

Abstract
Adverse drug reactions are a significant health care burden. Immune-mediated adverse drug reactions (IM-ADRs) contribute a disproportionately high amount of that burden. Variation in human leukocyte antigen (HLA) genes has emerged as a potential pre-prescription screening strategy for the prevention of previously unpredictable IM-ADRs. Immunopharmacogenomics combines the disciplines of immunogenomics and pharmacogenomics and focuses on the effects of immune-specific variation on drug disposition and IM-ADRs. In this review, we will present the latest evidence for HLA associations with IM-ADRs, ongoing research into biological mechanisms of IM-ADRs, and translation of clinical actionable biomarkers for IM-ADRs, with a focus on T-cell-mediated adverse drug reactions.
Introduction
Adverse drug reactions (ADRs) are estimated to be the 4–6th leading cause of death, are the cause of 3–6% of inpatient admissions, and cost up to $136 billion annually in the United States (US).1–3 ADRs may be caused by predictable effects based on pharmacologic activity (type A) or off target, typically immune-mediated effects independent of pharmacologic activity (type B) (Figure 1). Type B or immune mediated-ADRs (IM-ADRs) comprise approximately 20% of all ADRs, but are considerably more costly than pharmacologically predictable ADRs.4 IM-ADRs can be classified according to primary immune cell involvement, including B-cell mediated (antibody mediated, Gell-Coombs type I-III) and T-cell-mediated (delayed hypersensitivity, Gell-Coombs type IV) reactions. Historically, type A ADRs are considered predictable and type B ADRs unpredictable, but recent research has made prediction and prevention of IM-ADRs possible.
Figure 1:
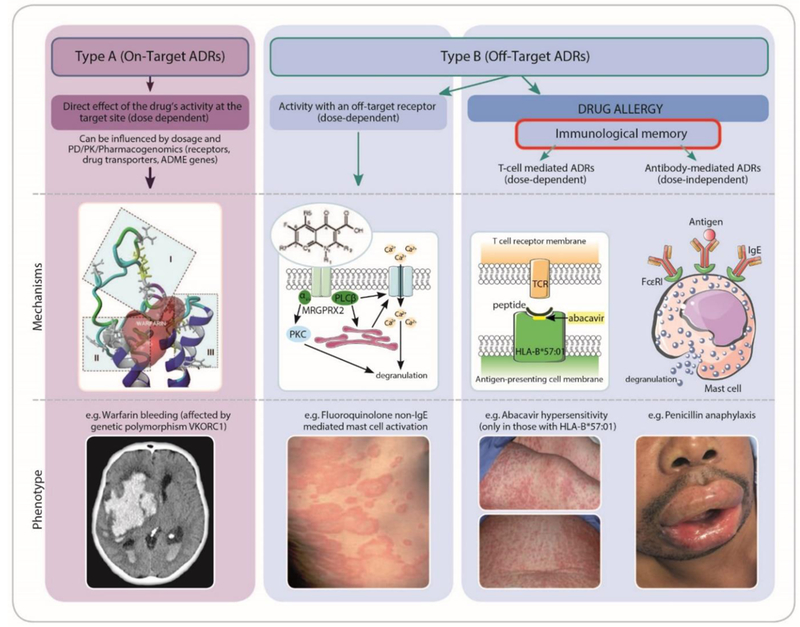
ADRs can be classified as either Type A (on target effects) or Type B (off target effects). Both types of ADRs can exhibit concentration dependent relationships with development and severity of the ADR. Type A reactions relate to the known primary therapeutic action of the drug (e.g. bleeding related to warfarin) whereas Type B reactions relate to mechanisms not directly related to the drugs intended pharmacological action. Off target reactions can occur without a direct immunological effect and have an immunological phenotype (e.g. non-IgE mediated mast-cell activation with fluoroquinolones.7 Off target reactions can also be immune-mediated, associated with immunological memory, and either dose dependent (T-cell-mediated reactions as in abacavir hypersensitivity) or non-dose dependent (IgE-mediated recognition and amplification of small amounts of antigen as in penicillin anaphylaxis). ADME indicates absorption, distribution, metabolism, and excretion; ADR, adverse drug reaction; HLA, human leukocyte antigen; PD, pharmacodynamics; PK, pharmacokinetics; PKC, protein kinase C; VKORC1, vitamin K epoxide reductase.
Immunogenomics is a broad scientific discipline focused on variation in immune-specific genes. Immunogenomics has facilitated a comprehensive catalogue of strong genomic associations with immune-mediated diseases and IM-ADRs.5 These typically manifest as associations with HLA alleles since HLA alleles capture the amino acid sequence and thus specific binding and signal transduction capacity of major histocompatibility complex (MHC) molecules. Immunogenomics also deals with the interplay of diseases and IM-ADRs with other immune-related genetic variation such as killer immunoglobulin-like receptors (KIRs), T-cell receptors (TCRs), and B-cell receptors (BCRs), as well as expression levels for immune-related molecules such as cytokines and chemokines.
Immunopharmacogenomics combines the disciplines of immunogenomics and pharmacogenomics. Pharmacogenomics focuses on the influence of genomic and other “omic” variation on drug response and ADRs. Immunopharmacogenomics focuses on the effects of immune-specific “omic” variation on drug disposition and development of the next generation of immunotherapy. Immunopharmacogenomics has the potential to predict and prevent IM-ADRs, including possibly catastrophic severe cutaneous adverse reactions (SCARs). Immunopharmacogenomics also has the potential to facilitate selection and monitoring of immunotherapy in autoimmunity and cancer, to predict and prevent reactions to antigenic foods, and to contribute to our understanding of autoimmune disease and transplant rejection, which could lead to new therapies.6 In this review, we will present the latest evidence for the prediction of IM-ADRs with a focus on T-cell-mediated IM-ADRs. We will also discuss progress in ongoing research into biological mechanisms of IM-ADRs. Finally, we will provide examples and perspectives on translational success stories such as implementation of genetic screening to prevent IM-ADRs.
Immune-mediated adverse drug reactions
Drug reactions that occur within one hour of dosing are called immediate ADRs. These can include dose independent reactions, which in their severest form manifest as anaphylaxis characterized by cardiovascular collapse, airway constriction, angioedema and urticarial rash or dose-related non-IgE mediated mast-cell activation. These reactions can present very similar to IgE-mediated reactions but are inconsistent in their onset and reproducibility and do not intensify with time. The mechanism of one form of non-IgE mediated mast cell activation associated with drugs such as opioids, fluoroquinolones and neuromuscular blocking agents has been recently elucidated to be secondary to activation of a specific G-protein coupled receptor (MRGPRX2) only present on mast cells and dorsal root ganglia.7 Immunity to IgE-mediated ADRs will wane over time. For instance, about 10% of patients per year will lose skin test reactivity to penicillin. The most common manifestation of allergic drug reactions are mild delayed cutaneous reactions that include both urticarial and maculopapular exanthems.8 The most severe IM-ADRs are classified as SCARs, which typically have multi-organ involvement and constitute three major T-cell-mediated syndromes.9 These include drug reaction with eosinophilia and systemic symptoms (DRESS), acute generalized exanthematous pustulosis (AGEP), and Stevens-Johnson syndrome (SJS)/Toxic epidermal necrosis (TEN). The most common SCARs are SJS, TEN, and DRESS with a global prevalence of 1/1,000,000, 7/1,000,000, and 4/10,000, respectively.10–13 More than 100 drugs have been associated with SCARs but the most common offending drugs for SCARs include allopurinol, aromatic anticonvulsants, antimicrobials, antiretrovirals, and oxicam nonsteroidal anti-inflammatory drugs.14
Stevens-Johnson syndrome/Toxic epidermal necrosis
SJS/TEN is the most severe IM-ADR with a mortality rate up to 50 percent.9 SJS/TEN risk is increased in patients who carry specific class I HLA alleles and may also be increased in patients with underlying co-morbidities and drug exposures such as in HIV, cancer and systemic lupus erythematosus (SLE).15,16 Clinical features include a prodrome of fever, sore throat and eyes (mucosal involvement) followed by blister formation, epidermal necrosis and sloughing and often secondary internal organ involvement. Mucocutaneous involvement and full thickness epidermal necrosis is a typical pathological feature of SJS/TEN. SJS and TEN represent a spectrum of disease defined primarily by total body surface area (TBSA), where SJS is diagnosed for affected TBSA less than 10%, SJS/TEN for 10–30% and TEN for more than 30%. The severity of SJS/TEN can be predicted from clinical characteristics using the validated measure SCORTEN.17 Long term complications of SJS/TEN are a source of significant disability and include scarring, blindness, and psychiatric illness. Clinical management of SJS/TEN consists primarily of cessation of offending medication, aggressive supportive care in a tertiary critical care center (usually a burn unit), early evaluation by subspecialists of all SJS/TEN-involved organ systems, and long term follow-up and management of SJS/TEN complications.18 Treatment for SCARs does not include a treatment guideline, highlighting the inadequacy of available evidence-based treatment regimens.19,20 Systemic corticosteroids continue to be a mainstay of treatment but there is the potential for more directed therapies that antagonize the effect of granulysin or TCR engagement. There is a need for evidence-based approaches to guide therapeutic intervention in SJS/TEN beyond aggressive supportive care.18
The first open-label randomized controlled clinical trial of 96 patients with probable or definitive SJS/TEN that had a positive treatment outcome was recently published that compared the TNF-alpha antagonist etanercept with corticosteroids.21 In this study etanercept decreased the overall mortality rate and the median time to healing significantly was shorter for etanercept versus corticosteroids (14 versus 19 days respectively, p=0.010). Etanercept was associated with significantly lower cytokine concentrations in blister fluid and plasma (TNF-alpha and granulysin) and a two-fold increase in the regulatory T cells (Treg) population. This study suggests promise for TNF-alpha antagonists in the treatment of SJS/TEN, particularly when utilized early in disease. It also highlights the need for comparative data with other drugs such as cyclosporine which has been shown to have an efficacy benefit in smaller observational studies.
Drug reaction with eosinophilia and systemic symptoms
In contrast with SJS/TEN, DRESS presents without skin detachment and mucocutaneous involvement. The mortality rate of DRESS is approximately 10 percent and symptoms as well as later complications may mimic viral illness, including lymphadenopathy, pneumonitis, encephalitis myocarditis, and nephritis.22 DRESS is characterized by fever, hematologic abnormalities (eosinophilia, atypical lymphocytosis), internal organ involvement, and a non-specific rash of varying severity.23 The onset of DRESS typically occurs 2–8 weeks after drug initiation and long term autoimmune sequelae are common, including type 1 diabetes, thyroiditis, and SLE.24
Other immune-mediated adverse drug reactions
Other T-cell mediated delayed IM-ADRs include abacavir hypersensitivity syndrome (HSN). Occuring in 5–8% of patients within the first 6 weeks of abacavir treatment, HSN is characterized by fever, malaise, GI and respiratory symptoms, and a generalized rash. IM-ADRs can also present as organ specific manifestations such as pancreatitis and drug-induced liver injury (DILI). DILI is a rare but life threatening hepatic failure accounting for 7–15% of the cases of acute liver failure in Europe and the US.25,26 Agranulocytosis involves a severe, life threatening decrease in white blood cell count and can be induced with antithyroid drugs in an HLA-dependent manner.27 Erythema multiforme majus (EMM) is a severe bullous disorder with raised lesions affecting less than 10% TBSA.28,29 This disorder can mimic SJS/TEN but is distinct in that erythema multiforme is associated with infections causes, such as herpes simplex virus (HSV) and Mycoplasma pneumoniae, rather than drug exposure. A significant proportion of cases diagnosed as SJS/TEN have detectable M. pneumoniae antibodies.28,30 SJS/TEN with eye involvement is more common in children with concurrent M. pneumoniae or HSV infection. Mycoplasma-induced rash and mucositis is a term that has been coined to highlight the disproportionate mucous membrane involvement that is seen in the M. pneumonia-associated phenyotype of EMM.
Emerging IM-ADRs have been observed with the recently approved immune checkpoint inhibitors ipilimumab, nivolumab, and pembrolizumab.31 These agents are used in the treatment of metastatic melanoma, head and neck cancer, lung cancer, bladder and kidney cancer, and lymphoma. Although mild immune reactions have been observed to occur in up to 50% of patients (typically after 3–6 months of treatment), an increasing number of severe IM-ADRs, such as fatal myositis, are being recognized with these agents.32 Severity of checkpoint inhibitor-induced IM-ADRs correlates with the improved tumor response and patient survival, underscoring the need to manage these toxicities to maximize drug benefit.33
Predicting Immune-Mediated ADRs: Genetic Associations with IM-ADRs
The HLA region has emerged as a preventive screening strategy for immune-mediated disease and IM-ADRs. HLA alleles are highly polymorphic and contain over 8000 class I and over 3000 class II β-chain variants with the most variability mapping to peptide-binding grooves.34 Variation in HLA is critical to prevent allograft rejection and plays a critical role in determining susceptibility to infection and autoimmune disease. A wealth of HLA alleles have been previously associated with immune-mediated phenotypes and, within the genome-wide association study (GWAS) catalogue, more phenotype associations have been identified in the HLA region than any other region of the genome.5,35 Immune response is modulated by the HLA system and amino acid sequences of each HLA molecule determine peptide binding and antigen presentation to T-lymphocytes.36 Class I MHC molecules (HLA-A, -B, and -C) are present on all nucleated cells and activate CD8+ cytotoxic lymphocytes. Class II MHC molecules (HLA-DP, -DQ, and –DR) are present only on antigen presenting cells, such as B cells, dendritic cells, and macrophages, and active CD4+ helper T lymphocytes. Since four digit HLA alleles characterize the amino acid sequence of class I and II MHC molecules, extremely strong associations with immunologic disease and IM-ADRs is common, even when limited case number are available.23,24 HLA allele associations not only have potential for clinical prediction and diagnosis of IM-ADRs, but also provide valuable insights into immunopathology.
HLA allele associations with IM-ADRs
One of the earliest examples of HLA-associated IM-ADRs was with abacavir HSN, for which a strong association was observed with HLA-B*57:01.37,38 The presence of at least one HLA-B*57:01 allele is necessary for the HSN reaction to occur, although the HLA-B*57:01 allele is not sufficient to predict HSN. Since 2008 screening for HLA-B*57:01 prior to abacavir prescription has been considered part of guideline based HIV practice in the developed world. Subsequently, HLA-B*15:02 was observed to be strongly associated with carbamazepine-induced SJS.39 In this study, 100% of patients (44/44) with carbamazepine-induced SJS had the HLA-B*15:02 allele compared to only 3% of control patients (3/101), resulting in an odds ratio of 2504 and p value of 3.13×10-27. These results illustrate the commonly observed strength of HLA associations with IM-ADRs despite limited case numbers, for which as few as 6 cases would have been sufficient to define this association. Similarly, HLA-B*57:01 was found to be a major determinant of drug-induced liver injury due to flucloxacillin (odds ratio=80.6, p=9.0×10−19) in a GWAS that included only 51 cases.40
Carbamazepine is associated with a number of IM-ADRs, including SJS/TEN, maculopapular erythema, and DRESS. The HLA-B*15:02 association is specific to carbamazepine-induced SJS/TEN, although other HLA alleles with B75 serotypes, most commonly HLA-B*15:21, but also HLA-B*15:08 and HLA-B*15:11, have been associated.41,42 HLA-B*15:02 has also been much more weakly associated with IM-ADRs to other aromatic amine anticonvulsants, including oxcarbazepine, phenytoin, and lamotrigine.43 The HLA-B*15:02 association with carbamazepine-induced SJS has been observed in Asian populations where the allele frequency is relatively high, but this allele is rare in Europeans. The HLA-B*31:01 allele has been associated with carbamazepine-induced SJS in Europeans in several studies.44–47 This example illustrates the strong influence of race/ethnicity on HLA associations and the unique challenges that race presents when implementing HLA alleles as predictors of IM-ADRs.
HLA allele associations with IM-ADRs and their characteristics are summarized in Table 1 and Supplementary Table S1. A notable example is the association of HLA-B*58:01 with SJS/TEN and DRESS induced by allopurinol, a xanthine oxidase inhibitor used for gout and hyperuricemia.48 This association has been observed in both Asian and European populations and the risk of allopurinol-induced SCAR is influenced by clearance of allopurinol’s main metabolite oxypurinol. However the negative predictive value of HLA-B*58:01 is 100% in Southeast Asians alone, and HLA-B*58:01 explains only 60% of allopurinol SCAR in other races.49,50 Other IM-ADRs associated with HLA alleles include dapsone hypersensitivity and HLA-B*13:01, amoxicillin-clavulanate-induced DILI and several HLA alleles (both risk and protective) and antithyroid-induced agranulocytosis and the HLA-B*38:02-HLA-DRB1*08:03 haplotype.51–58
Table 1.
Key HLA-associated immune-mediated adverse drug reactions
| Drug (references) | HLA allele | Adverse reaction | Prevalence of ADR | Carriage rate (%) of HLA allelea | OR | NPV (population) | PPV (population) | NNT |
|---|---|---|---|---|---|---|---|---|
| Abacavir (46, 47, 110, 137) | B∗57:01 | Hypersensitivity reaction | 8% of population (3% true, 2–7% false positive HSR) | 5–8
(European) <1 (Sub-Saharan African) <1 (Southeast Asian) 2–3 (African American) |
960 | 100% | 55% | 13 |
| Allopurinol (58, 129, 138–145) | B∗58:01 | SJS/TEN; DRESS/DIHS | 1–4/1,000 | 1–6 (European) 10 (Sub-Saharan African) 10–15 (Southeast/South Asian) 4 (African American) |
580 | 100% (Han Chinese) | 3% (Han Chinese) | 250 |
| Carbamazepine (50, 109, 117–120, 146–151) | B∗15:02 | SJS/TEN | <1–6/1,000 |
<0.1
(European) 10–15 (Southeast Asian) <1 (African) |
>1,000 | 100% (Southeast Asian) | 2–8% | 1,000 |
| Carbamazepine (54, 56, 112, 122, 152, 153) | A∗31:01 | DRESS/DIHS | 0.05% | ≤6
(European) <1 (Sub-Saharan African) |
57.6 | 99.9% | 0.89% | 3,334 |
| Dapsone (67) | B∗13:01 | DRESS/DIHS | 1–4/100 | 0 (European) 2–30 (Southeast Asian) |
20 | 99.8% | 7.8% | 84 |
| Flucloxacillin (43) | B∗57:01 | Drug-induced liver injury | 8.5/100,000 | 5–8
(European) <1 (Sub-Saharan African) <1 (Southeast Asian) 2–3 (African American) |
81 | 99.9% | 0.12% | 13,819 |
| Methimazole/ carbimazole (32, 65, 154) | B∗38:02 | Agranulocytosis | Unknown | 5–15 (China,
Taiwan) <1 (European) <1 (African) |
266–753 | 99.9% | 7–30% | 211–238 |
| B∗27:05 | 4–8
(European) <2 (China) <1 (Africa) |
|||||||
| Nevirapine (4, 155) | C∗04:01 | DRESS | Unknown | 15–30 (average prevalence across races) | 3–7 | 95–97% | 5–27% | Variable |
| Oxcarbazepine (156–158) | B∗15:02 | SJS/TEN | Unknown |
<0.1
(European) 10–15 (Southeast Asian) <1 African |
27.9 | 99.9% (Han Chinese) | 0.73% (Han Chinese) | >5,000 |
Carriage rates were obtained from http://allelefrequencies.net. Carriage rate describes the percentage of individuals who have the alleles in the population, including both homozygous and heterozygous carriers.
Abbreviations: ADR, adverse drug reaction; DIHS, drug-induced hypersensitivity syndrome; DRESS, drug reaction with eosinophilia and systemic symptoms; HLA, human leukocyte antigen; NNT, number needed to treat; NPV, negative predictive value; OR, odds ratio; PPV, positive predictive value; SJS, Stevens-Johnson syndrome; TEN, toxic epidermal necrolysis.
Challenges for Genetic Association Studies of IM-ADRs
The rarity of SCARs presents challenges for the study of IM-ADR risk factors as well as evidence-based treatment of IM-ADRs. The prevalence of SCARs makes prospective studies difficult and underscores the need for research and clinical networks to drive research, implementation, and evidence based treatment guidelines.18 Despite recent advance in understanding of mechanisms for SCARs, diagnosis and management continue to rely heavily on clinical case definitions and causality assessments supported by histology and laboratory results.59 IM-ADRs have a wide variety of clinical presentations, but no universal system exists to organize reactions into phenotypes. Classifications of reactions are often defined based on clinical observation such as time course (immediate versus delayed onset), a reaction after single or multiple dose, and organ involvement (isolated, single organ versus systemic, multiple organ involvement). IM-ADRs can be classified according to those with and without immunologic memory or based on mechanisms, such as with IgE-mediated and T-cell mediated hypersensitivity. Further research has the potential to improve phenotype classifications of IM-ADRs using biological data and emerging tests to re-taxonomize these reactions. Patient-derived induced pluripotent stem cells (IPSCs) have been proposed as a means of determining patient-specific drug hypersensitivity. This procedure would be akin to the basophil activation test for IgE-mediated ADRs, which has been used to identify sensitized patients for multiple drugs, including antibiotics, anesthetics and chemotherapy.60 Such advancements would address challenges related to phenotyping of IM-ADRs.
Emerging data suggests that interactions between HLA variation and other genomic variation play a critical role in IM-ADR development. This constitutes both a challenge for IM-ADR research and an opportunity to understand immunopathogenesis and discover non-HLA biomarkers. For instance, phenytoin-associated SCARs are associated with HLA-B*15:02 but are also increased by CYP2C9 loss of function alleles.61,62 These alleles reduce activity of the drug metabolizing enzyme CYP2C9, which is primarily response for phenytoin metabolism. Similarly, both CYP2B6 poor metabolizer genotypes and multiple HLA alleles have been associated with nevirapine-induced SCARs.63 These examples illustrate the influence of drug clearance and pharmacokinetics on the development of IM-ADRs. Allele by gene interactions have not only been observed in genes coding for drug metabolizing enzymes. SJS/TEN secondary to nevirapine is influenced by the interaction of HLA-C*04:01 with variation in the endoplasmic reticulum aminopeptidase 2 gene (ERAP2).64 Considering these observations, future investigations of genomic influences on IM-ADRs will need to consider non-HLA variation.
The highly polymorphic nature of the HLA region presents technical difficulties in HLA typing and significant expense and expertise is required to directly sequence HLA loci with four digit resolution. Rapidly available single allele tests are available for HLA-B*57:01, including sequence-specific oligonucleotide (SSO) assays, quantitative polymerase chain reaction (qPCR), and monoclonal antibody assays (identifying B17 serogroup alleles HLA-B*57:01, HLA-B*57:02, HLA-B*57:03, and HLA-B*58:01) and requiring confirmation with another assay).65 Although designed for HLA-B*57:01, this technology can be applied to other alleles. Because the HLA region is characterized by high linkage disequilibrium, a small number of single nucleotide polymorphisms (SNPs) can be used to tag the majority of HLA alleles.66,67 Consequently, HLA alleles are frequently imputed from SNP-level data due to the widespread availability of GWAS data and the large cohorts that can be interrogated without further expense. Although the accuracy rate is high (up to 98%), the reduced accuracy in class II alleles and diverse race/ethnic groups make clinical implementation of this data unlikely. The reduced accuracy in non-Caucasian populations underscores the limited availability of appropriate reference panels of HLA alleles and the need to accumulate immunogenomic data in diverse populations.
Understanding Immune-Mediated Drug Toxicities: Mechanisms of IM-ADRs
Proposed Mechanisms of IM-ADRs
Drugs that induce IM-ADRs are usually small molecules, which are too small to induce an immunogenomic response directly. Three major mechanisms have been proposed to describe how these small molecules elicit T-cell mediated immune responses and cause IM-ADRs (Figure 2). IM-ADRs can follow one of these mechanisms but likely include a combination of these mechanisms and/or other undescribed mechanisms. The hapten/prohapten model supposes that covalent bonds are formed between drugs and endogenous proteins and modified peptides are processed by APCs and presented on the MHC, resulting in a T-cell response.68 The pharmacological-Interaction (p-i) model stipulates that the drug forms a direct non-covalent bond to stimulate immune receptors such as MHC or TCR.69 The p-i model does not require antigen processing and could thereby explain how T-cell stimulation can occur immediately and after a single exposure to certain agents. In the altered peptide repertoire binding model, the drug non-covalently binds to the MHC, altering the MHC binding cleft so that an altered repertoire of self-peptides are recognized and presented on the MHC. Abacavir hypersensitivity is thought to follow the altered peptide repertoire binding model, based on the crystal structure of abacavir bound to HLA-B*57:01 and synthetic or self-peptides that was solved by two independent groups.70,71
Figure 2:
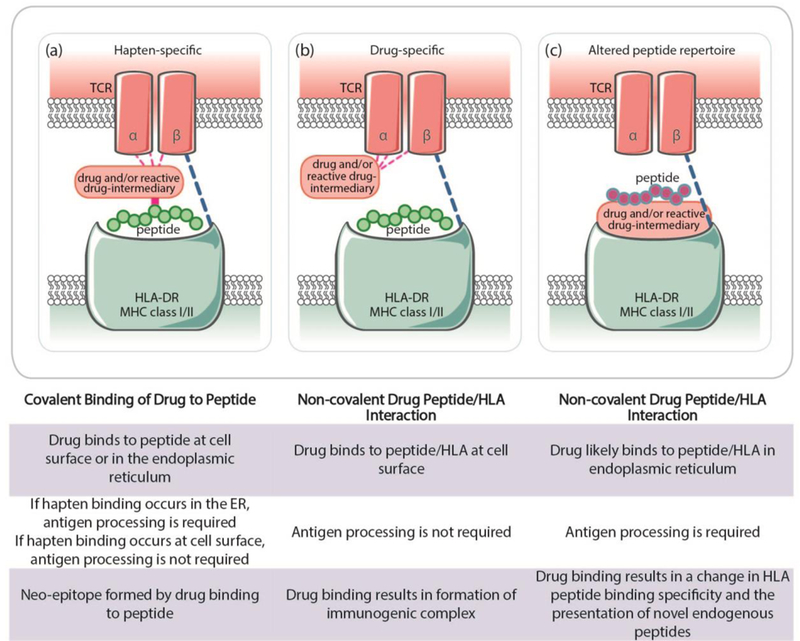
Proposed immunopathogenesis for T-cell-mediated ADRs: a) the hapten/prohapten model stipulates that drugs form covalent bonds with endogenous peptides, which are processed by antigen-presenting cells, presented on MHC molecules, and trigger a T-cell response; b) the pharmacological-interaction model proposes that a drug binds directly to receptors such as TCR or HLA triggering an immune response; c) the altered peptide repertoire model stipulates that the drug forms noncovalent bonds within the MHC binding pocket, altering the MHC such that endogenous peptides bind and trigger an immune response. HLA indicates human leukocyte antigen; MHC, major histocompatibility complex; TCR, T-cell receptor.
Immunopathogenesis of SCARs
The immunopathogenesis of drug-induced SCARs is incompletely understood. SCARs are T-cell mediated drug hypersensitivity reactions that mechanistically may be explained by non-mutually exclusive models including heterologous immunity in predisposed patients. The strong associations between MHC class I and II variation suggest that CD8+ and CD4+ T-cells immune response forms the basis of these syndromes.38,39,48 However, in up to 20% of SJS/TEN cases the causative drug is unidentified and, in many cases, drugs that are implicated in SJS/TEN do not yet have an associated HLA risk allele.
SJS/TEN is primarily mediated by granulysin-producing cytotoxic CD8+ T cells, natural killer (NK) cells, and natural killer T (NKT) cells (Figure 3).72,73 Cytotoxic T-cells, NK cells, and NKT-cells, have been isolated from the blister fluid from patients with SJS/TEN, from inflamed skin biopsies during acute IM-ADRs, and after intradermal administration of offending drugs in patients with a history of IM-ADRs.72–74 Furthermore, circulating T-cells from patients with IM-ADRs have been shown to response to the offending agent ex vivo.75,76 As granulysin is a key mediator of keratinocyte death and SJS/TEN severity, serum granulysin levels may prove a useful prognostic indicator as well as a potential drug target.21,77 Much of the focus in SJS/TEN is on effector memory CD8+ T-cell responses, but suppressor immune responses such as those conferred by Tregs play a key role in immunity in the skin. Treg cells are diminished in early SJS/TEN and augmenting Treg cell responses could provide complimentary or alternative treatments.18,21 Extensive research suggests that SJS/TEN is bolstered by an imbalance of effector/autoreactive and regulatory immune responses.
Figure 3:
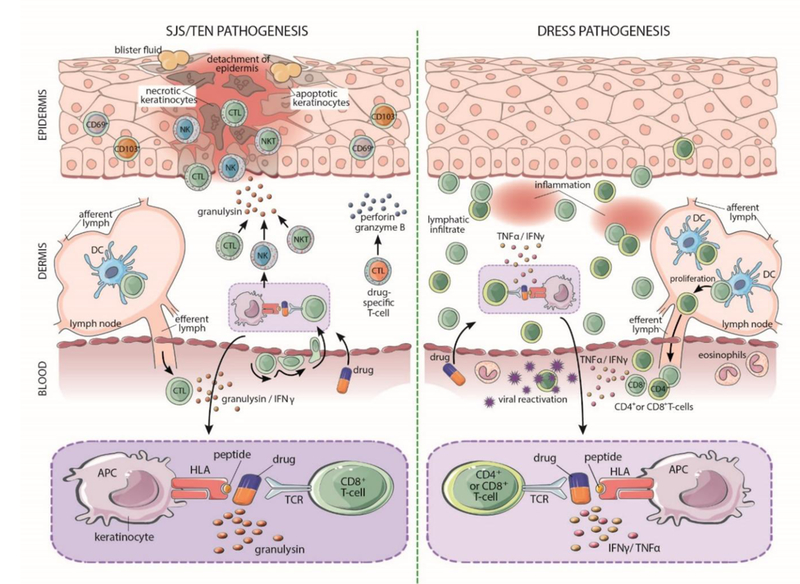
Pathogenic mechanisms for drug-induced SJS/TEN and DRESS. SJS/TEN primarily involves the epidermis. The drug likely interacts with HLA protein on keratinocytes, which act as antigen presenting cells to active CD8+ cytotoxic T-cells. Drug-specific CD8+ cytotoxic T-cells accumulate within blisters and release perforin and granzyme B that kills keratinocytes. CD8+ T-cells, NK cells, and NKT cells are also triggered to secrete granulysin, which is cytotoxic and the most powerful mediator of keratinocyte death. DRESS primarily involves the dermis. DRESS is characterized by a lymphocytic infiltrate of T-cells into the dermis and release of TNF-alpha and IFN-gamma. DRESS is also associated with replication of human herpesviruses, which have an unclear role in pathogenesis. APC indicates antigen presenting cell; CTL, cytotoxic lymphocyte; DC, dendritic cell; DRESS, drug reaction with eosinophilia and systemic symptoms; NK, natural killer cell; NKT, natural killer T-cell; SJS, Stevens-Johnson Syndrome; TCR, T-cell receptor; TEN; toxic epidermal necrolysis.
The immunopathogenesis of DRESS is thought to be mediated by both CD4+ and CD8+ effector T-cells, by reactivation of human herpes viruses (HHV), and by expansion of CD4+FoxP3+ T-cells.23,78,79 Reactivation of HHV, including HHV6,Epstein-Barr virus, and cytomegalovirus often occurs during acute and recovery phase DRESS, although the process by which this viral reactivation contributes to DRESS pathogenesis is under investigation. It is unclear whether viral replication is the result of general immune dysfunction or if this replication contributes to the development of DRESS. Virus-specific T-cell responses are likely to contribute to the prolonged and relapsing disease as well as multi-organ involvement in DRESS.80 Interestingly, autoimmune diseases often occur after the resolution of DRESS. The most common of these is autoimmune thyroid disease, which has been recently independently associated with HHV6 infection.81,82
Heterologous Immunity Models
Adaptive immunity has evolved to generate polyspecific TCRs capable of recognizing various epitopes in order to combat antigenic diversity. This ability to recognize multiple microbial epitopes with the same TCR is termed heterologous immunity.80,83 Heterologous immunity has been proposed as a potential mechanism of IM-ADRs such that prior exposure to a pathogen elicits a response from a cross-reactive effector memory T-cell when the offending drug is administrated. The heterologous immunity model could potentially explains why so many IM-ADRs have HLA allelic associations with 100% negative predictive value (NPV) but a low positive predictive value (PPV), since the HLA allele is necessary but not sufficient to trigger the IM-ADR. For some IM-ADRs, disease manifestation may occur within 1.5 days of drug exposure.84 Yet drug re-exposure is typically followed by rapid and enhanced toxicity.85 Heterologous immunity might also explain the diverse clinical phenotypes, widely variable latency periods, and immunologic memory characteristic of IM-ADRs. Using this model, one might predict IM-ADRs based on carriage of HLA allele combined with past pathogen exposure or existing T-cell repertoires.
Approaches to determine IM-ADR mechanisms
HLA associations with IM-ADRs have provided key insights into immunopathogenesis and allowed the development of models for T-cell mediated IM-ADRs. Carriage of an HLA is generally necessary but not sufficient to induce an IM-ADR and the low PPVs indicate that there are additional causative factors that can be identified. Identification of these factors is important for understanding of IM-ADR mechanisms as well as guiding pharmacogenomics screening to avoid IM-ADRs. Surface plasmon resonance experiments using site-directed mutagenesis have been fruitful in determining the nature of HLA-B*15:02-carbamazepine and HLA-B*57:01-abacavir drug binding.70,71,86,87 In some cases, the association of multiple HLA alleles can provide clues regarding critical amino acid residues in HLA binding clefts.88 More recently, in silico approaches have been employed to model drug binding affinities and to simulate and predict immunogenicity of new therapeutic agents. Other emerging approaches include TCR and BCR sequencing, HLA expression studies, and experiments interrogating the role of the microbiome in the immunopathogenesis of SCARs.18
Next generation TCR sequencing has emerged as an experimental approach to determine immunologic mechanisms as well as biomarkers for drug-induced SCARs. TCR clonotypes are produced by random somatic rearrangements of Vβ, Dβ, Jβ, Vα, and Jα genes on the β- and α-chain variable domains, which recombine to form the complimentary determining region 3 (CDR3), which recognizes an antigen.89 TCR β-chain DNA quantification and CDR3 characterization indicate early T-cell activation and clonal expansion in response to antigens.90,91 Clonotypic analysis can be used to measure disease-associated T-cells, evaluate expression profiles of pathogenic T-cells, and determine correlations between TCR clonotypes and disease severity.92 In studies of carbamazepine-induced SJS/TEN, T-cells from patients with HLA-B*15:02-associated carbamazepine SJS/TEN carried a shared CD8+ T-cell clonotype bearing a common CDR3 sequence not found in controls.86,93,94 This data suggests that development of immune-mediated ADRs can be dependent on the presence of both an HLA allele and a specific TCR clonotype. However, such clonotypic restriction was not observed in one study of HLA-B*58:01-restricted allopurinol-induced SJS/TEN.95 Although clonotypic analysis is a potential strategy for early diagnosis of immunologic processes, these approaches could be challenging due to the lack of knowledge of the specificity of these approaches and the rare presence of antigen-driven T-cells in the peripheral blood preceding and following the acute drug reaction.
Preventing Immune-Mediated Adverse Drug Reactions: Opportunities for Clinical Implementation with IM-ADRs
Clinical applications of immunopharmacogenomic biomarkers include the a priori exclusion of potentially allergic patients, re-classification of patients previously thought to be allergic, and identification of patients yielding potential benefit from desensitization procedures.96 HLA alleles have emerged as potentially powerful immunopharmacogenomic biomarkers for the prevention of previously unpredictable, immune-mediated diseases. Alleles that predict drug IM-ADRs may supplement limited clinical utility of in vivo biomarkers such as skin testing, which is unavailable for the majority of drug allergens and for which results may have unclear predictive values.97 HLA alleles are now genotyped to guide drug treatment and prevent IM-ADRs, moving personalized medicine toward reality. However, recent proceedings of an investigator-driven SJS/TEN meeting highlighted the need to broaden discoveries related to implementation of immunopharmacogenomic testing across diverse populations and causative drugs.18
Abacavir-induced HSN
A flagship example for implementation of genetic testing to prevent HLA-associated drug reactions is abacavir and HLA-B*57:01. Discovered in 2002, the HLA-B*57:01 biomarker is now recommended by multiple guidelines to avoid abacavir HSN in carriers of the HLA-B*57:01 allele.98–100 HLA-B*57:01 screening prior to abacavir is one of the few pharmacogenomic tests supported by a randomized controlled trial (RCT).98 HLA-B*57:01 screening prior to abacavir is supported by a 100% NPV, high PPV (55%) and hence a low number needed to test (<30) to prevent one case of true immunologically-mediated abacavir HSN. Structural and biochemical science further support this by defining the specificity of binding between HLA-B*57:01 and abacavir, which fails to interact with closely-related HLA alleles differing by only 2 amino acids. The accurate identification of true immunologically-mediated abacavir HSN using skin patch testing, which was used as a co-primary endpoint in the PREDICT-1 HLA-B*57:01 licensing study, also supports the specificity of HLA-B*57:01 for true immunologically-mediated abacavir HSN. The success of HLA-B*57:01 translation into clinical practice was highly dependent on the fact that abacavir is used by a small contingent of physicians treating HIV, making education and implementation more straightforward. The reduction in HSN and the increase in the use of abacavir in the armamentarium for HIV is a success story for personalized medicine. The success of a priori pharmacogenomic screening for abacavir highlights the importance of accurate HLA genotyping and IM-ADR phenotyping, a high NPV and PPV, adequate provider education, and rigorous evidential support. HLA-B*57:01 testing to prevent abacavir-induced HSN provides a model whereby prospective immunopharmacogenomic screening may help to reduce potentially fatal immunologic reactions.
Negative Predictive Value, Positive Predictive Value and Number Needed to Treat
A major reason for the success of HLA testing with abacavir is that it only occurs in individuals with the allele, giving the test a 100% NPV. The 100% NPV, coupled with the 55% PPV, indicates that the allele is necessary but not sufficient to incite the IM-ADR. A high NPV is of paramount importance to the physician, as this excludes the possibility of the reaction based on the absence of the HLA allele. A low NPV (less than 100%) limits the clinical utility of the biomarker. In addition, 55% of patients with HLA-B*57:01 who are exposed to abacavir will develop HSN, giving this test an unusually high PPV for an HLA allele associated with an IM-ADR. Other HLA-associated ADRs have much lower PPVs (≤8%) or NPVs (<100%) making screening less practical. The identification of other factors predisposing patients to the IM-ADRs is necessary for effective clinical implementation.
The low incidence of IM-ADRs and the low PPV means that many patients would need to be tested to prevent a single incident and many patients would have potentially useful treatments withheld due to being HLA positive. In the case of flucloxacillin-induced DILI, almost 14,000 patients would need to be screened to prevent a single incident. In such cases, implementation of HLA biomarkers is impractical due to a low NPV, PPV, and/or number needed to treat (NNT). The success of HLA-B*57:01 testing to prevent abacavir HSN is an exception, since only 13 patients need to be tested to prevent one clinically-diagnosed HSN reaction and only 30 patients need to be tested to prevent one case of true immunologically-mediated abacavir HSN. For many commonly used drugs such as antibiotics, the PPV is too low to use HLA as a screening test. In the future, HLA may have utility in combination with functional and other assays to aid in the diagnosis when multiple drugs or antibiotics are dosed concurrently.
Race-based implementation
Frequencies of HLA alleles differ substantially between populations.101 For instance, carriage of HLA-B*57:01 is essentially non-existent in many African populations but is as high as 11.2% in Southern Ireland and 17.8% in Sri Lanka. The common carriage rate of IM-ADR-associated HLA alleles in certain populations and the low frequency of IM-ADRs in non-carriers suggests that HLA screening need not be implemented in all populations. For instance, although HLA-B*58:01 is considered a genetic marker for allopurinol-induced SCARs in multiple populations worldwide, screening for HLA-B*58:01 before allopurinol is recommended by the American College of Rheumatology only for those with advanced renal failure and/or for high-risk populations such as persons of Southeast Asian ancestry where the NPV is 100%.102,103 Implementation of HLA alleles in preventing IM-ADRs has been met with much more enthusiasm in Asian countries where there is higher prevalence of pertinent alleles paired with use of key drugs such as aromatic anticonvulsants.104–106 Genotyping HLA-B*15:02 for new users of carbamazepine in Singapore has reduced the number of associated SJS/TEN cases from 18 per year to 1 case in the 4 years since implementation.
The association of HLA-B*15:02 with carbamazepine-induced SJS/TEN was initially discovered among Han Chinese in Taiwan and subsequently validated in other regions in Southeast Asian including Hong Kong, Malaysia, and Thailand.107–111 The US Food and Drug Administration (FDA) recommends HLA-B*15:02 testing prior to initiation of carbamazepine in high risk populations, namely those with Southeast Asian ancestry. In Taiwan, HLA-B*15:02 screening for carbamazepine in combination with restricted off-label use of carbamazepine has resulted in dramatic decreases in the incidence of carbamazepine-associated IM-ADRs. Similar programs have been successfully implemented in other parts of Southeast Asia. However, differences in allele frequencies have resulted in a relatively weak association in other populations such as Asian Indians, Koreans, Japanese and European groups.112–115 As reviewed above, the HLA-B*31:01 allele has been associated with carbamazepine-induced SJS in several populations, including Europeans and Japanese, illustrating the unique challenges that race presents when implementing HLA alleles as predictors of IM-ADRs.44–46,116 The differential clinical utility of HLA testing by race/ethnicity requires that patients know their ancestry accurately, which may be difficult in countries where admixture is common, such as in the US.
Threshold of evidence for genetic testing
A critical factor in translation of HLA testing to clinical practice is the endorsement by general medical guidelines. HLA-B*57:01 testing to prevent abacavir HSN was supported in several guidelines, which has facilitated widespread use of genetic testing in routine HIV care.98–100 Several other HLA screening tests have received support from specialty organizations such as the Clinical Pharmacogenetics Implementation Consortium (CPIC), but have not received support from relevant general medical guidelines.47,61,117 The endorsement of guidelines to support HLA testing is ultimately needed for widespread implementation, but relies on the availability of rigorous clinical trials evidence. While there are multiple reasons why HLA screening for abacavir has met with implementation success, a major reason is that it was possible to feasibly power a clinical trial to confirm the utility of HLA-B*57:01 screening for abacavir. This RCT was possible due to the high prevalence of abacavir HSN, the high NPV and PPV of HLA-B*57:01 screening, and the high carriage rate of HLA-B*57:01 in HIV treatment populations in the developed world where abacavir was being used. Screening for other IM-ADRs are typically supported by retrospective case control studies evaluations rather than an RCT, which is viewed as the gold standard methodological design to establish new treatments.118 However, the logistical, ethical and statistical barriers for RCTs are particularly difficult in pharmacogenomic research, considering sample size requirements to prove clinical benefits among genotype groups, the necessity of assessing interactions, and requirements for replication. A RCT for personalized medicine can be seen as a contradiction in terms, in so much as employing a randomized treatment structure to a treatment that is meant to be tailored to an individual.119
RCTs have traditionally been applied to support new drugs or new indications for drug treatment, but evidence supporting most contraindications to drug use, and currently-used genetic tests is not derived from RCTs.120,121 Since drugs being evaluated for HLA screening are not newly approved and not being used for a newly approved indication, an alternative threshold of evidence for implementation may be warranted. Cost effectiveness of HLA PGX testing has been proposed as a logical threshold of evidence for implementation. The experiences of the drug regulatory authority of Singapore, the Health Sciences Authority (HSA), are illustrative of these challenges and opportunities in implementation. Using an incremental cost effectiveness ratio of US $50,000 per quality-adjusted life year, HLA-B*15:02 testing to prevent carbamazepine-related IM-ADRs was considered cost effective whereas HLA-B*58:01 testing to prevent allopurinol-related IM-ADRs was not.122,123 In addition, multiple alternative drugs are available to treat epilepsy or neuropathic pain in place of carbamazepine, but cost effective options are not as abundant for allopurinol. Based on these and other considerations, the HSA and Singapore Ministry of Health require HLA genetic testing in new patients treated with carbamazepine but not with allopurinol.124 HLA-B*58:01 testing for new patient on allopurinol could be considered in patients with other pre-existing risk factors such as renal impairment. Neither HLA screening test is supported by a RCT, but genotyping HLA-B*15:02 for new users of carbamazepine in Singapore has reduced the number of associated SJS/TEN cases from 18 per year to 1 case in the 4 years since implementation. In other countries such as the US, HLA typing may already be available from bone marrow or organ transplant records, eliminating the cost burden associated with genotyping. Existing HLA types provide an opportunity to use electronic medical records for clinical decision support when new prescriptions are ordered for drugs with HLA-associated IM-ADRs.
Conclusions and Future Directions
IM-ADRs and particularly SCARs have considerable short and long-term mortality that poses a burden disproportionate to its prevalence. Although much progress has been made in IM-ADR research, many opportunities remain for predicting, preventing, and understanding IM-ADRs. The reason for low PPVs with so many IM-ADRs and the factors that combine with HLA alleles to elicit IM-ADRs are still largely unclear. Also unclear are the reasons for the tissue specificity of these reactions and the fact that they occur so rapidly yet show evidence of immunologic memory. The wealth of biological data generated from “omics” approaches has the potential to provide answers to some of these questions as well as to re-taxonomize IM-ADRs based on pathogenic mechanisms (Figure 4). Inadequate treatment regimens for SCARs remain a fruitful area of research with potentially paradigm shifting treatments such as use of genetically engineered T-cells on the horizon.
Figure 4:

Integrated “omics” approaches to drive personalized medicine in IM-ADRs. Approaches for prevention and treatment of IM-ADRs will include multiple omics platforms that link genomic, epigenomic, transcriptomic, metabolomic and other biological data. There is an opportunity to leverage this comprehensive “omic” data to estimate disease risk, facilitate rapid and accurate diagnosis, and predict the safety and efficacy of therapeutic interventions. Aggregation of this data is also likely to garner critical insights into the pathogenesis of IM-ADRs and lead to novel therapeutic interventions.
For a given T-cell mediated IM-ADR, one of the most important first steps is the identification of the correct HLA allelic association, which triggers downstream research with regard to mechanisms insights and genetic screening. Ultimately, understanding IM-ADR mechanisms will be informative for translating preventive strategies that maximize clinical utility of screening and for identifying new therapeutic targets. Large effect sizes and near perfect NPVs make these biomarkers potentially translatable into preventive medicine and clinical care. Other important factors for implementation include accurate genotyping and phenotyping, patient and provider education, cost effectiveness of screening, and the availability of evidence-based guideline recommendations. Because of the low prevalence of IM-ADRs, large global networks will likely be required to facilitate research, translation, and novel therapeutic interventions that are generalizable across diverse populations.
Supplementary Material
References Cited
- 1.Johnson JA, Bootman JL. Drug-related morbidity and mortality. A cost-of-illness model. Arch Intern Med 1995;155:1949–56. [PubMed] [Google Scholar]
- 2.Bond CA, Raehl CL. Adverse drug reactions in United States hospitals. Pharmacotherapy 2006;26:601–8. [DOI] [PubMed] [Google Scholar]
- 3.Lazarou J, Pomeranz BH, Corey PN. Incidence of adverse drug reactions in hospitalized patients: a meta-analysis of prospective studies. JAMA 1998;279:1200–5. [DOI] [PubMed] [Google Scholar]
- 4.Pavlos R, Mallal S, Ostrov D, et al. T cell-mediated hypersensitivity reactions to drugs. Annu Rev Med 2015;66:439–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Karnes JH, Bastarache L, Shaffer CM, et al. Phenome-wide scanning identifies multiple diseases and disease severity phenotypes associated with HLA variants. Sci Transl Med 2017;9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Nakamura Y The current and future applications of immunopharmacogenomics. Clin Adv Hematol Oncol 2015;13:815–7. [PubMed] [Google Scholar]
- 7.McNeil BD, Pundir P, Meeker S, et al. Identification of a mast-cell-specific receptor crucial for pseudo-allergic drug reactions. Nature 2015;519:237–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Khan DA. Cutaneous drug reactions. J Allergy Clin Immunol 2012;130:1225–e6. [DOI] [PubMed] [Google Scholar]
- 9.Pirmohamed M, Aithal GP, Behr E, Daly A, Roden D. The phenotype standardization project: improving pharmacogenetic studies of serious adverse drug reactions. Clin Pharmacol Ther 2011;89:784–5. [DOI] [PubMed] [Google Scholar]
- 10.Sun J, Liu J, Gong QL, et al. Stevens-Johnson Syndrome and toxic epidermal necrolysis: a multi-aspect comparative 7-year study from the People’s Republic of China. Drug Des Devel Ther 2014;8:2539–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Su P, Aw CW. Severe cutaneous adverse reactions in a local hospital setting: a 5-year retrospective study. Int J Dermatol 2014;53:1339–45. [DOI] [PubMed] [Google Scholar]
- 12.Lehloenya RJ, Kgokolo M. Clinical presentations of severe cutaneous drug reactions in HIV-infected Africans. Dermatol Clin 2014;32:227–35. [DOI] [PubMed] [Google Scholar]
- 13.Pavlos R, Mallal S, Ostrov D, Pompeu Y, Phillips E. Fever, rash, and systemic symptoms: understanding the role of virus and HLA in severe cutaneous drug allergy. J Allergy Clin Immunol Pract 2014;2:21–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Roujeau JC, Stern RS. Severe adverse cutaneous reactions to drugs. N Engl J Med 1994;331:1272–85. [DOI] [PubMed] [Google Scholar]
- 15.Zimmermann S, Sekula P, Venhoff M, et al. Systemic Immunomodulating Therapies for Stevens-Johnson Syndrome and Toxic Epidermal Necrolysis: A Systematic Review and Meta-analysis. JAMA Dermatol 2017;153:514–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mittmann N, Knowles SR, Koo M, Shear NH, Rachlis A, Rourke SB. Incidence of toxic epidermal necrolysis and Stevens-Johnson Syndrome in an HIV cohort: an observational, retrospective case series study. Am J Clin Dermatol 2012;13:49–54. [DOI] [PubMed] [Google Scholar]
- 17.Bastuji-Garin S, Fouchard N, Bertocchi M, Roujeau JC, Revuz J, Wolkenstein P. SCORTEN: a severity-of-illness score for toxic epidermal necrolysis. J Invest Dermatol 2000;115:149–53. [DOI] [PubMed] [Google Scholar]
- 18.White KD, Abe R, Ardern-Jones M, et al. SJS/TEN 2017: Building Multidisciplinary Networks to Drive Science and Translation. J Allergy Clin Immunol Pract 2018;6:38–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yacoub MR, Berti A, Campochiaro C, et al. Drug induced exfoliative dermatitis: state of the art. Clin Mol Allergy 2016;14:9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Su SC, Hung SI, Fan WL, Dao RL, Chung WH. Severe Cutaneous Adverse Reactions: The Pharmacogenomics from Research to Clinical Implementation. Int J Mol Sci 2016;17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wang CW, Yang LY, Chen CB, et al. Randomized, controlled trial of TNF-alpha antagonist in CTL-mediated severe cutaneous adverse reactions. J Clin Invest 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chen YC, Chiu HC, Chu CY. Drug reaction with eosinophilia and systemic symptoms: a retrospective study of 60 cases. Arch Dermatol 2010;146:1373–9. [DOI] [PubMed] [Google Scholar]
- 23.Shiohara T, Kano Y, Takahashi R, Ishida T, Mizukawa Y. Drug-induced hypersensitivity syndrome: recent advances in the diagnosis, pathogenesis and management. Chem Immunol Allergy 2012;97:122–38. [DOI] [PubMed] [Google Scholar]
- 24.Minegaki Y, Higashida Y, Ogawa M, Miyachi Y, Fujii H, Kabashima K. Drug-induced hypersensitivity syndrome complicated with concurrent fulminant type 1 diabetes mellitus and Hashimoto’s thyroiditis. Int J Dermatol 2013;52:355–7. [DOI] [PubMed] [Google Scholar]
- 25.Ostapowicz G, Fontana RJ, Schiodt FV, et al. Results of a prospective study of acute liver failure at 17 tertiary care centers in the United States. Ann Intern Med 2002;137:947–54. [DOI] [PubMed] [Google Scholar]
- 26.Russo MW, Galanko JA, Shrestha R, Fried MW, Watkins P. Liver transplantation for acute liver failure from drug induced liver injury in the United States. Liver Transpl 2004;10:1018–23. [DOI] [PubMed] [Google Scholar]
- 27.Chen PL, Shih SR, Wang PW, et al. Genetic determinants of antithyroid drug-induced agranulocytosis by human leukocyte antigen genotyping and genome-wide association study. Nat Commun 2015;6:7633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Moreau JF, Watson RS, Hartman ME, Linde-Zwirble WT, Ferris LK. Epidemiology of ophthalmologic disease associated with erythema multiforme, Stevens-Johnson syndrome, and toxic epidermal necrolysis in hospitalized children in the United States. Pediatr Dermatol 2014;31:163–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sokumbi O, Wetter DA. Clinical features, diagnosis, and treatment of erythema multiforme: a review for the practicing dermatologist. Int J Dermatol 2012;51:889–902. [DOI] [PubMed] [Google Scholar]
- 30.Finkelstein Y, Soon GS, Acuna P, et al. Recurrence and outcomes of Stevens-Johnson syndrome and toxic epidermal necrolysis in children. Pediatrics 2011;128:723–8. [DOI] [PubMed] [Google Scholar]
- 31.Michot JM, Bigenwald C, Champiat S, et al. Immune-related adverse events with immune checkpoint blockade: a comprehensive review. Eur J Cancer 2016;54:139–48. [DOI] [PubMed] [Google Scholar]
- 32.Johnson DB, Balko JM, Compton ML, et al. Fulminant Myocarditis with Combination Immune Checkpoint Blockade. N Engl J Med 2016;375:1749–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sanlorenzo M, Vujic I, Daud A, et al. Pembrolizumab Cutaneous Adverse Events and Their Association With Disease Progression. JAMA Dermatol 2015;151:1206–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Robinson J, Halliwell JA, McWilliam H, Lopez R, Parham P, Marsh SG. The IMGT/HLA database. Nucleic Acids Res 2013;41:D1222–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.MacArthur J, Bowler E, Cerezo M, et al. The new NHGRI-EBI Catalog of published genome-wide association studies (GWAS Catalog). Nucleic Acids Res 2017;45:D896–D901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Nepom GT, Erlich H. MHC class-II molecules and autoimmunity. Annu Rev Immunol 1991;9:493–525. [DOI] [PubMed] [Google Scholar]
- 37.Hetherington S, Hughes AR, Mosteller M, et al. Genetic variations in HLA-B region and hypersensitivity reactions to abacavir. Lancet 2002;359:1121–2. [DOI] [PubMed] [Google Scholar]
- 38.Mallal S, Nolan D, Witt C, et al. Association between presence of HLA-B*5701, HLA-DR7, and HLA-DQ3 and hypersensitivity to HIV-1 reverse-transcriptase inhibitor abacavir. Lancet 2002;359:727–32. [DOI] [PubMed] [Google Scholar]
- 39.Chung WH, Hung SI, Hong HS, et al. Medical genetics: a marker for Stevens-Johnson syndrome. Nature 2004;428:486. [DOI] [PubMed] [Google Scholar]
- 40.Daly AK, Donaldson PT, Bhatnagar P, et al. HLA-B*5701 genotype is a major determinant of drug-induced liver injury due to flucloxacillin. Nat Genet 2009;41:816–9. [DOI] [PubMed] [Google Scholar]
- 41.Phillips EJ, Chung WH, Mockenhaupt M, Roujeau JC, Mallal SA. Drug hypersensitivity: pharmacogenetics and clinical syndromes. J Allergy Clin Immunol 2011;127:S60–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kaniwa N, Saito Y, Aihara M, et al. HLA-B*1511 is a risk factor for carbamazepine-induced Stevens-Johnson syndrome and toxic epidermal necrolysis in Japanese patients. Epilepsia 2010;51:2461–5. [DOI] [PubMed] [Google Scholar]
- 43.Hung SI, Chung WH, Liu ZS, et al. Common risk allele in aromatic antiepileptic-drug induced Stevens-Johnson syndrome and toxic epidermal necrolysis in Han Chinese. Pharmacogenomics 2010;11:349–56. [DOI] [PubMed] [Google Scholar]
- 44.McCormack M, Alfirevic A, Bourgeois S, et al. HLA-A*3101 and carbamazepine-induced hypersensitivity reactions in Europeans. N Engl J Med 2011;364:1134–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Genin E, Chen DP, Hung SI, et al. HLA-A*31:01 and different types of carbamazepine-induced severe cutaneous adverse reactions: an international study and meta-analysis. Pharmacogenomics J 2014;14:281–8. [DOI] [PubMed] [Google Scholar]
- 46.Ozeki T, Mushiroda T, Yowang A, et al. Genome-wide association study identifies HLA-A*3101 allele as a genetic risk factor for carbamazepine-induced cutaneous adverse drug reactions in Japanese population. Hum Mol Genet 2011;20:1034–41. [DOI] [PubMed] [Google Scholar]
- 47.Phillips EJ, Sukasem C, Whirl-Carrillo M, et al. Clinical Pharmacogenetics Implementation Consortium Guideline for HLA Genotype and Use of Carbamazepine and Oxcarbazepine: 2017 Update. Clin Pharmacol Ther 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hung SI, Chung WH, Liou LB, et al. HLA-B*5801 allele as a genetic marker for severe cutaneous adverse reactions caused by allopurinol. Proc Natl Acad Sci U S A 2005;102:4134–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Somkrua R, Eickman EE, Saokaew S, Lohitnavy M, Chaiyakunapruk N. Association of HLA-B*5801 allele and allopurinol-induced Stevens Johnson syndrome and toxic epidermal necrolysis: a systematic review and meta-analysis. BMC Med Genet 2011;12:118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Carroll MB, Smith DM, Shaak TL. Genomic sequencing of uric acid metabolizing and clearing genes in relationship to xanthine oxidase inhibitor dose. Rheumatol Int 2017;37:445–53. [DOI] [PubMed] [Google Scholar]
- 51.Hautekeete ML, Horsmans Y, van Waeyenberge C, et al. HLA association of amoxicillin-clavulanate–induced hepatitis. Gastroenterology 1999;117:1181–6. [DOI] [PubMed] [Google Scholar]
- 52.O’Donohue J, Oien KA, Donaldson P, et al. Co-amoxiclav jaundice: clinical and histological features and HLA class II association. Gut 2000;47:717–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Lucena MI, Molokhia M, Shen Y, et al. Susceptibility to Amoxicillin-Clavulanate-Induced Liver Injury Is Influenced by Multiple HLA Class I and II Alleles. Gastroenterology 2011;141:338–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Chen P-L, Shih S-R, Wang P-W, et al. Genetic determinants of antithyroid drug-induced agranulocytosis by human leukocyte antigen genotyping and genome-wide association study. Nature Communications 2015;6:7633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Cheung CL, Sing CW, Tang CSM, et al. HLA-B*38:02:01 predicts carbimazole/methimazole-induced agranulocytosis. Clinical Pharmacology & Therapeutics 2016;99:555–61. [DOI] [PubMed] [Google Scholar]
- 56.He Y, Zheng J, Zhang Q, et al. Association of HLA-B and HLA-DRB1 polymorphisms with antithyroid drug-induced agranulocytosis in a Han population from northern China. Sci Rep 2017;7:11950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Zhang F-R, Liu H, Irwanto A, et al. HLA-B*13:01 and the Dapsone Hypersensitivity Syndrome. New England Journal of Medicine 2013;369:1620–8. [DOI] [PubMed] [Google Scholar]
- 58.Chen WT, Wang CW, Lu CW, et al. The function of HLA-B*13:01 involved in the pathomechanism of dapsone-induced severe cutaneous adverse reactions. J Invest Dermatol 2018. [DOI] [PubMed] [Google Scholar]
- 59.Peter JG, Lehloenya R, Dlamini S, et al. Severe Delayed Cutaneous and Systemic Reactions to Drugs: A Global Perspective on the Science and Art of Current Practice. J Allergy Clin Immunol Pract 2017;5:547–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Gonzalez-de-Olano D, Morgado JM, Juarez-Guerrero R, et al. Positive basophil activation test following anaphylaxis to pertuzumab and successful treatment with rapid desensitization. J Allergy Clin Immunol Pract 2016;4:338–40. [DOI] [PubMed] [Google Scholar]
- 61.Caudle KE, Rettie AE, Whirl-Carrillo M, et al. Clinical pharmacogenetics implementation consortium guidelines for CYP2C9 and HLA-B genotypes and phenytoin dosing. Clin Pharmacol Ther 2014;96:542–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Chung WH, Chang WC, Lee YS, et al. Genetic variants associated with phenytoin-related severe cutaneous adverse reactions. JAMA 2014;312:525–34. [DOI] [PubMed] [Google Scholar]
- 63.Yuan J, Guo S, Hall D, et al. Toxicogenomics of nevirapine-associated cutaneous and hepatic adverse events among populations of African, Asian, and European descent. AIDS 2011;25:1271–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Carr DF, Bourgeois S, Chaponda M, et al. Genome-wide association study of nevirapine hypersensitivity in a sub-Saharan African HIV-infected population. J Antimicrob Chemother 2017;72:1152–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Fan WL, Shiao MS, Hui RC, et al. HLA Association with Drug-Induced Adverse Reactions. J Immunol Res 2017;2017:3186328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Leslie S, Donnelly P, McVean G. A statistical method for predicting classical HLA alleles from SNP data. Am J Hum Genet 2008;82:48–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Karnes JH, Shaffer CM, Bastarache L, et al. Comparison of HLA allelic imputation programs. PLoS One 2017;12:e0172444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Pichler WJ. Delayed drug hypersensitivity reactions. Ann Intern Med 2003;139:683–93. [DOI] [PubMed] [Google Scholar]
- 69.Pichler WJ, Beeler A, Keller M, et al. Pharmacological interaction of drugs with immune receptors: the p-i concept. Allergol Int 2006;55:17–25. [DOI] [PubMed] [Google Scholar]
- 70.Ostrov DA, Grant BJ, Pompeu YA, et al. Drug hypersensitivity caused by alteration of the MHC-presented self-peptide repertoire. Proc Natl Acad Sci U S A 2012;109:9959–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Illing PT, Vivian JP, Dudek NL, et al. Immune self-reactivity triggered by drug-modified HLA-peptide repertoire. Nature 2012;486:554–8. [DOI] [PubMed] [Google Scholar]
- 72.Nassif A, Bensussan A, Boumsell L, et al. Toxic epidermal necrolysis: effector cells are drug-specific cytotoxic T cells. J Allergy Clin Immunol 2004;114:1209–15. [DOI] [PubMed] [Google Scholar]
- 73.Leyva L, Torres MJ, Posadas S, et al. Anticonvulsant-induced toxic epidermal necrolysis: monitoring the immunologic response. J Allergy Clin Immunol 2000;105:157–65. [DOI] [PubMed] [Google Scholar]
- 74.Garcia-Doval I, LeCleach L, Bocquet H, Otero XL, Roujeau JC. Toxic epidermal necrolysis and Stevens-Johnson syndrome: does early withdrawal of causative drugs decrease the risk of death? Arch Dermatol 2000;136:323–7. [DOI] [PubMed] [Google Scholar]
- 75.Chessman D, Kostenko L, Lethborg T, et al. Human leukocyte antigen class I-restricted activation of CD8+ T cells provides the immunogenetic basis of a systemic drug hypersensitivity. Immunity 2008;28:822–32. [DOI] [PubMed] [Google Scholar]
- 76.Naisbitt DJ, Farrell J, Wong G, et al. Characterization of drug-specific T cells in lamotrigine hypersensitivity. J Allergy Clin Immunol 2003;111:1393–403. [DOI] [PubMed] [Google Scholar]
- 77.Chung WH, Hung SI, Yang JY, et al. Granulysin is a key mediator for disseminated keratinocyte death in Stevens-Johnson syndrome and toxic epidermal necrolysis. Nat Med 2008;14:1343–50. [DOI] [PubMed] [Google Scholar]
- 78.Morito H, Ogawa K, Fukumoto T, et al. Increased ratio of FoxP3+ regulatory T cells/CD3+ T cells in skin lesions in drug-induced hypersensitivity syndrome/drug rash with eosinophilia and systemic symptoms. Clin Exp Dermatol 2014;39:284–91. [DOI] [PubMed] [Google Scholar]
- 79.Shiohara T, Ushigome Y, Kano Y, Takahashi R. Crucial Role of Viral Reactivation in the Development of Severe Drug Eruptions: a Comprehensive Review. Clin Rev Allergy Immunol 2015;49:192–202. [DOI] [PubMed] [Google Scholar]
- 80.Pavlos R, White KD, Wanjalla C, Mallal SA, Phillips EJ. Severe Delayed Drug Reactions: Role of Genetics and Viral Infections. Immunol Allergy Clin North Am 2017;37:785–815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Cookson H, Creamer D, Walsh S. Thyroid dysfunction in drug reaction with eosinophilia and systemic symptoms (DRESS): an unusual manifestation of systemic drug hypersensitivity. Br J Dermatol 2013;168:1130–2. [DOI] [PubMed] [Google Scholar]
- 82.Sultanova A, Cistjakovs M, Gravelsina S, et al. Association of active human herpesvirus-6 (HHV-6) infection with autoimmune thyroid gland diseases. Clin Microbiol Infect 2017;23:50 e1- e5. [DOI] [PubMed] [Google Scholar]
- 83.Welsh RM, Che JW, Brehm MA, Selin LK. Heterologous immunity between viruses. Immunol Rev 2010;235:244–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Lucas A, Lucas M, Strhyn A, et al. Abacavir-reactive memory T cells are present in drug naive individuals. PLoS One 2015;10:e0117160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Pichler W, Yawalkar N, Schmid S, Helbling A. Pathogenesis of drug-induced exanthems. Allergy 2002;57:884–93. [DOI] [PubMed] [Google Scholar]
- 86.Wei CY, Chung WH, Huang HW, Chen YT, Hung SI. Direct interaction between HLA-B and carbamazepine activates T cells in patients with Stevens-Johnson syndrome. J Allergy Clin Immunol 2012;129:1562–9 e5. [DOI] [PubMed] [Google Scholar]
- 87.Yang CW, Hung SI, Juo CG, et al. HLA-B*1502-bound peptides: implications for the pathogenesis of carbamazepine-induced Stevens-Johnson syndrome. J Allergy Clin Immunol 2007;120:870–7. [DOI] [PubMed] [Google Scholar]
- 88.Pavlos R, McKinnon EJ, Ostrov DA, et al. Shared peptide binding of HLA Class I and II alleles associate with cutaneous nevirapine hypersensitivity and identify novel risk alleles. Sci Rep 2017;7:8653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Rudolph MG, Stanfield RL, Wilson IA. How TCRs bind MHCs, peptides, and coreceptors. Annu Rev Immunol 2006;24:419–66. [DOI] [PubMed] [Google Scholar]
- 90.Robins HS, Campregher PV, Srivastava SK, et al. Comprehensive assessment of T-cell receptor beta-chain diversity in alphabeta T cells. Blood 2009;114:4099–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Cohen GB, Islam SA, Noble MS, et al. Clonotype tracking of TCR repertoires during chronic virus infections. Virology 2002;304:474–84. [DOI] [PubMed] [Google Scholar]
- 92.Hunder NN, Wallen H, Cao J, et al. Treatment of metastatic melanoma with autologous CD4+ T cells against NY-ESO-1. N Engl J Med 2008;358:2698–703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Ko TM, Chung WH, Wei CY, et al. Shared and restricted T-cell receptor use is crucial for carbamazepine-induced Stevens-Johnson syndrome. J Allergy Clin Immunol 2011;128:1266–76 e11. [DOI] [PubMed] [Google Scholar]
- 94.Ko TM, Chen YT. T-cell receptor and carbamazepine-induced Stevens-Johnson syndrome and toxic epidermal necrolysis: understanding a hypersensitivity reaction. Expert Rev Clin Immunol 2012;8:467–77. [DOI] [PubMed] [Google Scholar]
- 95.Chung WH, Pan RY, Chu MT, et al. Oxypurinol-Specific T Cells Possess Preferential TCR Clonotypes and Express Granulysin in Allopurinol-Induced Severe Cutaneous Adverse Reactions. J Invest Dermatol 2015;135:2237–48. [DOI] [PubMed] [Google Scholar]
- 96.Muraro A, Lemanske RF Jr, Castells M, et al. Precision medicine in allergic disease-food allergy, drug allergy, and anaphylaxis-PRACTALL document of the European Academy of Allergy and Clinical Immunology and the American Academy of Allergy, Asthma and Immunology. Allergy 2017;72:1006–21. [DOI] [PubMed] [Google Scholar]
- 97.Empedrad R, Darter AL, Earl HS, Gruchalla RS. Nonirritating intradermal skin test concentrations for commonly prescribed antibiotics. J Allergy Clin Immunol 2003;112:629–30. [DOI] [PubMed] [Google Scholar]
- 98.Mallal S, Phillips E, Carosi G, et al. HLA-B*5701 screening for hypersensitivity to abacavir. N Engl J Med 2008;358:568–79. [DOI] [PubMed] [Google Scholar]
- 99.Martin MA, Klein TE, Dong BJ, Pirmohamed M, Haas DW, Kroetz DL. Clinical pharmacogenetics implementation consortium guidelines for HLA-B genotype and abacavir dosing. Clin Pharmacol Ther 2012;91:734–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Churchill D, Waters L, Ahmed N, et al. British HIV Association guidelines for the treatment of HIV-1-positive adults with antiretroviral therapy 2015. HIV Med 2016;17 Suppl 4:s2–s104. [DOI] [PubMed] [Google Scholar]
- 101.Gonzalez-Galarza FF, Takeshita LY, Santos EJ, et al. Allele frequency net 2015 update: new features for HLA epitopes, KIR and disease and HLA adverse drug reaction associations. Nucleic Acids Res 2015;43:D784–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Yip VL, Alfirevic A, Pirmohamed M. Genetics of immune-mediated adverse drug reactions: a comprehensive and clinical review. Clin Rev Allergy Immunol 2015;48:165–75. [DOI] [PubMed] [Google Scholar]
- 103.Khanna D, Fitzgerald JD, Khanna PP, et al. 2012. American College of Rheumatology guidelines for management of gout. Part 1: systematic nonpharmacologic and pharmacologic therapeutic approaches to hyperuricemia. Arthritis Care Res (Hoboken) 2012;64:1431–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Chen P, Lin JJ, Lu CS, et al. Carbamazepine-induced toxic effects and HLA-B*1502 screening in Taiwan. N Engl J Med 2011;364:1126–33. [DOI] [PubMed] [Google Scholar]
- 105.Chen Z, Liew D, Kwan P. Effects of a HLA-B*15:02 screening policy on antiepileptic drug use and severe skin reactions. Neurology 2014;83:2077–84. [DOI] [PubMed] [Google Scholar]
- 106.Ko TM, Tsai CY, Chen SY, et al. Use of HLA-B*58:01 genotyping to prevent allopurinol induced severe cutaneous adverse reactions in Taiwan: national prospective cohort study. BMJ 2015;351:h4848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Chung W-H, Hung S-I, Hong H-S, et al. Medical genetics: A marker for Stevens-Johnson syndrome. Nature 2004;428:486-. [DOI] [PubMed] [Google Scholar]
- 108.Man CBL, Kwan P, Baum L, et al. Association between HLA-B*1502 Allele and Antiepileptic Drug-Induced Cutaneous Reactions in Han Chinese. Epilepsia 2007;48:1015–8. [DOI] [PubMed] [Google Scholar]
- 109.Locharernkul C, Loplumlert J, Limotai C, et al. Carbamazepine and phenytoin induced Stevens-Johnson syndrome is associated with HLA-B*1502 allele in Thai population. Epilepsia 2008;49:2087–91. [DOI] [PubMed] [Google Scholar]
- 110.Chang C-C, Too C-L, Murad S, Hussein SH. Association of HLA-B*1502 allele with carbamazepine-induced toxic epidermal necrolysis and Stevens–Johnson syndrome in the multi-ethnic Malaysian population. Int J Dermatol 2011;50:221–4. [DOI] [PubMed] [Google Scholar]
- 111.Wang Q, Zhou J-q, Zhou L-m, et al. Association between HLA-B*1502 allele and carbamazepine-induced severe cutaneous adverse reactions in Han people of southern China mainland. Seizure 2011;20:446–8. [DOI] [PubMed] [Google Scholar]
- 112.Kaniwa N, Saito Y, Aihara M, et al. HLA-B*1511 is a risk factor for carbamazepine-induced Stevens-Johnson syndrome and toxic epidermal necrolysis in Japanese patients. Epilepsia 2010;51:2461–5. [DOI] [PubMed] [Google Scholar]
- 113.Kim S-H, Lee KW, Song W-J, et al. Carbamazepine-induced severe cutaneous adverse reactions and HLA genotypes in Koreans. Epilepsy Research 2011;97:190–7. [DOI] [PubMed] [Google Scholar]
- 114.Mehta TY, Prajapati LM, Mittal B, et al. Association of HLA-B*1502 allele and carbamazepine-induced Stevens-Johnson syndrome among Indians. Indian J Dermatol Venereol Leprol 2009;75:579–82. [DOI] [PubMed] [Google Scholar]
- 115.Alfirevic A, Jorgensen AL, Williamson PR, Chadwick DW, Park BK, Pirmohamed M. HLA-B locus in Caucasian patients with carbamazepine hypersensitivity. Pharmacogenomics 2006;7:813–8. [DOI] [PubMed] [Google Scholar]
- 116.Ozeki T, Mushiroda T, Yowang A, et al. Genome-wide association study identifies HLA-A*3101 allele as a genetic risk factor for carbamazepine-induced cutaneous adverse drug reactions in Japanese population. Human Molecular Genetics 2011;20:1034–41. [DOI] [PubMed] [Google Scholar]
- 117.Saito Y, Stamp LK, Caudle KE, et al. Clinical Pharmacogenetics Implementation Consortium (CPIC) guidelines for human leukocyte antigen B (HLA-B) genotype and allopurinol dosing: 2015 update. Clin Pharmacol Ther 2016;99:36–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Mrazek DA, Lerman C. Facilitating clinical implementation of pharmacogenomics. JAMA 2011;306:304–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Karnes JH, Van Driest S, Bowton EA, et al. Using systems approaches to address challenges for clinical implementation of pharmacogenomics. Wiley Interdiscip Rev Syst Biol Med 2014;6:125–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Johnson JA. Pharmacogenetics in clinical practice: how far have we come and where are we going? Pharmacogenomics 2013;14:835–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Khoury MJ, Coates RJ, Evans JP. Evidence-based classification of recommendations on use of genomic tests in clinical practice: dealing with insufficient evidence. Genet Med 2010;12:680–3. [DOI] [PubMed] [Google Scholar]
- 122.Dong D, Sung C, Finkelstein EA. Cost-effectiveness of HLA-B*1502 genotyping in adult patients with newly diagnosed epilepsy in Singapore. Neurology 2012;79:1259–67. [DOI] [PubMed] [Google Scholar]
- 123.Dong D, Tan-Koi WC, Teng GG, Finkelstein E, Sung C. Cost-effectiveness analysis of genotyping for HLA-B*5801 and an enhanced safety program in gout patients starting allopurinol in Singapore. Pharmacogenomics 2015;16:1781–93. [DOI] [PubMed] [Google Scholar]
- 124.Toh DS, Tan LL, Aw DC, et al. Building pharmacogenetics into a pharmacovigilance program in Singapore: using serious skin rash as a pilot study. Pharmacogenomics J 2014;14:316–21. [DOI] [PubMed] [Google Scholar]
- 125.Saag M, Balu R, Phillips E, et al. High sensitivity of human leukocyte antigen-b*5701 as a marker for immunologically confirmed abacavir hypersensitivity in white and black patients. Clin Infect Dis 2008;46:1111–8. [DOI] [PubMed] [Google Scholar]
- 126.Mallal S, Nolan D, Witt C, et al. Association between presence of HLA-B*5701, HLA-DR7, and HLA-DQ3 and hypersensitivity to HIV-1 reverse-transcriptase inhibitor abacavir. The Lancet 2002;359:727–32. [DOI] [PubMed] [Google Scholar]
- 127.Mallal S, Phillips E, Carosi G, et al. HLA-B*5701 Screening for Hypersensitivity to Abacavir. New England Journal of Medicine 2008;358:568–79. [DOI] [PubMed] [Google Scholar]
- 128.Hetherington S, Hughes AR, Mosteller M, et al. Genetic variations in HLA-B region and hypersensitivity reactions to abacavir. The Lancet 2002;359:1121–2. [DOI] [PubMed] [Google Scholar]
- 129.Saag M, Balu R, Phillips E, et al. High Sensitivity of Human Leukocyte Antigen-B*5701 as a Marker for Immunologically Confirmed Abacavir Hypersensitivity in White and Black Patients. Clinical Infectious Diseases 2008;46:1111–8. [DOI] [PubMed] [Google Scholar]
- 130.Tassaneeyakul W, Jantararoungtong T, Chen P, et al. Strong association between HLA-B*5801 and allopurinol-induced Stevens–Johnson syndrome and toxic epidermal necrolysis in a Thai population. Pharmacogenetics and Genomics 2009;19:704–9. [DOI] [PubMed] [Google Scholar]
- 131.Chan SH, Tan T. HLA and allopurinol drug eruption. Dermatologica Sinica 1989;179:32–3. [DOI] [PubMed] [Google Scholar]
- 132.Hung S-I, Chung W-H, Liou L-B, et al. HLA-B*5801 allele as a genetic marker for severe cutaneous adverse reactions caused by allopurinol. Proc Natl Acad Sci U S A 2005;102:4134–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Lonjou C, Borot N, Sekula P, et al. A European study of HLA-B in Stevens-Johnson syndrome and toxic epidermal necrolysis related to five high-risk drugs. Pharmacogenet Genomics 2008;18. [DOI] [PubMed] [Google Scholar]
- 134.Génin E, Schumacher M, Roujeau J-C, et al. Genome-wide association study of Stevens-Johnson Syndrome and Toxic Epidermal Necrolysis in Europe. Orphanet Journal of Rare Diseases 2011;6:52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Kang H-R, Jee YK, Kim Y-S, et al. Positive and negative associations of HLA class I alleles with allopurinol-induced SCARs in Koreans. Pharmacogenetics and Genomics 2011;21:303–7. [DOI] [PubMed] [Google Scholar]
- 136.Somkrua R, Eickman EE, Saokaew S, Lohitnavy M, Chaiyakunapruk N. Association of HLA-B*5801 allele and allopurinol-induced stevens johnson syndrome and toxic epidermal necrolysis: a systematic review and meta-analysis. BMC Medical Genetics 2011;12:118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Sukasem C, Jantararoungtong T, Kuntawong P, et al. HLA-B (*) 58:01 for Allopurinol-Induced Cutaneous Adverse Drug Reactions: Implication for Clinical Interpretation in Thailand. Frontiers in pharmacology 2016;7:186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Leckband SG, Kelsoe JR, Dunnenberger HM, et al. Clinical Pharmacogenetics Implementation Consortium guidelines for HLA-B genotype and carbamazepine dosing. Clin Pharmacol Ther 2013;94:324–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Hung S-I, Chung W-H, Jee S-H, et al. Genetic susceptibility to carbamazepine-induced cutaneous adverse drug reactions. Pharmacogenetics and Genomics 2006;16:297–306. [DOI] [PubMed] [Google Scholar]
- 140.Tassaneeyakul W, Tiamkao S, Jantararoungtong T, et al. Association between HLA-B*1502 and carbamazepine-induced severe cutaneous adverse drug reactions in a Thai population. Epilepsia 2010;51:926–30. [DOI] [PubMed] [Google Scholar]
- 141.Wu XT, Hu FY, An DM, et al. Association between carbamazepine-induced cutaneous adverse drug reactions and the HLA-B*1502 allele among patients in central China. Epilepsy & Behavior 2010;19:405–8. [DOI] [PubMed] [Google Scholar]
- 142.Then SM, Rani ZZ, Raymond AA, Ratnaningrum S, Jamal R. Frequency of the HLA-B*1502 allele contributing to carbamazepine-induced hypersensitivity reactions in a cohort of Malaysian epilepsy patients. Asian Pacific Journal of Allergy and Immunology 2011;29:290–3. [PubMed] [Google Scholar]
- 143.Zhang Y, Wang J, Zhao L-M, et al. Strong association between HLA-B*1502 and carbamazepine-induced Stevens-Johnson syndrome and toxic epidermal necrolysis in mainland Han Chinese patients. Eur J Clin Pharmacol 2011;67:885. [DOI] [PubMed] [Google Scholar]
- 144.Kulkantrakorn K, Tassaneeyakul W, Tiamkao S, et al. HLA-B*1502 Strongly Predicts Carbamazepine-Induced Stevens–Johnson Syndrome and Toxic Epidermal Necrolysis in Thai Patients with Neuropathic Pain. Pain Practice 2012;12:202–8. [DOI] [PubMed] [Google Scholar]
- 145.McCormack M, Alfirevic A, Bourgeois S, et al. HLA-A*3101 and Carbamazepine-Induced Hypersensitivity Reactions in Europeans. New England Journal of Medicine 2011;364:1134–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Niihara H, Kakamu T, Fujita Y, Kaneko S, Morita E. HLA-A31 strongly associates with carbamazepine-induced adverse drug reactions but not with carbamazepine-induced lymphocyte proliferation in a Japanese population. The Journal of Dermatology 2012;39:594–601. [DOI] [PubMed] [Google Scholar]
- 147.Zhang FR, Liu H, Irwanto A, et al. HLA-B*13:01 and the dapsone hypersensitivity syndrome. N Engl J Med 2013;369:1620–8. [DOI] [PubMed] [Google Scholar]
- 148.Cheung CL, Sing CW, Tang CS, et al. HLA-B*38:02:01 predicts carbimazole/methimazole-induced agranulocytosis. Clin Pharmacol Ther 2016;99:555–61. [DOI] [PubMed] [Google Scholar]
- 149.Hallberg P, Eriksson N, Ibanez L, et al. Genetic variants associated with antithyroid drug-induced agranulocytosis: a genome-wide association study in a European population. Lancet Diabetes Endocrinol 2016;4:507–16. [DOI] [PubMed] [Google Scholar]
- 150.Keane NM, Pavlos RK, McKinnon E, et al. HLA Class I restricted CD8+ and Class II restricted CD4+ T cells are implicated in the pathogenesis of nevirapine hypersensitivity. AIDS 2014;28:1891–901. [DOI] [PubMed] [Google Scholar]
- 151.Lin L-C, Lai P-C, Yang S-F, Yang R-C. Oxcarbazepine-induced Stevens-Johnson Syndrome: A Case Report. The Kaohsiung Journal of Medical Sciences 2009;25:82–6. [DOI] [PubMed] [Google Scholar]
- 152.Hung SI, Chung WH, Liu ZS, et al. Common risk allele in aromatic antiepileptic-drug induced Stevens-Johnson syndrome and toxic epidermal necrolysis in Han Chinese. Pharmacogenomics 2010;11:349–56. [DOI] [PubMed] [Google Scholar]
- 153.Chen CB, Hsiao YH, Wu T, et al. Risk and association of HLA with oxcarbazepine-induced cutaneous adverse reactions in Asians. Neurology 2017;88:78–86 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.