

Summary
Episodic ataxia is a genetically heterogeneous neurological condition characterized by spells of incoordination and imbalance, often associated with progressive ataxia. Episodic ataxia type 2, caused by calcium voltage-gated channel subunit alpha1 A (CACNA1A MIM: 601011) mutation, is the most common form of episodic ataxia. It is characterized by recurrent attacks of imbalance associated with interictal nystagmus lasting hours to days and triggered by emotional stress or exercise. In this article, we report a novel heterozygous intronic variant c.5743+14A>G in the CACNA1A gene in a Saudi family. To the best of our knowledge, this variant has not been described in the literature or reported in public mutation databases. This report indicated that acetazolamide is not beneficial, and it may be even harmful to patients with episodic ataxia type 2 if used in later stages. In addition, treatment with 4-aminopyridine did not show any efficacy to improve walking or balance in our patient, which indicates the importance of early initiation of therapy before the later stages of the disease. Further research is needed to explore potential treatments for this challenging disease.
Keywords: CACNA1A, alpha-1A subunit, autosomal dominant cerebellar ataxia, novel Mutation, Saudi Arabia
1. Introduction
Episodic ataxia is a genetically heterogeneous neurological condition characterized by spells of incoordination and imbalance, often associated with progressive ataxia (1). Episodic ataxia type 2 has a wide range of expressivity among patients. The disease is characterized by paroxysmal attacks of ataxia, vertigo, and nausea that usually last hours to days. Associated symptoms of the attacks include dysarthria, dystonia, tinnitus, diplopia, headache, and hemiplegia (2). Approximately 50% of patients with this disease have migraine headaches without loss of consciousness. Other less commonly reported manifestations include torticollis, intellectual disability, and psychiatric disorders. Initially, patients are usually asymptomatic between attacks, but with disease progression, they develop permanent cerebellar symptoms (3).
Mutations in the CACNA1A gene are associated with autosomal dominant neurological disorders including episodic ataxia, familial hemiplegic migraine, early infantile epileptic encephalopathy 42, and spinocerebellar ataxia 6 (4). More than 170 disease-causing variants in CACNA1A are reported in the Human Gene Mutation Database (HGMD). This includes missense variants (51.5%), nonsense variants (11.7%), splice variants (7.0%), small deletions (11.7%), small insertions (5.8%), gross deletions (9.4%), complex rearrangements (0.6%), and repeat variations (2.3%). Among these variants, 128 are implicated in ataxia phenotypes, mainly episodic ataxia (5). Episodic ataxia type 2, caused by CACNA1A mutation, is the most common form of episodic ataxia. It is characterized by recurrent attacks of imbalance associated with interictal nystagmus lasting hours to days and triggered by emotional stress or exercise. The age of onset is usually in childhood or early adolescence (age range 2-32 years). Its prevalence has been estimated at lower than 1/100,000 (6).
The currently available treatments include acetazolamide and 4-aminopyridine (fampridine), which may reduce the frequency, severity, and duration of attacks but have no effect on the disease progression (6). In this article, we report a novel mutation in CACNA1A gene in a young Saudi lady with episodic ataxia type 2 with no response to acetazolamide or 4-aminopyridine.
2. Case Report
Proband III-2 is a 25-year-old Saudi woman presented with 9 years history of ataxia, imbalance, dysarthria, and difficulty swallowing. Initially, her symptoms were episodic with intermittent improvement between episodes. However, over the past five years, her disease became progressive. Her symptoms increased with time to the point that she needed a wheelchair due to inability to walk especially in dark rooms. There was no history of joint pain, skin rash, visual symptoms, mouth or genital ulcers, or gastrointestinal symptoms. The patient denied memory loss or psychiatric symptoms. Her past medical history was unremarkable including endocrine disorders, infections, sleep disorders, or trauma. Family history was positive for a similar condition. On examination, the patient was conscious and oriented with normal cognitive functions. She was dysarthric with scanning speech, and she had impaired saccadic eye movement. She also had impaired dorsal column structure and pectus excavatum. The rest of her cranial nerve examination was normal. Motor examination showed 3/5 power in the upper limbs and 4/5 power in the lower limbs, and her reflexes were absent. Cerebellar examination showed dysmetria and dysdiadokinesia. She had difficulty walking without assistance. There were no associated skeletal deformities, and her fundus examination was normal. Gait examination showed wide-based gait with a tendency to fall to either side.
Extensive blood work including both routine and specific tests to rule out the cause of ataxia in this age group was negative. These include vitamin E level, vitamin B-12 level, blood smear for acanthocytes, viral serology, thyroid function test, serology for celiac disease, and lipoprotein electrophoresis. Genetic testing was sent, and the sequence analysis identified a heterozygous intron variant c.5743+14A>G in the CACNA1A gene, which is inherited in an autosomal dominant manner (Figure 1). This intronic variant has been identified in four heterozygous individuals in the Genome Aggregation Database (gnomAD, n > 120,000 exomes and > 15,000 genomes). Database curators have made every effort to exclude individuals with severe pediatric diseases from these cohorts. This variant is absent in the Greater Middle East (GME) Variome Project with 2,497 individuals analyzed. This variant has not been reported in public mutation databases or the literature. It is classified as a variant of uncertain significance according to the ACMG guidelines. According to the Human Splicing Finder in silico tool, this variant probably has no impact on splicing.
Figure 1.
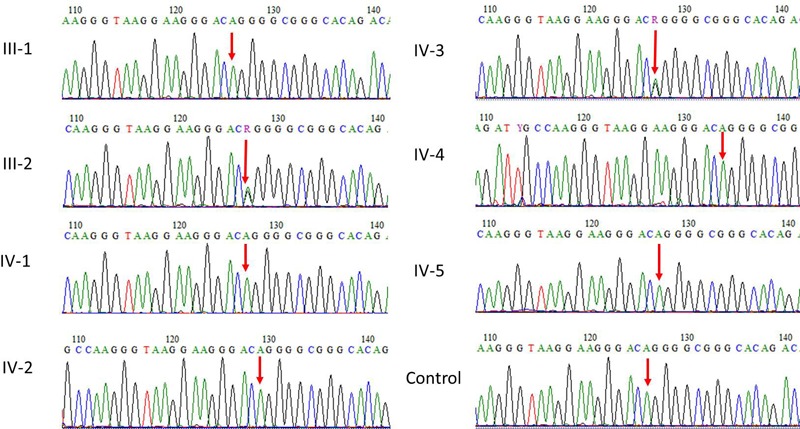
Sanger sequence analysis for validation of exome sequencing variant found in CACNA1A gene.
Additionally, our exome sequencing data showed a heterozygous carrier for three missense novel mutations in POLG gene (c.576G˃T, p.(Glu192Asp)), PNKP (c.1160C˃A, p.(Leu454Met)), and ABCB7 (c.14C˃T, p.(Ala5Val)). The PNKP (c.1160C˃A, p.(Leu454Met)) variant has previously been identified in clinical testing and classified as a variant of uncertain significance (ClinVar variation ID: 95478). To the best of our knowledge, the POLG gene (c.576G˃T, p.(Glu192Asp)) and ABCB7 (c.14C˃T, p.(Ala5Val)) variants have not been reported in the literature or public mutation databases. As there is not enough data to support or rule out pathogenicity, these variants are classified as variants of uncertain significance. It has to be noted that both autosomal recessive and dominant inheritance patterns have been identified in the diseases caused by POLG mutations. POLG-related mitochondrial ataxia syndrome and mitochondrial DNA depletion syndromes are inherited in an autosomal recessive manner, but POLG-related progressive external ophthalmoplegia can also be inherited in an autosomal dominant manner. The disease caused by PNKP mutations is inherited in an autosomal recessive manner, and the disease caused by ABCB7 mutations is inherited in an X-linked recessive manner. The patient is heterozygous for the identified variants.
The patient was also a heterozygous carrier for a pathogenic mutation in TCTN1 (c.342-2A>G). This variant affects the acceptor splice site of intron 2. This variant has been identified as homozygous in patients with Joubert syndrome or Joubert-like syndrome. Considering the current literature and the well-established role of the variant, it is classified as pathogenic. Our patient is heterozygous for the variant, and diseases caused by TCTN1 mutations are inherited in an autosomal recessive manner. Thus, the identified variant is not sufficient to cause disease without another disease-causing variant in the same gene.
Magnetic resonance imaging (MRI) of the brain revealed moderate atrophy of the midbrain, pontine tegmentum, and vermis of the cerebellum with a mild degree of atrophic changes within the cord (Figure 2). Nerve conduction studies showed axonal sensory neuropathy with reduced SNAP amplitude in the bilateral median and ulnar nerves with absent unrecordable responses from the sural and superficial peroneal nerves bilaterally. In addition, low CMAP amplitude in both peroneal nerves was observed. Cardiac echocardiography was unremarkable with normal left ventricular size and systolic function. A 24-hour Holter monitor showed sinus bradycardia with marked sinus arrhythmia and a heart rate ranging from 37 to 122 beats per minute (average 69 beats per minute).
Figure 2.
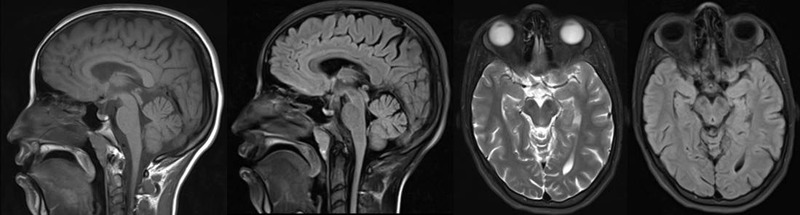
MRI of the brain showing moderate atrophy of the midbrain, pontine tegmentum, and vermis of the cerebellum with a mild degree of atrophic changes within the cord. MRI, magnetic resonance imaging.
The detailed family pedigree was drawn after getting the detailed information from the parents as shown in Figure 3. Family and genetic counseling were offered through a genetic consultant. The patient was tried on both acetazolamide (up to 1 gram daily in two divided doses) and 4-aminopyridine with no improvement in her condition. In fact, she developed several side effects due to acetazolamide including paresthesias, polyuria, and drowsiness.
Figure 3.
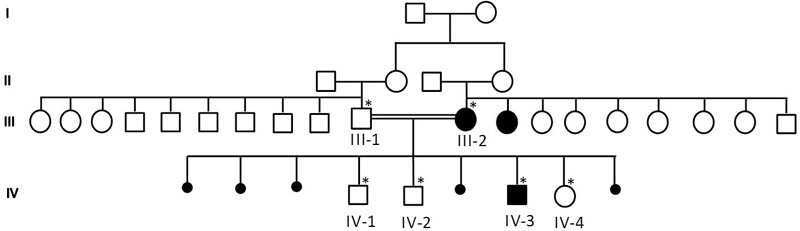
Family pedigree showing the details of the members of the family. The samples marked with asterisks were available for genetic testing.
This study was approved by the Institutional Review Board (IRB) of King Abdullah International Medical Research Center (KAIMRC).
3. Discussion
Voltage-dependent calcium channels mediate the entry of calcium ions into excitable cells, and are also involved in a variety of calcium-dependent processes including muscle contraction, hormone or neurotransmitter release, and gene expression (7). Calcium channels are multisubunit complexes composed of alpha-1, beta, alpa-2/delta, and gamma subunits. The channel activity is directed by the pore-forming alpha-1 subunit, whereas the others act as auxiliary subunits regulating this activity. The distinctive properties of the calcium channel types are related primarily to the expression of a variety of alpha-1 isoforms, alpha-1A, B, C, D, E, and S (8).
The CACNA1A gene on chromosome 19p13.13 encodes the alpha-1A subunit, which is the pore-forming subunit of the voltage-dependent calcium channels. It is predominantly expressed in neuronal tissue, particularly Purkinje and granule cells of the cerebellum (9). These calcium channels mediate entry of calcium into the cells and regulate the pacemaking precision. These channels regulate the whole-cell calcium current density and the intrinsic excitability of Purkinje cells and granule cells, and exert major control over glutamate release at the parallel fiber onto Purkinje-cell synapses (10). Targeted deletions of CACNA1A in cerebellar granule cells or Purkinje cells result in altered cerebellar output by decreasing the excitatory drive on Purkinje cells or their ability to release neurotransmitters (10).
Our patient had clinical features similar to other patients with episodic ataxia type 2 caused by CACNA1A gene mutation but differed in the time of diagnosis. Most of the reported cases in the literature were diagnosed early in childhood when the disease was still episodic. Although early diagnosis does not seem to alter the natural history of the disease, it allows appropriate family counseling and planning and also leads to a better response to the available symptomatic treatment. Our patient did not have genetic testing to identify the responsible gene for many years due to unavailability of advanced genetic testing in most of the hospitals. With the current availability of molecular genetics in Saudi Arabia, the identification of responsible genes associated with a wide variety of diseases will be feasible. This progress is hoped to eliminate delay in diagnosis of rare genetic diseases.
The whole exome sequencing performed on our patient identified several other variants in POLG, PNKP, ABCB7, and TCTN1 genes. These variants are not related to the clinical phenotype of the patient. This is because she is a carrier for those identified variants and most of the disease caused by them are inherited in an autosomal recessive manner. Our patient's clinical phenotype does not fit the phenotype of diseases caused by mutations in these genes.
History and physical examination are of paramount importance in indicating a diagnosis of cerebellar ataxia. MRI of the brain is important in evaluating structural abnormalities and typically demonstrates atrophy of the cerebellar vermis (11). Genetic testing remains the only definitive diagnostic method to identify the responsible gene. Nuclear magnetic spectroscopy may demonstrate abnormal cerebellar intracellular pH levels and low cerebellar creatine. EMG typically shows absent myokymia, and single fiber EMG may demonstrate jitter and blocking (12).
Management of these patients mainly includes occupational and physical therapy for gait dysfunction and speech therapy. There is no currently available treatment that can cure the disease or alter the disease progression (13). Acetazolamide is an effective medication for controlling or reducing the frequency and severity of attacks, but it does not prevent the progression of the disease (14). In addition, recent studies showed that 4-aminopyridine (fampridine) might also be effective in reducing the frequency and duration of attacks. Prognosis is variable in these patients, but improvement in their condition is unlikely (15). We have recently reported our experience in the use of 4-aminopyridine in a patient with a rare hereditary ataxia with no response. This indicates that the management of patients with hereditary ataxias remains challenging (16).
The rate of consanguinity in Saudi Arabia is high ranging from 25% to 65%. This consanguinity favors the occurrence of genetic diseases, especially the diseases inherited in an autosomal recessive manner. It is important to educate the Saudi population about this fact to hopefully reduce the occurrence of rare and devastating diseases. We have previously reported several novel and rare mutations causing a wide variety of genetic neurological diseases in the Saudi community (16-24). This paper may be considered as an urgent call for action to address the consanguinity issue in Saudi Arabia.
In conclusion, we report a novel heterozygous intron variant c.5743+14A>G in the CACNA1A gene in a Saudi family. This report indicated that acetazolamide is not beneficial, and it may be even harmful to patients with episodic ataxia type 2 if used in later stages. In addition, 4-aminopyridine did not show any efficacy to improve walking or balance in our patient, which indicates the importance of early initiation of therapy before the later stages of the disease. Further research is needed to explore potential treatments for this challenging condition.
References
- 1. Jen JC, Graves TD, Hess EJ, Hanna MG, Griggs RC, Baloh RW; CINCH investigators. Primary episodic ataxias: Diagnosis, pathogenesis and treatment. Brain. 2007; 130:2484-2493. [DOI] [PubMed] [Google Scholar]
- 2. Baloh RW. Episodic ataxias 1 and 2. Handb Clin Neurol. 2012; 103:595-602. [DOI] [PubMed] [Google Scholar]
- 3. Bertholon P, Chabrier S, Riant F, Tournier-lasserve E, Peyron R. Episodic ataxia type 2: Unusual aspects in clinical and genetic presentation. Special emphasis in childhood. J Neurol Neurosurg Psychiatry. 2009; 80:1289-1292. [DOI] [PubMed] [Google Scholar]
- 4. Barros J, Damásio J, Tuna A, Alves I, Silveira I, Pereira- Monteiro J, Sequeiros J, Alonso I, Sousa A, Coutinho P. Cerebellar ataxia, hemiplegic migraine, and related phenotypes due to a CACNA1A missense mutation: 12-year follow-up of a large Portuguese family. JAMA Neurol. 2013; 70:235-240. [DOI] [PubMed] [Google Scholar]
- 5. Human Gene Mutation Database Professional 2017.2. https://www.nihlibrary.nih.gov/resources/tools/humangene-mutation-database-professional (Accessed on December 6, 2018)
- 6. Spacey S. Episodic Ataxia Type 2. In: Pagon RA, Adam MP, Bird TD et al: (eds) Seattle (WA): GeneReviews, 1993. [Google Scholar]
- 7. Greenberg DA. Calcium channels in neurological disease. Ann Neurol. 1997; 42:275-282. [DOI] [PubMed] [Google Scholar]
- 8. Randall AD. The molecular basis of voltage-gated Ca2+ channel diversity: Is it time for T?. J Membr Biol. 1998; 161:207-213. [DOI] [PubMed] [Google Scholar]
- 9. R a j a k u l e n d r a n S, G r a v e s T D, L a b r u m RW, Kotzadimitriou D, Eunson L, Davis MB, Davies R, Wood NW, Kullmann DM, Hanna MG, Schorge S. Genetic and functional characterisation of the P/Q calcium channel in episodic ataxia with epilepsy. J Physiol. 2010; 588:1905-1913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Damaj L, Lupien-Meilleur A, Lortie A, Riou É, Ospina LH, Gagnon L, Vanasse C, Rossignol E. CACNA1A haploinsufficiency causes cognitive impairment, autism and epileptic encephalopathy with mild cerebellar symptoms. Eur J Hum Genet. 2015; 23:1505-1512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Mantuano E, Romano S, Veneziano L, et al. Identification of novel and recurrent CACNA1A gene mutations in fifteen patients with episodic ataxia type 2. J Neurol Sci. 2010; 291:30-36. [DOI] [PubMed] [Google Scholar]
- 12. Harno H, Heikkinen S, Kaunisto MA, Kallela M, Häkkinen AM, Wessman M, Färkkilä M, Lundbom N. Decreased cerebellar total creatine in episodic ataxia type 2: A 1H MRS study. Neurology. 2005; 64:542-544. [DOI] [PubMed] [Google Scholar]
- 13. Kotagal V. Acetazolamide-responsive ataxia. Semin Neurol. 2012; 32:533-537. [DOI] [PubMed] [Google Scholar]
- 14. Ilg W, Bastian AJ, Boesch S, Burciu RG, Celnik P, Claaßen J, Feil K, Kalla R, Miyai I, Nachbauer W, Schöls L, Strupp M, Synofzik M, Teufel J, Timmann D. Consensus paper: Management of degenerative cerebellar disorders. Cerebellum. 2014; 13:248-268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Hess E. A randomized trial of 4-aminopyridine in EA2 and related familial episodic ataxias. Neurology. 2011; 77:1996-1997. [DOI] [PubMed] [Google Scholar]
- 16. Algahtani H, Shirah B, Algahtani R, Naseer MI, Al- Qahtani MH, Abdulkareem AA. Ataxia with ocular apraxia type 2 not responding to 4-aminopyridine: A rare mutation in the SETX gene in a Saudi patient. Intractable Rare Dis Res. 2018; 7:275-279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Algahtani H, Shirah B. A novel mutation in the DNMT1 gene in a patient presenting with pure cerebellar ataxia. J Genet Med. 2017; 14:71-74. [Google Scholar]
- 18. Algahtani H, Naseer MI, Al-Qahtani M, Abdulrahman SA, Boker F, Shirah B. Congenital insensitivity to pain with anhidrosis: A report of two siblings with a novel mutation in (TrkA) NTRK1 gene in a Saudi family. J Neurol Sci. 2016; 370:35-38. [DOI] [PubMed] [Google Scholar]
- 19. Algahtani H, Marzouk Y, Algahtani R, Salman S, Shirah B. Autosomal recessive cerebellar ataxia type 1 mimicking multiple sclerosis: A report of two siblings with a novel mutation in SYNE1 gene in a Saudi family. J Neurol Sci. 2017; 372:97-100. [DOI] [PubMed] [Google Scholar]
- 20. Algahtani H, Ghamdi S, Shirah B, Alharbi B, Algahtani R, Bazaid A. Biotin-thiamine-responsive basal ganglia disease: Catastrophic consequences of delay in diagnosis and treatment. Neurol Res. 2017; 39:117-125. [DOI] [PubMed] [Google Scholar]
- 21. Algahtani H, Ibrahim B, Shirah B, Aldarmahi A, Abdullah A. More than a decade of misdiagnosis of alternating hemiplegia of childhood with catastrophic outcome. Case Rep Med. 2017; 2017:5769837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Algahtani H, Alameer S, Marzouk Y, Shirah B. Urea cycle disorder misdiagnosed as multiple sclerosis: A case report and review of the literature. Neuroradiol J. 2018; 31:213-217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Algahtani H, Absi A, Bassuni W, Shirah B. Adult-onset hemophagocytic lymphohistiocytosis type 2 presenting as a demyelinating disease. Mult Scler Relat Disord. 2018; 25:77-82. [DOI] [PubMed] [Google Scholar]
- 24. Algahtani H, Shirah B, Alassiri AH, Habib BA, Almuhanna R, Ahamed MF. Limb-girdle muscular dystrophy type 2B: An unusual cause of proximal muscular weakness in Saudi Arabia. J Back Musculoskelet Rehabil. 2018; 31:999-1004. [DOI] [PubMed] [Google Scholar]