

Abstract
Resistance to current HER2 inhibitors, such as trastuzumab (Ttzm), is a major unresolved clinical problem in HER2-positive BC (HER2-BC). Because HER2 remains overexpressed in drug-resistant HER2-BC cells, we investigated whether PEPDG278D can overcome the resistance. PEPDG278D is a recombinant enzymatically-inactive mutant of human peptidase D, which strongly inhibits HER2 in cancer cells by binding to its extracellular domain. Here we show that PEPDG278D is highly active in preclinical models of HER2-BC resistant to Ttzm and other HER2 inhibitors and also enhances the therapeutic efficacy of paclitaxel. The therapeutic activity is underscored by its ability to bind to HER2 and free it from protection by mucin 4 (MUC4), disrupt its interplay with other receptor tyrosine kinases, and subsequently direct HER2 for degradation. PEPDG278D also downregulates epidermal growth factor receptor (EGFR) which contributes to drug resistance in HER2-BC. In contrast, Ttzm, whose therapeutic activity also depends on its binding to the extracellular domain of HER2, cannot perform any of these functions of PEPDG278D. Indeed, PEPDG278D inhibits HER2-BC cells and tumors that carry clinically relevant molecular changes that confer resistance to Ttzm. Our results show that HER2 remains a critical target in drug-resistant HER2-BC and that PEPDG278D is a promising agent for overcoming drug resistance in this disease.
One Sentence Summary:
HER2 remains a therapeutic target in drug-resistant HER2-positive breast cancer, and a recombinant human protein overcomes the drug resistance.
Introduction
HER2 is an oncogenic receptor tyrosine kinase (RTK) implicated in several types of human cancer. It is strongly expressed in about 20% of breast cancer (BC), known as HER2-positive BC (HER2-BC), due to gene amplification (1, 2). HER2 amplification or overexpression is a strong predictor of poor disease prognosis (3, 4). HER2-targeting drugs are available for treating HER2-BC, including monoclonal antibodies Ttzm and pertuzumab, T-DM1 (Ttzm coupled to a microtubule inhibitor), and tyrosine kinase inhibitors (TKIs) lapatinib and neratinib. While these agents have greatly improved disease outcomes, primary and acquired drug resistance is common. Ttzm, the mainstay treatment for HER2-BC, achieves an overall response rate of about 25% as a single agent and about 50% when combined with chemotherapy in metastatic disease (5, 6). Most patients with advanced disease show disease progression after some time on treatment. Even the triple combination of Ttzm, pertuzumab and docetaxel produces median progression-free survival of only about 18 months (7). Many drug resistance mechanisms have been reported, including decreased drug binding to HER2 (8, 9), activation of compensatory signaling (10, 11), defects in apoptosis and cell cycle control (12, 13), and host factors (14, 15). However, the relative importance of these mechanisms is poorly understood, hampering development of better therapies.
One of the mechanisms of action of Ttzm is HER2 downregulation, but Ttzm is relatively weak or inactive in downregulating HER2 in tumors in vivo (11, 16, 17), which may be an important reason for its therapeutic limitation. We recently found that recombinant human peptidase D (PEPD), also known as prolidase, strongly downregulates HER2 and EGFR in cancer cells in vitro and in vivo. Whereas endogenous PEPD residing intracellularly has no effect on HER2 and EGFR, exogenously administered PEPD binds to the extracellular domains (ECDs) of the receptors, disrupting their signaling and downregulating their expression in cancer cells overexpressing the receptors, resulting in growth inhibition (18, 19). However, PEPD does not bind to other HER family members, including HER3 and HER4 (20). The enzymatic activity of PEPD plays no role in its modulation of HER2 and EGFR, and we subsequently focused on recombinant PEPDG278D, an enzymatically inactive mutant (point mutation at codon 278). PEPDG278D specifically binds to HER2 and EGFR, and cells and tumors lacking these receptors are insensitive to it (18, 19). Its ability to target both HER2 and EGFR is important, because EGFR is expressed in 35–40% of HER2-BC and its expression is associated with worse survival (21, 22). PEPDG278D differs from the clinically available TKIs of HER2 and EGFR, because the TKIs target the kinase domains of the receptors. Here we investigated the therapeutic activity and mechanism of action of PEPDG278D in cell lines and mouse models of HER2-BC resistant to Ttzm and other HER2 inhibitors.
Results
PEPDG278D inhibits drug-resistant HER2-BC cells
We compared PEPDG278D with Ttzm in seven HER2-BC cell lines, namely BT-474, BT-474R2, JIMT-1, HCC-1419, HCC-1569, HCC-1954, and UACC-893, along with MCF-7 BC cells. The HER2-BC cell lines overexpress HER2 and EGFR to varying degree, whereas MCF-7 cells, which are estrogen receptor-positive, have almost no HER2 or EGFR (Fig. 1A and fig. S1A). All the HER2-BC cell lines have HER2 amplification (8–66 copies per cell) (23–25). BT-474R2 cells, derived from BT-474 cells, show acquired resistance to Ttzm and exhibit cyclin E1 amplification, which was linked to poor response of HER2-BC tumors to Ttzm (13). The other HER2-BC cell lines show primary resistance to Ttzm. HCC-1569 and UACC-893 are also resistant to lapatinib, and JIMT-1 is resistant to both pertuzumab and lapatinib (26, 27). All the HER2-BC cell lines carry activating mutations in phosphatidylinositol 3-kinase catalytic subunit alpha (PIK3CA), including C420R, E545A, and/or H1047R (fig. S1, B to H). JIMT-1, HCC-1569 and HCC-1954 are also low in phosphatase and tensin homolog (PTEN) (fig. S1I). PIK3CA mutation and/or PTEN loss are associated with poor response of patients to Ttzm (28). These cell lines represent different BC subtypes, including luminal A (JIMT-1 and MCF-7), luminal B (BT-474 and BT-474R2), and HER2-enriched (others) (29). Despite PIK3CA mutations, PTEN loss, cyclin E overexpression, and difference in BC subtype, PEPDG278D caused concentration- and time-dependent growth inhibition in all the HER2-BC cell lines, whereas Ttzm was active only in BT-474, even at 40 fold higher concentration than PEPDG278D (Fig. 1B). PEPDG278D was moderately active in HCC-1569 cells, which show relatively low expression of HER2 and EGFR, and was inactive in MCF-7 cells, which lack both receptors; the result is consistent with PEPDG278D being a specific dual inhibitor of HER2 and EGFR.
Fig. 1. PEPDG278D targets HER2-BC cells resistant to Ttzm.
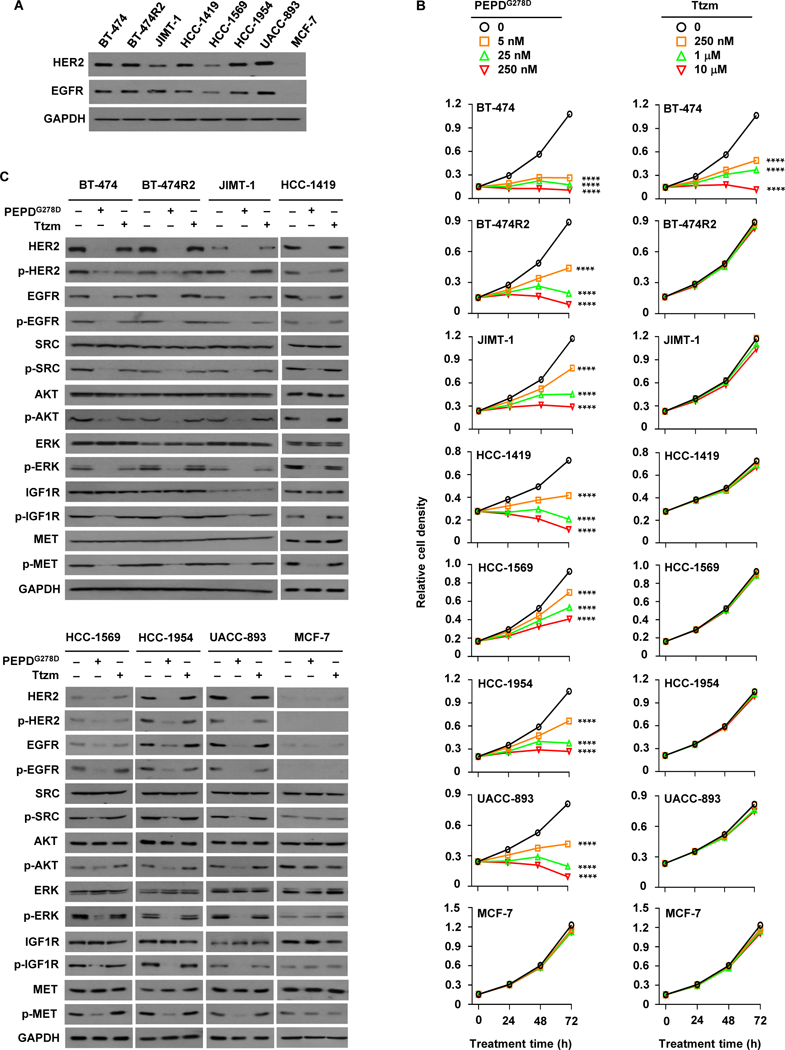
(A) IB analysis of whole cell lysates from untreated cells. (B) Effects of PEPDG278D and Ttzm on cell proliferation, measured by MTT assay. Each value is mean ± SD (n=3). ****P<0.0001 by one-way ANOVA, followed by Tukey test for comparison to the control. (C) IB analysis of whole cell lysates from cells treated with vehicle, PEPDG278D (25 nM) or Ttzm (1 μM) for 48 hours. The following phosphorylation sites were measured: pY1221/1222-HER2, pY1173-EGFR, pY416-SRC, pS473-AKT, pT202/Y204-ERK, pY1131-IGF1R, and pY1234/1235-MET. Glyceraldehyde 3-phosphate dehydrogenase (GAPDH) was measured as a loading control here and elsewhere.
Upon PEPDG278D treatment (25 nM, 48 hours), all the cell lines except MCF-7 showed profound loss of HER2 and EGFR (both expression and phosphorylation), loss of phosphorylation (inactivation) of all key downstream signaling partners analyzed, including SRC, AKT and ERK (extracellular signal-regulated kinase), and loss of phosphorylation of RTKs which heterodimerize with HER2, including IGF1R (insulin-like growth factor 1 receptor) and MET (c-MET) (Fig. 1C). SRC, AKT, ERK, IGF1R, and MET all play important roles in HER2-BC resistance to current HER2 inhibitors (10, 11, 30, 31). PEPDG278D-induced changes in SRC, AKT, ERK, IGF1R, and MET apparently resulted from the inhibition of HER2 and EGFR, because none of the changes occurred in MCF-7 cells (Fig. 1C). In BT-474, Ttzm (1 μM, 48 hours) had moderate effects on HER2, SRC, AKT and ERK, but no effect on EGFR, IGF1R and MET, and no effect on any of these proteins in any of the other cell lines (Fig. 1C).
Our results show that PEPDG278D is effective against HER2-BC cells resistant to Ttzm and other HER2 inhibitors. These results also show that HER2-BC cells that are resistant to current HER2 inhibitors still require HER2 for survival and growth. Notably, HER2 knockdown by siRNA also inhibits HER2-BC cells resistant to Ttzm and lapatinib (32, 33). PEPDG278D also targets EGFR, which likely contributes to its activity, because EGFR knockdown by siRNA decreased the survival of both BT-474R2 and JIMT-1 cells (fig. S1, J and K). Neratinib, a pan-HER TKI, was also active in JIMT-1 and BT-474R2 cells but appeared less potent than PEPDG278D (fig. S1L).
PEPDG278D induces internalization and lysosomal degradation of HER2 in drug-resistant HER2-BC cells
Because both PEPDG278D and Ttzm bind to HER2 ECD, at subdomain 3 for PEPDG278D (20) and subdomain 4 for Ttzm (34), we sought to address how PEPDG278D succeeded in inhibiting HER2 in cells that failed Ttzm. In both BT-474R2 and JIMT-1 cells, HER2 began to decrease after 2–3 hours of PEPDG278D treatment (25 nM), decreased markedly at 6 hours and was almost undetectable at 24 hours, accompanied by initial increase in p-HER2 (Fig. 2A), which is consistent with PEPDG278D-induced HER2 dimerization followed by internalization and degradation. In contrast, Ttzm was totally inactive even at 1 μM (Fig. 2B). PEPDG278D did not modulate HER2 gene transcription (Fig. 2C). Because RTK amounts are regulated by internalization and lysosomal degradation, we asked whether lysosome inhibition would rescue HER2 in PEPDG278D-treated cells. Chloroquine, a lysosome inhibitor, had no effect on baseline HER2 expression but completely blocked PEPDG278D-induced HER2 loss (Fig. 2D) and caused accumulation of internalized HER2 (Fig. 2E). Indeed, PEPDG278D appeared to bind to HER2 on cell surface before co-internalization (Fig. 2F). Chloroquine-induced accumulation of internalized HER2 co-localized with lysosomal-associated membrane protein 1 (LAMP1) (Fig. 2G).
Fig. 2. PEPDG278D, but not Ttzm, induces internalization and lysosomal degradation of HER2 in Ttzm-resistant HER2-BC cells.
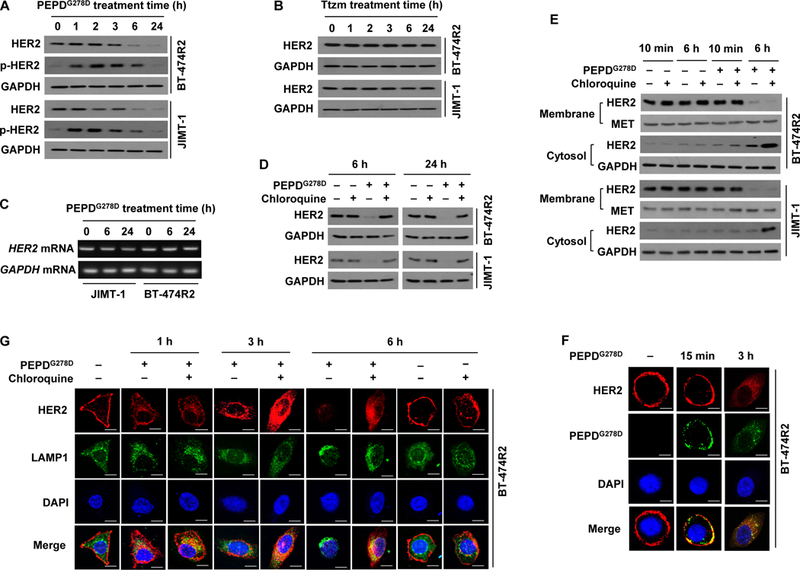
(A and B) IB analysis of whole cell lysates from cells treated with PEPDG278D (25 nM, A) or Ttzm (1 μM, B) for 0–24 hours. p-HER2: pY1221/1222-HER2. (C) RT-PCR analysis of gene expression in cells treated with PEPDG278D (25 nM) for 0–24 hours. (D) IB analysis of whole cell lysates from cells treated with vehicle, PEPDG278D (25 nM), and/or chloroquine (25 μM) for 6 or 24 hours. (E) IB analysis of membrane and cytosol fractions from cells treated with vehicle, PEPDG278D (25 nM) and/or chloroquine (25 μM) for 10 min or 6 hours, using MET and GAPDH as loading controls. (F) Confocal fluorescence staining images of HER2, PEPDG278D (staining its His tag) and nuclei (DAPI) in BT-474R2 cells treated with PEPDG278D (25 nM) for 0 or 15 min or 3 hours. Scale bar: 10 μm. (G) Confocal fluorescence staining images of HER2, LAMP1 (lysosome) and nuclei (DAPI) in BT-474R2 cells with or without treatment with PEPDG278D (25 nM) and/or chloroquine (25 μM) for 0–6 hours. Scale bar, 10 μm.
Because PEPDG278D is internalized with HER2, we investigated whether PEPDG278D can also modulate nuclear HER2 and EGFR, which are implicated in cancer cell growth and drug resistance (35, 36). However, treatment of BT-474R2 or JIMT-1 cells with PEPDG278D up to 250 nM had no effect on nuclear expression of HER2 and EGFR (fig. S2A). Endogenous PEPD was recently found to bind and regulate p53 (37). However, PEPDG278D showed no effect on p53, with no disruption of the endogenous PEPD-p53 complex, no binding of PEPDG278D to p53, and no change in p53 transcriptional activity (fig. S2, B to E). Thus, internalized PEPDG278D does not appear to be biologically active.
PEPDG278D binds and frees HER2 from MUC4
Membrane-associated MUC4 binds and stabilizes HER2 (38, 39) and prevents Ttzm from binding to HER2 (8, 38). We found that all Ttzm-resistant HER2-BC cell lines in this study strongly overexpress MUC4 (Fig. 3A). In BT-474R2 cells, approximately 10 times more PEPDG278D than Ttzm bound to HER2, even though the PEPDG278D concentration was 40-fold lower than that of Ttzm (fig. S3A). Pertuzumab bound to HER2 approximately 4 times better than Ttzm in BT-474R2 cells, showed negligible effect on cell proliferation, but antagonized PEPDG278D in both HER2 binding and inhibition of cell proliferation (fig. S3, B and C). Pertuzumab antagonism toward PEPDG278D is not surprising, however, since it binds to subdomain 2 of HER2 ECD to block HER2 dimerization (40), whereas PEPDG278D functions by causing HER2 dimerization. In both JIMT-1 and BT-474R2 cells, MUC4 knockdown by siRNA markedly decreased the amount of HER2 and cell survival (Fig. 3, B and C,). Conversely, both BT-474 and SKBR3 cells (HER2-positive BC cells) are MUC4-low and Ttzm-sensitive, and overexpressing MUC4 markedly reduced their sensitivity to Ttzm but not to PEPDG278D (Fig. 3, D and E). MUC4 overexpression did not completely protect the cells from Ttzm, probably because gene transfection efficiency is unlikely to be 100%. In both BT-474R2 and JIMT-1 cells, PEPDG278D binds to HER2 and frees it from MUC4 (Fig. 3F), without altering MUC4 expression (fig. S3D). Thus, although MUC4 overexpression may play a crucial role in resistance to Ttzm and other HER2 inhibitors in HER2-BC cells, it has no effect on HER2 targeting by PEPDG278D. In both BT-474R2 and JIMT-1 cells, PEPDG278D (homodimer) underwent two phases of HER2 binding: rapid binding to preexisting HER2 homodimer (no change in HER2 monomer content, decrease in HER2 dimer content and formation of heterotetramer of one HER2 dimer and one PEPDG278D dimer at 10 min), followed by binding to HER2 monomer, leading to decrease in HER2 monomer content, formation of heterotrimer (one HER2 monomer and one PEPDG278D dimer), and further increase in heterotetramer content at 60 min (Fig. 3G). The heterotrimer is presumably an intermediate. New HER2 dimers were detected after PEPDG278D treatment, which likely resulted from dissociation of some heterotetramers during sample preparation. In contrast, each molecule of Ttzm binds to a HER2 monomer (34).
Fig. 3. PEPDG278D breaks MUC4 protection of HER2 in Ttzm-resistant HER2-BC cells.
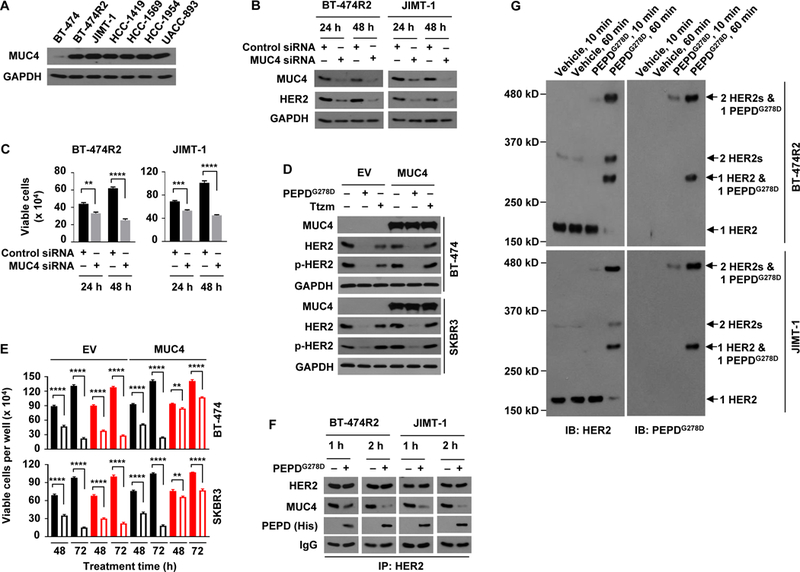
(A) IB analysis of whole cell lysates from untreated cells. (B and C) IB analysis of whole cell lysates (B) and trypan blue viability analysis (C) of cells after treatment with siRNA for 24 or 48 hours. (D and E) Cells were transfected with empty vector (EV) or MUC4, and 24 hours later treated with vehicle, PEPDG278D (25 nM) or Ttzm (1 μM), followed by IB analysis of whole cell lysates at 48 hours (D) and trypan blue viability analysis at 48 and 72 hours (E). Filled and unfilled black bars represent vehicle and PEPDG278D, respectively; filled and unfilled red bars represent vehicle and Ttzm, respectively. p-HER2: pY1221/1222-HER2. (F) IB analysis of anti-HER2 IP of whole cell lysates from cells treated with vehicle or PEPDG278D (25 nM) for 1 or 2 hours. (G) IB analysis of whole cell lysates from cells treated with vehicle or PEPDG278D (25 nM) for 10 or 60 min and then treated with BS3 (2 mM) for 30 min. Each value in (C and E) is mean ± SD (n=3), **P<0.01, ***P<0.001, P****<0.0001 by paired two-tailed t-test.
PEPDG278D disrupts HER2 heterodimers
Phosphorylation of IGF1R and MET was abolished in all the HER2-BC cell lines treated with PEPDG278D, whereas Ttzm was inactive (Fig. 1C). Both IGF1R and MET contribute to Ttzm resistance in HER2-BC (10, 31). Because HER2 forms heterodimeric signaling units, and PEPDG278D disrupts HER2-EGFR and HER2-HER3 heterodimers (18) but showed no effect on phosphorylation and expression of vascular endothelial growth factor receptor-1 (VEGFR-1) which is not known to dimerize with HER2 (fig. S4A), we asked whether PEPDG278D disrupts HER2 heterodimerization with IGF1R and MET. Chinese hamster ovary CHO-K1 cells express very low amounts of HER2 and none of its family members (20). We overexpressed human HER2 in these cells (CHO-K1/HER2). Neither IGF1R nor MET was affected by PEPDG278D in CHO-K1 cells, whereas phosphorylation of both proteins, but not their expression, was abolished in PEPDG278D-treated CHO-K1/HER2 cells, along with HER2 loss (Fig. 4A). Moreover, while insulin-like growth factor 1 (IGF1), a ligand of IGF1R, stimulated IGF1R phosphorylation in both CHO-K1 cells and CHO-K1/HER2 cells, PEPDG278D abolished IGF1R phosphorylation only in CHO-K1/HER2 cells (fig. S4B). Thus, the impact of PEPDG278D on MET and IGF1R appears to be mediated by HER2. Both BT-474R2 and JIMT-1 cells contained HER2-IGF1R and HER2-MET heterodimers, which were stimulated by IGF1 or MET ligand hepatocyte growth factor (HGF) (Fig. 4, B and C). Treatment with PEPDG278D (25 nM) for only 1 hour caused profound disruption of both heterodimers, regardless of ligand involvement (Fig. 4, B and C), whereas the expression of IGF1R and MET remained unchanged (fig. S4, C and D). PEPDG278D bound to HER2 but not to IGF1R or MET in both cell lines, and PEPDG278D appeared to disrupt HER2 association with IGF1R or MET even when the latter RTKs were bound by their ligands (Fig. 4, D and E). HER2 phosphorylation increased upon PEPDG278D treatment (Fig. 4, D and E), which is consistent with PEPDG278D-induced HER2 homodimerization before internalization and degradation (20). The ability of PEPDG278D to simultaneously suppress multiple oncogenic RTKs directly or indirectly further shows its therapeutic potential.
Fig. 4. PEPDG278D disrupts HER2-IGF1R and HER2-MET heterodimers.
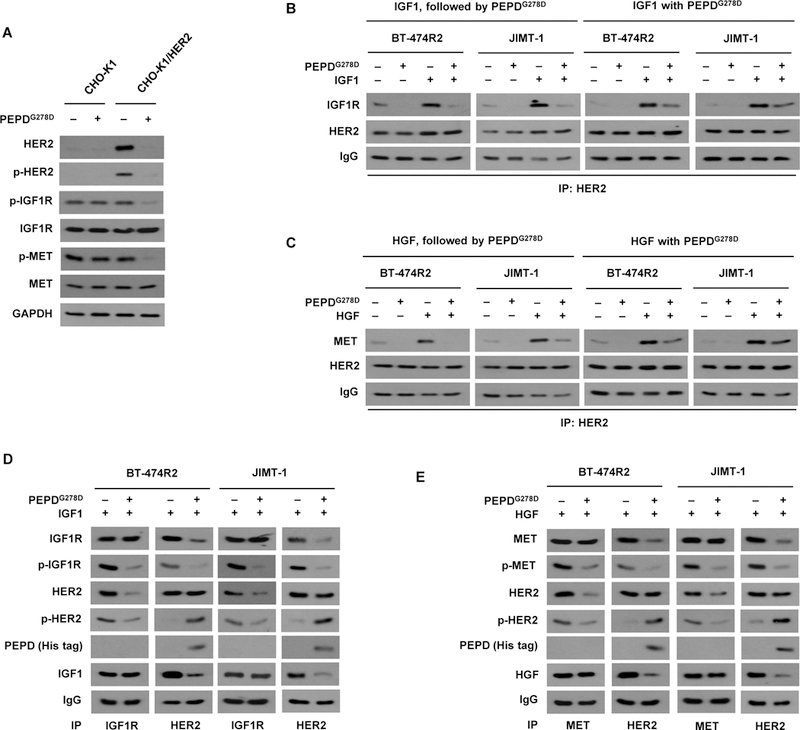
(A) IB analysis of whole cell lysates from cells which were transfected with an empty vector (CHO-K1) or human HER2 (CHO-K1/HER2) for 24 hours and then treated with vehicle or PEPDG278D (25 nM) for 48 hours. (B) IB analysis of anti-HER2 IP of whole cell lysates from cells treated first with IGF1 (12.5 nM) for 15 min and then PEPDG278D (25 nM) for 1 hour or treated simultaneously with IGF1 (12.5 nM) and PEPDG278D (25 nM) for 1 hour. (C) IB analysis of anti-HER2 IP of whole cell lysates from cells treated first with HGF (0.3 nM) for 15 min and then with PEPDG278D (25 nM) for 1 hour or treated simultaneously with HGF (0.3 nM) and PEPDG278D (25 nM) for 1 hour. (D) IB analysis of anti-IGF1R IP or anti-HER2 IP of whole cell lysates from cells treated with IGF1 (12.5 nM) with or without PEPDG278D (25 nM) for 1 hour. (E) IB analysis of anti-MET IP or anti-HER2 IP of whole cell lysates from cells treated with HGF (0.3 nM) with or without PEPDG278D (25 nM) for 1 hour. Protein phosphorylation sites are described in Fig. 1C legend.
PEPDG278D shows therapeutic activity in vivo
PEPDG278D was compared with Ttzm in two orthotopic models of HER2-BC. PEPDG278D was evaluated in combination with enoxaparin (EP), a clinically used anticoagulant, because PEPDG278D is degraded by coagulation proteases in vivo, and EP blocks the degradation (18, 41). Notably, EP does not interfere with modulation of EGFR and HER2 by PEPDG278D and does not impact tumor growth itself (18, 19).
JIMT-1 cells, which show primary drug resistance, were inoculated into the mammary fat pads of female severe combined immune deficiency (SCID) mice. The mice were randomized, and treated with EP, PEPDG278D plus EP, or Ttzm once tumors reached 100–150 mm3. EP was given by intraperitoneal injection (ip) at 0.5 mg/kg body weight daily, to inhibit PEPDG278D degradation (19). PEPDG278D was given ip at 4 mg/kg thrice weekly, starting 5 days after EP to ensure that the mice were fully primed by EP (19). Ttzm was given ip at 10 mg/kg weekly (25). EP is given to patients by subcutaneous injection (sc), and PEPDG278D, if advanced to clinical testing, will likely be given intravenously (iv), but similar plasma concentrations of PEPDG278D were obtained regardless of administration routes of the agents (fig. S5, A and B). Whereas tumors in mice treated by EP alone grew rapidly and Ttzm had no effect on tumor growth, PEPDG278D caused rapid tumor regression, followed by complete remission of all the tumors, with no recurrence after 2 months of follow-up (Fig. 5A). In contrast, a previous study showed that even combining Ttzm with pertuzumab (~5 mg/kg ip twice weekly for each agent) and starting the treatment at the time of cell inoculation did not cause tumor regression in this model (27), although pertuzumab has synergistic activity with Ttzm (42). To obtain tumor samples for molecular analysis, mice in the EP group were randomly divided either to continue EP treatment only or to receive PEPDG278D on days 27 and 29 while continuing EP treatment; PEPDG278D addition caused rapid tumor regression again (Fig. 5A). There was no sign of agent toxicity. In tumors obtained 24 hours after the last treatment, PEPDG278D caused almost total loss of HER2, p-HER2, EGFR, p-EGFR, p-HER3, p-IGF1R, p-MET, p-SRC, p-AKT, p-ERK, and strong activation of caspase 3, without altering the expression of HER3, neuregulin-1 (NRG-1, HER3 ligand), IGF1R, MET, SRC, AKT, or ERK, whereas Ttzm had no effect (Fig. 5B). NRG-1, which may contribute to Ttzm resistance (43), was not overexpressed in JIMT-1 and BT-4742R2 cells (fig. S6A). PEPDG278D blocked NRG-1-induced phosphorylation of both HER3 and AKT, and its inhibition of cell proliferation was not attenuated by NRG-1 (fig. S6, B-D). PEPDG278D disrupts HER3 heterodimerization with HER2 (18).
Fig. 5. PEPDG278D inhibits Ttzm-resistant HER2-BC in vivo.
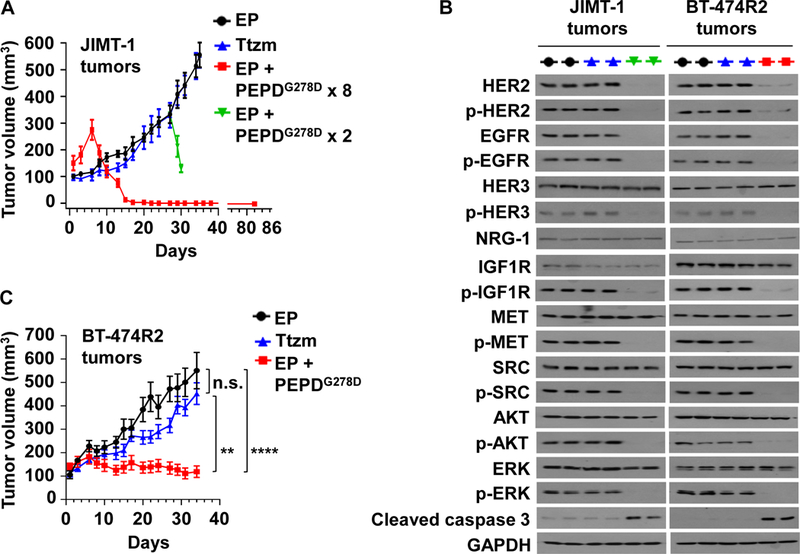
(A) JIMT-1 tumors in SCID mice, treated with EP (daily, days 1–34, n=10), Ttzm (weekly, days 6–34, n=8), or EP + PEPDG278D x 8 (EP daily, days 1–22; PEPDG278D thrice weekly, days 6–22, 8 doses; n=8). In the fourth group, PEPDG278D was added to the EP control on days 27 and 29 (PEPDG278D x 2; n=4). All tumors were harvested 24 hours after the last treatment, but one group of mice (EP + PEPDG278D x 8) became tumor-free and were observed without treatment for 60 days (days 23–82). (B) IB analysis of tumor homogenates (2 tumors/group). See Fig. 1C legend for protein phosphorylation sites. (C) BT-474R2 tumors in nude mice, treated with EP daily (days 1–34, n = 13), Ttzm weekly (days 6–34, n =15), or EP daily (days 1–34) plus PEPDG278D thrice weekly (days 6–34) (n = 12). All tumors were harvested 24 hours after the last treatment. EP, Ttzm and PEPDG278D were dosed ip at 0.5 mg/kg, 10 mg/kg and 4 mg/kg per dose, respectively. Each value in (A and C) is mean ± SEM **P<0.01, ****P<0.0001, by one-way ANOVA followed by Tukey test; n.s., not significant.
BT-474R2 cells, which show acquired drug resistance, grew poorly in SCID mice and were inoculated into the mammary fat pads of female athymic nude mice. The mice were randomized and treated with EP, PEPDG278D plus EP, or Ttzm, as described above. Ttzm was ineffective, whereas PEPDG278D rapidly arrested tumor growth (Fig. 5C). However, PEPDG278D did not induce tumor regression but rather induced a senescence-like state, since the treated tumors neither grew nor regressed and the remnants were tumor tissues, not fibrosis (fig. S7A). In BT-474R2 tumors obtained 24 hours after the last treatment, PEPDG278D caused the same molecular changes as in JIMT-1 tumors, whereas Ttzm was inactive (Fig. 5B). Thus, the inability of PEPDG278D to induce tumor regression is not due to inadequate targeting of HER2 and its signaling partners. Indeed, increasing PEPDG278D dose to 8 mg/kg did not induce tumor regression (Fig. 6, A and B). BT-474R2 cells overexpress cyclin E1, which confers resistance to Ttzm (13). PEPDG278D showed no effect on cyclin E1 (Fig. 6C). However, it is not clear whether cyclin E1 overexpression contributes to PEPDG278D’s inability to induce tumor regression. There was no sign of agent toxicity. Despite strong effect of PEPDG278D on HER2 and EGFR in the tumors, neither PEPDG278D nor Ttzm impacted the expression of the RTKs in vital organs (heart, kidney and liver), except a slight increase in HER2 phosphorylation in the heart by both agents (fig. S7B). PEPDG278D also had no effect on the histology of these organs in mice in our previous study (18). The expression levels of both EGFR and HER2 were very low in these organs, compared to that in the tumor tissues (fig. S7C), suggesting that PEPDG278D may primarily targets the RTKs overexpressed in cancer cells.
Fig. 6. ombination of PEPDG278D and paclitaxel enhances therapeutic outcome.
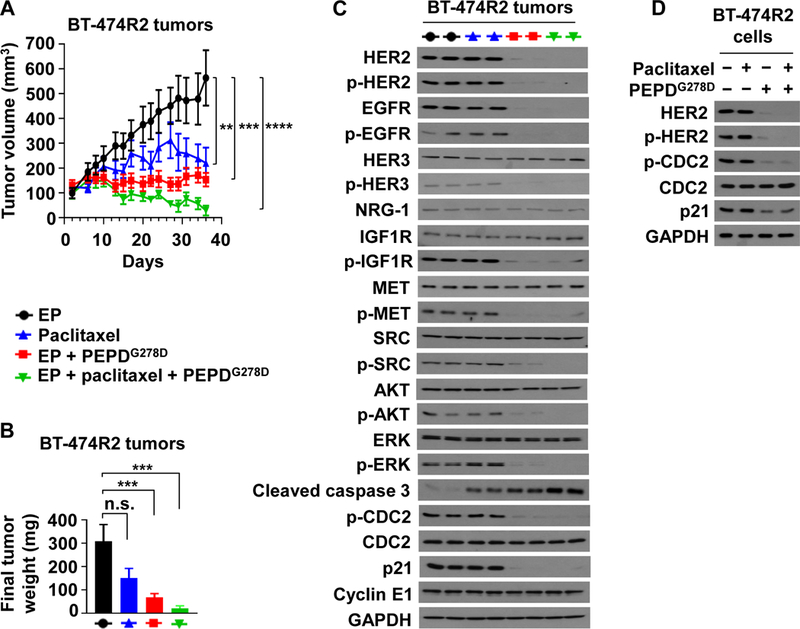
(A) BT-474R2 tumors in nude mice, treated with EP (0.5 mg/kg daily, days 2–35, n = 8), paclitaxel (20 mg/kg weekly, days 8–35, n = 7), EP plus PEPDG278D (EP at 0.5 mg/kg daily, days 2–35; PEPDG278D at 8 mg/kg thrice weekly, days 8–35; n = 10), or EP plus paclitaxel plus PEPDG278D (EP at 0.5 mg/kg daily, days 2–35; paclitaxel at 20 mg/kg weekly, days 8–35; PEPDG278D at 4 mg/kg thrice weekly, days 8–35; n = 8). (B) Weights of tumors harvested 24 hours after the last treatment. All agents were dosed ip. (C) IB analysis of tumors homogenates (2 tumors/group). See Fig. 1C legend for protein phosphorylation sites, except for pY15-CDC2. (D) IB analysis of whole cell lysates of cells after treatment with vehicle, paclitaxel (25 nM), and/or PEPDG278D (25 nM) for 24 hours. Each value in (A and B) is mean ± SEM. **P<0.01, ***P<0.001, ****P<0.0001, by one-way ANOVA followed by Tukey test; n.s., not significant.
Combination of PEPDG278D with paclitaxel enhances therapeutic outcome
Paclitaxel is commonly used to treat HER2-BC. However, HER2 inhibits paclitaxel-induced apoptosis in BC cells by inducing Y15 phosphorylation of CDC2 and p21 upregulation (44, 45). To assess the therapeutic benefit of PEPDG278D combination with paclitaxel, mice bearing BT-474R2 tumors were treated with EP as described before, paclitaxel (20 mg/kg ip weekly), PEPDG278D (8 mg/kg ip thrice weekly) plus EP, or PEPDG278D (4 mg/kg ip thrice weekly) plus EP and paclitaxel. Both paclitaxel and PEPDG278D inhibited tumor growth as single agents (Fig. 6A), but the final tumor weights between EP and paclitaxel was not statistically different (Fig. 6B). Whereas PEPDG278D did not induce tumor regression even at 8 mg/kg, combining PEPDG278D with paclitaxel resulted in tumor regression, and despite the short treatment time selected to obtain an adequate amount of tumor tissue for molecular analysis, 60% of the tumors already achieved complete remission. There was no detectable treatment toxicity. Paclitaxel, which targets microtubules, had no effect on any of the molecules measured in the tumors except for caspase 3 activation, but tumors treated by PEPDG278D with or without paclitaxel showed profound loss of HER2, p-HER2, EGFR, p-EGFR, p-HER3, p-IGF1R, p-MET, p-SRC, p-AKT, p-ERK, p-CDC2 and p21, whereas the expression of cyclin E1, HER3, NRG-1, IGF1R, MET, SRC, AKT, ERK and CDC2 remained unchanged (Fig. 6C). Caspase 3 activation was more pronounced in tumors treated by PEPDG278D plus paclitaxel than by a single agent (Fig. 6C), which is consistent with their tumor inhibitory efficacies. PEPDG278D with or without paclitaxel also markedly decreased p-CDC2 and p21 in cultured BT-474R2 and JIMT-1 cells (Fig. 6D and fig. S8).
Discussion
In this study, we demonstrate the therapeutic activity of PEPDG278D against HER2-BC resistant to current HER2 inhibitors. PEPDG278D exerts its action in two phases (fig. S9). In phase 1, which is well underway at 1 hour of treatment, PEPDG278D frees HER2 from MUC4 and disrupts the interaction of HER2 with other RTKs, including MET and IGF1R. PEPDG278D also disrupts HER2 interaction with EGFR and HER3 (18), whose heterodimerization with HER2 contributes to HER2-BC aggressiveness (21, 46). PEPDG278D apparently disrupts HER2 heterodimerization by forcing its homodimerization. PEPDG278D also binds to preformed HER2 homodimers and disassembles their signaling unit, thereby causing SRC to dissociate from the dimer (20). Thus, PEPDG278D rapidly suppresses multiple RTKs either directly or indirectly in HER2-BC cells. In phase 2, which begins at 2–3 hours of PEPDG278D treatment, PEPDG278D induces persistent downregulation of HER2 via internalization and lysosomal degradation. In addition, PEPDG278D downregulates EGFR, apparently by binding to the receptor to induce its internalization and degradation (18, 19).
Our results also provide insight into the drug resistance mechanism in HER2-BC. All Ttzm-resistant HER2-BC cell lines tested were exquisitely sensitive to PEPDG278D, despite carrying activating PIK3CA mutations, low expression of PTEN, and/or cyclin E overexpression. The therapeutic activity of PEPDG278D highlights that HER2 remains a critical target in drug-resistant HER2-BC. All Ttzm-resistant cell lines overexpressed MUC4, and overexpressing MUC4 in Ttzm-sensitive cells converted them to Ttzm resistance. In addition to shielding HER2 from Ttzm, MUC4 also stabilizes HER2 and therefore may confer resistance to other HER2 inhibitors. MUC4 overexpression may be the key factor responsible for drug resistance in HER2-BC, whereas increased signaling downstream of HER2 may confer resistance to therapy if HER2 is not strongly inhibited. For example, studies have shown that the gain of function induced by helical domain mutations of PI3KCA such as E545A requires interaction with RAS, and the gain of function induced by kinase domain mutations of PI3KCA such as H1047R is modulated by the regulatory subunit p85 and phosphorylated RTKs (47, 48). In PEPDG278D-treated HER2-BC cells, phosphorylation of multiple RTKs was lost, and RAS was likely inhibited in view of loss of ERK phosphorylation, thereby limiting the impact of PI3KCA mutation. The reason why MUC4 shields HER2 from Ttzm but not PEPDG278D is unknown but may be related to how Ttzm and PEPDG278D bind to HER2. Each subunit of homodimeric PEPDG278D binds to a HER2 monomer, forming a tetra-complex, which may be relatively stable and may disrupt MUC4 binding to HER2 by steric hindrance or inducing conformation change of HER2. Each molecule of Ttzm binds to one HER2 monomer (34). The finding that MUC4 has no effect on PEPDG278D targeting of HER2 is important, because a recent study shows that MUC4 is positive in 60% of human HER2-BCs and is an independent predictor of poor disease-free survival in patients treated with Ttzm (38).
Our finding that combining paclitaxel with PEPDG278D improves antitumor outcome offers a potential combination approach. In the present study, PEPDG278D was used in combination with EP in order for PEPDG278D to escape degradation in the plasma in vivo. It will be important to investigate whether PEPDG278D can be structurally modified to resist degradation without EP, while retaining its antitumor activity. However, the EP dose for inhibition of PEPDG278D degradation is much lower than for anticoagulation (49, 50). Moreover, because increased coagulation and thromboembolic disease affect a high percentage of cancer patients (51, 52), combination of PEPDG278D and EP may be beneficial.
There are limitations to this study. First, because PEPDG278D is a human protein, it could not be evaluated in tumor models in immunocompetent mice. Second, the tumor models used in our study did not show metastasis and therefore did not allow evaluation of potential effect of PEPDG278D on tumor metastasis. Nevertheless, given the strong therapeutic activity of PEPDG278D shown in the present study, it may also be effective against metastatic HER2-BC. Indeed, JIMT-1 cells, which were derived from pleural effusion of a patient (25), were exceedingly sensitive to PEPDG278D. Third, p95HER2, a truncated form of HER2 lacking its ECD, confers resistance to Ttzm (9) and would be insensitive to PEPDG278D, potentially limiting the effectiveness of PEPDG278D. Finally, we did not have access to patient-derived xenograft (PDX) models of drug-resistant HER2-BC. Because PDX models are recognized to conserve the heterogeneity typical of the original patient tumors and to reliably predict disease response to therapies, evaluation of PEPDG278D in such a model, once becoming available, would be worthwhile.
Additional preclinical evaluation of both therapeutic efficacy and safety of PEPDG278D produced in compliance with Good Manufacturing Practices regulations, will be needed before PEPDG278D can be investigated clinically. Particularly, given the cardiotoxicity of current HER2 inhibitors, evaluation of potential effects of PEPDG278D on cardiac function is needed. However, our present finding that PEPDG278D is active in drug-resistant HER2-BC with a clear mechanism, together with the fact that PEPDG278D is a recombinant human protein and does not show adverse effects in mouse studies, provides a strong scientific premise for its evaluation in patients with HER2-BC that does not respond to current HER2 inhibitors.
Materials and Methods
Study design
This study aimed to determine whether PEPDG278D is active in preclinical models of HER2-BC resistant to clinically available HER2 inhibitors and if so, its mechanism of action. We first evaluated PEPDG278D in human HER2-BC cell lines that show either primary or acquired resistance to one or more current HER2 inhibitors. PEPDG278D was compared to Ttzm which is the mainstay treatment for HER2-BC. As positive and negative controls, we included a HER2-BC cell line sensitive to Ttzm and a BC cell line lacking HER2. The cell lines were characterized for key molecular features, including expression of HER2, MUC4, and PTEN, as well as PIK3CA mutation. We measured the effect of each agent on cell proliferation and survival. Each experiment was performed three times. We also measured the modulation of HER2 and other molecules in all the cell lines. The endpoints were predetermined. To elucidate the mechanism of action of PEPDG278D, we focused on two HER2-BC cell lines, with one showing primary drug resistance (JIMT-1 cells) and the other showing acquired drug resistance (BT-474R2 cells). In addition, CHO-K1 cells with or without expression of human HER2 were used to aid the mechanistic investigation. Once the cell line experiments showed promising activity of PEPDG278D, we proceeded to in vivo experiments, comparing the therapeutic effects of PEPDG278D and Ttzm in two orthotopic mouse models of drug-resistant HER2-BC. We established tumors by inoculating JIMT-1 cells or BT-474R2 cells into the mammary fat pads of female mice. The cell lines were selected to represent both primary and acquired drug resistance in HER2-BC. We used two strains of mice: SCID mice and athymic nude mice. In addition to comparing PEPDG278D with Ttzm, we also evaluated the therapeutic benefit of combining PEPDG278D with paclitaxel, because paclitaxel is commonly used to treat patients with HER2-BC. We did not use statistics to predetermine sample size. Sample size in the mouse experiments was estimated based on our recent studies (18, 19). Randomization was used in assignment of mice to different treatment groups. Sample size varied slightly among treatment groups in each experiment due to xenograft failure in some mice. Investigators were not blinded during experimental treatment and data collection. All experimental treatments were stopped either because all tumors in a treatment group had disappeared completely or in order to obtain an adequate amount of tumor tissue from all treatment groups for molecular analysis. No data were excluded.
PEPDG278D and other materials
PEPDG278D was generated in E.coli using pBAD/TOPO-PEPDG278D-His, purified by nickel nitrilotriacetic acid-agarose chromatography, concentrated in phosphate-buffered saline (PBS) using Ultracel YM-30 Centricon (Millipore), and quantified by the bicinchoninic acid (BCA) assay, as previously reported (53). All other materials were purchased commercially and are described in the Supplementary Materials and Methods.
Cell lines and cell culture
Catalog numbers are listed in parentheses. BT-474 (HTB-20), CHO-K1 (CCL-61), HCC-1419 (CRL-2326), HCC-1569 (CRL-2330), HCC-1954 (CRL-2338), MCF-7 (HTB-22), SKBR3 (HTB-30) and UACC-893 (CRL-1902) were from American Type Culture Collection (ATCC). JIMT-1 (ACC-589) was from Deutsche Sammlung von Mikroorganismen und Zellkulturen (DSMZ). BT-474R2 was provided by Maurizio Scaltriti of Memorial Sloan Kettering Cancer Center. All cell lines were mycoplasma-free and were authenticated using short tandem repeat analysis. Cells were cultured with suitable medium containing 10% fetal bovine serum in humidified incubators with 5% CO2 at 37 oC. Additional information is provided in the Supplementary Materials and Methods.
Measurement of cell proliferation and cell viability
Cell proliferation was measured by the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay in 96-well plates. Cells were seeded in each well with 150 μl medium and after overnight growth, treated with vehicle or test agents in 200 μl medium for up to 72 hours and then incubated with medium containing 9.2 mM MTT (200 μl/well) for 3 hours at 37 oC. The cells were then washed once with PBS and mixed with dimethyl sulfoxide (150 μl/well). Cell density in each well was determined by measuring MTT reduction to formazan spectroscopically at 570 nm using a SPECTRAmax microtiter plate reader (Molecular Devices).Cell viability was measured by trypan blue exclusion assay. Cells were grown in 6-well plates and after experimental treatments were trypsinized and suspended in fresh medium. Approximately 550 cells in 10 μl suspension was mixed with 10 μl of 0.4% trypan blue solution, and viable cells (unstained cells) were counted using a hemocytometer under an inverted microscope. Additional information is provided in the Supplementary Materials and Methods.
Immunoblotting (IB) and immunoprecipitation (IP)
Protein concentrations in whole cell lysates, tissue homogenates and other samples were measured by the BCA assay. For IB, each sample was resolved by sodium dodecyl sulfate – polyacrylamide gel electrophoresis (SDS-PAGE, 8–12.5%). Proteins were transferred to polyvinylidene fluoride membrane, probed with specific antibodies, and detected using either Luminata Classico or Luminata Crescendo. Certain IB bands were quantified by ImageJ (NIH Image). For IP, whole cell lysates (0.5 mg protein/sample) were incubated with the required antibody in 500 μl volume overnight at 4 oC, followed by incubation with 30 μl Protein G Sepharose beads (2 mg/ml) for 1 hour at room temperature (RT). The beads were washed with IP buffer, suspended in 2x SDS loading buffer, boiled for 5 min, and analyzed by IB. Information about the antibodies is provided in the Supplementary Materials and Methods.
Measurement of PEPDG278D-induced dimerization of HER2
BT-474R2 and JIMT-1 cells were seeded at 1 × 106 cells per well with 0.2 ml medium in 6-well plates and 24 hours later treated with vehicle or PEPDG278D (25 nM) for 10 or 60 min. The cells were then washed with ice-cold PBS and incubated with 1 ml/well of 2 mM cross linker bis(sulfosuccinimidyl) suberate (BS3) for 30 min at RT. The cross linking reaction was terminated by adding 1 ml of 100 mM Tris (pH7.5) to each well, followed by incubation at RT for 15 min. Whole cell lysates were prepared and analyzed by IB (3.5% SDS-PAGE).
Immunofluorescence staining and confocal microscopy
Cells were grown in 8-well chamber slides (1.5 × 104 cells/well with 0.3 ml medium) overnight, followed by treatment with chloroquine, PEPDG278D or vehicle. The cells were then washed with ice-cold PBS, fixed with 4% paraformaldehyde for 15 min at RT, washed again with ice-cold PBS and blocked with 1% bovine serum albumin in PBS for 45 min at RT. The cells were then incubated with a HER2 antibody and a LAMP1 antibody for 1 hour at RT, washed with PBS, incubated with goat anti-mouse IgG Alexa-Fluor 488 conjugate (for LAMP1 detection) and goat anti-rabbit IgG Alexa-Fluor Plus 647 conjugate (for HER2 detection) for 1 hour at RT and washed again with PBS. The cells were mounted with ProLong Gold Antifade Mountant containing 4’,6-diamidino-2-phenylindole (DAPI) and then examined using a LSM 510 confocal microscope with a 63 × 1.4 Plan –Apochromat oil immersion objective (Zeiss). To assess PEPDG278D and HER2 co-localization, cells were treated with PEPDG278D for 0, 15 min or 3 hours. After fixing and blocking as described above, the cells were incubated with a HER2 antibody and a His antibody (for the His tag on PEPDG278D) for 1 hour at RT, washed with PBS, incubated with goat anti-mouse IgG Alexa-Fluor 488 conjugate (for PEPDG278D detection) and goat anti-rabbit IgG Alexa-Fluor Plus 647 (for HER2 detection) for 1 hour at RT and washed again with PBS. The cells were mounted using ProLong Gold Antifade Mountant containing DAPI and then examined using a Zeiss LSM 510 confocal microscope with a 63 × 1.4 Plan –Apochromat oil immersion objective. Merged images from Z-stack were organized using the ImageJ software. All antibodies are listed in Supplementary Materials and Methods.
RT-PCR
Total RNA was isolated using the RNeasy mini kit; 500 ng RNA per sample was reverse transcribed into cDNA in 25 μl reaction using the TaqMan Reverse Transcription Reagents. The RT reaction was performed at 25 oC for 10 min, followed by heating at 48 oC for 30 min, and then 95 oC for 5 min. Each PCR amplification was carried out in 20 μl volume, containing 10 μl GoTaq Master Mix (2x), 0.5–1 μl of the reverse-transcribed mixture (cDNA), 0.25 μM each of specific forward and reverse primers. The primers were as follows: for human HER2, forward, 5’- CTGTTTGCCGTGCCACCCTGAGT-3’, reverse, 5’-CTTCTGCTGCCGTCGCTTGATGAG-3’; for human GAPDH, forward, 5’-CCAGGGCTGCTTTTAACTC-3’, reverse, 5’-GCTCCCCCCTGCAAATGA-3’. The PCR conditions used for all reactions are as follows: 94 oC for 3 min, 28 cycles (HER2) / 25 cycles (GAPDH) at 94 oC (denaturation) for 30 sec, 63 oC (HER2) / 60 oC (GAPDH) for 30 sec (annealing), and 72 oC for 30 sec (extension); the final extension was performed at 72 oC for 5 min. The PCR products were analyzed by electrophoresis with 1% agarose gel, stained by ethidium bromide, and visualized under UV light.
Mouse study
SCID mice (C.B-17 SCID) were bred by the Laboratory Animal Shared Resource at Roswell Park Comprehensive Cancer Center (RPCCC). Athymic nude mice (Hsd:Athymic Nude-Foxn1nu) were purchased from Envigo. All mouse experiments were approved by the Institutional Animal Care and Use Committee at RPCCC under protocol 1022M. We established orthotopic BC tumors in mice by inoculating JIMT-1 cells or BT-474R2 cells into the mammary fat pads of female mice at 6–7 weeks of age. JIMT-1 cells were inoculated into the mammary fat pads of SCID mice (2 × 106 cells in 0.1 ml serum-free DMEM per site). BT-474R2 cells were inoculated into the mammary fat pads of athymic nude mice (5 × 106 cells in 0.1 ml of 50% Matrigel/50% serum-free DMEM per site). Because BT-474R2 tumor growth is estrogen-dependent, a 0.72 mg 17β-estradiol pellet for 60-day release was implanted in each mouse subcutaneously 1 day before cell inoculation. Another pellet was implanted to the mice after 60 days. Mice in each tumor model were randomized cage-wise into treatment groups using Research Randomizer (www.randomizer.org). Tumor size was measured using length x width2 ÷ 2. Tumor volume was measured three times each week. Drug treatment was started once tumor size reached 100–150 mm3. EP (0.5 mg/kg) was administered to mice ip once daily. PEPDG278D (4 or 8 mg/kg) was administered to mice ip thrice weekly (Monday, Wednesday, Friday) unless indicated otherwise. Ttzm (10 mg/kg) and paclitaxel (20 mg/kg) were administered to mice ip once weekly. EP and PEPDG278D were each prepared and administered to mice in PBS. Ttzm was first prepared in water at 21 mg/ml and diluted with PBS for administration. Paclitaxel was first prepared at 6 mg/ml containing approximately 50% alcohol and 50% polyoxyl 35 castor oil (v/v), which was diluted with PBS for administration. Each agent was administered to mice at 0.1 ml volume per 20 g body weight. When a mouse was given multiple agents on the same day, the agents were dosed at approximately 1 hour intervals. The mice were closely monitored for sign of adverse effects, weighed three times each week, and were sacrificed 24 hours after the last treatment, at which point the tumors were promptly removed, snap frozen and stored at −80 oC for later analysis. One group of mice in the JIMT-1 model showed complete tumor remission after treatment with EP plus PEPDG278D and were maintained for observation without treatment for 60 days. In one experiment using the BT-474R2 model, heart, kidney and liver were also removed from each mouse when it was sacrificed and weighed, and the organs were stored at −80 oC for later molecular analysis. Some tumors were fixed in 10% buffered formalin, paraffin embedded, cut at 4 μm, and stained with hematoxylin and eosin (H/E) for histological analysis.
Statistical analysis
All statistical analyses were carried out using Prizm GraphPad. For two-group comparison, we used paired two-tailed t-test. For multi-group comparisons, we used analysis of variance (ANOVA) followed by Tukey test. P value of 0.05 or lower was considered statistically significant. Additional information is provided in figure legends. Original data are provided in table S1.
Supplementary Material
Acknowledgments:
We thank Boyko S. Atanassov of RPCCC for critical reading of the manuscript and valuable comments. We thank Maurizio Scaltriti of Memorial Sloan Kettering Cancer Center for providing BT-474R2 cells.
Funding: The National Cancer Institute (R01CA164574, R01CA215093 and P30CA016056), and Roswell Park Alliance Foundation (Developmental Funds).
Footnotes
Competing interests:The authors declare that they have no competing interests.
Data and materials availability:All data associated with this study are present in the paper or supplementary materials. Materials are available by request from the corresponding author through an MTA.
REFERENCES AND NOTES:
- 1.Choritz H, Busche G, Kreipe H, On behalf of the Study Group HER2 Monitor, Quality assessment of HER2 testing by monitoring of positivity rates. Virchows Arch. 459, 283–289 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Witton CJ, Reeves JR, Going JJ, Cooke TG, Bartlett JM, Expression of the HER1–4 family of receptor tyrosine kinases in breast cancer. J. Pathol. 200, 290–297 (2003). [DOI] [PubMed] [Google Scholar]
- 3.Paik S, Hazan R, Fisher ER, Sass RE, Fisher B, Redmond C, Schlessinger J, Lippman ME, King CR, Pathologic findings from the National Surgical Adjuvant Breast and Bowel Project: prognostic significance of erbB-2 protein overexpression in primary breast cancer. J. Clin. Oncol. 8, 103–112 (1990). [DOI] [PubMed] [Google Scholar]
- 4.Slamon DJ, Clark GM, Wong SG, Levin WJ, Ullrich A, McGuire WL, Human breast cancer: correlation of relapse and survival with amplification of the HER-2/neu oncogene. Science 235, 177–182 (1987). [DOI] [PubMed] [Google Scholar]
- 5.Romond EH, Perez EA, Bryant J, Suman VJ, Geyer CE Jr., Davidson NE, Tan-Chiu E, Martino S, Paik S, Kaufman PA, Swain SM, Pisansky TM, Fehrenbacher L, Kutteh LA, Vogel VG, Visscher DW, Yothers G, Jenkins RB, Brown AM, Dakhil SR, Mamounas EP, Lingle WL, Klein PM, Ingle JN, Wolmark N, Trastuzumab plus adjuvant chemotherapy for operable HER2-positive breast cancer. N. Engl. J. Med. 353, 1673–1684 (2005). [DOI] [PubMed] [Google Scholar]
- 6.Vogel CL, Cobleigh MA, Tripathy D, Gutheil JC, Harris LN, Fehrenbacher L, Slamon DJ, Murphy M, Novotny WF, Burchmore M, Shak S, Stewart SJ, Press M, Efficacy and safety of trastuzumab as a single agent in first-line treatment of HER2-overexpressing metastatic breast cancer. J. Clin. Oncol. 20, 719–726 (2002). [DOI] [PubMed] [Google Scholar]
- 7.Baselga J, Cortes J, Kim SB, Im SA, Hegg R, Im YH, Roman L, Pedrini JL, Pienkowski T, Knott A, Clark E, Benyunes MC, Ross G, Swain SM, Pertuzumab plus trastuzumab plus docetaxel for metastatic breast cancer. N. Engl. J. Med. 366, 109–119 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nagy P, Friedlander E, Tanner M, Kapanen AI, Carraway KL, Isola J, Jovin TM, Decreased accessibility and lack of activation of ErbB2 in JIMT-1, a herceptin-resistant, MUC4-expressing breast cancer cell line. Cancer Res. 65, 473–482 (2005). [PubMed] [Google Scholar]
- 9.Scaltriti M, Rojo F, Ocana A, Anido J, Guzman M, Cortes J, Di Cosimo S, Matias-Guiu X, Ramon y Cajal S, Arribas J, Baselga J, Expression of p95HER2, a truncated form of the HER2 receptor, and response to anti-HER2 therapies in breast cancer. J. Natl. Cancer Insti. 99, 628–638 (2007). [DOI] [PubMed] [Google Scholar]
- 10.Gallardo A, Lerma E, Escuin D, Tibau A, Munoz J, Ojeda B, Barnadas A, Adrover E, Sanchez-Tejada L, Giner D, Ortiz-Martinez F, Peiro G, Increased signalling of EGFR and IGF1R, and deregulation of PTEN/PI3K/Akt pathway are related with trastuzumab resistance in HER2 breast carcinomas. Br. J. Cancer 106, 1367–1373 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhang S, Huang WC, Li P, Guo H, Poh SB, Brady SW, Xiong Y, Tseng LM, Li SH, Ding Z, Sahin AA, Esteva FJ, Hortobagyi GN, Yu D, Combating trastuzumab resistance by targeting SRC, a common node downstream of multiple resistance pathways. Nat. Med. 17, 461–469 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Oliveras-Ferraros C, Vazquez-Martin A, Cufi S, Torres-Garcia VZ, Sauri-Nadal T, Barco SD, Lopez-Bonet E, Brunet J, Martin-Castillo B, Menendez JA, Inhibitor of Apoptosis (IAP) survivin is indispensable for survival of HER2 gene-amplified breast cancer cells with primary resistance to HER1/2-targeted therapies. Biochem. Biophys. Res. Commun. 407, 412–419 (2011). [DOI] [PubMed] [Google Scholar]
- 13.Scaltriti M, Eichhorn PJ, Cortes J, Prudkin L, Aura C, Jimenez J, Chandarlapaty S, Serra V, Prat A, Ibrahim YH, Guzman M, Gili M, Rodriguez O, Rodriguez S, Perez J, Green SR, Mai S, Rosen N, Hudis C, Baselga J, Cyclin E amplification/overexpression is a mechanism of trastuzumab resistance in HER2+ breast cancer patients. Proc. Natl. Acad. Sci. U S A 108, 3761–3766 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Clynes RA, Towers TL, Presta LG, Ravetch JV, Inhibitory Fc receptors modulate in vivo cytotoxicity against tumor targets. Nat. Med. 6, 443–446 (2000). [DOI] [PubMed] [Google Scholar]
- 15.Musolino A, Naldi N, Bortesi B, Pezzuolo D, Capelletti M, Missale G, Laccabue D, Zerbini A, Camisa R, Bisagni G, Neri TM, Ardizzoni A, Immunoglobulin G fragment C receptor polymorphisms and clinical efficacy of trastuzumab-based therapy in patients with HER-2/neu-positive metastatic breast cancer. J. Clin. Oncol. 26, 1789–1796 (2008). [DOI] [PubMed] [Google Scholar]
- 16.Mohsin SK, Weiss HL, Gutierrez MC, Chamness GC, Schiff R, Digiovanna MP, Wang CX, Hilsenbeck SG, Osborne CK, Allred DC, Elledge R, Chang JC, Neoadjuvant trastuzumab induces apoptosis in primary breast cancers. J. Clin. Oncol. 23, 2460–2468 (2005). [DOI] [PubMed] [Google Scholar]
- 17.Moulder SL, Yakes FM, Muthuswamy SK, Bianco R, Simpson JF, Arteaga CL, Epidermal growth factor receptor (HER1) tyrosine kinase inhibitor ZD1839 (Iressa) inhibits HER2/neu (erbB2)-overexpressing breast cancer cells in vitro and in vivo. Cancer Res. 61, 8887–8895 (2001). [PubMed] [Google Scholar]
- 18.Yang L, Li Y, Bhattacharya A, Zhang Y, Inhibition of ERBB2-overexpressing tumors by recombinant human prolidase and its enzymatically inactive mutant. EBioMedicine 2, 396–405 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yang L, Li Y, Bhattacharya A, Zhang Y, Dual inhibition of ErbB1 and ErbB2 in cancer by recombinant human prolidase mutant hPEPD-G278D. Oncotarget 7, 42340–42352 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yang L, Li Y, Zhang Y, Identification of prolidase as a high affinity ligand of the ErbB2 receptor and its regulation of ErbB2 signaling and cell growth. Cell Death Dis. 5, e1211 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.DiGiovanna MP, Stern DF, Edgerton SM, Whalen SG, Moore D 2nd, Thor AD, Relationship of epidermal growth factor receptor expression to ErbB-2 signaling activity and prognosis in breast cancer patients. J. Clin. Oncol. 23, 1152–1160 (2005). [DOI] [PubMed] [Google Scholar]
- 22.Tsutsui S, Ohno S, Murakami S, Kataoka A, Kinoshita J, Hachitanda Y, Prognostic value of the combination of epidermal growth factor receptor and c-erbB-2 in breast cancer. Surgery 133, 219–221 (2003). [DOI] [PubMed] [Google Scholar]
- 23.Jarvinen TA, Tanner M, Rantanen V, Barlund M, Borg A, Grenman S, Isola J, Amplification and deletion of topoisomerase IIalpha associate with ErbB-2 amplification and affect sensitivity to topoisomerase II inhibitor doxorubicin in breast cancer. Am. J. Pathol. 156, 839–847 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kao J, Salari K, Bocanegra M, Choi YL, Girard L, Gandhi J, Kwei KA, Hernandez-Boussard T, Wang P, Gazdar AF, Minna JD, Pollack JR, Molecular profiling of breast cancer cell lines defines relevant tumor models and provides a resource for cancer gene discovery. PLoS One 4, e6146 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tanner M, Kapanen AI, Junttila T, Raheem O, Grenman S, Elo J, Elenius K, Isola J, Characterization of a novel cell line established from a patient with Herceptin-resistant breast cancer. Mol. Cancer Ther. 3, 1585–1592 (2004). [PubMed] [Google Scholar]
- 26.O’Brien NA, Browne BC, Chow L, Wang Y, Ginther C, Arboleda J, Duffy MJ, Crown J, O’Donovan N, Slamon DJ, Activated phosphoinositide 3-kinase/AKT signaling confers resistance to trastuzumab but not lapatinib. Mol. Cancer Ther. 9, 1489–1502 (2010). [DOI] [PubMed] [Google Scholar]
- 27.Toth G, Szoor A, Simon L, Yarden Y, Szollosi J, Vereb G, The combination of trastuzumab and pertuzumab administered at approved doses may delay development of trastuzumab resistance by additively enhancing antibody-dependent cell-mediated cytotoxicity. MABS 8, 1361–1370 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Esteva FJ, Guo H, Zhang S, Santa-Maria C, Stone S, Lanchbury JS, Sahin AA, Hortobagyi GN, Yu D, PTEN, PIK3CA, p-AKT, and p-p70S6K status: association with trastuzumab response and survival in patients with HER2-positive metastatic breast cancer. Am. J. Pathol. 177, 1647–1656 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jiang G, Zhang S, Yazdanparast A, Li M, Pawar AV, Liu Y, Inavolu SM, Cheng L, Comprehensive comparison of molecular portraits between cell lines and tumors in breast cancer. BMC Genomics 17 Suppl 7, 525 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gayle SS, Castellino RC, Buss MC, Nahta R, MEK inhibition increases lapatinib sensitivity via modulation of FOXM1. Curr. Med. Chem. 20, 2486–2499 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Shattuck DL, Miller JK, Carraway KL 3rd, Sweeney C, Met receptor contributes to trastuzumab resistance of Her2-overexpressing breast cancer cells. Cancer Res. 68, 1471–1477 (2008). [DOI] [PubMed] [Google Scholar]
- 32.Wang YC, Morrison G, Gillihan R, Guo J, Ward RM, Fu X, Botero MF, Healy NA, Hilsenbeck SG, Phillips GL, Chamness GC, Rimawi MF, Osborne CK, Schiff R, Different mechanisms for resistance to trastuzumab versus lapatinib in HER2-positive breast cancers--role of estrogen receptor and HER2 reactivation. Breast Cancer Res. 13, R121 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gu S, Hu Z, Ngamcherdtrakul W, Castro DJ, Morry J, Reda MM, Gray JW, Yantasee W, Therapeutic siRNA for drug-resistant HER2-positive breast cancer. Oncotarget 7, 14727–14741 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cho HS, Mason K, Ramyar KX, Stanley AM, Gabelli SB, Denney DW Jr., Leahy DJ, Structure of the extracellular region of HER2 alone and in complex with the Herceptin Fab. Nature 421, 756–760 (2003). [DOI] [PubMed] [Google Scholar]
- 35.Li C, Iida M, Dunn EF, Ghia AJ, Wheeler DL, Nuclear EGFR contributes to acquired resistance to cetuximab. Oncogene 28, 3801–3813 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Li LY, Chen H, Hsieh YH, Wang YN, Chu HJ, Chen YH, Chen HY, Chien PJ, Ma HT, Tsai HC, Lai CC, Sher YP, Lien HC, Tsai CH, Hung MC, Nuclear ErbB2 enhances translation and cell growth by activating transcription of ribosomal RNA genes. Cancer Res. 71, 4269–4279 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yang L, Li Y, Bhattacharya A, Zhang Y, PEPD is a pivotal regulator of p53 tumor suppressor. Nat. Commun. 8, 2052 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mercogliano MF, De Martino M, Venturutti L, Rivas MA, Proietti CJ, Inurrigarro G, Frahm I, Allemand DH, Deza EG, Ares S, Gercovich FG, Guzman P, Roa JC, Elizalde PV, Schillaci R, TNFalpha-induced mucin 4 expression elicits trastuzumab resistance in HER2-positive breast cancer. Clin. Cancer Res. 23, 636–648 (2017). [DOI] [PubMed] [Google Scholar]
- 39.Ponnusamy MP, Seshacharyulu P, Vaz A, Dey P, Batra SK, MUC4 stabilizes HER2 expression and maintains the cancer stem cell population in ovarian cancer cells. J Ovarian Res. 4, 7 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Franklin MC, Carey KD, Vajdos FF, Leahy DJ, de Vos AM, Sliwkowski MX, Insights into ErbB signaling from the structure of the ErbB2-pertuzumab complex. Cancer Cell 5, 317–328 (2004). [DOI] [PubMed] [Google Scholar]
- 41.Yang L, Li Y, Bhattacharya A, Zhang Y, A plasma proteolysis pathway comprising blood coagulation proteases. Oncotarget 7, 40919–40938 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Scheuer W, Friess T, Burtscher H, Bossenmaier B, Endl J, Hasmann M, Strongly enhanced antitumor activity of trastuzumab and pertuzumab combination treatment on HER2-positive human xenograft tumor models. Cancer Res. 69, 9330–9336 (2009). [DOI] [PubMed] [Google Scholar]
- 43.Yang L, Li Y, Shen E, Cao F, Li L, Li X, Wang X, Kariminia S, Chang B, Li H, Li Q, NRG1-dependent activation of HER3 induces primary resistance to trastuzumab in HER2-overexpressing breast cancer cells. Int. J. Oncol. 51, 1553–1562 (2017). [DOI] [PubMed] [Google Scholar]
- 44.Tan M, Jing T, Lan KH, Neal CL, Li P, Lee S, Fang D, Nagata Y, Liu J, Arlinghaus R, Hung MC, Yu D, Phosphorylation on tyrosine-15 of p34(Cdc2) by ErbB2 inhibits p34(Cdc2) activation and is involved in resistance to taxol-induced apoptosis. Mol. Cell 9, 993–1004 (2002). [DOI] [PubMed] [Google Scholar]
- 45.Yu D, Jing T, Liu B, Yao J, Tan M, McDonnell TJ, Hung MC, Overexpression of ErbB2 blocks Taxol-induced apoptosis by upregulation of p21Cip1, which inhibits p34Cdc2 kinase. Mol. Cell 2, 581–591 (1998). [DOI] [PubMed] [Google Scholar]
- 46.Lee-Hoeflich ST, Crocker L, Yao E, Pham T, Munroe X, Hoeflich KP, Sliwkowski MX, Stern HM, A central role for HER3 in HER2-amplified breast cancer: implications for targeted therapy. Cancer Res. 68, 5878–5887 (2008). [DOI] [PubMed] [Google Scholar]
- 47.Zhao L, Vogt PK, Helical domain and kinase domain mutations in p110alpha of phosphatidylinositol 3-kinase induce gain of function by different mechanisms. Proc. Natl. Acad. Sci. U S A 105, 2652–2657 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hon WC, Berndt A, Williams RL, Regulation of lipid binding underlies the activation mechanism of class IA PI3-kinases. Oncogene 31, 3655–3666 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bergamaschini L, Rossi E, Storini C, Pizzimenti S, Distaso M, Perego C, De Luigi A, Vergani C, De Simoni MG, Peripheral treatment with enoxaparin, a low molecular weight heparin, reduces plaques and beta-amyloid accumulation in a mouse model of Alzheimer’s disease. J. Neurosci. 24, 4181–4186 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Caliskan A, Yavuz C, Karahan O, Yazici S, Guclu O, Demirtas S, Mavitas B, Factor-Xa inhibitors protect against systemic oxidant damage induced by peripheral-ischemia reperfusion. J. Thromb. Thrombolysis 37, 464–468 (2014). [DOI] [PubMed] [Google Scholar]
- 51.Donati MB, Cancer and thrombosis. Haemostasis 24, 128–131 (1994). [DOI] [PubMed] [Google Scholar]
- 52.Green KB, Silverstein RL, Hypercoagulability in cancer. Hematol. Oncol. Clin. North Am. 10, 499–530 (1996). [DOI] [PubMed] [Google Scholar]
- 53.Yang L, Li Y, Ding Y, Choi KS, Kazim AL, Zhang Y, Prolidase directly binds and activates epidermal growth factor receptor and stimulates downstream signaling. J. Biol. Chem. 288, 2365–2375 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Muller P, Ceskova P, Vojtesek B, Hsp90 is essential for restoring cellular functions of temperature-sensitive p53 mutant protein but not for stabilization and activation of wild-type p53: implications for cancer therapy. J. Biol. Chem. 280, 6682–6691 (2005). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.