

Abstract
Objective
This research aimed to determine biological pathways and protein-protein interaction (PPI) networks for 305-d milk yield (MY), 305-d fat yield (FY), and age at first calving (AFC) in the Thai multibreed dairy population.
Methods
Genotypic information contained 75,776 imputed and actual single nucleotide polymorphisms (SNP) from 2,661 animals. Single-step genomic best linear unbiased predictions were utilized to estimate SNP genetic variances for MY, FY, and AFC. Fixed effects included herd-year-season, breed regression and heterosis regression effects. Random effects were animal additive genetic and residual. Individual SNP explaining at least 0.001% of the genetic variance for each trait were used to identify nearby genes in the National Center for Biotechnology Information database. Pathway enrichment analysis was performed. The PPI of genes were identified and visualized of the PPI network.
Results
Identified genes were involved in 16 enriched pathways related to MY, FY, and AFC. Most genes had two or more connections with other genes in the PPI network. Genes associated with MY, FY, and AFC based on the biological pathways and PPI were primarily involved in cellular processes. The percent of the genetic variance explained by genes in enriched pathways (303) was 2.63% for MY, 2.59% for FY, and 2.49% for AFC. Genes in the PPI network (265) explained 2.28% of the genetic variance for MY, 2.26% for FY, and 2.12% for AFC.
Conclusion
These sets of SNP associated with genes in the set enriched pathways and the PPI network could be used as genomic selection targets in the Thai multibreed dairy population. This study should be continued both in this and other populations subject to a variety of environmental conditions because predicted SNP values will likely differ across populations subject to different environmental conditions and changes over time.
Keywords: Cattle, Gene Network, Multibreed, Single Nucleotide Polymorphism Marker, Tropical
INTRODUCTION
The Thai multibreed dairy population is primarily composed of crossbred animals with over 75% Holstein (91%) and the remainder comes from various Bos indicus (Red Sindhi, Sahiwal, Brahman, and Thai Native) and Bos taurus (Brown Swiss, Red Danish, and Jersey) breeds [1]. Recent genome-wide association studies (GWAS) in Thailand found sets of significant single nucleotide polymorphism (SNP) markers from GeneSeek 9K chip associated with genes affecting lactation characteristics, milk yield (MY), fat yield (FY), and age at first calving (AFC) that were mostly different from those found in Bos taurus breeds in temperate regions [2,3]. Use of low-density in the Thai studies (9K) and high density in the studies in temperate regions (50K to 770K) may have been largely responsible for these differences. Unfortunately, budgetary restrictions have allowed only a small fraction of the animals in the Thai multibreed dairy population to be genotyped with GeneSeek 80K. An efficient alternative to increase the numbers of SNP per animal without increasing the cost of genotyping SNP is genomic imputation. Jattawa et al [4] found that program FImpute was more accurate than Findhap and Beagle software when imputing from GeneSeek 9K, 20K, and 26K to 80K in the Thai multibreed dairy population. Thus, imputation with FImpute of all genotyped animals with low-density chips could help increase the accuracy of estimation of SNP marker effects and the likelihood of identifying SNP markers associated with genes affecting dairy traits in this population. Further, because only a fraction of animals with phenotypes have genotypes, computation of SNP marker effects and explained genomic variation could be accomplished by utilizing the single-step genomic best linear unbiased prediction (ssGBLUP) developed at the University of Georgia [5].
The GWAS for milk production and reproductive traits in Holstein in temperate regions identified regions associated with MY, FY, and AFC in all autosomes [6–8]. Similarly, GWAS in Thailand found a largely different set of significant SNP distributed across all 29 autosomes and the X chromosome associated with milk production and reproductive traits in the Holstein upgraded Thai multibreed dairy population [2,3]. However, GWAS provide limited information on relationships among genes affecting quantitative traits. Analysis of gene networks and biological pathways would provide a more comprehensive understanding of the sets of genes affecting multiple milk production and reproduction traits in dairy cattle. Biological pathway research in Holstein indicated that most sets of genes associated with milk production in these studies were involved in metabolic pathways, fat digestion and absorption, arginine and proline metabolism and tight junctions [7]. However, sets of genes involved in biological pathways related to milk production may be influenced by population structure and selection [8]. As with differences in sets of SNP associated with milk production and reproduction traits between the multibreed cattle in Thailand and Holstein cattle in temperate zones [2,3], biological pathways and gene networks associated with these traits may also differ in Thai multibreed and purebred Holstein dairy populations. Thus, the objectives of this study were to determine biological pathways and protein-protein interaction (PPI) gene networks associated with MY, FY, and AFC in the Thai multibreed dairy population under tropical environmental conditions.
MATERIALS AND METHODS
Animals, management and traits
This research utilized 8,361 first-lactation cows from 810 farms located in the Northern, Northeastern, Central, Western, and Southern regions of Thailand. These cows were the progeny of 1,210 sires and 6,992 dams. Eighty-eight percent of animals in the database were Holstein (H) crossbreds (75% H and above); the remaining 25% belonged to other breeds (O) including Jersey, Brown Swiss, Red Danish, Sahiwal, Red Sindhi, Brahman, and Thai Native.
Cows were housed in open barns where they had access to roughage, concentrate and a mineral supplement. Green roughage consisted of freshly cut grasses (cut and carry) including Napier grass (Pennisetum purpureum), Guinea grass (Panicum maximum), Ruzi grass (Brachiaria ruziziensis), and Para grass (Brachiaria mutica). Cows were fed approximately 30 to 40 kg/d of roughage and 5 to 10 kg/d of concentrate, or equivalently, 1 kg of concentrate per 2 kg/milk produced. The concentrate (14% to 22% of crude protein and 63% to 83% of nitrogen-free extract) was provided twice per day during milking (morning: 4:30 to 7:00 am and afternoon: 2:30 to 4:30 pm). Agricultural byproducts (rice straw, pineapple waste and sweet corn cob or husk), hay, and (or) silage were used as supplements as green roughage decreased in winter and summer [1].
Traits in this research were 305-d MY, 305-d FY, and AFC. Test-day MY and fat percentage were collected monthly from individual first-lactation cows between 1989 and 2014. Test-day FY was computed as the product of fat percentage and MY. Subsequently, monthly test-day MY and FY were used to compute MY and FY using the test-interval procedure [9,10].
Genomic DNA and genotypic data
Blood and semen samples were collected from 2,661 animals (89 sires and 2,572 dams) of the Thai multibreed dairy population. Genomic DNA was extracted from blood using a MasterPure DNA Purification kit for blood version II (EPICENTRE Biotechnologies, Madison, WI, USA) and from semen using a GenElute Mammalian Genomic DNA Miniprep Kit (Sigma, Ronkonkoma, NY, USA). The DNA quality was assessed with a NanoDrop 2000 spectrophotometer (Thermo Fisher Scientific Inc., Wilmington, DE, USA). DNA samples from all animals (n = 2,661) were ensured to contain sufficient DNA for genotyping (absorbance ratio of approximately 1.8 at 260/280 nm and DNA concentration higher than 15 ng/μL). DNA samples were genotyped with GeneSeek Genomic Profiler (GGP) 9K, 20K, 26K, and 80K chips (GeneSeek Inc., Lincoln, NE, USA).
Animals genotyped with GGP9K, GGP20K, and GGP26K were imputed to GGP80K using program FImpute version 2.2 [4,11]. The imputed markers were subjected to quality control prior to further analysis. Quality control consisted of removing imputed markers with call rates lower than 90% and minor allele frequencies lower than 0.01. The resulting edited file contained 75,776 SNP markers per genotyped animal.
Genome-wide association analysis
A GWAS for MY, FY, and AFC was performed using ssGBLUP [5]. Animals with phenotypes and genotypes as well as animals with only phenotypes were included in this analysis. A 3-trait genomic-polygenic model was used to obtain genetic variances for and covariances between MY, FY, and AFC. Fixed effects included contemporary group (herd-year-season), breed regression effect (as a linear function of expected O fraction in each animal, where O = other breeds, including Brown Swiss, Red Danish, Jersey, Red Sindhi, Sahiwal, Brahman, and Thai Native), and heterosis regression effect as a linear function of heterozygosity (expected H fraction in the sire times expected O fraction in the dam plus expected O fraction in the sire times expected H fraction in the dam). Random effects were animal additive genetic and residual. The mean for random animal additive genetic and residual effects was assumed to be zero. The variance-covariance matrix among animal additive genetic effects for MY, FY, and AFC was equal to H⨂Va where H was the genomic-polygenic relationship matrix, Va was variance-covariance matrix among additive genetic effects for these traits, and ⨂ was the Kronecker product. Residual variance-covariance matrix was equal to I⨂Ve where I was identity matrix and Ve was variance-covariance matrix among residual effects. The H equal to , where A11 was the submatrix of additive relationships between non-genotyped animals, A12 was the submatrix of additive relationships between genotyped animals, was the inverse of the matrix of additive genetic relationships between genotyped animals, and G22 was the matrix of genomic relationships among genotyped animals [12]. Matrix G22 = ZZ′/2 ∑ pj (1 – pj), where pj was the frequency of the second allele in locus j and Z was the incidence matrix of SNP effects whose elements were defined as zij = (0 – 2pj) if the genotype for locus j was homozygous 11, zij = (1 – 2pj) if the genotype for locus j was heterozygous 12 or 21 and zij = (2 – 2pj) if the genotype for locus j was homozygous 22. Matrix G22 was scaled using the default parameters of the BLUPF90 Family of Programs [13], i.e., the default scaling of matrix required the mean of the diagonal elements of G22 to be equal to the mean of the diagonal elements of A22 and the mean of the off-diagonal elements of G22 to be equal to the mean of the off-diagonal elements of A22.
Variance and covariance components for MY, FY, and AFC were estimated using restricted maximum likelihood procedures and computed via program AIREMLF90 [14] using an average information algorithm. Program POSTGSF90 was used to calculate the proportion of genetic variance explained by each SNP, additive SNP marker effect and construct Manhattan plots of percentages of the genetic variance explained by individual SNP. The percentage of the genetic variance explained by each SNP was calculated as the ratio of the variance explained by that SNP divided by the total genetic variance [15]. The predicted value of SNP associated with genes was calculated as sum of the additive SNP markers effect for each gene.
Identification of genes associated with milk yield, fat yield, and age at first calving
Individual SNP that explained at least 0.001% of the genetic variance for MY, FY, and AFC were selected to determine potential genes associated with these traits. The position of these SNP markers in base pairs was used to locate genes or nearby genes in the UMD Bos taurus 3.1 assembly of the bovine genome at the National Center for Biotechnology Information (NCBI) using R package Map2NCBI [16]. Only SNP inside or within 2,500 bp of genes in the NCBI database were utilized for the pathway enrichment and (PPI) network analyses.
Pathway enrichment analysis
Genes associated with MY, FY, and AFC were used to identify biological pathways in Bos taurus at the Kyoto encyclopedia of genes and genomes database using the ClueGo plugin of Cytoscape [17]. The statistical test used for the pathway enrichment analysis by ClueGo was a right-sided test based on the hypergeometric distribution corrected for multiple testing with the Bonferroni step-down method. Significantly enriched pathways for these traits were defined to be those with p<0.05.
Protein-protein interaction network analysis
The name of genes for MY, FY, and AFC was used to identify PPI from neighborhood, co-occurrence, gene fusions, co-expression, experiments, databases, and text mining using program STRING [18]. The STRING defined PPI as a probabilistic confidence score. A high confidence score implied that interactions between proteins from the database could be considered as valid edges in a network. Thus, only PPI with a high confidence score (>0.7) were used to construct the PPI network. The PPI network was visualized using Cytoscape [19]. The CytoNCA plugin for Cytoscape was used to analyze the number connections between genes in the PPI network [20].
RESULTS AND DISCUSSION
Genetic variance explained by individual single nucleotide polymorphism and chromosomes
The percentage of genetic variance explained by each SNP are shown in Figure 1. Most SNP markers (65%) explained less than 0.001% of the genetic variance each and together they accounted for 13% of the genetic variance. Conversely, 35% of SNP markers that explained 0.001% or more of the genetic variance and accounted for the largest fraction (87%) of the total genetic variance for MY, FY, and AFC. SNP markers were located inside genes, within 2,500 bp, between 2,500 and 5,000 bp, between 5,000 and 25,000 bp and beyond 25,000 bp of genes in the NCBI database (Supplementary Table S1). The percent of SNP inside genes or within 2,500 bp of genes explaining at least 0.001% of the genetic variance was 44% for MY, and FY, 43% for AFC, and accounted for 38% of the genetic variance for these traits.
Figure 1.
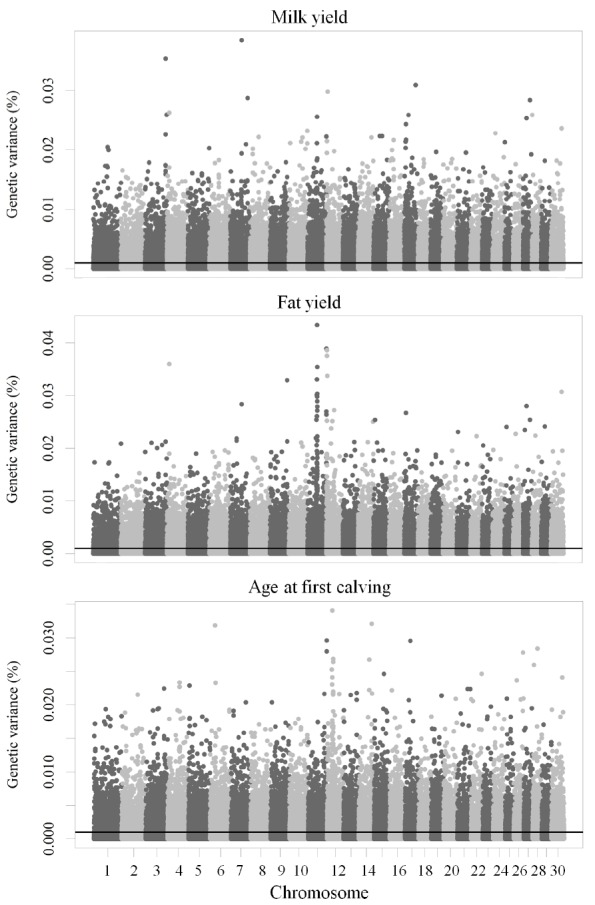
Manhattan plot of the percent of the genetic variance for milk yield, fat yield, and age at first calving explained by each SNP. The black line is the threshold for SNP accounting for more than 0.001% of the genetic variance for these traits. SNP, single nucleotide polymorphism.
Numbers of SNP per gene ranged from 1 to 37 for MY, 1 to 25 for FY, and 1 to 29 for AFC (Figure 2). Seventy one percent of SNP associated with these traits had a one to one correspondence with genes in the NCBI database indicating that the vast majority of SNP markers in this population pointed to a single gene within the genome.
Figure 2.
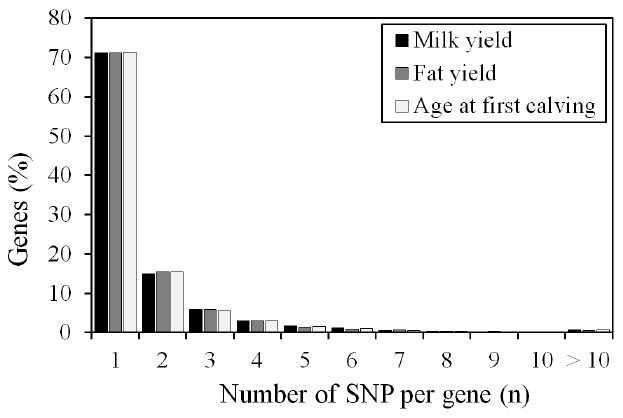
Distribution of genes associated with milk yield, fat yield and age at first calving by number of SNP per gene. SNP, single nucleotide polymorphism.
Numbers of genes and total genetic variance per chromo some for MY, FY, and AFC identified by SNP genotypes inside or within 2,500 bp of genes in the NCBI database are shown in Supplementary Table S2. The genetic variance explained by each chromosome ranged from 0.66% (chromosome 27) to 2.02% (chromosome 5) for MY, 0.58% (chromosome 27) to 2.09% (chromosome 11) for FY, and 0.58% (chromosome 27) to 2.01% (chromosome 4) for AFC. These low percentages of explained genetic variance indicated that MY, FY, and AFC were influenced by large numbers of genes accounting for small amounts of genetic variation scattered throughout the genome.
Figure 3 shows numbers of genes associated with only one trait (dark gray), two traits (bright gray), and all three traits (white) based on Map2NCBI allocations. Numbers of single-trait gene associations (861 for MY, 774 for FY and, 1806 for AFC) were lower than two-trait gene associations (1,851 for MY and FY, 782 for MY and AFC, and 898 for FY and AFC) and three-trait gene associations (3,436 for MY, FY, and AFC). This indicated that genes were likely to be involved in multiple-trait associations than single-trait associations. These associations offer a biological rationale for the existence of genetic correlations among these traits. All genes associated with all three traits were located in the 29 autosomes and the X chromosome. The percentage of the genetic variance explained by these genes across all chromosomes was 26.2% for MY, 26.3% for FY, and 24.7% for AFC (Supplementary Table S3). These results provide additional evidence for these three quantitative traits (MY, FY, and AFC) to be determined by sets of genes spread across the genome in the Thai multibreed [2,3] and Holstein populations [7].
Figure 3.
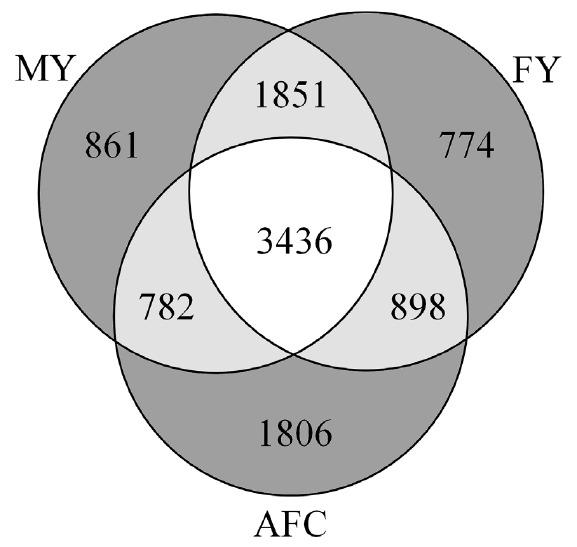
Number genes associated with one (dark gray), two (light gray) and three (white) traits in the Thai multibreed population. MY, milk yield; FY, fat yield; AFC, age at first calving.
Pathway enrichment analysis
Enriched pathways were classified into four categories: cellular processes, nervous system, digestive system, and environment adaptation are shown in Table 1. The genetic variance explained by the genes involved in these 16 significantly enriched pathways was 2.63% for MY, 2.59% for FY, and 2.49% for AFC (Table 1). The total predicted value of the SNP associated with these genes (as deviations from the second allele at each locus) were second allele at each locus had a larger effect than the first allele at most loci for MY and FY, and that the opposite occurred for AFC.
Table 1.
Percent of genetic variance for milk yield (MY), fat yield (FY), and age at first calving (AFC) explained by single nucleotide polymorphism located inside or within 2,500 bp of genes present in significantly enriched pathways
| Category | Pathway | p-value | Number of genes (n) | Genetic variance (%) | ||
|---|---|---|---|---|---|---|
|
| ||||||
| MY | FY | AFC | ||||
| Cellular processes | 266 | 2.2408 | 2.2867 | 2.1220 | ||
| Rap1 signaling | 8.9×10−8 | 63 | 0.5113 | 0.6292 | 0.6437 | |
| Calcium signaling | 1.3×10−7 | 58 | 0.5018 | 0.5157 | 0.5381 | |
| Phospholipase D signaling | 2.8×10−6 | 47 | 0.4691 | 0.4361 | 0.4657 | |
| Focal adhesion | 1.6×10−5 | 55 | 0.4087 | 0.3868 | 0.4621 | |
| MAPK signaling | 0.0002 | 62 | 0.4847 | 0.523 | 0.4807 | |
| Ras signaling | 0.0075 | 55 | 0.3982 | 0.4333 | 0.4639 | |
| Wnt signaling | 0.0077 | 38 | 0.3062 | 0.3016 | 0.2848 | |
| cGMP-PKG signaling | 0.0085 | 41 | 0.4702 | 0.483 | 0.4668 | |
| Sphingolipid signaling | 0.014 | 32 | 0.2295 | 0.2561 | 0.3181 | |
| Oxytocin signaling | 0.016 | 38 | 0.3528 | 0.3538 | 0.376 | |
| Gap junction | 0.021 | 26 | 0.3223 | 0.2973 | 0.3667 | |
| Nervous system | 70 | 0.7829 | 0.6567 | 0.7825 | ||
| Glutamatergic synapse | 1.9×10−8 | 42 | 0.4857 | 0.4409 | 0.4886 | |
| Dopaminergic synapse | 0.0036 | 36 | 0.4004 | 0.3415 | 0.4037 | |
| GABAergic synapse | 0.011 | 26 | 0.3182 | 0.2633 | 0.2692 | |
| Digestive system | 27 | 0.2748 | 0.2902 | 0.3216 | ||
| Pancreatic secretion | 0.03 | 27 | 0.2748 | 0.2902 | 0.3216 | |
| Environmental adaptation | 30 | 0.3818 | 0.3593 | 0.3672 | ||
| Circadian entrainment | 0.0018 | 30 | 0.3818 | 0.3593 | 0.3672 | |
| Total | 303 | 2.6282 | 2.5916 | 2.4893 | ||
Cellular processes
Cellular process pathways related to cell proliferation, differentiation, migration, survival and apoptosis are essential for physiological changes in the ovarian follicle and mammary gland [21]. Therefore, cellular processes pathways contained the largest number of genes (266) and accounted for the largest percentage of the genetic variance for MY (2.24%), FY (2.29%), and AFC (2.12%) of all categories of significantly enriched pathways (Table 1). The sum of the predicted values of the SNP associated genes in enriched cellular pathways were −3.7513 for MY, −0.1524 for MY, and 0.0128 for AFC (Table 2), indicating that allele 2 at each locus had a larger effect than allele 1 for MY and FY, but a smaller effect than allele 1 for AFC.
Table 2.
Predicted value of SNP located inside or within 2,500 bp of genes associated with milk yield (MY), fat yield (FY), and age at first calving (AFC) present in significantly enriched pathways
| Category | Pathway | p-value | Number of genes (n) | Predicted SNP value | ||
|---|---|---|---|---|---|---|
|
| ||||||
| MY | FY | AFC | ||||
| Cellular processes | 266 | −3.7513 | −0.1524 | 0.0128 | ||
| Rap1 signaling | 8.9×10−8 | 63 | −0.9683 | −0.0715 | 0.0036 | |
| Calcium signaling | 1.3×10−7 | 58 | −2.5956 | −0.0520 | 0.0005 | |
| Phospholipase D signaling | 2.8×10−6 | 47 | −2.7299 | −0.0919 | 0.0072 | |
| Focal adhesion | 1.6×10−5 | 55 | −3.5352 | −0.0795 | −0.0027 | |
| MAPK signaling | 0.0002 | 62 | −0.0853 | −0.0382 | 0.0033 | |
| Ras signaling | 0.0075 | 55 | −1.1262 | −0.0448 | 0.0025 | |
| Wnt signaling | 0.0077 | 38 | −0.2874 | −0.0133 | 0.0017 | |
| cGMP-PKG signaling | 0.0085 | 41 | −0.5336 | −0.0534 | 0.0037 | |
| Sphingolipid signaling | 0.014 | 32 | 0.5767 | 0.0287 | 0.0056 | |
| Oxytocin signaling | 0.016 | 38 | −0.9332 | −0.0438 | −0.0022 | |
| Gap junction | 0.021 | 26 | −1.0156 | −0.0477 | 0.0017 | |
| Nervous system | 70 | −4.1918 | −0.0938 | 0.0039 | ||
| Glutamatergic synapse | 1.9×10−8 | 42 | −2.0444 | −0.0804 | −0.0032 | |
| Dopaminergic synapse | 0.0036 | 36 | −1.2675 | −0.0328 | 0.0002 | |
| GABAergic synapse | 0.011 | 26 | −2.5585 | −0.0434 | 0.0041 | |
| Digestive system | 27 | −0.9985 | −0.0260 | −0.0006 | ||
| Pancreatic secretion | 0.03 | 27 | −0.9985 | −0.0260 | −0.0006 | |
| Environmental adaptation | 30 | −1.0546 | −0.0425 | −0.0020 | ||
| Circadian entrainment | 0.0018 | 30 | −1.0546 | −0.0425 | −0.0020 | |
| Total | 303 | −5.0430 | −0.1573 | 0.0077 | ||
SNP, single nucleotide polymorphism.
Ovarian follicle and mammary gland development are influenced by the calcium-signaling pathway, which in turn is regulated by growth factors through changes in the concentration of free calcium ions (Ca2+). Specifically, Ca2+ acts as an activator in the mitogen-activated protein kinase (MAPK) signaling pathway in ovarian follicle and mammary gland cells [22]. The MAPK links extracellular signals to the machinery that controls many fundamental cellular processes such as cell inflammation, proliferation, metabolism, motility, and apoptosis [23]. Extracellular signal-regulated kinase 5, a member of the MAPK family, mediates the production of prolactin [24], a regulator in the development of the mammary gland. The MAPK signaling pathway was found to be essential for the development of ovarian follicles in heifers [21] and the mammary gland during lactation in Holstein [6,24] and Jersey [6]. The MAPK pathway is regulated by proteins from three associated pathways: Ras-related protein 1 from the Rap1 signaling pathway (http://www.genome.jp/dbgetbin/www_bget?pathway:bta04015), Ras proteins from the Ras signaling pathway (http://www.genome.jp/dbget-bin/www_bget?pathway:bta04014), and Wnt proteins from the Wnt signaling pathway (http://www.genome.jp/dbget-bin/www_bget?pathway:bta04310).
Phospholipase D from the phospholipase D signaling path way is an essential enzyme for the production of phosphatidic acid (http://www.genome.jp/dbget-bin/www_bget?pathway:bta04310), a key intermediate in milk fat synthesis during lactation [25]. The Focal adhesion and Gap junction pathways receive and send signals that affect the motility, proliferation, differentiation, metabolic transport, apoptosis, and tissue homeostasis of ovarian follicle and mammary gland cells [21]. The cyclic guanosine monophosphate from the cGMP-PKG signaling pathway involved in the activation and regulation of protein kinase G in smooth muscle cells to promote their relaxation (http://www.genome.jp/dbget-bin/www_bget?pathway:bta04022). The ceramide and sphingosine-1-phosphate from the sphingolipid signaling pathway acts an as a regulator of cell responses to stress (http://www.genome.jp/dbget-bin/www_bget?pathway:bta04071). The oxytocin hormone (oxytocin signaling pathway), produced by the hypothalamus, stimulates the contraction of mammary gland myoepithelial cells, causing milk to be ejected into the ducts, and cisterns during milking (http://www.genome.jp/dbget-bin/www_bget?pathway:bta04921).
Cellular processes influenced MY, FY, and AFC through a large number of genes located in multiple pathways, each having a small effect and explaining a small percentage of genetic variation for these traits. However, the combined effect of all genes in all enriched cellular pathways explained a noticeable amount of genetic variation. Therefore, the combined effect of all cellular processes for MY, FY, and AFC could potentially be considered as a functional genomic selection target within each trait in this population.
Nervous system
The nervous system pathways include glutamatergic synapse, GABAergic synapse, and dopaminergic synapse pathways involved in brain remodeling. There were 70 genes in these three pathways, and they together explained 0.78% of the genetic variance for MY, 0.66% for FY, and 0.78% for AFC (Table 1), and their associated SNP had a total predicted value of −4.1918 for MY, −0.0938 for FY, and 0.0039 for AFC (Table 2). These three pathways are involved in the onset of puberty, which in turn determines AFC. Changes in the concentration of gonadotropin-releasing hormone (GnRH) trigger of the onset of puberty. Glutamate from the glutamatergic synapse pathway and gamma-aminobutiyric acid from the GABAergic synapse pathway stimulate the production of GnRH whereas dopamine from the dopaminergic synapse inhibits it [26,27]. The GnRH stimulates the secretion of gonadotropins from the pituitary gland (luteinizing and follicle-stimulating hormones) involved in the development of follicles, ovulation and the production of estrogen and progesterone. Fortes et al [28] provided evidence for the involvement of genes from the glutamatergic synapse and GABAergic synapse pathways in the attainment of puberty in beef cattle.
Digestive system
The only significant pathway in the digestive system category was the pancreatic secretion pathway. This pathway was 27 genes and they together explained 0.27% of the genetic variance for MY, 0.29% for FY, and 0.32% for AFC (Table 1), and the total predicted value of the associated SNP were −0.9985 for MY, −0.0260 for FY, and −0.0006 for AFC (Table 2). The digestive system pathway was also found to be associated with milk production traits in Holstein [7]. Pancreatic enzymes (lipases, amylases, proteases) from the pancreatic secretion pathway are important for the digestion and absorption of nutrients (carbohydrates, proteins, fats, vitamins) in the small intestine. Thus, genes involved in the pancreatic pathway likely influenced differences in MY, FY, and AFC among animals in the Thai multibreed dairy population. Heifers that digested and absorbed nutrients more efficiently would be expected to have faster growth rates, achieve puberty earlier, have higher conception rates and produce more milk than less efficient heifers. Thus, heifers in the Thai multibreed dairy population that digested and absorbed nutrients from local roughages, concentrate and byproducts of agricultural efficiently likely had lower AFC than less efficient heifers.
Environmental adaptation
The circadian entrainment pathway was the only significant pathway in the environmental adaptation category. This pathway contained 30 genes that explained 0.38% of the genetic variance for MY, 0.36% for FY, and 0.37% for AFC (Table 1), and the sum of the predicted values of the SNP associated with these genes was −1.0546 for MY, −0.0425 for FY, and −0.0020 for AFC (Table 2). The circadian entrainment pathway is important for animal adaptation to number of daylight hours, temperature and humidity [29]. The cyclic adenosine monophosphate (cAMP) response element-binding protein from the circadian entrainment pathway regulates the circadian clock (http://www.genome.jp/dbget-bin/www_bget?pathway:bta04713). The circadian clock is influenced by the length of photoperiod [30], which in turn influences the activity of multiple hormones (estrogen, progesterone, placental lactogen, prolactin, leptin, cortisol) that affect metabolites (glucose, amino acids, free fatty acids, triglycerides) received by cells of the mammary gland [31]. Dairy cows in Thailand are exposed directly to day-length changes because farmers house cows in open barns. Thus, it is not surprising that genes involved in the circadian entrainment pathway explained a significant portion of the genetic variation for MY and FY in the Thai multibreed dairy population.
Protein-protein interaction network analysis
The PPI network for MY, FY, and AFC contained 265 nodes (i.e., genes) connected via 1,158 edges (Figure 4). Approximately 90% of the genes had two or more connections (Figure 5). The preponderance of multiple interactions among genes in the PPI network indicated that this was a highly interconnected network where most genes affected the expression of other genes relevant to MY, FY, and AFC. The number of connections per node ranged from 1 to 44 and the number of pathways fluctuated between 1 and 15 (Supplementary File S1). The PPI network for MY, FY, and AFC showed a dense center with highly interconnected genes (Figure 4). Genes in the PPI network explained 2.28% of the genetic variance for MY, 2.26% for FY, and 2.12% for AFC (Supplementary File S1). Thus, genes in the PPI network explained an average of 86.3% of the genetic variation as genes present in significantly enriched pathways (86.7% for MY, 87.2% for FY, and 85.2% for AFC). The sum of the predicted SNP values of the 265 genes in the PPI network was −5.6150 for MY, −0.1404 for FY, and 0.0067 for AFC (Supplementary File S2). As with explained genetic variances, these sums of predicted values for PPI genes were similar to those obtained for the 303 genes in the significantly enriched pathways (Table 2). All genes in the PPI network were also involved in one or more enriched pathways in Table 1. Thus, the 265 genes in the PPI network were a subset of the 303 genes in the set of enriched pathways, meaning that 87.5 of enriched pathway genes were represented in the PPI network. Thus, the 14% lower amount of genetic variation explained by PPI genes was due to a 12.5% lower number of genes than those present in the enriched pathways in Table 1.
Figure 4.

Protein-protein interaction (PPI) network of genes involved in significant pathways. Gray nodes represent genes with large numbers of connections with other genes in the PPI network.
Figure 5.
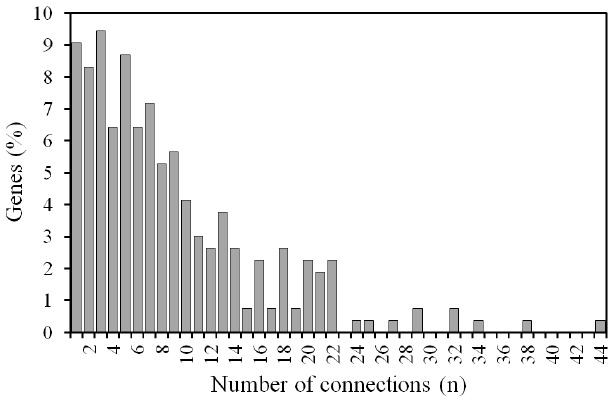
Distribution of genes associated with milk yield, fat yield, and age at first calving by number of connections in the protein-protein interaction network.
Figure 6 show a subset of the most represented genes in the PPI network for MY, FY, and AFC (16 genes with a minimum of 22 connections and 1 pathway). The protein kinase C beta (PRKCB) gene had the largest number of significantly enriched pathways for MY, FY, and AFC (14) and accounted for 0.012% of the genetic variance for MY, 0.009% for FY and 0.016% for AFC (Supplementary File S1). This gene participated in 9 biological process pathways, 3 nervous system pathways, digestive system pathway and environmental adaptation pathway. PRKCB codes for protein kinase C beta that is involved in diverse cellular signaling pathways (http://www.genecards.org/cgi-bin/carddisp.pl?gene=PRKCB&keywords=PRKCB). Further, PRKCB is also involved in the circadian entrainment pathway. This pathway contributes to the adaptation of organisms to their environment [29]. A positive influence of PRKCB on body temperature regulation during climate stress was reported in Angus and Simmental cattle [32]. Higher MY and fat percentages were observed in Holstein that were better adapted to climatic heat stress [33]. The predicted value of the set of SNP associated with PRKCB was 0.226 for MY, 0.005 for FY, and −0.002 for AFC (Supplementary File S2). These predicted SNP values indicate that the second PRKCB allele would result in higher MY and FY as well as shorter AFC in cows from the Thai multibreed dairy population, whereas the first PRKCB allele would have the opposite effect.
Figure 6.
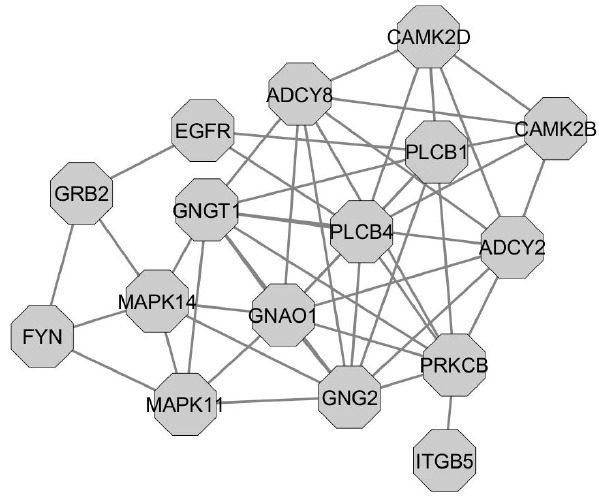
Genes with large numbers of connections in the protein-protein interaction network of the Thai multibreed population.
The phospholipase C beta 1 ( PLCB1), PLCB4, adenylate cyclase 2 (ADCY2), ADCY8, calcium/calmodulin dependent protein kinase II beta (CAMK2B), CAMK2D, mitogen-activated protein kinase 11 (MAPK11), MAPK14, epidermal growth factor receptor (EGFR), growth factor receptor bound protein 2 (GRB2), Fyn proto-oncogene, Src family tyrosine kinase (FYN), and integrin subunit beta 5 (ITGB5) genes were involved in 12 significantly enriched pathways and accounted for 0.126% of the genetic variance for MY, 0.109% for FY, and 0.197% for AFC (Supplementary File S1). The predicted values of the set of SNP associated with these genes was −0.739 for MY, −0.026 for FY, and 0.001 for AFC. These predicted SNP values indicated that the subset of 16 PPI second alleles would decrease MY, FY, and increase AFC, whereas the subset of 16 PPI first alleles would increase MY and FY, but decrease AFC. These genes participated in 8 cellular process pathways (such as MAPK signaling, Ras signaling, Wnt signaling) related to the development of ovarian follicles and cells from the mammary gland [6,21]. PLCB1 and PLCB4 code for phospholipase C beta 1 to 4 that function as signal transducers for the transmission of extracellular signals to multiple intracellular targets (http://www.genecards.org/cgi-bin/carddisp.pl?gene=PLCB1&keywords=PLCB1; http://www.genecards.org/cgi-bin/carddisp.pl?gene=PLCB4&keywords=PLCB4). ADCY2 and ADCY8 code for adenylyl cyclase type 2 and 8 that act as catalysts for the formation of cAMP which is involved in many cellular processes (http://www.genecards.org/cgi-bin/carddisp.pl?gene=ADCY2&keywords=ADCY2; http://www.genecards.org/cgi-bin/carddisp.pl?gene=ADCY8&keywords=ADCY8). CAMK2B and CAMK2D code for calcium/calmodulin-dependent protein kinases that function as mediators of calcium signaling in cells (http://www.genecards.org/cgi-bin/carddisp.pl?gene=CAMK2B&keywords=CAMK2B; http://www.genecards.org/cgi-bin/carddisp.pl?gene=CAMK2D&keywords=CAMK2D). MAPK11 and MAPK14 code for p38 mitogen-activated protein kinases 11 to 14 that function as mediators of the cellular response to external signals [34]. EGFR codes for epidermal growth factor receptor that act as a receptor for the growth factor (http://www.genecards.org/cgi-bin/carddisp.pl?gene=EGFR&keywords=EGFR). GRB2 codes for a growth factor receptor-bound protein that functions as a signal transducer (http://www.genecards.org/cgi-bin/carddisp.pl?gene=GRB2&keywords=GRB2). FYN codes for protein-tyrosine kinase that acts as an activator of molecular signals (http://www.genecards.org/cgi-bin/carddisp.pl?gene=FYN&keywords=FYN). ITGB5 codes for integrin beta type 5 that functions as a receptor for fibronectin (http://www.genecards.org/cgi-bin/carddisp.pl?gene=ITGB5&keywords=ITGB5), which regulates cell proliferation and differentiation during the development of ovarian follicles and mammary gland cells.
The G protein subunit gamma 2 ( GNG2), G protein subunit gamma transducin 1 (GNGT1), and G protein subunit alpha O1 (GNAO1) genes were involved in 6 significantly enriched pathways and accounted for 0.040% of the genetic variance for MY, 0.027% for FY, and 0.020% for AFC (Supplementary File S1). The predicted values of the set of SNP associated with these genes were −0.177 for MY, 0.001 for FY, and −0.002 for AFC (Supplementary File S2). Thus, the combined effect of the three second alleles from these genes would decrease MY, increase FY, and decrease AFC, and the set of first alleles of these genes would have the opposite effect. These three genes involved in the glutamatergic, GABAergic and dopaminergic synapse pathways. These three pathways are involved in the onset of puberty [28]. Lastly, GNAO1 participates in the development of ovarian follicles [21].
Pathway enrichment and PPI network analyses indicated that MY, FY, and AFC of animals in the Thai multibreed dairy population were influenced by sets of genes that were important for cellular processes, nervous and digestive systems and environmental adaptation. Cellular processes were involved with the largest number of biological pathways and PPI among genes associated with MY, FY, and AFC. This likely occurred because cellular processes are important for fundamental cell activities related to the development of cells from the mammary gland and the development of ovarian follicles. Although individual genes or biological pathways explained a small fraction of the genetic variance for MY, FY, and AFC, the combined effect of all genes in all enriched biological pathways and the PPI network explained a substantially larger amount of the genetic variance for these traits. Thus, the set of SNP associated with the enriched pathways and the PPI network in this study could be considered as specific genomic selection targets to help increase MY, FY, and decrease AFC in the Thai multibreed dairy population. However, because the amount of explained genetic variation for each trait was a minor fraction of their total, these studies need to continue with the ultimate goal of accounting for most of the genetic variation due to biological processes in the Thai multibreed dairy population. It should be kept in mind that size and direction of the predicted SNP values here will likely differ in other dairy populations due to breed composition and environmental conditions (climate, management, nutrition, and health care) and will also likely differ over time as population characteristics and environmental conditions change.
ACKNOWLEDGMENTS
The authors gratefully thank the Royal Golden Jubilee project (PHD/0040/2558) of the Thailand Research Fund for the financial support and the University of Florida for supporting the training of the first author as a research scholar. The authors thank the National Science and Technology Development Agency, the Kasetsart University and the Dairy Farming Promotion Organization of Thailand for supporting the genomic dataset for this research as part of project SK(KS)1.58. We also thank Thai dairy farmers, dairy cooperatives and private organizations for their participation and support.
Footnotes
CONFLICT OF INTEREST
We certify that there is no conflict of interest with any financial organization regarding the material discussed in the manuscript.
Supplementary Data
REFERENCES
- 1.Koonawootrittriron S, Elzo MA, Thongprapi T. Genetic trends in a Holstein × other breeds multibreed dairy population in central Thailand. Livest Sci. 2009;122:186–92. [Google Scholar]
- 2.Laodim T, Elzo MA, Koonawootrittriron S, et al. Identification of SNP markers associated with milk and fat yields in multibreed dairy cattle using two genetic group structures. Livest Sci. 2017;206:95–104. [Google Scholar]
- 3.Yodklaew P, Koonawootrittriron S, Elzo MA, et al. Genome-wide association study for lactation characteristics, milk yield and age at first calving in a Thai multibreed dairy cattle population. Agric Nat Res. 2017;51:223–30. [Google Scholar]
- 4.Jattawa D, Elzo MA, Koonawootrittriron S, et al. Imputation accuracy from low to moderate density single nucleotide polymorphism chips in a Thai multibreed dairy cattle population. Asian-Australas J Anim Sci. 2016;29:464–70. doi: 10.5713/ajas.15.0291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Aguilar I, Misztal I, Johnson DL, et al. Hot topic: a unified approach to utilize phenotypic, full pedigree, and genomic information for genetic evaluation of Holstein final score. J Dairy Sci. 2010;93:743–752. doi: 10.3168/jds.2009-2730. [DOI] [PubMed] [Google Scholar]
- 6.Raven LA, Cocks BG, Goddard ME, et al. Genetic variants in mammary development, prolactin signalling and involution pathways explain considerable variation in bovine milk production and milk composition. Genet Sel Evol. 2014;46:29. doi: 10.1186/1297-9686-46-29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Edwards SM, Thomsen B, Madsen P, et al. Partitioning of genomic variance reveals biological pathways associated with udder health and milk production traits in dairy cattle. Genet Sel Evol. 2015;47:60. doi: 10.1186/s12711-015-0132-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Purfield DC, Bradley DG, Evans RD, et al. Genome-wide association study for calving performance using high-density genotypes in dairy and beef cattle. Genet Sel Evol. 2015;47:47. doi: 10.1186/s12711-015-0126-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sargent FD, Lytton VH, Wall JROG. Test interval method of calculating dairy herd improvement association records. J Dairy Sci. 1968;51:170–9. [Google Scholar]
- 10.Koonawootrittriron S, Elzo MA, Tumwasorn S. Multibreed genetic parameters and predicted genetic values for first lactation 305-d milk yield, fat yield, and fat percentage in a Bos taurus ×Bos indicus multibreed dairy population in Thailand. Thai J Agric Sci. 2002;35:339–60. [Google Scholar]
- 11.Sargolzaei M, Chesnais JP, Schenkel FS. A new approach for efficient genotype imputation using information from relatives. BMC Genomics. 2014;15:478. doi: 10.1186/1471-2164-15-478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Legarra A, Aguilar I, Misztal I. A relationship matrix including full pedigree and genomic information. J Dairy Sci. 2009;92:4656–63. doi: 10.3168/jds.2009-2061. [DOI] [PubMed] [Google Scholar]
- 13.Misztal I, Tsuruta S, Lourenco D, et al. Manual for BLUPF90 family of programs [Internet] Athens, GA, USA: University of Georgia; 2015. [cited 2017 Aug 10]. Available from: http://nce.ads.uga.edu/wiki/lib/exe/fetch.php?media=blupf90_all2.pdf. [Google Scholar]
- 14.Tsuruta S. Average Information REML with several options including EM-REML and heterogeneous residual variances [Internet] Athens, GA, USA: University of Georgia, USA; 2016. [cited 2017 Aug 25], Available from: http://nce.ads.uga.edu/wiki/lib/exe/fetch.php?media=blupf90_all4.pdf. [Google Scholar]
- 15.Wang H, Misztal I, Aguilar I, et al. Genome-wide association mapping including phenotypes from relatives without genotypes in a single-step (ssGWAS) for 6-week body weight in broiler chickens. Front Genet. 2014;5:134. doi: 10.3389/fgene.2014.00134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hanna LLH, Riley DG. Mapping genomic markers to closest feature using the R package Map2NCBI. Livest Sci. 2014;162:59–65. [Google Scholar]
- 17.Bindea G, Mlecnik B, Hackl H, et al. ClueGO: a Cytoscape plug-in to decipher functionally grouped gene ontology and pathway annotation networks. Bioinformatics. 2009;25:1091–3. doi: 10.1093/bioinformatics/btp101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Szklarczyk D, Franceschini A, Wyder S, et al. STRING v10: protein-protein interaction networks, integrated over the tree of life. Nucleic Acids Res. 2015;43:D447–52. doi: 10.1093/nar/gku1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Shannon P, Markiel A, Ozier O, et al. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 2003;13:2498–504. doi: 10.1101/gr.1239303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tang Y, Li M, Wang J, et al. CytoNCA: a cytoscape plugin for centrality analysis and evaluation of protein interaction networks. Biosystems. 2015;127:67–72. doi: 10.1016/j.biosystems.2014.11.005. [DOI] [PubMed] [Google Scholar]
- 21.Zielak-Steciwko AE, Browne JA, McGettigan PA, et al. Expression of microRNAs and their target genes and pathways associated with ovarian follicle development in cattle. Physiol Genomics. 2014;46:735–45. doi: 10.1152/physiolgenomics.00036.2014. [DOI] [PubMed] [Google Scholar]
- 22.Haisenleder D, Farris HA, Shapnik MA. The calcium component of gonadotropin-releasing hormone-stimulated luteinizing hormone subunit gene transcription is mediated by calcium/calmodulin-dependent protein kinase type II. Endocrinology. 2003;144:2409–16. doi: 10.1210/en.2002-0013. [DOI] [PubMed] [Google Scholar]
- 23.Munshi A, Ramesh R. Mitogen-activated protein kinases and their role in radiation response. Genes Cancer. 2013;4:401–8. doi: 10.1177/1947601913485414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wang W, Pan YW, Wietecha T, et al. Extracellular signal-regulated kinase 5 (ERK5) mediates prolactin-stimulated adult neurogenesis in the subventricular zone and olfactory bulb. J Biol Chem. 2013;288:2623–31. doi: 10.1074/jbc.M112.401091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bionaz M, Loor JJ. Gene networks driving bovine milk fat synthesis during the lactation cycle. BMC Genomics. 2008;9:366. doi: 10.1186/1471-2164-9-366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Clarkson J, Herbison AE. Development of GABA and glutamate signaling at the GnRH neuron in relation to puberty. Mol Cell Endocrinol. 2006;254–255:32–8. doi: 10.1016/j.mce.2006.04.036. [DOI] [PubMed] [Google Scholar]
- 27.Liu X, Herbison AE. Dopamine regulation of gonadotropin-releasing hormone neuron excitability in male and female mice. Endocrinology. 2013;154:340–50. doi: 10.1210/en.2012-1602. [DOI] [PubMed] [Google Scholar]
- 28.Fortes MR, Reverter A, Zhang Y, et al. Association weight matrix for the genetic dissection of puberty in beef cattle. Proc Natl Acad Sci USA. 2010;107:13642–7. doi: 10.1073/pnas.1002044107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Golombek DA, Rosenstein RE. Physiology of circadian entrainment. Physiol Rev. 2010;90:1063–102. doi: 10.1152/physrev.00009.2009. [DOI] [PubMed] [Google Scholar]
- 30.Plaut K, Casey T. Does the circadian system regulate lactation? Animal. 2012;6:394–402. doi: 10.1017/S1751731111002187. [DOI] [PubMed] [Google Scholar]
- 31.Fu M, Zhang L, Ahmed A, et al. Does circadian disruption play a role in the metabolic-hormonal link to delayed lactogenesis II? Front Nutr. 2015;2:4. doi: 10.3389/fnut.2015.00004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Howard JT, Kachman SD, Snelling WM, et al. Beef cattle body temperature during climatic stress: a genome-wide association study. Int J Biometeorol. 2014;58:1665–72. doi: 10.1007/s00484-013-0773-5. [DOI] [PubMed] [Google Scholar]
- 33.West JW. Effects of heat-stress on production in dairy cattle. J Dairy Sci. 2003;86:2131–44. doi: 10.3168/jds.S0022-0302(03)73803-X. [DOI] [PubMed] [Google Scholar]
- 34.Johnstone ED, Sibley CP, Lowen B, et al. Epidermal growth factor stimulation of trophoblast differentiation requires MAPK11/14 (p38 MAP kinase) activation. Biol Reprod. 2005;73:1282–8. doi: 10.1095/biolreprod.105.044206. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.