

Abstract
The introduction and advances on next-generation sequencing have led to novel ways to integrate simultaneous assessment of multiple target genes in routine laboratory analysis. Assessment of myeloid neoplasms with targeted next-generation sequencing panels shows evidence to improve diagnosis, assist therapeutic decisions, provide better information about prognosis, and better detection of minimal residual disease. Herein, we provide information for application and utilization of next-generation sequencing studies with a focus on the most important mutations in acute myeloid leukemia, myelodysplastic syndrome, myeloproliferative neoplasms, and other myelodysplastic / myeloproliferative neoplasms in order to integrate them into the daily clinical practice.
Keywords: Myeloid neoplasia, next-generation sequencing, somatic mutation
Next-generation sequencing (NGS) technology can be of great utility in the clinical assessment of various neoplasms. NGS can be rapidly integrated into the diagnostic laboratories and merged into current clinical practice. However, bioinformatics and genetic code technology and language are difficult to understand for practitioners who are not familiar with molecular biology. We will review the recent developments in NGS, technical progress, and focus on the approach of diseases affecting the myeloid lineage, including acute myeloid leukemia (AML), myelodysplastic syndrome (MDS), chronic myeloproliferative diseases (CMPDs) such as polycythemia vera (PV), essential thrombocythemia (ET), primary myelofibrosis (PMF), atypical CMPD, chronic myelomonocytic leukemia (CMML) and chronic eosinophilic leukemia (CEL) and mast cell disease.
Due to the increasing number of mutations that are necessary for diagnosis, it is becoming impractical to do a single gene testing analysis. Furthermore, multigene assessment is providing guidance for determining prognosis in many diseases. Since many of the diseases encountered in clinical practice show more than one molecular abnormality, we could adapt NGS based assays to have simultaneous detection of multiple somatic mutations in tens or hundreds of target genes that are associated with specific diseases. NGS assays are becoming cheaper, faster, and rapidly adopted by many laboratories and since there is an increasing number of actionable targets that could benefit from these assays. However, NGS technology requires highly complex bioinformatics and laboratory technology to develop a diagnostic pipeline. Currently, there are two commonly used NGS. Sequencing differs in these technologies but they use similar bioinformatics principles for determining abnormal sequences that are not matching reference sequences.
There are also three different enrichment methods used for NGS sequencing: Hybrid capture, Amplicons, and Anchored Multiplex polymerase chain reaction (1). However, until now there is no single database available for clinical annotations to determine the significance of variants. Moreover, these assays are expensive and reimbursement remains low.
The introduction of NGS into clinical laboratories has significantly changed the use of molecular techniques. Testing for multiple genes in a panel using NGS platforms is more practical and economical (2,3). Furthermore, the cost of adding a new gene to the panel is relatively inexpensive, in comparison to performing multiple single-gene tests. The presence of specific genetic mutations could aid clinicians in 4 ways: 1) diagnostic algorithms, 2) prognosis assessment, 3) use of targeted therapy, and 4) monitoring treatment response and residual disease. Despite increased utilization, NGS assays remain challenging particularly for the identifications of variants without a known clinical significance. To address these concerns, new terms in NGS reports have been adopted: “actionable”, “potentially actionable”, “Variant of Undetermined Significance”, “likely benign” or “benign”.
Mutation profiles in acute myeloid leukemia
The Cancer Genome Atlas Network has many studies that report recurrent mutations and show a complex network of genetic mutations (4,5). Despite these exhaustive studies, there is no gene-specific diagnostic entity. However, there are some mutations leading to an aberrant activation of proteins that have a crucial effect on hematopoietic progenitor cell proliferation and differentiation. The mutations in AML involve epigenetic modifiers (TET2, IDH1/IDH2, DNMT3A, ASXL1, KMT2A, EZH2), activated signaling pathway (FLT3, KRAS, NRAS, KIT), tumor suppressor genes (TP53, WT1), RNA splicing (SF3B1), nucleophosmin mutation (NPM1), and genes coding for transcription-differentiation (CEBPA, RUNX1) (Figure 1) (6,7,8). Recurrently mutated genes in AML and MDS are listed in Table 1.
Figure 1.
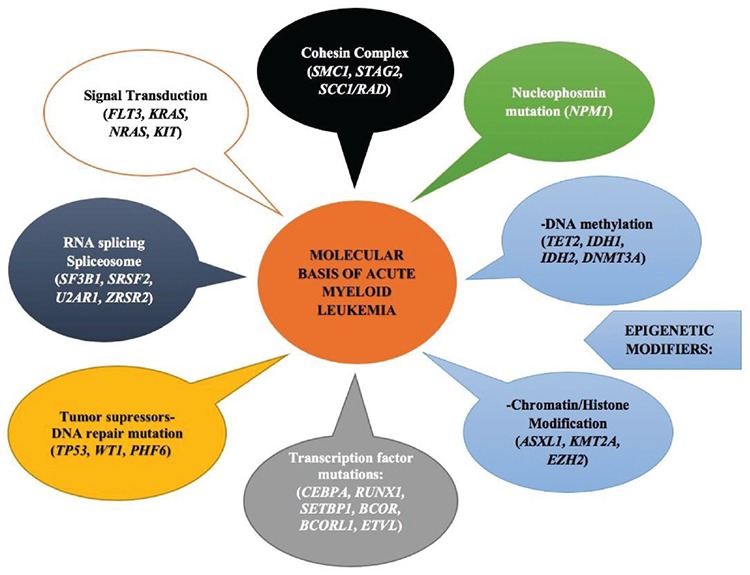
The most frequent gene mutations and cellular pathways in acute myeloid leukemia.
Table 1. Common gene mutations in AML, MDS, and their clinical significance.
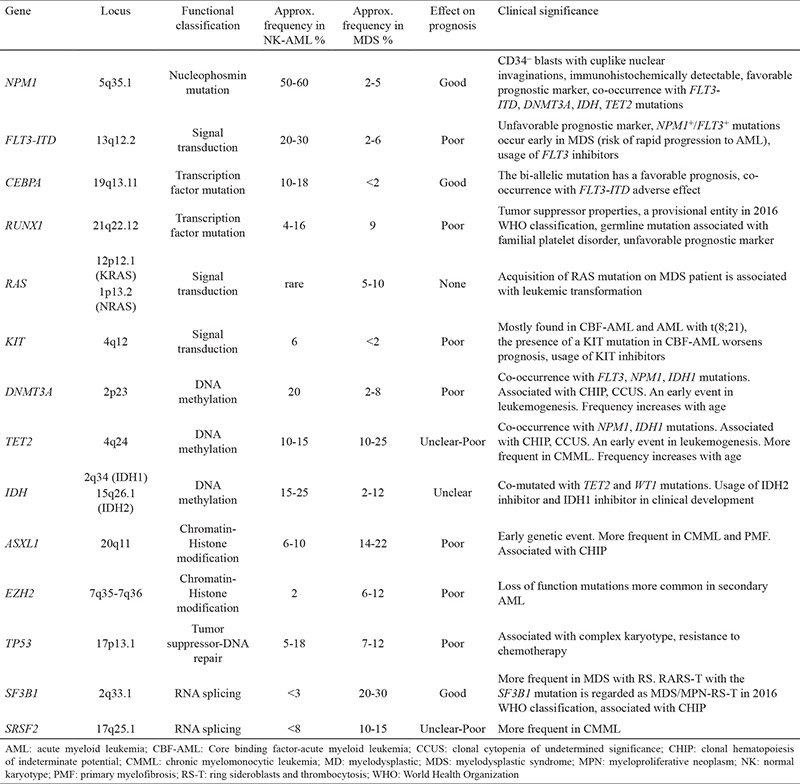
Recent studies have demonstrated that genes mutated in AML with a normal karyotype AML (NK-AML) share significant overlap with genes mutated in MDS (9,10). The World Health Organization (WHO) 2016 classification recognizes AML with mutated NPM1 and AML with bi-allelic mutation of CEBPA as specific to AML classification categories and AML with mutated RUNX1 as a provisional entity (8,9,10). The most frequently mutated genes in NK-AML occur in the following genes: nucleophosmin (NPM1), Fms-related tyrosine kinase 3 (FLT3) and DNA methyltransferase 3A (DNMT3A), Ten-eleven translocation-2 (TET2), isocitrate dehydrogenase 1 and 2 (IDH1 and IDH2), CCAAT/enhancer binding protein alpha (CEBPA), NRAS, Additional sex comb-like 1 (ASXL1), WT1 and runt-related transcription factor-1 (RUNX1). Mutations of FLT3, DNMT3A, and NPM1 are often present concurrently, while other mutations of NPM1, RUNX1, CEBPA, and TP53 are almost always mutually exclusive both at diagnosis and during disease transformation (11,12,13,14,15,16,17,18,19).
The WHO classification of AML includes many specific chromosomal abnormalities determined by karyotyping (11). The gene mutations recognized as specific entities by the 2016 WHO classification include NPM1, CEBPA, and RUNX1 mutations. However, since the publication of this classification, it became very important to clinically recognize other pathogenic variants such as those involving FLT3 (ITD and TKD) and IDH1/2. This can drastically change the clinical management of AML patients since the US Food and Drug Administration (FDA) has approved specific inhibitors for FLT3 and IDH2 positive AML (20,21).
The gene NPM1 is the most frequently mutated gene in the adult type AML. It occurs approximately in 50 to 60% of cytogenetically normal AMLs and in 2 to 5% of MDS cases. It is often characterized by CD34 antigen negativity with a bright expression of CD33 and monocytic differentiation. NPM1 mutations include small insertions (4-11bp in size), resulting in an aberrant cytoplasmic localization of the mutant protein nucleophosmin which can be detected immunohistochemically (21,22). NPM1 mutations increase disease-free and overall survival in patients with AML. Secondary mutations [FLT3-ITD, DNMT3A (21), IDH, TET2 mutations] are frequently accompanying these patients. FLT3-ITD mutations are observed in 40% of NPM1 mutated AML patients. In general, FLT3 ITD positive patients without other molecular abnormalities show poor clinical outcome while NPM1 positive patients have a better clinical response. Coexistence of FLT3-ITD mutation in NPM1 positive patients is associated with a poor prognosis in young AML patients compared with patients with NPM1 mut /FLT3 wild type (4,11,17,23).
Among transcription factor mutations in myeloid neoplasia, CEBPA and RUX1 mutations are deemed important by the WHO due to their prognostic implications. CCAAT Enhancer Binding Protein-alpha (CEBPA) is a transcription factor responsible for promoting granulocytic maturation in regulating cellular function. CEBPA mutations are observed in approximately 10% of de novo AML patients and are commonly bi-allelic. The bi-allelic mutation is a favorable prognostic marker. Germline mutations of CEBPA predispose to the development of AML in young patients. The concurrence of FLT3-ITD mutation is an adverse prognostic finding in CEBPA mutated-AML patients (23). RUNX1 encodes the DNA binding alpha subunit of the core binding factor (CBFalpha) that is involved in the normal differentiation of hematopoietic cells. RUNX1 has also tumor suppressor properties and loss of function of the gene that contributes to tumorigenesis in myeloid cancers. De novo AML with mutated RUNX1 in the absence of MDS related cytogenetic abnormalities appears to be a distinct entity with adverse outcome. RUNX1 mutations are detected in approximately 4% to 16% of AML and 9% of MDS (11). They are commonly seen in older patients with NK-AML and MDS-associated AML. In several multivariate analyses, they have been associated with worse overall survival (23,24,25).
FLT3 is a class III receptor tyrosine kinase coding for signal transduction gene. This receptor is important for proliferation and differentiation of hematopoietic stem cells. Its expression is downregulated during differentiation. FLT3 mutations occur in 30% to 40% of AML patients with normal cytogenetics and 2% to 6% of MDS cases. Two types of mutations are observed in FLT3 gene: FLT3-ITD (internal tandem duplications) mutations (with a frequency of 75% to 80%) and FLT3-TKD (tyrosine kinase domain) (with a frequency of 20% to 25%). FLT3 mutated patients show higher white blood cell count, loss of HLA-DR and CD34 antigen negativity, and impaired survival (26). AML patients with FLT3-ITD mutation appear to benefit from allogeneic stem cell transplantation. However, the prognostic effect of FLT3-TKD remains controversial (27). In 2017, the FDA approved treatment with midostaurin for newly diagnosed patients with FLT3-mutated AML due to the observation of a significant beneficial outcome (28,29). In MDS, FLT3 mutations are observed in high-risk subgroups and are associated with complex karyotype (30,31). NPM1 and FLT3 mutations are found primarily in AML; however, the presence of these mutations in an MDS patient should alert the clinician about the risk of rapid progression to AML (22). Alterations on FLT3 signaling pathway seems to be the most important prognostic factor for overall survival in AML patients younger than 60 years.
The other signal transduction genes are KIT mutations that are frequent in gastrointestinal stromal tumors, germ cell tumors, melanomas, and systemic mast cell diseases. They can be detected in approximately 6% of patients with newly diagnosed AML and 20% of AML patients with t(8;21) RUNX1-RUNXT1 and 30% of AML patients with inv(16) or t(16;16) CBFB-MYH11 (the core binding factor AMLs-CBF-AML). KIT mutation in AML increases the risk of relapse and worsens the good prognosis of CBF-AML (32,33,34,35,36).
There are a number of other genes playing a role in the pathogenesis of AML and appear to be important for overall survival despite not being recognized as specific entities. DNA methylation mutations and chromatin/histone modifications belong to epigenetic modifiers. DNMT3A encodes an epigenetic regulator and mediates de novo methylation of CpG dinucleotides and are seen in approximately 20% of AML patients with normal karyotype. This mutation tends to shorten overall survival (14,37). TET2 is also involved in the epigenetic regulation of DNA methylation. TET2 mutations occur in approximately 10% to 15% of AMLs. As TET2 is a key regulator in hematopoietic stem cell renewal and differentiation, their mutations result in increased stem cell renewal and myeloid hyperplasia with impaired differentiation. These tend to have a poorer prognosis in NK-AML. TET2 mutations predict the response to hypomethylating agents (4,38).
IDH1/IDH2 mutations prevent the conversion of isocitrate to alpha-ketoglutarate in the Krebs cycle; create an oncometabolite that inhibits the function of TET2. IDH1/IDH2 mutations often mutate with TET2 and WT1 mutations (39). Between 15% to 25% of AML and approximately 2% to 12% of MDS cases display IDH mutations (40). The prognostic significance of IDH mutations in AML is currently unclear. IDH2 inhibitor had been successful in the management of IDH2 positive AML patients while IDH1 inhibitor is showing promising results in ongoing clinical trials (4,20,34,40).
Chromatin/Histone Modification mutations such as Additional Sex Coms Like Transcriptional Regulator 1 (ASXL1) mutations and Enhancer of Zeste 2 Polycomb Repressive Complex 2 Subunit (EZH2) mutations are a part of the epigenetic modifiers. ASXL1 gene is involved in the epigenetic regulation of gene expression. They are uncommon in de novo AML (6.5%), but more frequent in AML arising from a prior myeloid neoplasm (up to 30%) (41,42,43). These mutations are associated with shorter overall survival and resistance to chemotherapy. KMT2A: lysine(K)-specific methyltransferase 2A (KMT2A/MLL) gene plays a role in hematopoiesis and cell differentiation. KMT2A mutations co-occur with an additional gene mutation (IDH2/DNMT3A/U2AF1/TET2). This mutation is frequently associated with trisomy 11 (90%) and a poorer prognosis (17,44,45).
TP53 protein is a transcription factor and tumor suppressor gene that determines whether the cell undergoes repair, senescence or apoptosis. Although somatic TP53 mutations are frequently seen in 50% of solid tumors, they are uncommon in de novo AML (5% to 18%) and they are associated with secondary or therapy-related AMLs. TP53 mutations often coexist with complex karyotypes, chemotherapy resistance, and reduced overall survival compared with TP53 wild AML patients. TP53 mut AML patients have also high relapse rates after stem cell transplantation (46,47,48). Despite these findings, a recent study indicated that AML patients with cytogenetic abnormalities associated with unfavorable risk, TP53 mutations, or both had a favorable clinical response to decitabine (49,50).
Figure 2 represents these risk categories associated with distinct molecular mutations that should be screened by NGS in AML patients.
Figure 2.
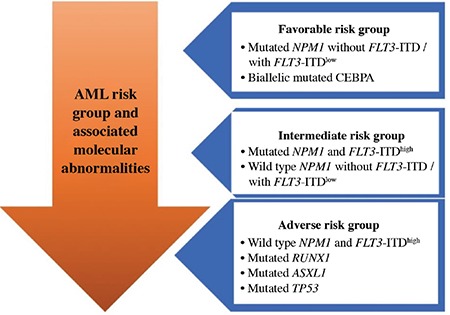
AML risk categories and associated molecular abnormalities. (FLT3-ITD low : FLT3-ITD/wild type allelic ratio <0.5; FLT3-ITD high : FLT3-ITD/wild type allelic ratio >0.5)
AML: acute myeloid leukemia
For the monitoring of minimal residual disease (MRD) in AML; NGS based technologies can be used besides multiparameter flow cytometry (MFC). MFC is a more reliable method for monitoring remission than conventional morphology-based assessment in AML patients; however, MFC lacks sensitivity for detecting residual disease. Therefore, a combined result of sequencing and flow cytometry detects residual disease more specifically. A recent study about molecular MRD in AML showed that the detection of molecular MRD is associated with a significantly higher relapse rate (51). During the complete remission, persistent mutations associated with clonal hematopoiesis (DNMT3A, TET2, and ASXL1 referred as DTA mutations) and non-DTA mutations can be detected by NGS. While DTA mutations do not have prognostic value for MRD, non-DTA mutations (such as mutations in TP53, IDH1, IDH2 and genes related to the Ras pathway) with a higher allele frequency can predict relapse rates (51,52).
Among AML with myelodysplasia-related changes (AML-MRC) and therapy-related AML (t-AML), mutations in splicing factors genes such as SRSF2, SF3B1, U2AF1, ZRSR2, mutations on chromatin/histone modifications like ASXL1, EZH2, BCOR, STAG2, and TP53 mutations are common (53). NPM1 mutations in the absence of FLT3-ITD and double CEBPA mutations are fairly uncommon in AML-MRC and are associated with favorable overall survival. Patients in this category with TP53 mutations had a poor prognosis regardless of age (46,54,55,56). After treating patients with alkylating agents/ionizing radiation, patients often have increased blasts with associated multilineage dysplasia that cause similar mutation profiles like in AML-MRC. Mutations of TP53 can be seen in as many as 50% of t-AML cases with worse survival (11,53,57,58). Following a topoisomerase II inhibitor therapy, 20 to 30% of patients develop overt acute leukemia without a preceding myelodysplastic phase. These patients often show balanced chromosomal translocations involving 11q23 (KMT2A/MLL) or 21q22 (RUNX1) and are associated with monoblastic or myelomonocytic morphology (11,53,59,60). Mutations in TP53, TET2, and PTPN11 (protein tyrosine phosphatase, non-receptor type 11), IDH1/2, NRAS are frequent in t-AML, but FLT3 and NPM1 mutations are less frequent than de novo AML (58,61).
Mutation profiles in Myelodysplastic Syndrome
Approximately half of the myelodysplastic syndrome is associated with recurrent cytogenetic abnormalities such as -5/del(5q), -7/del(7q), +8del(20q) or complex karyotypes (62). Targeted panels and whole-genome NGS assay detect somatic mutations in up to 90% of MDS patients (63,64,65). Some of the mutations overlap with those observed in AML, none of these mutations are specific for MDS diagnosis. However, having multiple genetic abnormalities with high variant allele frequency is starting to be recognized for its supportive evidence of evolving MDS (66).
The most frequently mutated genes in MDS are SF3B1 (~25%), TET2 (~25%-20%), ASXL1 (~15%), SRSF2 (~15%), RUNX1 (~15%-10%), DNMT3A (~15%-10%) (23). Mutations in ASXL1, TP53, EZH2, ETV6, and RUNX1 are also found to be recurrent in MDS patients and predict a poor overall survival (41). DNMT3A mutations occur early in the course of MDS and suggest an early genetic event in leukemogenesis. Patients with DNMT3A mutations have a worse overall survival and more rapid progression to AML. They are associated with mutations in FLT3 (ITD or TKD), NPM1, IDH1 genes (67). TP53 mutations in MDS are associated with the very aggressive disease. Even in low-risk MDS with del(5q), the presence of a TP53 mutation at low frequency is correlated with a higher rate of leukemic transformation and poor response to lenalidomide (11,68).
Mutations in splicing factors are commonly detected in MDS patients with a frequency of 50%. The most common of these mutations are SF3B1, SRSF2, U2AF1, and ZSR2 mutations. SF3B1 mutations are the most common among splicing mutations in MDS. These mutations are highly correlated with the presence of ring sideroblast (RS). They are observed in 20% to 28% of MDS, more frequent in MDS with RSS and the frequency of SF3B1 mutation in AML is <5% (11,23,69,70,71). Mutations in Cohesin Complex genes (SMC1, SMC3, SCC1/RAD, STAG2) can be germline resulting in Congenital Malformation syndrome. These mutations can be detected in 10% to 25% of MDS and 10% to 15% of de novo AML (17,69).
Nearly all cases of MDS show at least one mutation and demonstrate that specific mutation with a targeted NGS panel is highly useful for the diagnosis of MDS. However, determination of mutational burden/variant allele frequency (VAF) (usually >25% for MDS) is critical for the diagnostic assessment to distinguish MDS from clonal hematopoiesis of indeterminate potential (CHIP) or clonal cytopenia of undetermined significance (CCUS). CHIP is the proposed term for healthy individuals, lacking hematological malignancy or clonal disorder, but carrying a hematological somatic mutation. Key findings in CHIP are single gene mutations (mostly DNMT3A, TET2 or ASXL1 mutation), increased risk of developing hematological malignancy, and increased rate of cardiovascular mortality regardless of their cancer risk. The single mutation burden in CHIP has a VAF more than 2% but typically not exceeding 20%-30%, which allows the distinction from underlying dysplastic process (3). Patients who are not meeting the WHO defined criteria for a hematologic neoplasm, but show a clonal mutation with unexplained cytopenia, are referred to as CCUS. The mutation spectrum of CCUS patients is similar to mutations seen in MDS. Mutations in spliceosome genes (SF3B1, SRSF2, U2AF1, ZRSR2) have the highest predictive value for CCUS (72). Co-mutations of DNMT3A, TET2, and ASXL1 genes remain also common in CCUS. There are no clear recommendations to follow up CCUS patients. However, cytopenia, mutation burden, number and type of the mutations or the detection of additional mutations in the follow-up of a patient with suspected MDS appear to have a course similar to MDS (72,73,74,75,76).
Mutation profiles in myeloproliferative and myelodysplastic/myeloproliferative neoplasms
Disorders altering the myeloid elements include the following disorders: Polycythemia vera (PV), essential thrombocythemia (ET), primary myelofibrosis (PMF), chronic myelogenous leukemia (CML), chronic neutrophilic leukemia (CNL), hypereosinophilic leukemia/CEL, CMML, and mast cell disease. Among CMPD (PV, ET, PMF, and CML), molecular markers have been essential for diagnosis. Recurrent somatic mutations in BCR-ABL2 negative MPNs and MDS/MPNs are listed in Table 2.
Table 2. Common gene mutations in MPN, MDS/MPN, and their clinical significance.
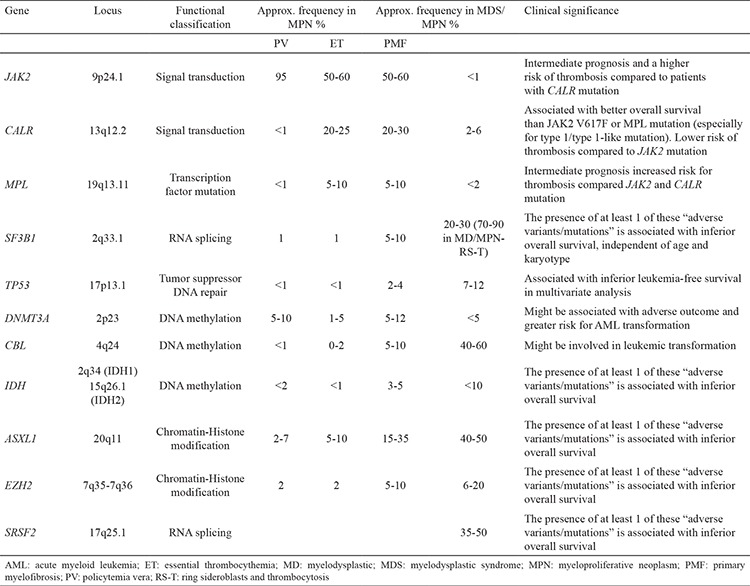
The most frequent mutation in BCR-ABL2 negative MPN, JAK2 exon 14 (V617F) mutations are observed in 95% of PV patients (77). The majority of remaining PV patients may harbor a mutation in JAK2 exon 12 (missense mutation, deletion, insertion). JAK2 (V617F) mutation occurs in approximately 50-60% of ET or PMF patients. Homozygous mutations of JAK2 (V617F) (with a VAF value >50%) are more common in PV than in ET (78,79). JAK2 (V617F) mutation may not be present in half of the MPN patients. This type of mutation has rarely been reported in patients with MDS/MPN; however, they are found in patients with MDS, CMML, atypical CML, and de novo AML (approx. <5%) and MDS/MPN-RS-T (approx. 50%). In PV and ET; JAK2 (V617F) allele burden is also associated with a more aggressive behavior. Thrombotic events and fibrotic transformation are more common in patients with a high JAK2 (V617F) allele burden (higher JAK2 VAF). These findings suggest that monitoring JAK2 (V617F) allele burden could be useful to identify patients at higher risk of myelofibrotic transformation. JAK2 mutation in PMF is associated with intermediate prognosis when compared with CALR mutation (3,11,17,23,80).
Patients with ET have mutations in the CALR gene that encodes the endoplasmic reticulum and are associated with chaperone calreticulin at a frequency of 20% to 25%. CALR mutations are frequently seen in ET and PMF patients with JAK2 wild and MPL wild type. They are rarely detected in PV, CMML, MDS/MPN patients. CALR mutations are not seen in AML, mastocytosis, lymphoid neoplasia and solid tumors (81,82). CALR mutated MPNs demonstrate distinct features; such as younger age group, lower hemoglobin levels, higher platelet counts, decreased risk of thrombosis, and improved overall survival compared to patients with JAK2 or MPL mutations (83,84). In ET and PMF patients, CALR show 2 different mutations: type-I that typically has 52-bp deletion (p.L367fs*46), and type-II characterized by 5-bp TTGTC insertion (p.K385fs*47). Type I CALR mutation is detected more frequently in PMF than ET. Type II CALR mutations tend to show leukemic transformation (namely increased leucocyte count and higher circulating blasts) than type I CALR mutation (81,85,86). Platelet count is significantly higher in type II vs type I CALR mut ET patients, but no difference was noted in thrombosis-free survival in a study with a large cohort of ET patients. However, there is a significant survival difference in the setting of PMF, as CARL type-I positive PMF patients do much better than CALR type-II or JAK2 (V617F) positive PMF patients (84).
The activating mutation on codon W515 of MPL gene is seen in 5-10% of ET and PMF patients who are negative for JAK2 (V617F) (87). Patients that are JAK2 wild and CALR wild , but harboring ET or PMF clinical features should be screened for MPL mutation. MPL mutated patients have increased the risk for thrombotic episodes and transfusion requirement compared with JAK2 mut and CALR mut patients (80,88,89). Patients with PMF have mutations for JAK2 in 60%, CALR in 20-30% and MPL in5-10% of the cases (Figure 3). The remaining 10% to 15% of patients with ET or PMF patients have none of the above-mutated genes and these are named “triple negative” (85,90,91). This is a heterogeneous category and some of the ET patients may have rare non-canonical mutations in JAK2, MPL, and SH2B3; however, the large majority remain without a specific mutation. For “triple negative” PMF patients, a detailed search for the mutation profiles regarded as a marker of clonality (such as ASXL1, EZH2, TET2, IDH1/IDH2, SRSF2, SF3B1) is currently recommended (77). “Triple negative” patients tend to show inferior outcome compared to JAK2 mut /CALR must and MPL mut CMPD patients. The presence of at least one of the 3 variants/mutations (ASXL1, SRSF2, and IDH2) is associated with inferior overall and myelofibrosis-free survival in patients with PV. SH2B3, IDH2, U2AF1, SF3B1, EZH2, and TP53 mutations are identified as significant risk factors for inferior overall, myelofibrosis-free survival, and leukemia-free survival in patients with ET (85).
Figure 3.
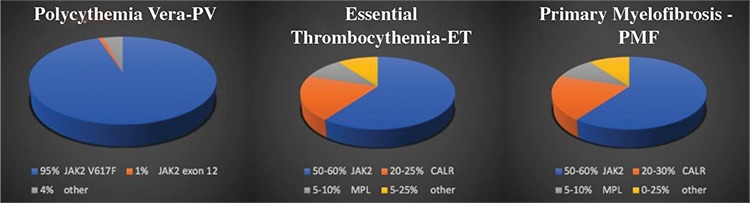
Distribution of mutation profiles in myeloproliferative neoplasm.
ET: essential thrombocythemia; PMF: primary myelofibrosis; PV: policytemia vera
Recent studies about leukemic transformation in MPNs show ASXL1, SRSF2, IDH1, IDH2, RUNX1 mutations have been associated with leukemic transformation in PMF, whereas SRSF2, IDH2 mutations in PV and TP53, EZH2 mutations in ET predicts more leukemic transformation (92). These findings show that the mutation spectrum in primary NK-AML is different from secondary AML. Recurrent somatic gene mutations are observed in up to 90% of CMML patients. TET2 (~60%), SRSF2 (~50%), ASXL1 (~50%), RUX1, NRAS, and TP53 are the most frequent mutations. However, these mutations can also be encountered in healthy aging people such as CHIP with a single mutation of a VAF <20-30%. Therefore, a low VAF of these mutations should not be used as a definitive diagnosis for CMML (23). ASXL1 mutations are associated with a higher white blood cell count, lower hemoglobin, extramedullary disease, and an abnormal karyotype. ASXL1 mutations lead to the aggressive clinical course in CMML patients (93,94). The JAK2 mutation also occurs in CMML patients sharing some mutual features with JAK2 mut MPNs. These cases predict more reticulin fibrosis, occasional megakaryocytic clustering with atypia, and erythroid and megakaryocytic hyperplasia (95). In case of presence, NPM1 mut AML with monocytic differentiation should be suspected. Variable results are also expected on JAK mut CMML patients treated with JAK2 inhibitors (11,96,97).
Juvenile myelomonocytic leukemia (JMML) patients harbor also clonal cytogenetic abnormalities. The commonly mutated genes are KRAS/NRAS, NF1, PTPN11, CBL, which encode proteins of the Ras oncogene pathway. PTPN11 mutations are the most frequent alterations (~35%). Somatic NRAS and KRAS mutations occur in 20-25% of JMML cases. Germline mutations in CBL and NF1 genes are also frequent and are associated with Noonan Syndrome-like disorder and neurofibromatosis type-I on JMML patients, respectively (98,99).
SETBP1 and ethanolamine kinase 1 (ETNK1) mutations are relatively common in atypical CML (aCML). CSF3R mutation is frequently associated with CNL, this type of mutation is uncommon (<10%) in aCML and rarely encountered in CMML and AML cases (100,101). Correlation of CSF3R mutation with CNL has been included in the diagnostic criteria in the revised 2016 WHO classification (10,21). In myelodysplastic/myeloproliferative neoplasm with ring sideroblasts and thrombocytosis (MDS/MPN-RS-T), SF3B1 mutations are pesent in 70-90% of cases. They are frequently commutated with JAK2 (V617F) (50-65%) and less frequently with CALR/MPL (11,23,70,71).
Among myeloid neoplasms with eosinophilia; CEL-NOS, idiopathic hypereosinophilic syndrome, myeloid/lymphoid neoplasms with eosinophilia, and gene rearrangement are considered in the revised 2016 WHO classification. However, hypereosinophilia can be encountered in systemic mastocytosis, CML, other types of MPNs, MDS/MPNs, AML, MDS and T cell lymphoproliferative disorders. Once, cytogenetically PDGFRA, PDGFRB, FGFR1 rearrangements, and PCM1-JAK2 fusion and other causes of hypereosinophilia are excluded, NGS should be performed for the patients with eosinophilia.
Lastly, mast cell diseases show point mutations of the KIT gene (D816V) in 95% of cases. Although the presence of D816V mutations is important at diagnosis for systemic mastocytosis, a minority of patients harbor mutations on exon 17 or other exons (102) Additionally; TET2, ASXL1, RUX1, SRSF2, and JAK2 mutations can be seen on the patients with mastocytosis (103,104).
We outlined some of the common genes that are important to assess in various hematologic neoplasms with emphasis on myeloid neoplasia. Despite the significant progress in lymphoid neoplasms, particularly for understanding its pathogenesis, NGS is no ready to be integrated into routine clinical testing. Most of the NGS panels offered for assessment of myeloid neoplasms contain between 30-60 targeted genes. The market for NGS-based diagnostic procedures is rapidly growing, therefore it is impossible to list all the available tests. However, based on current literature, a panel with the following genes will most likely be informative in clinical assessment for most of the myeloid neoplasms: FLT3, NPM1, CEBPA, TP53, IDH1/2, DNMT3A, TET2, CSF3R, SRSF2, KIT, NRAS, RUNX1, WT1, ASXL1, SF3B1 for AML; NRAS, KRAS, IDH1/2, TET2, EZH2, ASXL1, RUNX1, TP53, DNMT3A, SF3B1, U2AF1, ETV6 for MDS; JAK2, CALR, MPL, IDH1/2, ASXL1, TET2, EZH2, SRSF2, SF3B1, TP53for MPN; TET2, SRSF2, ASXL1, RUNX1, NRAS, TP53 for CMML; SF3B1 for MD/MPN RS-T; CSF3R for CNL, SETBP1 for aCML, KIT (D816V) for mast cell disease.
In conclusion; the progress of NGS and widen utilization on hematologic malignancies will facilitate a more precise diagnosis and treatment for patients with hematologic neoplasms. AML, MDS, and MPNs are characterized by morphologic or phenotypic similarities but these disorders harbor different mutations types with different prognostic implications. Myeloid neoplasms that lack a cytogenetic alteration but show a somatic mutation can be diagnosed by the above-mentioned mutation profiles. These mutations can also facilitate new novel therapeutic agents. In the coming years, it appears that major efforts with more comprehensive high throughput NGS sequencing and inclusion of genomic data in clinical management will enable the delivery of precision medicine.
Footnotes
Conflict of Interest: No conflict of interest was declared by the authors.
Financial Disclosure: No financial disclosure was declared by the authors.
References
- 1.Kuo FC. Next generation sequencing in hematolymphoid neoplasia. Seminars in Hematology. 2018. doi: 10.1053/j.seminhematol.2018.05.006. [DOI] [PubMed] [Google Scholar]
- 2.Yohe S, Thyagarajan B. Review of Clinical Next-Generation Sequencing. Arch Pathol Lab Med. 2017;141:1544–57. doi: 10.5858/arpa.2016-0501-RA. [DOI] [PubMed] [Google Scholar]
- 3.Sallman DA, Padron E. Integrating mutation variant allele frequency into clinical practice in myeloid malignancies. Hematol Oncol Stem Cell Ther. 2016;9:89–95. doi: 10.1016/j.hemonc.2016.04.003. [DOI] [PubMed] [Google Scholar]
- 4.Jaffe E AD, Campo E, Harris NL, Quintanilla-Martinez L. Hematopathology Molecular Diagnosis in Hematopathology. Molecular Diagnosis in Hematopathology. Philadelphia PA: Elsevier. 2017:1–1216. [Google Scholar]
- 5.Cancer Genome Atlas Research Network, Ley TJ, Miller C, Ding L, Raphael BJ, Mungall AJ, et al. Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. N Engl J Med. 2013;368:2059–74. doi: 10.1056/NEJMoa1301689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mrozek K, Marcucci G, Paschka P, Whitman SP, Bloomfield CD. Clinical relevance of mutations and gene-expression changes in adult acute myeloid leukemia with normal cytogenetics: are we ready for a prognostically prioritized molecular classification? Blood. 2007;109:431–48. doi: 10.1182/blood-2006-06-001149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dohner H, Weisdorf DJ, Bloomfield CD. Acute Myeloid Leukemia. N Engl J Med. 2015;373:1136–52. doi: 10.1056/NEJMra1406184. [DOI] [PubMed] [Google Scholar]
- 8.Arber DA, Orazi A, Hasserjian R, Thiele J, Borowitz MJ, Le Beau MM, et al. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood. 2016;127:2391–405. doi: 10.1182/blood-2016-03-643544. [DOI] [PubMed] [Google Scholar]
- 9.Duncavage EJ, Tandon B. The utility of next-generation sequencing in diagnosis and monitoring of acute myeloid leukemia and myelodysplastic syndromes. Int J Lab Hematol. 2015;37(Suppl 1):115–21. doi: 10.1111/ijlh.12361. [DOI] [PubMed] [Google Scholar]
- 10.Sperling AS, Gibson CJ, Ebert BL. The genetics of myelodysplastic syndrome: from clonal haematopoiesis to secondary leukaemia. Nat Rev Cancer. 2017;17:5–19. doi: 10.1038/nrc.2016.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Swerdlow SH CE, Harris NL, Jaffe ES, Pileri SA, Stein H, Thiele J. WHO Classification of Tumours of the Haematopoietic. and Lymphoid Tissues. 2017. [Google Scholar]
- 12.Dang L, Jin S, Su SM. IDH mutations in glioma and acute myeloid leukemia. Trends Mol Med. 2010;16:387–97. doi: 10.1016/j.molmed.2010.07.002. [DOI] [PubMed] [Google Scholar]
- 13.Falini B, Mecucci C, Tiacci E, Alcalay M, Rosati R, Pasqualucci L, et al. Cytoplasmic nucleophosmin in acute myelogenous leukemia with a normal karyotype. N Engl J Med. 2005;352:254–66. doi: 10.1056/NEJMoa041974. [DOI] [PubMed] [Google Scholar]
- 14.Ley TJ, Ding L, Walter MJ, McLellan MD, Lamprecht T, Larson DE, et al. DNMT3A mutations in acute myeloid leukemia. N Engl J Med. 2010;363:2424–33. doi: 10.1056/NEJMoa1005143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Marcucci G, Haferlach T, Döhner H. Molecular genetics of adult acute myeloid leukemia: Prognostic and therapeutic implications. J Clin Oncol. 2011;29:475–86. doi: 10.1200/JCO.2010.30.2554. [DOI] [PubMed] [Google Scholar]
- 16.Metzeler KH, Maharry K, Radmacher MD, Mrozek K, Margeson D, Becker H, et al. TET2 mutations improve the new European LeukemiaNet risk classification of acute myeloid leukemia: A cancer and leukemia group B study. J Clin Oncol. 2011;29:1373–81. doi: 10.1200/JCO.2010.32.7742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Patel U, Luthra R, Medeiros LJ, Patel KP. Diagnostic, Prognostic, and Predictive Utility of Recurrent Somatic Mutations in Myeloid Neoplasms. Clin Lymphoma Myeloma Leuk. 2017;17:62–74. doi: 10.1016/j.clml.2017.02.015. [DOI] [PubMed] [Google Scholar]
- 18.Paschka P, Schlenk RF, Gaidzik VI, Habdank M, Kronke J, Bullinger L, et al. IDH1 and IDH2 mutations are frequent genetic alterations in acute myeloid leukemia and confer adverse prognosis in cytogenetically normal acute myeloid leukemia with NPM1 mutation without FLT3 internal tandem duplication. J Clin Oncol. 2010;28:3636–43. doi: 10.1200/JCO.2010.28.3762. [DOI] [PubMed] [Google Scholar]
- 19.Chang C-C OR. Precision Molecular Pathology of Myeloid Neoplasms. In: PT C, editor. Precision Molecular Pathology of Myeloid Neoplasms. Acute Myeloid Leukemia with Recurrent Genetic Abnormalities, Part II: Mutations Involving CEBPA, NPM1, and RUNX1: Springer International Publishing AG. 2018;427. [Google Scholar]
- 20.Perl AE. The role of targeted therapy in the management of patients with AML. Hematology Am Soc Hematol Educ Program. 2017;2017:54–65. doi: 10.1182/asheducation-2017.1.54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.DiNardo CD, Stein EM, de Botton S, Roboz GJ, Altman JK, Mims AS, et al. Durable Remissions with Ivosidenib in IDH1-Mutated Relapsed or Refractory AML. N Engl J Med. 2018;378:2386–98. doi: 10.1056/NEJMoa1716984. [DOI] [PubMed] [Google Scholar]
- 22.Bains A, Luthra R, Medeiros LJ, Zuo Z. FLT3 and NPM1 mutations in myelodysplastic syndromes: Frequency and potential value for predicting progression to acute myeloid leukemia. Am J Clin Pathol. 2011;135:62–9. doi: 10.1309/AJCPEI9XU8PYBCIO. [DOI] [PubMed] [Google Scholar]
- 23.E. H. Hematopathology: A Volume in the Series: Foundations in Diagnostic Pathology. In: JR G, editor. Hematopathology. Molecular Hematopathology. 3rd ed. Philadelphia, PA: Elsevier. 2018;800. [Google Scholar]
- 24.Mendler JH, Maharry K, Radmacher MD, Mrozek K, Becker H, Metzeler KH, et al. RUNX1 mutations are associated with poor outcome in younger and older patients with cytogenetically normal acute myeloid leukemia and with distinct gene and MicroRNA expression signatures. J Clin Oncol. 2012;30:3109–18. doi: 10.1200/JCO.2011.40.6652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gaidzik VI, Bullinger L, Schlenk RF, Zimmermann AS, Röck J, Paschka P, et al. RUNX1 mutations in acute myeloid leukemia: results from a comprehensive genetic and clinical analysis from the AML study group. J Clin Oncol. 2011;29:1364–72. doi: 10.1200/JCO.2010.30.7926. [DOI] [PubMed] [Google Scholar]
- 26.Kussick SJ, Stirewalt DL, Yi HS, Sheets KM, Pogosova-Agadjanyan E, Braswell S, et al. A distinctive nuclear morphology in acute myeloid leukemia is strongly associated with loss of HLA-DR expression and FLT3 internal tandem duplication. Leukemia. 2004;18:1591–8. doi: 10.1038/sj.leu.2403458. [DOI] [PubMed] [Google Scholar]
- 27.O’Donnell MR, Tallman MS, Abboud CN, Altman JK, Appelbaum FR, Arber DA, et al. Acute Myeloid Leukemia, Version 3. 2017, NCCN Clinical Practice Guidelines in Oncology. J Natl Compr Canc Netw. 2017;15:926–57. doi: 10.6004/jnccn.2017.0116. [DOI] [PubMed] [Google Scholar]
- 28.Stone RM, Mandrekar SJ, Sanford BL, Laumann K, Geyer S, Bloomfield CD, et al. Midostaurin plus Chemotherapy for Acute Myeloid Leukemia with a FLT3 Mutation. N Engl J Med. 2017;377:454–64. doi: 10.1056/NEJMoa1614359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Assi R, Ravandi F. FLT3 inhibitors in acute myeloid leukemia: Choosing the best when the optimal does not exist. Am J Hematol. 2018;93:553–63. doi: 10.1002/ajh.25027. [DOI] [PubMed] [Google Scholar]
- 30.Kayser S, Levis MJ. FLT3 tyrosine kinase inhibitors in acute myeloid leukemia: clinical implications and limitations. Leuk Lymphoma. 2014;55:243–55. doi: 10.3109/10428194.2013.800198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Nardi V, Hasserjian RP. Genetic Testing in Acute Myeloid Leukemia and Myelodysplastic Syndromes. Surg Pathol Clin. 2016;9:143–63. doi: 10.1016/j.path.2015.10.004. [DOI] [PubMed] [Google Scholar]
- 32.Kiyoi H, Naoe T. FLT3 mutations in acute myeloid leukemia. Methods Mol Med. 2006;125:189–97. doi: 10.1385/1-59745-017-0:189. [DOI] [PubMed] [Google Scholar]
- 33.Papaemmanuil E, Gerstung M, Bullinger L, Gaidzik VI, Paschka P, Roberts ND, et al. Genomic classification and prognosis in acute myeloid leukemia. N Engl J Med. 2016;374:2209–21. doi: 10.1056/NEJMoa1516192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Patel JP, Gonen M, Figueroa ME, Fernandez H, Sun Z, Racevskis J, et al. Prognostic relevance of integrated genetic profiling in acute myeloid leukemia. N Engl J Med. 2012;366:1079–89. doi: 10.1056/NEJMoa1112304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Allen C, Hills RK, Lamb K, Evans C, Tinsley S, Sellar R, et al. The importance of relative mutant level for evaluating impact on outcome of KIT, FLT3 and CBL mutations in core-binding factor acute myeloid leukemia. Leukemia. 2013;27:1891–901. doi: 10.1038/leu.2013.186. [DOI] [PubMed] [Google Scholar]
- 36.Boissel N, Leroy H, Brethon B, Philippe N, de Botton S, Auvrignon A, et al. Incidence and prognostic impact of c-Kit, FLT3, and Ras gene mutations in core binding factor acute myeloid leukemia (CBF-AML) Leukemia. 2006;20:965–70. doi: 10.1038/sj.leu.2404188. [DOI] [PubMed] [Google Scholar]
- 37.Balsat M, Renneville A, Thomas X, de Botton S, Caillot D, Marceau A, et al. Postinduction Minimal Residual Disease Predicts Outcome and Benefit From Allogeneic Stem Cell Transplantation in Acute Myeloid Leukemia With NPM1 Mutation: A Study by the Acute Leukemia French Association Group. J Clin Oncol. 2017;35:185–93. doi: 10.1200/JCO.2016.67.1875. [DOI] [PubMed] [Google Scholar]
- 38.Bejar R, Lord A, Stevenson K, Bar-Natan M, Pérez-Ladaga A, Zaneveld J. TET2 mutations predict response to hypomethylating agents in myelodysplastic syndrome patients. Blood. 2014;124:2705–12. doi: 10.1182/blood-2014-06-582809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Medeiros BC, Fathi AT, DiNardo CD, Pollyea DA, Chan SM, Swords R. Isocitrate dehydrogenase mutations in myeloid malignancies. Leukemia. 2017;31:272–81. doi: 10.1038/leu.2016.275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Patnaik MM, Hanson CA, Hodnefield JM, Lasho TL, Finke CM, Knudson RA, et al. Differential prognostic effect of IDH1 versus IDH2 mutations in myelodysplastic syndromes: A Mayo Clinic Study of 277 patients. Leukemia. 2012;26:101–5. doi: 10.1038/leu.2011.298. [DOI] [PubMed] [Google Scholar]
- 41.Bejar R, Stevenson K, Abdel-Wahab O, Galili N, Nilsson B, Garcia-Manero G, et al. Clinical effect of point mutations in myelodysplastic syndromes. N Engl J Med. 2011;364:2496–506. doi: 10.1056/NEJMoa1013343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gelsi-Boyer V, Brecqueville M, Devillier R, Murati A, Mozziconacci MJ, Birnbaum D. Mutations in ASXL1 are associated with poor prognosis across the spectrum of malignant myeloid diseases. J Hematol Oncol. 2012;5:12. doi: 10.1186/1756-8722-5-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gelsi-Boyer V, Trouplin V, Adelaide J, Bonansea J, Cervera N, Carbuccia N, et al. Mutations of polycomb-associated gene ASXL1 in myelodysplastic syndromes and chronic myelomonocytic leukaemia. Br J Haematol. 2009;145:788–800. doi: 10.1111/j.1365-2141.2009.07697.x. [DOI] [PubMed] [Google Scholar]
- 44.Bacher U, Haferlach T, Kern W, Haferlach C, Schnittger S. A comparative study of molecular mutations in 381 patients with myelodysplastic syndrome and in 4130 patients with acute myeloid leukemia. Haematologica. 2007;92:744–52. doi: 10.3324/haematol.10869. [DOI] [PubMed] [Google Scholar]
- 45.Sun QY, Ding LW, Tan KT, Chien W, Mayakonda A, Lin DC, et al. Ordering of mutations in acute myeloid leukemia with partial tandem duplication of MLL (MLL-PTD) Leukemia. 2017;31:1–10. doi: 10.1038/leu.2016.160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Devillier R, Mansat-De Mas V, Gelsi-Boyer V, Demur C, Murati A, Corre J, et al. Role of ASXL1 and TP53 mutations in the molecular classification and prognosis of acute myeloid leukemias with myelodysplasia-related changes. Oncotarget. 2015;6:8388–96. doi: 10.18632/oncotarget.3460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lindsley RC, Mar BG, Mazzola E, Grauman PV, Shareef S, Allen SL, et al. Acute myeloid leukemia ontogeny is defined by distinct somatic mutations. Blood. 2015;125:1367–76. doi: 10.1182/blood-2014-11-610543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Rucker FG, Schlenk RF, Bullinger L, Kayser S, Teleanu V, Kett H, et al. TP53 alterations in acute myeloid leukemia with complex karyotype correlate with specific copy number alterations, monosomal karyotype, and dismal outcome. Blood. 2012;119:2114–21. doi: 10.1182/blood-2011-08-375758. [DOI] [PubMed] [Google Scholar]
- 49.Welch JS, Petti AA, Miller CA, Fronick CC, O’Laughlin M, Fulton RS, et al. TP53 and Decitabine in Acute Myeloid Leukemia and Myelodysplastic Syndromes. N Engl J Med. 2016;375:2023–36. doi: 10.1056/NEJMoa1605949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Welch JS, Petti AA, Ley TJ. Decitabine in TP53-Mutated AML. N Engl J Med. 2017;376:797–8. doi: 10.1056/NEJMc1616062. [DOI] [PubMed] [Google Scholar]
- 51.Jongen-Lavrencic M, Grob T, Hanekamp D, Kavelaars FG, Al Hinai A, Zeilemaker A, et al. Molecular Minimal Residual Disease in Acute Myeloid Leukemia. N Engl J Med. 2018;378:1189–99. doi: 10.1056/NEJMoa1716863. [DOI] [PubMed] [Google Scholar]
- 52.Dohner H, Estey E, Grimwade D, Amadori S, Appelbaum FR, Buchner T, et al. Diagnosis and management of AML in adults: 2017 ELN recommendations from an international expert panel. Blood. 2017;129:424–47. doi: 10.1182/blood-2016-08-733196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Li P. ORS. Acute Myeloid Leukemia with Myelodysplasia-Related Changes, Therapy-Related Myeloid Neoplasms, and Acute Myeloid Leukemia, Not Otherwise Specified. In: Chang CC, Ohgami R, editors. Precision Molecular Pathology of Myeloid Neoplasms. Springer, Cham. 2018. [Google Scholar]
- 54.Metzeler KH, Herold T, Rothenberg-Thurley M, Amler S, Sauerland MC, Görlich D, et al. Spectrum and prognostic relevance of driver gene mutations in acute myeloid leukemia. Blood. 2016;128:686–98. doi: 10.1182/blood-2016-01-693879. [DOI] [PubMed] [Google Scholar]
- 55.Devillier R, Gelsi-Boyer V, Brecqueville M, Carbuccia N, Murati A, Vey N, et al. Acute myeloid leukemia with myelodysplasia-related changes are characterized by a specific molecular pattern with high frequency of ASXL1 mutations. Am J Hematol. 2012;87:659–62. doi: 10.1002/ajh.23211. [DOI] [PubMed] [Google Scholar]
- 56.Ohgami RS, Ma L, Merker JD, Gotlib JR, Schrijver I, Zehnder JL, et al. Next-generation sequencing of acute myeloid leukemia identifies the significance of TP53, U2AF1, ASXL1, and TET2 mutations. Mod Pathol. 2015;28:706–14. doi: 10.1038/modpathol.2014.160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Bhatia S. Therapy-related myelodysplasia and acute myeloid leukemia. Semin Oncol. 2013;40:666–75. doi: 10.1053/j.seminoncol.2013.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ok CY, Patel KP, Garcia-Manero G, Routbort MJ, Fu B, Tang G, et al. Mutational profiling of therapy-related myelodysplastic syndromes and acute myeloid leukemia by next generation sequencing, a comparison with de novo diseases. Leuk Res. 2015;39:348–54. doi: 10.1016/j.leukres.2014.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Arber DA, Slovak ML, Popplewell L, Bedell V, Ikle D, Rowley JD, et al. Therapy-related acute myeloid leukemia/myelodysplasia with balanced 21q22 translocations. Am J Clin Pathol. 2002;117:306–13. doi: 10.1309/C3G2-CXA0-HE9J-TKDR. [DOI] [PubMed] [Google Scholar]
- 60.Czader M, Orazi A. Acute Myeloid Leukemia and Other Types of Disease Progression in Myeloproliferative Neoplasms. Am J Clin Pathol. 2015;144:188–206. doi: 10.1309/AJCPZQK40JOZZZCC. [DOI] [PubMed] [Google Scholar]
- 61.Shih AH, Chung SS, Dolezal EK, Zhang SJ, Abdel-Wahab OI, Park CY, et al. Mutational analysis of therapy-related myelodysplastic syndromes and acute myelogenous leukemia. Haematologica. 2013;98:908–12. doi: 10.3324/haematol.2012.076729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Tiu RV, Visconte V, Traina F, Schwandt A, Maciejewski JP. Updates in cytogenetics and molecular markers in MDS. Curr Hematol Malig Rep. 2011;6:126–35. doi: 10.1007/s11899-011-0081-2. [DOI] [PubMed] [Google Scholar]
- 63.Haase D, Germing U, Schanz J, Pfeilstöcker M, Nösslinger T, Hildebrandt B, et al. New insights into the prognostic impact of the karyotype in MDS and correlation with subtypes: evidence from a core dataset of 2124 patients. Blood. 2007;110:4385–95. doi: 10.1182/blood-2007-03-082404. [DOI] [PubMed] [Google Scholar]
- 64.Haferlach T, Nagata Y, Grossmann V, Okuno Y, Bacher U, Nagae G, et al. Landscape of genetic lesions in 944 patients with myelodysplastic syndromes. Leukemia. 2014;28:241–7. doi: 10.1038/leu.2013.336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Papaemmanuil E, Gerstung M, Malcovati L, Tauro S, Gundem G, Van Loo P, et al. Clinical and biological implications of driver mutations in myelodysplastic syndromes. Blood. 2013;122:3616–27. doi: 10.1182/blood-2013-08-518886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Bartels S, Schipper E, Hasemeier B, Kreipe H, Lehmann U. Routine clinical mutation profiling using next generation sequencing and a customized gene panel improves diagnostic precision in myeloid neoplasms. Oncotarget. 2016;7:30084–93. doi: 10.18632/oncotarget.8310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Walter MJ, Ding L, Shen D, Shao J, Grillot M, McLellan M, et al. Recurrent DNMT3A mutations in patients with myelodysplastic syndromes. Leukemia. 2011;25:1153–8. doi: 10.1038/leu.2011.44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Jadersten M, Saft L, Smith A, Kulasekararaj A, Pomplun S, Göhring G, et al. TP53 mutations in low-risk myelodysplastic syndromes with del (5q) predict disease progression. J Clin Oncol. 2011;29:1971–9. doi: 10.1200/JCO.2010.31.8576. [DOI] [PubMed] [Google Scholar]
- 69.Kuo FC, Dong F. Next-generation sequencing-based panel testing for myeloid neoplasms. Curr Hematol Malig Rep. 2015;10:104–11. doi: 10.1007/s11899-015-0256-3. [DOI] [PubMed] [Google Scholar]
- 70.Papaemmanuil E, Cazzola M, Boultwood J, Malcovati L, Vyas P, Bowen D, et al. Somatic SF3B1 mutation in myelodysplasia with ring sideroblasts. N Engl J Med. 2011;365:1384–95. doi: 10.1056/NEJMoa1103283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Brecqueville M, Rey J, Bertucci F, Coppin E, Finetti P, Carbuccia N, et al. Mutation analysis of ASXL1, CBL, DNMT3A, IDH1, IDH2, JAK2, MPL, NF1, SF3B1, SUZ12, and TET2 in myeloproliferative neoplasms. Genes Chromosomes Cancer. 2012;51:743–55. doi: 10.1002/gcc.21960. [DOI] [PubMed] [Google Scholar]
- 72.Malcovati L, Galli A, Travaglino E, Ambaglio I, Rizzo E, Molteni E, et al. Clinical significance of somatic mutation in unexplained blood cytopenia. Blood. 2017;129:3371–8. doi: 10.1182/blood-2017-01-763425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Jain M, Tripathi A. ICUS/CCUS/CHIP: basics & beyond. Expert Rev Hematol. 2017;10:915–20. doi: 10.1080/17474086.2017.1371588. [DOI] [PubMed] [Google Scholar]
- 74.Jaiswal S, Fontanillas P, Flannick J, Manning A, Grauman PV, Mar BG, et al. Age-related clonal hematopoiesis associated with adverse outcomes. N Engl J Med. 2014;371:2488–98. doi: 10.1056/NEJMoa1408617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Zink F, Stacey SN, Norddahl GL, Frigge ML, Magnusson OT, Jonsdottir I, et al. Clonal hematopoiesis, with and without candidate driver mutations, is common in the elderly. Blood. 2017;130:742–52. doi: 10.1182/blood-2017-02-769869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Brauninger A, Blau W, Kunze K, Desch AK, Brobeil A, Tur MK, et al. Targeted Next-Generation Sequencing Is a Sensitive Tool for Differential Diagnosis of Myelodysplastic Syndromes in Bone Marrow Trephines. J Mol Diagn. 2018;20:344–54. doi: 10.1016/j.jmoldx.2018.01.006. [DOI] [PubMed] [Google Scholar]
- 77.Barbui T, Thiele J, Gisslinger H, Kvasnicka HM, Vannucchi AM, Guglielmelli P, et al. The 2016 WHO classification and diagnostic criteria for myeloproliferative neoplasms: document summary and in-depth discussion. Blood Cancer J. 2018;8:15. doi: 10.1038/s41408-018-0054-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Tefferi A, Lasho TL, Schwager SM, Steensma DP, Mesa RA, Li CY, et al. The JAK2(V617F) tyrosine kinase mutation in myelofibrosis with myeloid metaplasia: lineage specificity and clinical correlates. Br J Haematol. 2005;131:320–8. doi: 10.1111/j.1365-2141.2005.05776.x. [DOI] [PubMed] [Google Scholar]
- 79.Baxter EJ, Scott LM, Campbell PJ, East C, Fourouclas N, Swanton S, et al. Acquired mutation of the tyrosine kinase JAK2 in human myeloproliferative disorders. Lancet. 2005;365:1054–61. doi: 10.1016/S0140-6736(05)71142-9. [DOI] [PubMed] [Google Scholar]
- 80.Vannucchi AM, Antonioli E, Guglielmelli P, Rambaldi A, Barosi G, Marchioli R, et al. Clinical profile of homozygous JAK2 617V>F mutation in patients with polycythemia vera or essential thrombocythemia. Blood. 2007;110:840–6. doi: 10.1182/blood-2006-12-064287. [DOI] [PubMed] [Google Scholar]
- 81.Klampfl T, Gisslinger H, Harutyunyan AS, Nivarthi H, Rumi E, Milosevic JD, et al. Somatic mutations of calreticulin in myeloproliferative neoplasms. N Engl J Med. 2013;369:2379–90. doi: 10.1056/NEJMoa1311347. [DOI] [PubMed] [Google Scholar]
- 82.Nangalia J, Massie CE, Baxter EJ, Nice FL, Gundem G, Wedge DC, et al. Somatic CALR mutations in myeloproliferative neoplasms with nonmutated JAK2. N Engl J Med. 2013;369:2391–405. doi: 10.1056/NEJMoa1312542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Rotunno G, Mannarelli C, Guglielmelli P, Pacilli A, Pancrazzi A, Pieri L, et al. Impact of calreticulin mutations on clinical and hematological phenotype and outcome in essential thrombocythemia. Blood. 2014;123:1552–5. doi: 10.1182/blood-2013-11-538983. [DOI] [PubMed] [Google Scholar]
- 84.Tefferi A, Lasho TL, Finke C, Belachew AA, Wassie EA, Ketterling RP, et al. Type 1 vs type 2 calreticulin mutations in primary myelofibrosis: differences in phenotype and prognostic impact. Leukemia. 2014;28:1568–70. doi: 10.1038/leu.2014.83. [DOI] [PubMed] [Google Scholar]
- 85.Mesa RA, Jamieson C, Bhatia R, Deininger MW, Fletcher CD, Gerds AT, et al. NCCN Guidelines Insights: Myeloproliferative Neoplasms, Version 2. 2018. J Natl Compr Canc Netw. 2017;15:1193–207. doi: 10.6004/jnccn.2017.0157. [DOI] [PubMed] [Google Scholar]
- 86.Tefferi A, Guglielmelli P, Larson DR, Finke C, Wassie EA, Pieri L, et al. Long-term survival and blast transformation in molecularly annotated essential thrombocythemia, polycythemia vera, and myelofibrosis. Blood. 2014;124:2507–13. doi: 10.1182/blood-2014-05-579136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Pikman Y, Lee BH, Mercher T, McDowell E, Ebert BL, Gozo M, et al. MPLW515L is a novel somatic activating mutation in myelofibrosis with myeloid metaplasia. PLoS Med. 2006;3:270. doi: 10.1371/journal.pmed.0030270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Langabeer SE, Andrikovics H, Asp J, Bellosillo B, Carillo S, Haslam K, et al. Molecular diagnostics of myeloproliferative neoplasms. Eur J Haematol. 2015;95:270–9. doi: 10.1111/ejh.12578. [DOI] [PubMed] [Google Scholar]
- 89.Rumi E, Pietra D, Pascutto C, Guglielmelli P, Martínez-Trillos A, Casetti I, et al. Clinical effect of driver mutations of JAK2, CALR, or MPL in primary myelofibrosis. Blood. 2014;124:1062–9. doi: 10.1182/blood-2014-05-578435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Tefferi A. Novel mutations and their functional and clinical relevance in myeloproliferative neoplasms: JAK2, MPL, TET2, ASXL1, CBL, IDH and IKZF1. Leukemia. 2010;24:1128–38. doi: 10.1038/leu.2010.69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Tefferi A, Vainchenker W. Myeloproliferative neoplasms: molecular pathophysiology, essential clinical understanding, and treatment strategies. J Clin Oncol. 2011;29:573–82. doi: 10.1200/JCO.2010.29.8711. [DOI] [PubMed] [Google Scholar]
- 92.Yogarajah M, Tefferi A. Leukemic Transformation in Myeloproliferative Neoplasms: A Literature Review on Risk, Characteristics, and Outcome. Mayo Clin Proc. 2017;92:1118–28. doi: 10.1016/j.mayocp.2017.05.010. [DOI] [PubMed] [Google Scholar]
- 93.Itzykson R, Kosmider O, Renneville A, Gelsi-Boyer V, Meggendorfer M, Morabito M, et al. Prognostic score including gene mutations in chronic myelomonocytic leukemia. J Clin Oncol. 2013;31:2428–36. doi: 10.1200/JCO.2012.47.3314. [DOI] [PubMed] [Google Scholar]
- 94.Wassie EA, Itzykson R, Lasho TL, Kosmider O, Finke CM, Hanson CA, et al. Molecular and prognostic correlates of cytogenetic abnormalities in chronic myelomonocytic leukemia: a Mayo Clinic-French Consortium Study. Am J Hematol. 2014;89:1111–5. doi: 10.1002/ajh.23846. [DOI] [PubMed] [Google Scholar]
- 95.Pich A, Riera L, Sismondi F, Godio L, Davico Bonino L, Marmont F, et al. JAK2V617F activating mutation is associated with the myeloproliferative type of chronic myelomonocytic leukaemia. J Clin Pathol. 2009;62:798–801. doi: 10.1136/jcp.2009.065904. [DOI] [PubMed] [Google Scholar]
- 96.Padron E, Dezern A, Andrade-Campos M, Vaddi K, Scherle P, Zhang Q, et al. A Multi-Institution Phase I Trial of Ruxolitinib in Patients with Chronic Myelomonocytic Leukemia (CMML) Clin Cancer Res. 2016;22:3746–54. doi: 10.1158/1078-0432.CCR-15-2781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.E.D. GMH. Chronic Myelomonocytic Leukemia: Clinical and Pathologic Features. In: Chung-Che (Jeff) Chang RSO, editor. Precision Molecular Pathology of Myeloid Neoplasms. VOL 12: Springer International Publishing AG. 2018;2018. [Google Scholar]
- 98.Caye A, Strullu M, Guidez F, Cassinat B, Gazal S, Fenneteau O, et al. Juvenile myelomonocytic leukemia displays mutations in components of the RAS pathway and the PRC2 network. Nat Genet. 2015;47:1334–40. doi: 10.1038/ng.3420. [DOI] [PubMed] [Google Scholar]
- 99.Niemeyer CM, Kang MW, Shin DH, Furlan I, Erlacher M, Bunin NJ, et al. Germline CBL mutations cause developmental abnormalities and predispose to juvenile myelomonocytic leukemia. Nat Genet. 2010;42:794–800. doi: 10.1038/ng.641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Gambacorti-Passerini CB, Donadoni C, Parmiani A, Pirola A, Redaelli S, Signore G, et al. Recurrent ETNK1 mutations in atypical chronic myeloid leukemia. Blood. 2015;125:499–503. doi: 10.1182/blood-2014-06-579466. [DOI] [PubMed] [Google Scholar]
- 101.Meggendorfer M, Bacher U, Alpermann T, Haferlach C, Kern W, Gambacorti-Passerini C, et al. SETBP1 mutations occur in 9% of MDS/MPN and in 4% of MPN cases and are strongly associated with atypical CML, monosomy 7, isochromosome i(17)(q10), ASXL1 and CBL mutations. Leukemia. 2013;27:1852–60. doi: 10.1038/leu.2013.133. [DOI] [PubMed] [Google Scholar]
- 102.Wang SA, Tam W, Tsai AG, Arber DA, Hasserjian RP, Geyer JT, et al. Targeted next-generation sequencing identifies a subset of idiopathic hypereosinophilic syndrome with features similar to chronic eosinophilic leukemia, not otherwise specified. Mod Pathol. 2016;29:854–64. doi: 10.1038/modpathol.2016.75. [DOI] [PubMed] [Google Scholar]
- 103.Arock M, Sotlar K, Akin C, Broesby-Olsen S, Hoermann G, Escribano L, et al. KIT mutation analysis in mast cell neoplasms: recommendations of the European Competence Network on Mastocytosis. Leukemia. 2015;29:1223–32. doi: 10.1038/leu.2015.24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Schwaab J, Schnittger S, Sotlar K, Walz C, Fabarius A, Pfirrmann M, et al. Comprehensive mutational profiling in advanced systemic mastocytosis. Blood. 2013;122:2460–6. doi: 10.1182/blood-2013-04-496448. [DOI] [PubMed] [Google Scholar]