modifications in the protocol. Briefly, in situ Hi-C consists of 7 steps: (i) crosslinking cells with formaldehyde, (ii) digesting the DNA using a 4-cutter restriction enzyme (e.g., MboI) within intact permeabilized nuclei, (iii) filling in and biotinylating the resulting 5′-overhangs, (iv) ligating the blunt ends, (v) shearing the DNA, (vi) pulling down the biotinylated ligation junctions with streptavidin beads, and (vii) analyzing these fragments using paired end sequencing. As quality control (QC) steps, we checked for efficient restriction with an agarose DNA gel and for appropriate size selection in using the Agilent Bioanalyzer after steps (v) and (vi). For the final QC, we performed superficial sequencing on the Illumina MiSeq (~2–3M reads/sample) to assess quality of the libraries using metrics such as percent of reads passing filter, percent of chimeric reads, and percent of forward-reverse pairs (supplementary materials, table S1). For the forebrain directed differentiation neuronal library from subject S1, the Arima Hi-C kit (Arima Genomics, San Diego) was used according to the manufacturer’s instructions.
Hi-C read mapping and matrix generation
The Hi-C libraries were sequenced on the Illumina HiSeq1000 platform (125bp paired-end) (New York Genome Center). Technical replicates of subject S2 NPCs, neurons, and glia were also sequenced to enhance resolution. Initial processing of the raw 2 ×125 bp read pair FASTQ files was performed using the HiC-Pro analysis pipeline (51). In brief, HiC-Pro performs four major tasks: aligning short reads, filtering for valid pairs, binning, and normalizing contact matrices. HiCPro implements the truncation-based alignment strategy using Bowtie v2.2.3 (52), mapping full reads end-to-end or the 5′ portion of reads preceding a GATCGATC ligation site that results from restriction enzyme digestion with MboI followed by end ligation. Invalid interactions such as same-strand, dangling-end, self-cycle, and single-end pairs are not retained. Binning was performed in 10kb, 40 kb and 100 kb non-overlapping, adjacent windows across the genome and resulting contact matrices were normalized using iterative correction and eigenvector decomposition (ICE) as previously described (53), using HiC-Pro’s default settings of 100 maximum iterations, filtering of the sparse bins (lowest 2%), and a relative result increment of 0.1 before declaring convergence (http://nservant.github.io/HiC-Pro/MANUAL.html). Data are reported in browser-extensible-data-like (BED) format and visualized in the Washington University Epigenome Browser (http://epigenomegateway.wustl.edu). Hierarchical clustering was performed on the ICE-corrected intrachromosomal contact matrices after the bins with the 1% most extreme interaction values were excluded as largely artifactual. Clustering was performed using Ward’s method on the 1, 5, 10, 25, 50, and 100% most variable remaining bins using (1-correlation) as a distance metric. The results using the 10% most variable interaction bins, shown here in a cluster dendrogram and a Pearson correlation matrix, are representative of these results.
Hi-C loop calls using Juicer
Loop calling was performed using the software HiCCUPS (3). To format data for HiCCUPS input, we remapped reads from Hi-C libraries using the Juicer pipeline (54). Similar to HiC-Pro, the Juicer pipeline performs read alignment, filtering, binning, and matrix normalization. Samples were pooled for each cell type (S1 and 2 technical replicates from S2) to generate the maximum amount of coverage required for accurate loop calling. The resulting .hic matrix files (MAPQ > 0) were then used as input to HiCCUPS. The following parameters were set for HiCCUPS following the analysis in (3): FDR threshold (f) = 0.10, 0.10; peak width (p) = 4, 2; window width (i) = 7, 5; merge distance (d) = 20 kb, 20 kb. Values for parameters correspond to calls made at 5kb and 10kb, respectively. Representative neuronal and non-neuronal loops are presented in fig. S3. As the number of loops called is dependent upon the number of Hi-C contacts in the matrix (55), we also generated matrices with equivalent total Hi-C contacts via subsampling. hiPSC-derived Hi-C interaction matrices were randomly subsampled to 372,787,143 cis only contacts (the lowest number of cis contacts across all cell types) and HiCCUPS was rerun on the subsampled matrices. After loops were called for each cell type, we performed a reevaluation on this union set of loop loci. HiCCUPS was rerun using the union set of loop loci as input to produce q-values for each loop in the union set for every cell type. By default, HiCCUPS does not output a q-value for every pixel. Hence, this reevaluation produced q-values for pixels in cells that did not pass the significance threshold. We then defined any pixel from the union set with a q-value < 0.10 with respect to the donut neighborhood surrounding the pixel to be a loop and defined the loop to be shared with any cell types having a q-value < 0.10 for the same pixel.
These loop calls were used for comparing loop calls between cell types. Loops were also called and subsampled as above for the GM12878 cell line using the processed data from (3) found here: www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE63525. Loop calls were overlapped with compartment calls (supplementary materials, materials and methods), such that AA, BB, and AB refer to loops with both anchors in A, both anchors in B, and one anchor in A and other anchor in B, respectively. Loops in chromosomes 4, 18, 19, and X were removed from this compartment analysis since the first principle component most likely corresponded to p versus q arm distinctions and not A versus B compartments.
Hi-C interactions at risk loci
To approach 3DG conformation in context of the disease-relevant sequences, we adapted the binomial statistics based mapping strategy previously described by Won et al. (7). The set of schizophrenia risk loci used in this study included the original (PGC2, Psychiatric Genomics Consortium) (2) risk sequences, or 108 physically distinct association loci defined by 128 index SNPs (corrected P 10−8) and an additional 37 loci from the CLOZUK (a series of UK cases registered for clozapine treatment with a clinical diagnosis of schizophrenia) study for a total of 145 loci defined by 179 independent genome-wide significant SNPs (corrected P < 5 × 10–8), determined by GWAS in 40,675 cases and 64,643 controls (23). A risk locus is defined as a collection of (SNPs) existing in linkage dis-equilibrium, ranging from 1bp to 8.9Mb (average 256.2 kb) in length and in total equivalent to approximately 0.012% of human genomic sequence.
To identify significantly enriched interactions involving a bin of interest with another bin, our principal approach was to first estimate the expected interaction counts for each interaction distance by calculating the mean of all intrachromosomal bin-bin interactions of the same separation distance throughout the raw intrachromosomal contact matrix. We used the R package, HiTC (56), to facilitate manipulation of our HiC-Pro-produced raw contact matrices and estimation of the expected counts at various interaction distances. The probability of observing an interaction between a bin-of-interest and another bin was then defined as the expected interaction between those two bins divided by the sum of all expected interactions between the bin-of-interest and all other intrachromosomal bins. A P value was then calculated as binomial probability of observing the number of interaction counts or more between the bin-of-interest and some other bin where the number of successes was defined as the observed interaction count, the number of tries as the total number of observed interactions between the bin-of-interest and all other intrachromosomal bins, and the success probability as the probability of observing the bin-bin interaction estimated from the expected mean interaction counts. The Benjamini-Hochberg method was used to control false discovery rate (FDR) for P values determined for all interactions with a bin-of-interest (includes all bins 1Mb up and downstream in our tests).
Generation of stable selected dCas9-VP64/VPR and Cas9 NPCs
All CRISPR-based epigenomic editing assays were performed on antibiotic-selected dCas9-VP64 (VP64 as the tetrameric VP16 activator domain) and dCas9-VPR (VPR as the tripartite activator, VP64-p65-Rta) NPCs derived as described in (34). For generation of Cas9 stable, selected NPCs, we used a plasmid of lentiCRISPR v2 gifted by Feng Zhang (Addgene plasmid # 52961). DNA sequencing with a U6 primer confirmed the identity. Lentiviral production and titration were performed as described previously (14). Control S1 and S2 NPCs were spinfected with lentiCRISPR v2 virus as described (34). 48 hours post-transduction, cells were selected by exposure to puromycin at 0.3 μg/mL. Without transduction, all control cells died within around 5 days after the antibiotic addition. The puromycin-selected NPCs were subject to Western blot analysis of Cas9 expression. 30 μg of proteins were electrophoresed in NuPAGE 4–12% Bis-Tris Protein Gels (NP0323PK2, Life Technologies) in 1× MES running buffer, 200 V constant, 35 min. Proteins were transferred onto nitrocellulose membrane (IB23002, Life Technologies) on the iBlot® 2 Dry Blotting System (program P3, 7:00 min). The membranes were incubated with primary antibodies against Cas9 (1:250, monoclonal, clone 7A9, Millipore) and β-Actin (1:10,000, mouse, 1406030, Ambion) overnight at 4°C. Then, membranes were incubated with the IRDye-labeled secondary antibodies for 45 min at RT in the dark on the rocker. Fluorescence was visualized using a Li-Cor Odyssey Imaging System.
In vitro transcription and transfection of gRNAs
Guide RNAs (gRNAs) were designed on Benchling (www.benchling.com) using the CRISPR tool. gRNAs were generated via in vitro transcription (IVT) with the GeneArt Precision gRNA Synthesis Kit (Thermo Fisher Scientific, A29377) as per manufacturer instructions. Five gRNAs were designed per condition (i.e., “loop-SNP”, negative control, and positive control) and pooled for transfection. The genomic ranges within which loop-SNP gRNAs were designed (i.e., region spanning the SNP of interest and all gRNAs in the condition) were roughly 600 bp for ASCL1, 550 bp for MATR3, 460 bp for EFNB1 (with 2/5 gRNAs directly overlapping the SNP), 300 bp for SOX2. Puromycin-selected (1μg/mL in NPC media; Sigma, #P7255) dCas9-VP64 and dCas9-VPR NPCs (34) were seeded at a density of ~400,000 per well on Matrigel-coated (BD Biosciences) 24-well plates. Pooled IVT gRNAs (500 ng total RNA/well) and 2 μL EditPro Stem lipofectamine (MTI-GlobalStem, #GST-2174; now, ThermoFisher, STEM00003) were diluted in 50 μL Opti-MEM (Thermo Fisher Scientific, #31985062) and added dropwise to each well. Cells were harvested with TRIzol for total RNA extraction 48 hours later. All experiments were conducted with 3 to 6 biological replicates from 1 donor (subject S1), generated in parallel, with the donor contributing isogenic dCas9-VP64 and dCas9-VPR effector cells. Each data point in Fig. 2, D to F, represents one biological replicate within each condition. For each target gene promoter and candidate loop, control gRNAs were strategically placed into the middle third of the (linear) genome portion bypassed by the candidate loop. CRISPRa results were analyzed on PRISM with a one-way ANOVA across 3 conditions with a Dunnett’s test for multiple comparisons. Cas9 mutagenesis was also performed as described above with the exception of the negative control, which in these experiments consisted of an empty transfection (i.e., lipofectamine + Opti-MEM without any gRNA). Cas9 results were analyzed with an unpaired t test comparing the loop-SNP and negative control conditions.
RNA transcriptomic correlation heatmaps
Pearson correlation coefficient matrices were calculated for gene expression in the childhood onset schizophrenia data set (16) using R from lists of genes that are located in cell-type-specific loops anchored at schizophrenia risk loci and, as a subset of this list, sets of genes whose proteins participate in an association network for each of the three cell types (see below). Significance was computed calculating the absolute mean correlation coefficient of each correlation matrix (“organization score”) as a test statistic against a null distribution generated by random gene sampling. Randomized gene lists were drawn only from the pool of genes with over 1 count per million (CPM) in at least 30% of the experiments described in (16). To generate a null distribution of organization scores for a given cell type that accounted for genomic distance and neighborhood effects, we began by randomly selecting a significant PGC interaction for that cell type (e.g., random selection from table S12). Using the bp genomic distance of this interaction we randomly selected two 10kb bins from the genome separated by the same distance. All genes overlapping these bins were then added to the list of genes with which to calculate the organization score. This process was iterated until enough genes were added to the list to match the number of genes used in the original cell-type-specific organization score. Finally, this protocol was repeated 1000 times to generate the null distribution of random organization scores. This distribution was then used to calculate significance of co-regulation (i.e., P = number of times |r|avg of the null exceeded that of the test heatmap / 1000). Note that STRING gene network transcriptomic analyses (Fig. 4B) were performed with 1000 random permutations of genes sampled from the full schizophrenia risk connectome (i.e., risk locus + risk locus-connect genes) for each cell type.
Supplementary Material
ACKNOWLEDGMENTS
The authors thank all members of the Brennand and Akbarian laboratories for constructive comments and discussions; C. O’Shea, S. R. Shannon, I. Silverberg, and A. Austin for valuable assistance with PCR assays; E. Xia, C. Rosenbluh, and A. Chess for generous access to sequencing equipment; L. Winterkorn and staff at the New York Genome Center for logistical support; and M. Peters for help and support as it pertains to data accessibility.
Funding:
This work was supported by National Institute of Mental Health PsychENCODE grants U01MH103392 (S.A.) and R01MH106056 (S.A. and K.J.B.) and a predoctoral Ruth L. Kirschstein fellowship 1F30MH113330 (P.R.).
Footnotes
Competing interests:
The authors declare no conflicts of interest.
Data and materials accessibility:
Scripts used for bioinformatics analyses can be found at https://github.com/tborrman/Schahram-project and https://github.com/will-NYGC/sigHicInteractions. Hi-C data files and metadata are accessible through an open source platform: www.synapse.org/#!Synapse:syn12979101 (registration required; Data Download—Study “iPSC-HiC”). The code for this paper has been submitted to Zenodo, and the respective links are https://zenodo.org/badge/latestdoi/68036139 and https://zenodo.org/badge/latestdoi/151779353.
REFERENCES AND NOTES
- 1.Rajarajan P, Gil SE, Brennand KJ, Akbarian S, Spatial genome organization and cognition. Nat. Rev. Neurosci 17, 681–691 (2016). doi: 10.1038/nrn.2016.124; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Schizophrenia Working Group of the Psychiatric Genomics Consortium, Biological insights from 108 schizophrenia-associated genetic loci. Nature 511, 421–427 (2014). doi: 10.1038/nature13595; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rao SS et al. , A 3D map of the human genome at kilobase resolution reveals principles of chromatin looping. Cell 159, 1665–1680 (2014). doi: 10.1016/j.cell.2014.11.021; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Jiang Y et al. , The methyltransferase SETDB1 regulates a large neuron-specific topological chromatin domain. Nat. Genet 49, 1239–1250 (2017). doi: 10.1038/ng.3906; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bharadwaj R et al. , Conserved higher-order chromatin regulates NMDA receptor gene expression and cognition. Neuron 84, 997–1008 (2014). doi: 10.1016/j.neuron.2014.10.032; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bharadwaj R et al. , Conserved chromosome 2q31 conformations are associated with transcriptional regulation of GAD1 GABA synthesis enzyme and altered in prefrontal cortex of subjects with schizophrenia. J. Neurosci 33, 11839–11851 (2013). doi: 10.1523/JNEUROSCI.1252-13.2013; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Won H et al. , Chromosome conformation elucidates regulatory relationships in developing human brain. Nature 538, 523–527 (2016). doi: 10.1038/nature19847; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lieberman-Aiden E et al. , Comprehensive mapping of long-range interactions reveals folding principles of the human genome. Science 326, 289–293 (2009). doi: 10.1126/science.1181369; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Schmitt AD et al. , A compendium of chromatin contact maps reveals spatially active regions in the human genome. Cell Reports 17, 2042–2059 (2016). doi: 10.1016/j.celrep.2016.10.061; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dixon JR et al. , Chromatin architecture reorganization during stem cell differentiation. Nature 518, 331–336 (2015). doi: 10.1038/nature14222; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.de la Torre-Ubieta L et al. , The dynamic landscape of open chromatin during human cortical neurogenesis. Cell 172, 289–304.e18 (2018). doi: 10.1016/j.cell.2017.12.014; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mitchell AC et al. , The genome in three dimensions: A new frontier in human brain research. Biol. Psychiatry 75, 961–969 (2014). doi: 10.1016/j.biopsych.2013.07.015; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Topol A et al. , Dysregulation of miRNA-9 in a subset of schizophrenia patient-derived neural progenitor cells. Cell Reports 15, 1024–1036 (2016). doi: 10.1016/j.celrep.2016.03.090; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ho SM et al. , Rapid Ngn2-induction of excitatory neurons from hiPSC-derived neural progenitor cells. Methods 101, 113–124 (2016). doi: 10.1016/j.ymeth.2015.11.019; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tcw J et al. , An efficient platform for astrocyte differentiation from human induced pluripotent stem cells. Stem Cell Reports 9, 600–614 (2017). doi: 10.1016/j.stemcr.2017.06.018; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hoffman GE et al. , Transcriptional signatures of schizophrenia in hiPSC-derived NPCs and neurons are concordant with post-mortem adult brains. Nat. Commun 8, 2225 (2017). doi: 10.1038/s41467-017-02330-5; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Brennand KJ et al. , Modelling schizophrenia using human induced pluripotent stem cells. Nature 473, 221–225 (2011). doi: 10.1038/nature09915; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gil-Sanz C et al. , Lineage tracing using Cux2-Cre and Cux2-CreERT2 mice. Neuron 86, 1091–1099 (2015). doi: 10.1016/j.neuron.2015.04.019; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Charlesworth G et al. , Mutations in ANO3 cause dominant craniocervical dystonia: Ion channel implicated in pathogenesis. Am. J. Hum. Genet 91, 1041–1050 (2012). doi: 10.1016/j.ajhg.2012.10.024; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wang J et al. , Single-cell gene expression analysis reveals regulators of distinct cell subpopulations among developing human neurons. Genome Res. 27, 1783–1794 (2017). doi: 10.1101/gr.223313.117; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bonev B et al. , Multiscale 3D genome rewiring during mouse neural development. Cell 171, 557–572.e24 (2017). doi: 10.1016/j.cell.2017.09.043; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dixon JR et al. , Topological domains in mammalian genomes identified by analysis of chromatin interactions. Nature 485, 376–380 (2012). doi: 10.1038/nature11082; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pardiñas AF et al. , Common schizophrenia alleles are enriched in mutation-intolerant genes and in regions under strong background selection. Nat. Genet 50, 381–389 (2018). doi: 10.1038/s41588-018-0059-2; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dobbyn A et al. , Landscape of conditional eQTL in dorsolateral prefrontal cortex and co-localization with schizophrenia GWAS. Am. J. Hum. Genet 102, 1169–1184 (2018). doi: 10.1016/j.ajhg.2018.04.011; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chen WV, Maniatis T, Clustered protocadherins. Development 140, 3297–3302 (2013). doi: 10.1242/dev.090621; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yagi T, Molecular codes for neuronal individuality and cell assembly in the brain. Front. Mol. Neurosci 5, 45 (2012). doi: 10.3389/fnmol.2012.00045; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Durand NC et al. , Juicebox provides a visualization system for Hi-C contact maps with unlimited zoom. Cell Syst. 3, 99–101 (2016). doi: 10.1016/j.cels.2015.07.012; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Fromer M et al. , Gene expression elucidates functional impact of polygenic risk for schizophrenia. Nat. Neurosci 19, 1442–1453 (2016). doi: 10.1038/nn.4399; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Brennand K et al. , Phenotypic differences in hiPSC NPCs derived from patients with schizophrenia. Mol. Psychiatry 20, 361–368 (2015). doi: 10.1038/mp.2014.22; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gulsuner S et al. , Spatial and temporal mapping of de novo mutations in schizophrenia to a fetal prefrontal cortical network. Cell 154, 518–529 (2013). doi: 10.1016/j.cell.2013.06.049; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Perez-Pinera P et al. , RNA-guided gene activation by CRISPRCas9-based transcription factors. Nat. Methods 10, 973–976 (2013). doi: 10.1038/nmeth.2600; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Maeder ML et al. , CRISPR RNA-guided activation of endogenous human genes. Nat. Methods 10, 977–979 (2013). doi: 10.1038/nmeth.2598; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chavez A et al. , Comparison of Cas9 activators in multiple species. Nat. Methods 13, 563–567 (2016). doi: 10.1038/nmeth.3871; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ho SM et al. , Evaluating synthetic activation and repression of neuropsychiatric-related genes in hiPSC-derived NPCs, neurons, and astrocytes. Stem Cell Reports 9, 615–628 (2017). doi: 10.1016/j.stemcr.2017.06.012; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Fulco CP et al. , Systematic mapping of functional enhancer-promoter connections with CRISPR interference. Science 354, 769–773 (2016). doi: 10.1126/science.aag2445; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Girdhar K et al. , Cell-specific histone modification maps in the human frontal lobe link schizophrenia risk to the neuronal epigenome. Nat. Neurosci 21, 1126–1136 (2018). doi: 10.1038/s41593-018-0187-0; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kustatscher G, Grabowski P, Rappsilber J, Pervasive coexpression of spatially proximal genes is buffered at the protein level. Mol. Syst. Biol 13, 937 (2017). doi: 10.15252/msb.20177548; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Szklarczyk D et al. , STRING v10: Protein-protein interaction networks, integrated over the tree of life. Nucleic Acids Res. 43 (D1), D447–D452 (2015). doi: 10.1093/nar/gku1003; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Szklarczyk D et al. , The STRING database in 2017: Quality-controlled protein-protein association networks, made broadly accessible. Nucleic Acids Res. 45 (D1), D362–D368 (2017). doi: 10.1093/nar/gkw937; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Watanabe Y, Khodosevich K, Monyer H, Dendrite development regulated by the schizophrenia-associated gene FEZ1 involves the ubiquitin proteasome system. Cell Reports 7, 552–564 (2014). doi: 10.1016/j.celrep.2014.03.022; [DOI] [PubMed] [Google Scholar]
- 41.Puram SV et al. , A CaMKIIb signaling pathway at the centrosome regulates dendrite patterning in the brain. Nat. Neurosci 14, 973–983 (2011). doi: 10.1038/nn.2857; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ma P et al. , The ubiquitin ligase RNF220 enhances canonical Wnt signaling through USP7-mediated deubiquitination of b-catenin. Mol. Cell. Biol 34, 4355–4366 (2014). doi: 10.1128/MCB.00731-14; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hahn CG et al. , The post-synaptic density of human postmortem brain tissues: An experimental study paradigm for neuropsychiatric illnesses. PLOS ONE 4, e5251 (2009). doi: 10.1371/journal.pone.0005251; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lomvardas S et al. , Interchromosomal interactions and olfactory receptor choice. Cell 126, 403–413 (2006). doi: 10.1016/j.cell.2006.06.035; [DOI] [PubMed] [Google Scholar]
- 45.Quinodoz SA et al. , Higher-order inter-chromosomal hubs shape 3D genome organization in the nucleus. Cell 174, 744–757.e24 (2018). doi: 10.1016/j.cell.2018.05.024; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Khanna N, Hu Y, Belmont AS, HSP70 transgene directed motion to nuclear speckles facilitates heat shock activation. Curr. Biol 24, 1138–1144 (2014). doi: 10.1016/j.cub.2014.03.053; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Network and Pathway Analysis Subgroup of Psychiatric Genomics Consortium, Psychiatric genome-wide association study analyses implicate neuronal, immune and histone pathways. Nat. Neurosci 18, 199–209 (2015). doi: 10.1038/nn.3922; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Gilman SR et al. , Diverse types of genetic variation converge on functional gene networks involved in schizophrenia. Nat. Neurosci 15, 1723–1728 (2012). doi: 10.1038/nn.3261; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bogdan R, Baranger DAA, Agrawal A, Polygenic Risk Scores in Clinical Psychology: Bridging Genomic Risk to Individual Differences. Annu. Rev. Clin. Psychol 14, 119–157 (2018). doi: 10.1146/annurev-clinpsy-050817-084847; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Grubert F et al. , Genetic control of chromatin states in humans involves local and distal chromosomal interactions. Cell 162, 1051–1065 (2015). doi: 10.1016/j.cell.2015.07.048; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Servant N et al. , HiC-Pro: An optimized and flexible pipeline for Hi-C data processing. Genome Biol. 16, 259 (2015). doi: 10.1186/s13059-015-0831-x; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Langmead B, Trapnell C, Pop M, Salzberg SL, Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 10, R25 (2009). doi: 10.1186/gb-2009-10-3-r25; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Imakaev M et al. , Iterative correction of Hi-C data reveals hallmarks of chromosome organization. Nat. Methods 9, 999–1003 (2012). doi: 10.1038/nmeth.2148; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Durand NC et al. , Juicer provides a one-click system for analyzing loop-resolution Hi-C experiments. Cell Syst. 3, 95–98 (2016). doi: 10.1016/j.cels.2016.07.002; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Forcato M et al. , Comparison of computational methods for Hi-C data analysis. Nat. Methods 14, 679–685 (2017). doi: 10.1038/nmeth.4325; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Servant N et al. , HiTC: Exploration of high-throughput ‘C’ experiments. Bioinformatics 28, 2843–2844 (2012). doi: 10.1093/bioinformatics/bts521; [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.