

Abstract
INTRODUCTION:
The genetic architecture of Alzheimer disease (AD) is only partially understood.
METHODS:
We conducted an association study for AD using whole sequence data from 507 genetically enriched AD cases (i.e., cases having close relatives affected by AD) and 4,917 cognitively healthy controls of European Ancestry (EA) and 172 enriched cases and 179 controls of Caribbean Hispanic (CH) ancestry. Confirmation of top findings from Stage 1 was sought in two family-based GWAS datasets and in a whole genome sequencing dataset comprising members from 42 EA and 115 CH families.
RESULTS:
We identified associations in EAs with variants in 12 novel loci. The most robust finding is a rare CASP7 missense variant (rs116437863; p=2.44×10−10) which improved when combined with results from the Stage 2 datasets (p=1.92 ×10−10).
DISCUSSION:
Our study demonstrated that an enriched cases design can strengthen genetic signals, thus allowing detection of associations that would otherwise be missed in a traditional case-control study.
Keywords: enriched case-control, whole exome sequencing, association study, genome-wide association studies, gene-based analyses
1. Introduction
Late-onset Alzheimer disease (AD) is a progressive neurodegenerative disorder in persons ages 65 years and older characterized by memory loss and dementia. AD risk increases exponentially with age with a prevalence of 30–40% among 85–89 year-olds [1]. As average life expectancy has increased, the number of AD cases will increase to 11–16 million by 2050 with nearly 1 million new cases per year unless measures are identified to delay or prevent the disease [2, 3].
Risk of AD is modulated by variants in multiple genes, most notably the APOE ɛ2 and ɛ4 alleles, in combination with lifestyle and environmental factors. AD has a substantial genetic component with an estimated heritability of 58–79% [4]. Genome-wide association studies (GWAS) have identified >20 common susceptibility variant loci showing robust evidence for association with AD [5–7]. Recently, studies that performed whole exome sequencing (WES), targeting gene sequencing and rapid throughput genotyping using SNP microarray chips with high exome content have reported associations with rare risk variants in multiple novel loci including TREM2 (R47H) [8–10], PLD3 [11], AKAP9 [12], UNC5C [13], PLCG2 [14], ABI3 [14] as well as with rare risk and protective variants in several previously known AD genes (APP [15, 16], APOE (p.V236E) [17], SORL1 [18] and ABCA7 [19, 20]).
The Alzheimer’s Disease Sequencing Project (ADSP) is an NIH-funded initiative to identify novel genes and rare risk and protective variants using WES and whole genome sequencing (WGS) approaches. In this study, we analyzed a subset of the ADSP WES cohort including “enriched” AD cases (i.e., cases who have close relatives also affected by AD and thus more likely to have a high burden of AD risk alleles compared to cases not ascertained on the basis of a positive family history) and all controls to identify novel associations with single nucleotide variants (SNVs) and short insertions and deletions (indels).
2. Methods
2.1. Participants, Sequencing and Data Processing
The Alzheimer’s Disease Sequencing Project (ADSP) performed WES of DNA specimens from 5,778 AD cases and 5,136 controls at three NHGRI Genome Centers (Broad Institute, Baylor College of Medicine, and McDonnell Genome Institute at the Washington University). Detailed description of the ADSP WES study design has been published elsewhere [21]. In brief, subjects for this study were selected from datasets assembled by the Alzheimer’s Disease Genetics Consortium (ADGC), Cohorts for Heart and Aging Research in Genomic Epidemiology (CHARGE) consortium, and the Rotterdam Study [5, 22]. In the ADSP WES EA cohort, ~5,000 AD cases that were not ascertained on the basis of family history of AD were selected because they have the lowest risk explained by age and APOE genotypes (young onset, APOE ɛ2/ɛ2, ɛ2/ɛ3, or ɛ3/ɛ3). ~5,000 cognitively normal controls were selected as controls least likely to convert to a case based on age, APOE, and autopsy data (old, APOE ɛ2/ɛ2, ɛ2/ɛ3, or ɛ3/ɛ3, and little or no AD neuropathology). In addition, ~700 unrelated cases were selected from additional multiplex families with >2 close relatives affected by AD in each family, but only one case was selected from each family (i.e., “enriched-cases”). To enhance discovery of novel AD-related variants, enriched cases which could be explained by cosegregation of APOE ɛ4 were excluded.
After performing a series of filtering steps to identify duplicate samples and subjects with low genotype call rates, there remained a sample containing 10,441 individuals of European ancestry (EA) and 395 Caribbean Hispanics (CH). Subjects for this study included 679 unrelated AD cases (507 EA and 172 CH) from families containing at least three members affected by AD and 5,094 unrelated controls (4,917 EA and 177 CH).Characteristics of the 5,773 subjects included in this study are shown in Supplementary Table 1. Compared to the overall ADSP case-control study design, EA enriched cases are older (age at onset = 83.6 years) and similar in age to the controls (age at last examination = 86.5 years). The mean age at onset of CH enriched cases (75.4 years) is similar to the mean age at last examination (73.5 years) of the CH controls.
2.2. Whole Exome Sequencing and Quality Control
Details of library preparation, sequencing protocols, and variant calling pipelines are described in Supplementary Methods. After sequencing, 100 bp paired-end reads were mapped to human reference genome (GRCh37/hg19) using the Burrows-Wheeler Aligner (BWA) [23]. The ADSP Quality Control (QC) Working Group applied QC protocols to autosomal bi-allelic single nucleotide variants (SNVs) and short insertions and deletions (indels) to generate a high-quality variant call set. After QC, there remained 1,454,483 SNVs and 69,931 indels for association analyses.
2.3. Single-variant Association Analyses
In Stage 1, association of AD with each variant having a minor allele count (MAC) ≥10 (100,338 variants for EA and 64,691 for CH; Figure 1 and Supplementary Table 2) was tested in each population using score tests in seqMeta (https://github.com/DavisBrian/seqMeta) with three additive logistic regression models. A MAC cutoff of 10 is the minimum number of alleles to achieve statistical significance in this sample. A minimal adjustment model (Model 0) included covariates for sequencing center and principal components (PCs) of ancestry (the first 10 PCs for EAs and the first 3 PCs for CHs) in order to identify variants whose effects on AD risk are confounded by age and sex. This model was previously shown to increase detection of associations in this sample in which the mean age is substantially different between cases and controls [24]. A second model (Model 1) also included terms for age and sex, and a third model (Model 2) included all covariates from model 2 plus terms for the number of APOE ɛ4 and ɛ2 alleles. Results from analyses of 46,425 variants that were successfully called and passed criteria for single-variant analysis in the EA and CH data sets were combined using an inverse variance–weighted meta-analysis approach implemented in seqMeta after applying genomic control. A Bonferroni correction was applied to define study-wide significance (SWS) in each group (EA: p< 4.98×10−7, CH: p< 7.73×10−7, and meta p< 1.08×10−6; Supplementary Table 2).
Figure 1.
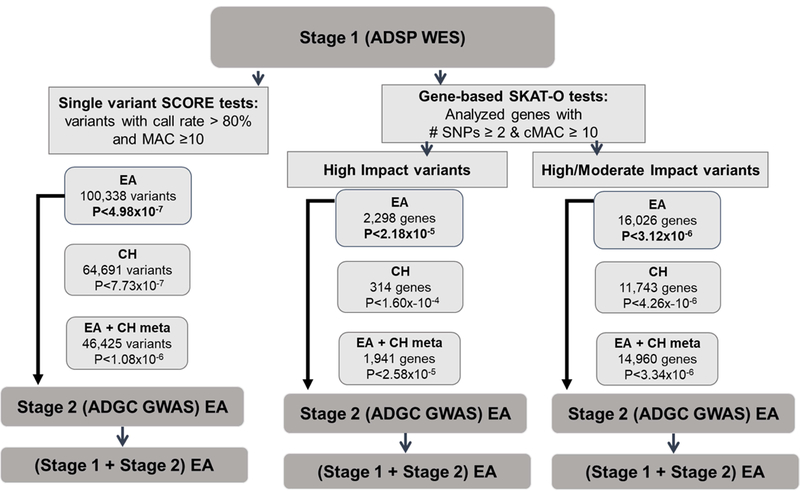
Analysis design. MAC = minor allele account; cMAC = cumulative minor allele count; EA = European ancestry; CH = Caribbean Hispanic; WES = whole exome sequencing; ADSP= Alzheimer’s Disease Sequencing Project; ADGC = Alzheimer’s Disease Genetics Consortium; GWAS = genome-wide association study.
It is well-known that the standard maximum likelihood estimation of the logistic model can suffer from small-sample bias [25]. We applied a penalized likelihood method (i.e., the Firth logistic regression test [25–27]) to evaluate association of the odds ratio (OR) and confidence intervals for all top single variants using Heinze’s “logistf” package in R (http://cemsiis.meduniwien.ac.at/en/kb/science-research/software/statistical-software/ fllogistf/).
2.4. Gene-based Association Analysis
In an attempt to improve power by removing non-functional variants, we selected variants on the basis of annotated function using the Ensembl Variant Effect Predictor (VEP) [28] and SnpEff [29] software as follows: a) HIGH IMPACT: variants classified as splice acceptor, splice donor, stop gained, frameshift, stop lost, start lost, or transcript amplification; b) HIGH or MODERATE IMPACT: included the categories above plus variants annotated as in-frame insertion, in-frame deletion, missense variant, or protein altering. Association was tested for genes with ≥ 2 variants and a cumulative MAC (cMAC) ≥ 10 after excluding variants with a minor allele frequency (MAF) >0.05 using the same three models tested in the individual variant analyses and the SKAT-O program in seqMeta [30]. Separate analyses were performed for high impact variants only (2,298 genes in EAs and 314 genes in CHs) and for high and moderate impact variants (16,026 genes in EAs and 11,743 genes in CHs). Analyses of the combined populations included 1,941 genes with high-impact variants and 14,960 genes with high/moderate impact variants. The ethnic-specific gene-based results were combined by meta-analysis of Z-scores weighted by the number of subjects using seqMeta, assuming the same direction of effect on a gene in both populations. Significance thresholds for each analysis were determined based on the number of genes tested (Supplementary Table 3).
2.5. Stage 2 Analyses in GWAS datasets
We attempted to confirm the top-ranked discovery stage results from single variant analysis (p<1.0×10−5) and gene-based tests (p<1.0×10−4) obtained from the best-model (i.e. the smallest P-value among the three models tested for each individual variant or gene) using ADGC GWAS datasets in which genotypes for ~39M variants as rare as MAF=0.0004 were imputed with the Haplotype Reference Consortium (HRC) r1.1 reference panel [31] using MiniMac3 (see Supplementary Methods for additional details of imputation procedures). In order to be consistent with enriched cases design of the discovery analyses, we evaluated the two ADGC family-based cohorts, MIRAGE (449 AD cases and 704 controls) and NIA-LOAD (1,568 cases and 1,457 controls), after excluding subjects who were included in the ADSP WES dataset (Table 1). Models 1 and 2 only were evaluated in each data set using imputed allele dosages for each variant, generalized estimating equations (GEE) implemented in geepack R package for single variant tests, and ‘F-SKAT’ [32] for gene-based tests to account for the family structure. Model 0 was not evaluated in the Stage 2 datasets because these samples did not have unique ascertainment schemes for AD cases and controls on the basis of age and sex. Results from the Stage 2 datasets were combined using a fixed-effects inverse variance-weighted method in METAL [33] applied to single variant results and the sample-size weighted Z-score method applied to gene-based results. Successful replication was determined using a nominal significance threshold (p<0.05) and, for single variants, if the effect direction was the same in the Stage 1 and Stage 2 datasets. Results from Stages 1 and 2 were combined using the same meta-analysis approach.Results for Models 0 and 1 in the Stage 1 dataset were each meta-analyzed with those obtained from Model 1 in the Stage 2 datasets.
Table 1.
Number of AD cases and controls in the Stage 1 WES dataset and Stage 2 ADGC family-based GWAS datasets.
| Stage | Cohort | Number controls |
Number AD cases |
Total |
|---|---|---|---|---|
| Stage 1 (ADSP WES) |
EA | 4,917 | 507 | 5,424 |
| CH | 177 | 172 | 349 | |
| Total | 5,094 | 679 | 5,773 | |
| Stage 2 (ADGC GWAS) |
MIRAGE (EA) | 704 | 449 | 1,153 |
| NIA-LOAD (EA) | 1,457 | 1,568 | 3,025 | |
| Total | 2,161 | 2,017 | 4,178 | |
| Combined | 7,255 | 2,696 | 9,951 | |
AD = Alzheimer’s Disease; EA = European Ancestry; CH = Caribbean Hispanic; WES = whole exome sequencing; ADSP= Alzheimer’s Disease Sequencing Project; ADGC = Alzheimer’s Disease Genetics Consortium; GWAS = genome-wide association study; MIRAGE = Multi Institutional Research in Alzheimer’s Genetic Epidemiology Study; NIA- LOAD = National Institute on Aging - Late Onset Alzheimer’s Disease Family Study
2.6. Stage 2 Analyses in the ADSP Family-based WGS Dataset
We further examined the top-ranked discovery stage results (52 individual variants and eight genes) in the ADSP WGS family-based dataset [21, 34]. This dataset includes 197 individuals sequenced in 42 EA families and 501 individuals in 115 CH families. Association of individual variants was evaluated by inspecting their segregation within families. Gene-based tests were conducted separately for EA and CH families using F- SKAT.
3. Results
There was little evidence for genomic inflation in single variant based exome-wide results in the EA (λ=0.92), CH (λ=1.05), or combined populations (λ=1.07; Supplementary Fig. 1).
3.1. .Single-variant Results in EAs
In Model 0, rare variants in TREM2 (p=4.56×10−12), NPC1 (p=5.78×10−9), CASP7 (p=2.44×10−10) and KCNK13 (p=1.55×10−7) were significantly associated with AD (Table 2, Supplementary Fig. 2). After adjusting for age and sex (Model 1), the evidence for association with all four genes was reduced but the TREM2 and NPC1 variants were still significant (P<5.0×10−7). Additional adjustment for APOE genotype (Model 2) further diminished associations with all genes but NPC1 (p<9.41×10−5; Supplementary Table 4). Variants in HOXB-AS1/HOXB2 (p=7.63×10−8), HTR3A (p=1.28×10−7), ZNF333(p=1.28×10−7) and STAB1 (p=3.58×10−7) surpassed the SWS threshold for Model 1, and the association with the HOXB-AS1/HOXB2 variant was also significant in Model 2 (p=1.30×10−7). For Model 2, SWS associations were identified with rare variants in six additional novel gene regions including SCN4A (p=6.30×10-14), MUC17 (p=1.63×10−9), AKNAD1 (two variants in complete LD, p=2.71×10−8 for both), KANSL3 (p=6.40×10−8), TMEM87A (p=2.79×10−7) and OTOG (p=4.16×10−7). Suggestive evidence for association (p<5.0×10−6) was obtained for variants in seven additional genes (Supplementary Table 4). In addition to the previously known TREM2 R47H variant [8–10], 52 variants including two indels from 50 novel loci met criteria for follow up in the Stage 2 datasets. Analyses of these variants yielded nominally significant (P<0.05) results, however only the CASP7 variant showed the same effect direction, and this association was slightly more significant (p=1.92×10−10) in the combined Stage 1+2 sample (Table 2, Supplementary Table 5). This CASP7 variant is a previously identified missense mutation (rs116437863) that results in an amino acid substitution of glycine for arginine and is predicted to be deleterious and probably damaging by SIFT [35] (score =0) and PolyPhen [36] (score = 0.99).
Table 2.
Single-variant association results in European ancestry individuals excluding the APOE region *
| Chr: Map Position : Effect Allele : Reference Allele | dbSNP ID | Function | Gene Symbol | Stage 1 | Stage 2 Datasets P-value | Stage 1 + 2 | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| MAC (EA) | MAC (CH) | MAC Total | OR** (95% CI) | Minimum P-value | Best Model | meta | MIRAGE† | NIA-LOAD† | P-value | Direction | ||||
| 6:41129252:C:T | rs75932628 (R47H) | missense | TREM2 | 34 | 0 | 34 | 11.8(5.0–29.1) | 4.56E-12 | M0 | 1.49E-01 | 7.68E-01 | 1.06E-01 | 5.23E-05 | +++ |
| 18:21118536:G:A | rs150334966 | missense | NPC1 | 11 | 0 | 11 | 38.2(6.7–506.4) | 5.78E-09 | M0 | 8.94E-02 | 9.09E-02 | 5.83E-01 | 4.68E-08 | +-- |
| 10:115489177:A:G | rs116437863 | missense | CASP7 | 10 | 0 | 10 | 19.6(5.6–68.2) | 2.44E-10 | M0 | 1.28E-01 | 3.51E-01 | 6.08E-02 | 1.92E-10 | +++ |
| 14:90651236:C:T | rs112153420 | synonymous | KCNK13 | 59 | 2 | 61 | 4.8(2.4–9.3) | 1.55E-07 | M0 | 5.86E-01 | 4.64E-01 | 3.56E-01 | 0.0024 | +-+ |
| 17:46620725:T:A | rs183316427 | missense | HOXB2 | 78 | 5 | 83 | 5.1(2.6–10.2) | 7.63E-08 | M1 | 7.92E-01 | 6.85E-01 | 5.05E-01 | 0.0026 | ++- |
| 11:113846023:C:T | rs62625041 | 5’UTR | HTR3A | 12 | 0 | 12 | 114.9(5.1–24,6) | 1.28E-07 | M1 | 1.23E-01 | 1.33E-01 | 5.23E-01 | 0.012 | +-- |
| 19:14829543:C:T | rs79724046 # | synonymous | ZNF333 | 35 | 26 | 61 | 8.8(3.4–22.0) | 1.28E-07 | M1 | 1.23E-01 | 1.33E-01 | 5.23E-01 | 0.00033 | +++ |
| 3:52552876:C:T | rs372601175 | synonymous | STAB1 | 10 | 0 | 10 | 13.9(3.5–62.7) | 3.58E-07 | M1 | 2.01E-01 | 2.96E-01 | 2.02E-01 | 8.68E-05 | +++ |
| 17:62026823:G:A | rs73992419 # | synonymous | SCN4A | 10 | 19 | 29 | 48.2(9.1–242.1) | 6.30E-14 | M2 | 3.76E-01 | 6.84E-01 | 4.13E-01 | 0.10 | +-- |
| 7:100679240:G:C | rs144027969 | missense | MUC17 | 35 | 0 | 35 | 191.2(11.4–28,0) | 1.63E-09 | M2 | NA | NA | NA | NA | NA |
| 1:109394718:G:T | rs141968711 | missense | AKNAD1 | 11 | 0 | 11 | 22.8(4.6–113.1) | 2.71E-08 | M2 | 1.26E-01 | 2.73E-02 | 2.22E-02 | 0.30 | ++- |
| 1:109394720:C:G | rs747976210 | synonymous | AKNAD1 | 11 | 0 | 11 | 22.8(4.6–113.1) | 2.71E-08 | M2 | 1.26E-01 | 2.73E-02 | 2.22E-02 | 0.30 | ++- |
| 2:97270095:C:T | rs34406082 | missense | KANSL3 | 12 | 7 | 19 | 47.7(6.6–366.3) | 6.40E-08 | M2 | 9.33E-01 | 6.83E-01 | 7.61E-01 | 0.21 | +-+ |
| 15:42565552: CAGA:C | rs550307753 | Inframe deletion | TMEM87A | 11 | 2 | 13 | 36.9(7.4–232.6) | 2.79E-07 | M2 | NA | NA | NA | NA | NA |
| 11:17581152:C:T | rs199968574 | missense | OTOG | 11 | 1 | 12 | 29.3(4.4–161.4) | 4.16E-07 | M2 | 9.90E-02 | 1.51E-01 | 1.07E-01 | 0.78 | +-- |
Table shows variants with p<5.0×10−7, bonferroni significance threshold (i.e., SWS);
OR was based on Firth logistic regression tests;
Family-based ADGC GWAS dataset excluding subjects included in the Stage 1 sample.
variant co-segregated with AD in ≥ 2 CH Stage 2 families;
MAC = minor allele account; EA = European ancestry; CH = Caribbean Hispanic;
OR = Odds Ratio for best model (OR and 95% CI for all three model are provided in Supplementary Table 4) M0 = Model 0: adjustment for PCs and sequencing center;
M1 = Model 1: same adjustments as Model 0 + age and sex;
M2 = Model 2: same adjustments as Model 1 + APOE ɛ4 status and APOE ɛ2 status Study-wide significant results highlighted in bold
3.2. .Single-variant results in Caribbean Hispanics
No variants reached SWS (p<7.73×10−7) in the CH group. However, notable novel association signals were observed with a SNV in LDB3 (p=5.11 ×10−6), a previously known 6 bp frameshift deletion (rs782084513) in ORAI1 (p=5.34×10−5), and a 3 bp deletion in KLHL40 (p=7.98×10−5). The strength of these associations was similar in all three models (Supplementary Table 6) indicating they are independent of age, sex and APOE genotype. The LDB3 and ORAI1 variants were also observed in EAs but were not associated with AD risk (p>0.25). Conversely, none of the top-ranked rare variants in EAs had a MAC≥10 in the CH group, except for the variants in ZNF333 (MAC=26) and SCN4A (MAC=19) which were not associated with AD risk in the CH dataset (p>0.20) (Supplementary Table 7). However, a rare variant SLAIN1 showed mild evidence of association in both EA (p=0.00015) and CH (p=0.0023) groups, and the Model 0 result from the two groups combined approached SWS (meta p=4.68×10−6; Supplementary Table 8).
3.3. .Gene-based association results
In analyses focused on high-impact variants, associations with six novel loci were SWS (p<2.18×10−5) including JMJD4, C1orf173, ANXA5, AARD, ASCC1, and ASB13 (Table 3). Results for two additional novel genes (DTYMK and IGHJ6) were SWS (p<3.12×10−6) in analyses that included high and moderate-impact variants. Among these findings, only AARD showed evidence for association in the Stage 2 datasets (p=6.16×10−3). However, associations were strengthened for C1orf173 (p=1.50×10−5), ANXA5 (p=2.27×10−5) and AARD (p=1.01×10−6) in the combined Stage 1+2 dataset. Although none of the gene-based tests were SWS in the CH group, analyses of high and moderate impact variants revealed a significant association with KLHL40 (p=9.98×10−5 in model 0 and p=1.01×10−4 in models 1 and 2).
Table 3.
Gene-based association results identified from EA individuals
| Stage 1 | Stage 2 Cohorts (MIRAGE + NIA-LOAD) | Stage 1 + 2 | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| High Impact Variants (SWS threshold - p< 2.18×10−5) | ||||||||||||
| Gene Symbol | Chr | Start | End | cMAC | # SNPs | cMAF | p-value | Best Model | # SNPs MIRAGE | # SNPs LOAD | Meta p-value | Meta p-value |
| JMJD4 | 1 | 227,920,244 | 227,922,975 | 16 | 10 | 0.00148 | 3.56E-05 | M0 | 7 | 6 | 9.36E-01 | 0.0019 |
| ANXA5 | 4 | 122,593,712 | 122,605,898 | 13 | 6 | 0.0012 | 4.66E-05 | M0 | 6 | 2 | 1.24E-01 | 2.27E-05 |
| ASB13 | 10 | 5,682,665 | 5,695,015 | 12 | 2 | 0.00111 | 4.32E-05 | M1 | 3 | 0 | 1.83E-01 | 5.20E-05 |
| ASCC1 | 10 | 73,857,161 | 73,970,584 | 122 | 5 | 0.01126 | 2.51E-06 | M2 | 6 | 5 | 7.89E-01 | 0.00019 |
| C1orf173 | 1 | 75,037,039 | 75,102,074 | 11 | 10 | 0.00102 | 4.41E-05 | M2 | 15 | 12 | 1.38E-01 | 1.50E-05 |
| AARD | 8 | 117,954,812 | 117,954,935 | 16 | 2 | 0.00148 | 4.84E-05 | M2 | 3 | 0 | 6.16E-03 | 1.01E-06 |
| High/Moderate Impact Variants (SWS threshold - p< 3.12×10−6) | ||||||||||||
| IGHJ6 | 14 | 106,329,417 | 106,329,468 | 10 | 9 | 0.00093 | 3.46E-07 | M0 | 0 | 0 | NA | NA |
| DTYMK | 2 | 242,615,564 | 242,625,278 | 15 | 13 | 0.00138 | 2.95E-11 | M2 | 0 | 3 | 6.60E-01 | 3.40E-08 |
Chr = chromosome; cMAC = cumulative minor allele account; cMAF = cumulative minor allele frequency; EA = European Ancestry; CH = Caribbean Hispanic
M0 = Model 0: adjustment for PCs and sequencing center; M1 = Model 1: same adjustments as Model 0 + age and sex; M2 = Model 2: same adjustments as Model 1 + APOE ɛ4 status and APOE ɛ2 status
Study-wide significant (SWS) results highlighted in bold
3.4. .Stage 2 results in the ADSP Family-based WGS data
SWS and suggestive associations for 52 individual variants (Supplementary Table 4) and eight genes (Table 3) were further examined in the ADSP WGS family-based dataset. Low-frequency missense variants in AIM1L (rs80177817, MAF=0.02, Discovery p=6.14×10−7) were observed and segregated nearly perfectly with disease in two EA families and one CH family. A previously identified rare frameshift mutation in DHX37 (rs779974893, MAF=0.001, Stage 1 p=7.68×10−6), and the known AD-associated rare missense mutation in TREM2 (R47H, MAF=0.003) each occurred and segregated with disease in one EA family (Supplementary Table 9). A rare missense variant in ENGASE (rs11871357, MAF=0.001, Stage 1 p=7.47×10−6) and a rare synonymous variant in ZNF333 (rs79724046, MAF=0.003, Stage 1 p=1.28×10−7) each occurred and were observed predominantly among affected members with disease in three CH families. A rare variant in KANSL3 (rs34406082, Stage 1 p=6.40×10−8) perfectly co- segregated with disease in two CH families. Low-frequency variants in SCN4A (rs73992419, Stage 1 p=6.30×10−14) and PTGIS (rs61322884, Stage 1 p=4.55×10−6) each showed a high degree of co-segregation with disease in two CH families. None of the gene-based tests in EA or CH families were nominally significant (P>0.20; Supplementary Table 10), noting that none of the rare variants that primarily accounted for the gene associations in the Stage 1 sample were observed in the WGS families (Supplementary Table 11).
3.5. .Findings for previously reported AD-associated rare variants
Of 15 rare variants previously reported to be associated with AD, two non-exonic ABCA7 variants not included in the WES capture. Three of the tested seven variants (i.e., those with MAC ≥ 10) were significantly associated with AD after correcting for the number of tests (P< 0.007) in a model adjusting for age, sex and APOE dosage: PLD3- V232M, ABI3-S209F, and SORL1-A528T (Supplementary Table 12).
4. Discussion
We identified novel associations for AD with a single rare variant in CASP7 and gene- based tests of aggregated rare variants in C1orf173, ANXA5, and AARD in 5,094 controls and a subset of 679 unrelated familial AD cases from the ADSP. These findings were study-wide significant and improved when combined with results obtained from HRC-imputed data from 2,161 AD cases and 2,017 controls in two family-based ADGC GWAS datasets. Study-wide significant findings at the variant or gene level were observed for several other loci (NPC1, KCNK13, HOXB2, HTR3A, ZNF333, STAB1, SCN4A, MUC17, AKNAD1, KANSL3, TMEM87A, OTOG, DTYMK, and IGHJ6), but these findings were not bolstered by the Stage 2 datasets. A previous study by the ADSP of the entire WES dataset including 5,740 AD cases reported associations with one rare variant in AC099552.4 and nine high-impact aggregated variants in ZNF655 [35]. The greatly improved detection of associations with rare variants in this smaller sample of AD cases is likely due to the enriched case design. For example, the result for the established rare TREM2 R47H variant (p=4.56×10−12) is virtually identical to the result for this variant in the prior study [35] which is consistent with the observation of the R47H variant only among enriched AD cases in the entire dataset. None of the other top association findings in this study were remarkable in the analysis of the enlarged ADSP sample including cases that were not ascertained on the basis of family history of AD; the strongest signals were observed for KCNK13 (P=0.0063) and HOXB2 (P=0.0019) (Supplementary Table 13).
CASP7 encodes a member of the cysteine-aspartic acid protease (caspase) family. Sequential activation of caspases plays a central role in the execution-phase of cell apoptosis. Caspase-7 is a protease involved in apoptosis and inflammation [37]. Activation of caspase apoptotic pathways involves apoptosome assembly [38] and multiple recent studies link this process to aging and AD neuropathology, including caspase cleavage of amyloid precursor protein [39–41] and tau [40, 42] (Figure 2). Roles for other caspases in AD pathogenesis have been described, including caspase-6 in cognitive impairment [43] and tau cleavage [44], caspase-8 in amyloid processing, synaptic plasticity, learning, memory and control of microglia pro-inflammatory activation and associated neurotoxicity [45, 46], and caspase-9 activation in tau cleavage [47]. Su et al. reported that activated caspase-3 expression correlates with Alzheimer pathology [48]. Caspase 3 is processed by caspases 8, 9, and 10, and is the predominant caspase involved in the cleavage of amyloid precursor protein, which is associated with neuronal death and plaque formation in AD brain [49]. A recent targeted sequencing study of genes involved in amyloid metabolism found association of AD with two CASP8 rare variants [46]. Our finding of a rare missense variant in CASP7 provides additional evidence for the role of caspase apoptotic pathways in AD. Further functional studies of CASP7, and the rs116437863 missense variant in particular, are needed to define its role in AD pathogenesis and evaluate the potential of caspase 7 inhibition as an AD treatment strategy.
Figure 2.
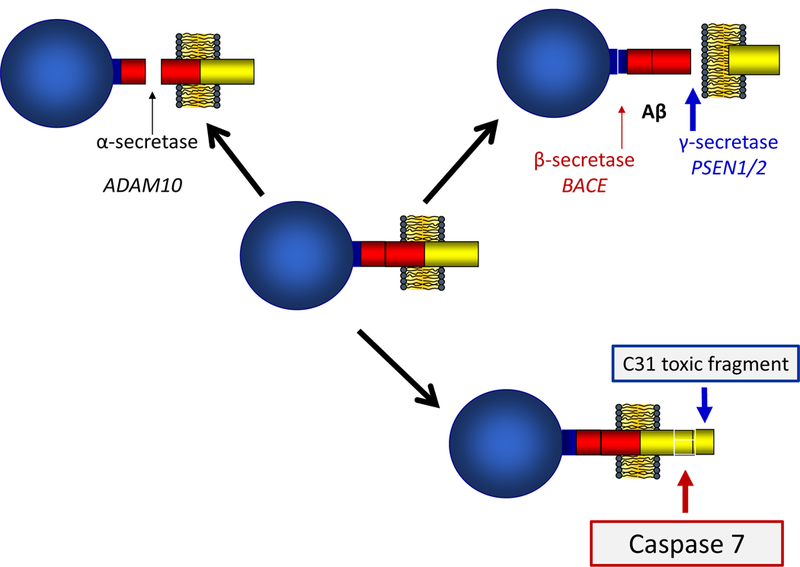
Processing of the amyloid precursor protein (APP) by α (ADAM10), β (BACE), and γ (PSEN1/2) secretases. Alternate processing of APP may result in cleavage of the C31 fragment by the protease encoded by CASP7 [41]. C31 is one of several C- terminal fragments produced from APP and there is some evidence that it is toxic [54].
The association with a rare 3 bp in-frame deletion (rs550307753) in TMEM87A reached the study-wide significance in EAs (p=2.79×10−7), but this finding could not be replicated because this variant was not genotyped or imputed well in the ADGC GWAS datasets.
Gene-based tests considering only highly deleterious SNVs and indels yielded highly significant associations with three novel genes (C1orf173, ANXA5, and AARD) which were strengthened by meta analysis with results from the Stage 2 datasets. ANXA5 is phosphoIipase A2 kinase C inhibitory protein that has been implicated in membrane- related events along exocytotic and endocytotic pathways, and in AD [50]. The function of Corf173 (alias ERICH3 - glutamate rich 3) is largely unknown. AARD has no obvious connection to AD or brain. IGHJ6 was another SWS gene-based finding that did not replicate because none of the high or moderate impact rare variants each occurring only once or twice in the Stage 1 sample were observed in the Stage 2 datasets. This gene encodes one of the immunoglobulin heavy gamma variable chains and is a very good candidate given its functional similarity to IGHG3, one of the top associations in the entire ADSP WES sample (unpublished result) and evidence that antibodies to IgG cross-react with fibril and oligomer amyloid-β aggregates [51].
In comparison with the EA cohort, the CH cohort is very small with only 172 enriched cases and 177 controls. Nonetheless, we observed suggestive CH-specific association signals with three infrequent (2%<MAF<5%) previously known variants including a SNV (rs76615432) in LDB3 (p=5.11×10−6), a small deletion causing a frameshift (rs782084513) in ORAI1 (p=5.34×10−5), and a small deletion in KLHL40 (p=7.98×10−5). Notably, KLHL40 is the only gene which yielded top-ranked results from individual variant and gene-based tests, due largely to the fact that among nine distinct KLH40 variants that were observed one SWS variant (rs34020089) accounted for 21 of the 36 aggregated rare variants (i.e., MAC=36) in the gene-based test (Supplementary Table 6, Supplementary Table 14).
Several strengths and limitations of our study warrant discussion. One of the major strengths lies in the careful clinical and genetic characterization of all individuals enrolled in the ADSP. Another strength of the study is the enriched cases design which strengthened the genetic signals thus allowing detection of associations that would otherwise be missed in a larger traditional case-control study.
A major limitation of the study is that the sample sizes are relatively small especially for enriched AD cases and the CH group overall. In the Stage 1 EA group, the enriched cases represent approximately 10% of the total sample which had sufficient power to detect association with moderate effect variants [24]. It has been shown that one can reduce the sample size of cases approximately four-fold using cases who have at least two affected relatives to have the same power as a sample of cases that are not ascertained on the basis of family history [52]. Although our case sample was not large enough to benefit fully from the enriched cases design, it was sufficient to detect association with highly penetrant variants whose effects would be diluted in a sample not ascertained on the basis of family history [52]. This idea is exemplified by the TREM2 R47H variant for which we observed similar p values but a higher odds ratio in the current study (p=4.56×10−12, OR=11.82, Supplementary Table 13) compared to the finding for this variant in the total group of ADSP cases (p=4.8×10−12, OR=3.61) [35].
Another limitation is that coding variants were identified directly in the Stage 1 dataset, but imputed in the Stage 2 GWAS datasets with varying degrees of confidence for those with MAF<0.5%−1.0%. In order to be consistent with the study design of the discovery stage analysis, we included only family-based cohorts from the ADGC HRC-imputed GWAS dataset, MIRAGE and NIA-LOAD, in Stage 2 and thus had little power to confirm our findings. The relatively small size and unreliable imputation of very rare variants in the Stage 2 sample particularly limited our ability to replicate findings from gene-based tests. Also, the ADSP WES study design, for which AD cases were selected to have relatively early onset and a lower frequency of the APOE ε4 allele and controls were selected to be as old as possible with preference given to those having at least one APOE ε4 allele to enrich this group for protective variants, introduced confounding between age and AD status which reduced power for detecting associations. To overcome this limitation, we included a model without age adjustment. These limitations underscore the need to replicate our findings in other datasets containing enriched cases. Finally, our primary analysis relied on the score test which is prone to increased type-I errors for rare variants in unbalanced samples [53]. For this reason, we re- evaluated our top results using the Firth test. This analysis showed that the finding for the rare CASP7 variant is attenuated (p=2.21×10−5). Recognizing that the Firth test over- corrects for bias in very sparse data [24], the true p-value may be between those obtained using the score and Firth tests.
In summary, we identified multiple novel associations for AD with individual and aggregated rare variants using an enriched case-control study design. A better understanding of the molecular mechanisms underlying these associations will require functional experiments and in silico studies of the connections of genetic variants to gene expression and processing of AD-related proteins.
Supplementary Material
RESEARCH IN CONTEXT.
Systematic review: The authors are members of the Alzheimer’s Disease Sequencing Project and therefore are familiar with emerging pertinent literature. PubMed searches were conducted to identify other relevant publications. References that support the significance of the identified risk loci are cited.
Interpretation: Although both common and rare variants in >30 late-onset Alzheimer’s disease (LOAD) risk genes have been identified from genome-wide association and whole exome sequencing studies, this report identifies associations with rare variants in several novel loci for LOAD using a design focused on LOAD cases that are likely genetically enriched because they are members of families with multiple affected members. CASP7 provides further evidence for the role of caspase apoptotic pathways in AD.
Future directions: A better understanding of the molecular mechanisms underlying these associations will require functional experiments and in silico studies of the connections of genetic variants to gene expression and processing of AD-related proteins. Further studies are also needed to determine whether CASP7 is a suitable target for development of novel therapies.
Acknowledgments
This work was supported in part by NIA grants R01AG048927, P30-AG13846 and RF1AG057519. The Alzheimer’s Disease Sequencing Project (ADSP) is comprised of two Alzheimer disease genetics consortia and three National Human Genome Research Institute (NHGRI) funded Large Scale Sequencing and Analysis Centers (LSAC). The two AD genetics consortia are the Alzheimer’s Disease Genetics Consortium (ADGC) funded by NIA (U01 AG032984), and the Cohorts for Heart and Aging Research in Genomic Epidemiology (CHARGE) funded by NIA (R01 AG033193), the National Heart, Lung, and Blood Institute (NHLBI), other National Institute of Health (NIH) institutes and other foreign governmental and non-governmental organizations. The Discovery Phase analysis of sequence data is supported through UF1AG047133 (to Drs. Farrer, Haines, Mayeux, Pericak-Vance and Schellenberg); U01AG049505 to Dr. Sudha Seshadri; U01AG049506 to Dr. Eric Boerwinkle; U01AG049507 to Dr. Ellen Wijsman; and U01AG049508 to Dr. Alison Goate and the Discovery Extension Phase analysis is supported through U01AG052411 to Dr. Goate and U01AG052410 to Dr. Pericak- Vance, and U01 AG052409 to Drs. Seshadri and Myriam Fornage. Data generation and harmonization in the Follow-up Phases is supported by U54AG052427 to Drs. Schellenberg and Li-San Wang. We thank the investigators who assembled and characterized participants of cohorts included in this study:
Abbreviations
- AD
Alzheimer disease
- ADGC
Alzheimer’s Disease Genetics Consortium
- ADSP
Alzheimer’s Disease Sequencing Project
- CH
Caribbean Hispanic
- cMAC
cumulative minor allele count
- EA
European ancestry
- GWAS
genome-wide association study
- HRC
Haplotype reference Consortium
- MAC
minor allele count
- MAF
minor allele frequency
- OR
odds ratio
- PC
principle component (of ancestry)
- QC
quality control
- SNV
single nucleotide variant
- SWS
study-wide significant
- VEP
Ensembl Variant Effect Predictor
- WES
whole exome sequencing
- WGS
whole genome sequencing
Appendix
The members of the Alzheimer’s Disease Sequencing Project are:
Baylor College of Medicine: Michelle Bellair, Huyen Dinh, Harsha Doddapeneni, Shannon Dugan-Perez, Adam English, Richard A. Gibbs, Yi Han, Jianhong Hu, Joy Jayaseelan, Divya Kalra, Ziad Khan, Viktoriya Korchina, Sandra Lee, Yue Liu, Xiuping Liu, Donna Muzny, Waleed Nasser, William Salerno, Jireh Santibanez, Evette Skinner, Simon White, Kim Worley, Yiming Zhu
Boston University: Alexa Beiser, Yuning Chen, Jaeyoon Chung, L. Adrienne Cupples, Anita DeStefano, Josee Dupuis, John Farrell, Lindsay Farrer, Daniel Lancour, Honghuang Lin, Ching Ti Liu, Kathy Lunetta, Yiyi Ma, Devanshi Patel, Chloe Sarnowski, Claudia Satizabal, Sudha Seshadri, Fangui Jenny Sun, Xiaoling Zhang
Broad Institute: Seung Hoan Choi, Eric Banks, Stacey Gabriel, Namrata Gupta
Case Western Reserve University: William Bush, Mariusz Butkiewicz, Jonathan Haines, Sandra Smieszek, Yeunjoo Song
Columbia University: Sandra Barral, Phillip L De Jager, Richard Mayeux, Christiane Reitz, Dolly Reyes, Giuseppe Tosto, Badri Vardarajan
Erasmus Medical University: Shahzad Amad, Najaf Amin, M Afran Ikram, Sven van der Lee, Cornelia van Duijn, Ashley Vanderspek
Medical University Graz: Helena Schmidt, Reinhold Schmidt
Mount Sinai School of Medicine: Alison Goate, Manav Kapoor, Edoardo Marcora, Alan Renton
Indiana University: Kelley Faber, Tatiana Foroud
National Center Biotechnology Information: Michael Feolo ,Adam Stine
National Institute on Aging: Lenore Launer Rush University: David A Bennett
Stanford University: Li Charlie Xia
University of Miami: Gary Beecham, Kara Hamilton-Nelson, James Jaworski, Brian Kunkle, Eden Martin, Margaret Pericak-Vance, Farid Rajabli, Michael Schmidt
University of Mississippi: Thomas H. Mosley
University of Pennsylvania: Laura Cantwell, Micah Childress, Yi-Fan Chou, Rebecca Cweibel, Prabhakaran Gangadharan, Amanda Kuzma, Yuk Yee Leung, Han-Jen Lin, John Malamon, Elisabeth Mlynarski, Adam Naj, Liming Qu, Gerard Schellenberg, Otto Valladares, Li-San Wang, Weixin Wang, Nancy Zhang
University of Texas Houston: Jennifer E. Below, Eric Boerwinkle, Jan Bressler, Myriam Fornage, Xueqiu Jian, Xiaoming Liu
University of Washington: Joshua C. Bis, Elizabeth Blue, Lisa Brown, Tyler Day, Michael Dorschner, Andrea R Horimoto, Rafael Nafikov, Alejandro Q Nato Jr., Pat Navas, Hiep Nguyen, Bruce Psaty, Kenneth Rice, Mohamad Saad, Harkirat Sohi, Timothy Thornton, Debby Tsuang, Bowen Wang, Ellen Wijsman, Daniela Witten
Washington University: Lucinda Antonacci-Fulton, Elizabeth Appelbaum, Carlos Cruchaga, Robert S. Fulton, Daniel C. Koboldt, David E. Larson, Jason Waligorski, Richard K. Wilson
Adult Changes in Thought: James D. Bowen, Paul K. Crane, Gail P. Jarvik, C. Dirk Keene, Eric B. Larson, W. William Lee, Wayne C. McCormick, Susan M. McCurry, Shubhabrata Mukherjee, Katie Rose Richmire
Atherosclerosis Risk in Communities Study: Rebecca Gottesman, David Knopman, Thomas H. Mosley, B. Gwen Windham,
Austrian Stroke Prevention Study: Thomas Benke, Peter Dal-Bianco, Edith Hofer, Gerhard Ransmayr, Yasaman Saba
Cardiovascular Health Study: James T. Becker, Joshua C. Bis, Annette L. Fitzpatrick, M. Ilyas Kamboh, Lewis H. Kuller, WT Longstreth, Jr, Oscar L. Lopez, Bruce M. Psaty, Jerome I. Rotter,
Chicago Health and Aging Project: Philip L. De Jager, Denis A. Evans
Erasmus Rucphen Family Study: Hieab H. Adams, Hata Comic, Albert Hofman, Peter J. Koudstaal, Fernando Rivadeneira, Andre G. Uitterlinden, Dina Voijnovic
Estudio Familiar de la Influencia Genetica en Alzheimer: Sandra Barral, Rafael Lantigua, Richard Mayeux, Martin Medrano, Dolly Reyes-Dumeyer, Badri Vardarajan
Framingham Heart Study: Alexa S. Beiser, Vincent Chouraki, Jayanadra J. Himali, Charles C. White
Genetic Differences: Duane Beekly, James Bowen, Walter A. Kukull, Eric B. Larson, Wayne McCormick, Gerard D. Schellenberg, Linda Teri
Mayo Clinic: Minerva M. Carrasquillo, Dennis W. Dickson, Nilufer Ertekin-Taner, Neill R. Graff-Radford, Joseph E. Parisi, Ronald C. Petersen, Steven G. Younkin
Mayo PD: Gary W. Beecham, Dennis W. Dickson, Ranjan Duara, Nilufer Ertekin-Taner, Tatiana M. Foroud, Neill R. Graff-Radford, Richard B. Lipton, Joseph E. Parisi, Ronald C. Petersen, Bill Scott, Jeffery M. Vance
Memory and Aging Project: David A. Bennett, Philip L. De Jager
Multi-Institutional Research in Alzheimer’s Genetic Epidemiology Study: Sanford Auerbach, Helan Chui, Jaeyoon Chung, L. Adrienne Cupples, Charles DeCarli, Ranjan Duara, Martin Farlow, Lindsay A. Farrer, Robert Friedland, Rodney C.P. Go, Robert C. Green, Patrick Griffith, John Growdon, Gyungah R. Jun, Walter Kukull, Alexander Kurz, Mark Logue, Kathryn L. Lunetta, Thomas Obisesan, Helen Petrovitch, Marwan Sabbagh, A. Dessa Sadovnick, Magda Tsolaki
National Cell Repository for Alzheimer’s Disease: Kelley M. Faber, Tatiana M. Foroud
National Institute on Aging (NIA) Late Onset Alzheimer’s Disease Family Study: David A. Bennett, Sarah Bertelsen, Thomas D. Bird, Bradley F. Boeve, Carlos Cruchaga, Kelley Faber, Martin Farlow, Tatiana M Foroud, Alison M Goate, Neill R. Graff-Radford, Richard Mayeux, Ruth Ottman, Dolly Reyes-Dumeyer, Roger Rosenberg, Daniel Schaid, Robert A Sweet, Giuseppe Tosto, Debby Tsuang, Badri Vardarajan
NIA Alzheimer Disease Centers: Erin Abner, Marilyn S. Albert, Roger L. Albin, Liana G. Apostolova, Sanjay Asthana, Craig S. Atwood, Lisa L. Barnes, Thomas G. Beach, David A. Bennett, Eileen H. Bigio, Thomas D. Bird, Deborah Blacker, Adam Boxer, James B. Brewer, James R. Burke, Jeffrey M. Burns, Joseph D. Buxbaum, Nigel J. Cairns, Chuanhai Cao, Cynthia M. Carlsson, Richard J. Caselli, Helena C. Chui, Carlos Cruchaga, Mony de Leon, Charles DeCarli, Malcolm Dick, Dennis W. Dickson, Nilufer Ertekin-Taner, David W. Fardo, Martin R. Farlow, Lindsay A. Farrer, Steven Ferris, Tatiana M. Foroud, Matthew P. Frosch, Douglas R. Galasko, Marla Gearing, David S. Geldmacher, Daniel H. Geschwind, Bernardino Ghetti, Carey Gleason, Alison M. Goate, Teresa Gomez-Isla, Thomas Grabowski, Neill R. Graff-Radford, John H. Growdon, Lawrence S. Honig, Ryan M. Huebinger, Matthew J. Huentelman, Christine M. Hulette, Bradley T. Hyman, Suman Jayadev, Lee-Way Jin, Sterling Johnson, M. Ilyas Kamboh, Anna Karydas, Jeffrey A. Kaye, C. Dirk Keene, Ronald Kim, Neil W Kowall, Joel H. Kramer, Frank M. LaFerla, James J. Lah, Allan I. Levey, Ge Li, Andrew P. Lieberman, Oscar L. Lopez, Constantine G. Lyketsos, Daniel C. Marson, Ann C. McKee, Marsel Mesulam, Jesse Mez, Bruce L. Miller, Carol A. Miller, Abhay Moghekar, John C. Morris, John M. Olichney, Joseph E. Parisi, Henry L. Paulson, Elaine Peskind, Ronald C. Petersen, Aimee Pierce, Wayne W. Poon, Luigi Puglielli, Joseph F. Quinn, Ashok Raj, Murray Raskind, Eric M. Reiman, Barry Reisberg, Robert A. Rissman, Erik D. Roberson, Howard J. Rosen, Roger N. Rosenberg, Martin Sadowski, Mark A. Sager, David P. Salmon, Mary Sano, Andrew J. Saykin, Julie A. Schneider, Lon S. Schneider, William W. Seeley, Scott Small, Amanda G. Smith, Robert A. Stern, Russell H. Swerdlow, Rudolph E. Tanzi, Sarah E Tomaszewski Farias, John Q. Trojanowski, Juan C. Troncoso, Debby W. Tsuang, Vivianna M. Van Deerlin, Linda J. Van Eldik, Harry V. Vinters, Jean Paul Vonsattel, Jen Chyong Wang, Sandra Weintraub, Kathleen A. Welsh-Bohmer, Shawn Westaway, Thomas S. Wingo, Thomas Wisniewski, David A. Wolk, Randall L. Woltjer, Steven G. Younkin, Lei Yu, Chang-En Yu
Religious Orders Study: David A. Bennett, Philip L. De Jager
Rotterdam Study: Kamran Ikram, Frank J Wolters
Texas Alzheimer’s Research and Care Consortium: Perrie Adams, Alyssa Aguirre, Lisa Alvarez, Gayle Ayres, Robert C. Barber, John Bertelson, Sarah Brisebois, Scott Chasse, Munro Culum, Eveleen Darby, John C. DeToledo, Thomas J. Fairchild, James R. Hall, John Hart, Michelle Hernandez, Ryan Huebinger, Leigh Johnson, Kim Johnson, Aisha Khaleeq, Janice Knebl, Laura J. Lacritz, Douglas Mains, Paul Massman, Trung Nguyen, Sid O’Bryant, Marcia Ory, Raymond Palmer, Valory Pavlik, David Paydarfar, Victoria Perez, Marsha Polk, Mary Quiceno, Joan S. Reisch, Monica Rodriguear, Roger Rosenberg, Donald R. Royall, Janet Smith, Alan Stevens, Jeffrey L. Tilson, April Wiechmann, Kirk C. Wilhelmsen, Benjamin Williams, Henrick Wilms, Martin Woon
University of Miami: Larry D Adams, Gary W. Beecham, Regina M Carney, Katrina Celis, Michael L Cuccaro, Kara L. Hamilton-Nelson, James Jaworski, Brian W. Kunkle, Eden R. Martin, Margaret A. Pericak-Vance, Farid Rajabli, Michael Schmidt, Jeffery M Vance
University of Toronto: Ekaterina Rogaeva, Peter St. George-Hyslop
University of Washington Families: Thomas D. Bird, Olena Korvatska, Wendy Raskind, Chang-En Yu
Vanderbilt University: John H. Dougherty, Harry E. Gwirtsman, Jonathan L. Haines
Washington Heights-Inwood Columbia Aging Project: Adam Brickman, Rafael Lantigua, Jennifer Manly, Richard Mayeux, Christiane Reitz, Nicole Schupf, Yaakov Stern, Giuseppe Tosto, Badri Vardarajan.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- [1].Hebert LE, Weuve J, Scherr PA, Evans DA. Alzheimer disease in the United States (2010–2050) estimated using the 2010 census. Neurology 2013;80:1778–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Alzheimer’s A 2015 Alzheimer’s disease facts and figures. Alzheimers Dement 2015;11:332–84. [DOI] [PubMed] [Google Scholar]
- [3].Brookmeyer R, Johnson E, Ziegler-Graham K, Arrighi HM. Forecasting the global burden of Alzheimer’s disease. Alzheimers Dement 2007;3:186–91. [DOI] [PubMed] [Google Scholar]
- [4].Gatz M, Reynolds CA, Fratiglioni L, Johansson B, Mortimer JA, Berg S, et al. Role of genes and environments for explaining Alzheimer disease. Arch Gen Psychiatry 2006;63:168–74. [DOI] [PubMed] [Google Scholar]
- [5].Lambert JC, Ibrahim-Verbaas CA, Harold D, Naj AC, Sims R, Bellenguez C, et al. Meta-analysis of 74,046 individuals identifies 11 new susceptibility loci for Alzheimer’s disease. Nat Genet 2013;45:1452–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Jun G, Ibrahim-Verbaas CA, Vronskaya M, Lambert JC, Chung J, Naj AC, et al. A novel Alzheimer disease locus located near the gene encoding tau protein. Mol Psychiatry 2016;21:108–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Jun G, Asai H, Zeldich E, Drapeau E, Chen C, Chung J, et al. PLXNA4 is associated with Alzheimer disease and modulates tau phosphorylation. Ann Neurol 2014;76:379– 92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Guerreiro R, Wojtas A, Bras J, Carrasquillo M, Rogaeva E, Majounie E, et al. TREM2 variants in Alzheimer’s disease. N Engl J Med 2013;368:117–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Jonsson T, Stefansson H, Steinberg S, Jonsdottir I, Jonsson PV, Snaedal J, et al. Variant of TREM2 associated with the risk of Alzheimer’s disease. N Engl J Med 2013;368:107–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Reitz C, Mayeux R, Alzheimer’s Disease Genetics C. TREM2 and neurodegenerative disease. N Engl J Med 2013;369:1564–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Cruchaga C, Karch CM, Jin SC, Benitez BA, Cai Y, Guerreiro R, et al. Rare coding variants in the phospholipase D3 gene confer risk for Alzheimer’s disease. Nature 2014;505:550–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Logue MW, Schu M, Vardarajan BN, Farrell J, Bennett DA, Buxbaum JD, et al. Two rare AKAP9 variants are associated with Alzheimer’s disease in African Americans. Alzheimers Dement 2014;10:609–18 e11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Wetzel-Smith MK, Hunkapiller J, Bhangale TR, Srinivasan K, Maloney JA, Atwal JK, et al. A rare mutation in UNC5C predisposes to late-onset Alzheimer’s disease and increases neuronal cell death. Nat Med 2014;20:1452–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Sims R, van der Lee SJ, Naj AC, Bellenguez C, Badarinarayan N, Jakobsdottir J, et al. Rare coding variants in PLCG2, ABI3, and TREM2 implicate microglial-mediated innate immunity in Alzheimer’s disease. Nat Genet 2017;49:1373–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Jonsson T, Atwal JK, Steinberg S, Snaedal J, Jonsson PV, Bjornsson S, et al. A mutation in APP protects against Alzheimer’s disease and age-related cognitive decline. Nature 2012;488:96–9. [DOI] [PubMed] [Google Scholar]
- [16].Wang LS, Naj AC, Graham RR, Crane PK, Kunkle BW, Cruchaga C, et al. Rarity of the Alzheimer disease-protective APP A673T variant in the United States. JAMA Neurol 2015;72:209–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Medway CW, Abdul-Hay S, Mims T, Ma L, Bisceglio G, Zou F, et al. ApoE variant p.V236E is associated with markedly reduced risk of Alzheimer’s disease. Mol Neurodegener 2014;9:11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Vardarajan BN, Zhang Y, Lee JH, Cheng R, Bohm C, Ghani M, et al. Coding mutations in SORL1 and Alzheimer disease. Ann Neurol 2015;77:215–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Steinberg S, Stefansson H, Jonsson T, Johannsdottir H, Ingason A, Helgason H, et al. Loss-of-function variants in ABCA7 confer risk of Alzheimer’s disease. Nat Genet 2015;47:445–7. [DOI] [PubMed] [Google Scholar]
- [20].Sassi C, Nalls MA, Ridge PG, Gibbs JR, Ding J, Lupton MK, et al. ABCA7 p.G215S as potential protective factor for Alzheimer’s disease. Neurobiol Aging 2016;46:235 e1- 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Beecham GW, Bis JC, Martin ER, Choi SH, DeStefano AL, van Duijn CM, et al. The Alzheimer’s Disease Sequencing Project: Study design and sample selection. Neurol Genet 2017;3:e194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Ahmad S, Bannister C, van der Lee SJ, Vojinovic D, Adams HHH, Ramirez A, et al. Disentangling the biological pathways involved in early features of Alzheimer’s disease in the Rotterdam Study. Alzheimers Dement 2018;14:848–57. [DOI] [PubMed] [Google Scholar]
- [23].Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009;25:1754–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Bis JC, Jian X, Kunkle BW, Chen Y, Hamilton-Nelson KL, Bush WS, et al. Whole exome sequencing study identifies novel rare and common Alzheimer’s-Associated variants involved in immune response and transcriptional regulation. Mol Psychiatry 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Firth D Bias reduction of maximum likelihood estimates. Biometrika 1993;80:27– 38. [Google Scholar]
- [26].Heinze G, Schemper M. A solution to the problem of separation in logistic regression. Stat Med 2002;21:2409–19. [DOI] [PubMed] [Google Scholar]
- [27].Heinze G A comparative investigation of methods for logistic regression with separated or nearly separated data. Stat Med 2006;25:4216–26. [DOI] [PubMed] [Google Scholar]
- [28].McLaren W, Gil L, Hunt SE, Riat HS, Ritchie GR, Thormann A, et al. The Ensembl Variant Effect Predictor. Genome Biol 2016;17:122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Cingolani P, Platts A, Wang le L, Coon M, Nguyen T, Wang L, et al. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly (Austin) 2012;6:80–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Lee S, Teslovich TM, Boehnke M, Lin X. General framework for meta-analysis of rare variants in sequencing association studies. Am J Hum Genet 2013;93:42–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].McCarthy S, Das S, Kretzschmar W, Delaneau O, Wood AR, Teumer A, et al. A reference panel of 64,976 haplotypes for genotype imputation. Nat Genet 2016;48:1279–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Yan Q, Tiwari HK, Yi N, Gao G, Zhang K, Lin WY, et al. A Sequence Kernel Association Test for Dichotomous Traits in Family Samples under a Generalized Linear Mixed Model. Hum Hered 2015;79:60–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Willer CJ, Li Y, Abecasis GR. METAL: fast and efficient meta-analysis of genomewide association scans. Bioinformatics 2010;26:2190–1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Blue EE BJ, Dorschner MO, Tsuang D, Barral SM, Beecham G, Below JE, Bush WS, Butkiewicz M, Cruchaga C, DeStefano A, Farrer LA, Goate A, Haines J, Jaworski J, Jun G, Kunkle B, Kuzma A, Lee JJ, Lunetta K, Ma Y, Martin E, Naj A, Nato AG Jr, Navas P, Nguyen H, Reitz C, Reyes D, Salerno W, Schellenberg G, Seshadri S, Sohi H, Thornton TA, Valladares O, van Duijn C, Vardarajan BN, Wang L- S, Boerwinkle E, Dupuis J, Pericak-Vance MA, Mayeux R, Wijsman EM, on behalf of the Alzheimer’s Disease Sequencing Project. Genetic variation in genes underlying diverse dementias may explain a small proportion of cases in the Alzheimer’s Disease Sequencing Project. . Dement Ger Cog Disorders 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Vaser R, Adusumalli S, Leng SN, Sikic M, Ng PC. SIFT missense predictions for genomes. Nat Protoc 2016;11:1–9. [DOI] [PubMed] [Google Scholar]
- [36].Adzhubei IA, Schmidt S, Peshkin L, Ramensky VE, Gerasimova A, Bork P, et al. A method and server for predicting damaging missense mutations. Nat Methods 2010;7:248–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Lamkanfi M, Kanneganti TD. Caspase-7: a protease involved in apoptosis and inflammation. Int J Biochem Cell Biol 2010;42:21–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Cullen SP, Martin SJ. Caspase activation pathways: some recent progress. Cell Death Differ 2009;16:935–8. [DOI] [PubMed] [Google Scholar]
- [39].Zhao M, Su J, Head E, Cotman CW. Accumulation of caspase cleaved amyloid precursor protein represents an early neurodegenerative event in aging and in Alzheimer’s disease. Neurobiol Dis 2003;14:391–403. [DOI] [PubMed] [Google Scholar]
- [40].Rohn TT, Kokoulina P, Eaton CR, Poon WW. Caspase activation in transgenic mice with Alzheimer-like pathology: results from a pilot study utilizing the caspase inhibitor, Q-VD-OPh. Int J Clin Exp Med 2009;2:300–8. [PMC free article] [PubMed] [Google Scholar]
- [41].Fiorelli T, Kirouac L, Padmanabhan J. Altered processing of amyloid precursor protein in cells undergoing apoptosis. PLoS One 2013;8:e57979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Cotman CW, Poon WW, Rissman RA, Blurton-Jones M. The role of caspase cleavage of tau in Alzheimer disease neuropathology. J Neuropathol Exp Neurol 2005;64:104–12. [DOI] [PubMed] [Google Scholar]
- [43].Albrecht S, Bourdeau M, Bennett D, Mufson EJ, Bhattacharjee M, LeBlanc AC. Activation of caspase-6 in aging and mild cognitive impairment. Am J Pathol 2007;170:1200–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Ramcharitar J, Albrecht S, Afonso VM, Kaushal V, Bennett DA, Leblanc AC. Cerebrospinal fluid tau cleaved by caspase-6 reflects brain levels and cognition in aging and Alzheimer disease. J Neuropathol Exp Neurol 2013;72:824–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Rohn TT, Head E, Nesse WH, Cotman CW, Cribbs DH. Activation of caspase-8 in the Alzheimer’s disease brain. Neurobiol Dis 2001;8:1006–16. [DOI] [PubMed] [Google Scholar]
- [46].Rehker J, Rodhe J, Nesbitt RR, Boyle EA, Martin BK, Lord J, et al. Caspase-8, association with Alzheimer’s Disease and functional analysis of rare variants. PLoS One 2017;12:e0185777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Rohn TT, Rissman RA, Davis MC, Kim YE, Cotman CW, Head E. Caspase-9 activation and caspase cleavage of tau in the Alzheimer’s disease brain. Neurobiol Dis 2002;11:341–54. [DOI] [PubMed] [Google Scholar]
- [48].Su JH, Zhao M, Anderson AJ, Srinivasan A, Cotman CW. Activated caspase-3 expression in Alzheimer’s and aged control brain: correlation with Alzheimer pathology. Brain Res 2001;898:350–7. [DOI] [PubMed] [Google Scholar]
- [49].Gervais FG, Xu D, Robertson GS, Vaillancourt JP, Zhu Y, Huang J, et al. Involvement of caspases in proteolytic cleavage of Alzheimer’s amyloid-beta precursor protein and amyloidogenic A beta peptide formation. Cell 1999;97:395–406. [DOI] [PubMed] [Google Scholar]
- [50].Arispe N, Diaz JC, Simakova O. Abeta ion channels. Prospects for treating Alzheimer’s disease with Abeta channel blockers. Biochim Biophys Acta 2007;1768:1952–65. [DOI] [PubMed] [Google Scholar]
- [51].O’Nuallain B, Acero L, Williams AD, Koeppen HP, Weber A, Schwarz HP, et al. Human plasma contains cross-reactive Abeta conformer-specific IgG antibodies. Biochemistry 2008;47:12254–6. [DOI] [PubMed] [Google Scholar]
- [52].Antoniou AC, Easton DF. Polygenic inheritance of breast cancer: Implications for design of association studies. Genet Epidemiol 2003;25:190–202. [DOI] [PubMed] [Google Scholar]
- [53].Ma C, Blackwell T, Boehnke M, Scott LJ, Go TDi. Recommended joint and meta- analysis strategies for case-control association testing of single low-count variants. Genet Epidemiol 2013;37:539–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Nhan HS, Chiang K, Koo EH. The multifaceted nature of amyloid precursor protein and its proteolytic fragments: friends and foes. Acta Neuropathol 2015;129:1–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.