

Abstract
Objective
To determine histopathologic, exome, and transcriptome nucleic acid material yield from prospectively collected metastatic tissue biopsy specimens in patients with metastatic castration-resistant prostate cancer (mCRPC).
Patients and Methods
Patients with mCRPC initiating abiraterone acetate therapy underwent 2 serial metastatic site core needle biopsies after study activation on May 17, 2013. Multiple cores were obtained, and from each core, 1- to 2-mm segments were separated and formalin fixed for histopathologic examination. Tumor purity was determined for DNA and RNA from the rest of the biopsy specimen. RNA quality was assessed by calculation of an RNA integrity number and a DV200 score.
Results
A total of 89 patients underwent 172 uniformly processed core needle biopsies (89 on visit 1 and 83 on visit 2) between May 30, 2013, and September 10, 2015. Metastatic sites biopsied included bone (131), lymph nodes (31), liver (5), lung (3), and pelvic soft tissues (2). Of the 172 biopsy specimens, 85 (49%) had at least one of the multiple cores positive for tumor on histopathologic examination (53 of 88 [60%] from visit 1 and 32 of 83 [39%] from visit 2; P=.006). Metastatic carcinoma was observed in 50 of 130 bone lesion specimens (38%), compared to 35 of 41 nonbone specimens (85%) (P<.001). More than 10% tumoral DNA purity was observed in 89% and 80% of visit 1 and visit 2 biopsy specimens, respectively. Similarly, more than 10% tumor RNA purity was observed in 79% of visit 1 vs 59% for visit 2 (P=.008). In all, 134 of 172 procedures (78%) yielded tumor material either by histopathologic or nucleic acid purity analysis.
Conclusion
This study found that biopsy specimens from mCRPC sites yield adequate histopathologic, exome, and transcriptome material in most, but not all, cases. This finding has relevance for future genome sequencing studies on the introduction of targeted therapeutic agents.
Trial Registration
clinicaltrials.gov Identifier: 01953640.
Abbreviations and Acronyms: CN, copy number; MAF, mutant allele frequency; mCRPC, metastatic castration-resistant prostate cancer; RIN, RNA integrity number
Prostate cancer is the second leading cause of cancer-related mortality in males in the United States, with an estimated 29,430 deaths in 2018.1 Despite advances in our understanding of the mutational landscapes in primary and metastatic prostate cancer,2, 3, 4 molecular biomarkers for predicting drug treatment effects are incompletely understood. Impediments for molecular profiling in advanced prostate cancer include obtaining adequate tissue from sclerotic skeletal metastasis, which is the predominant site of spread in advanced prostate cancer states (hormone sensitive and castration resistant). Biopsy of sclerotic lesions is technically challenging, with a limited amount of bone tissue obtained for the concomitant histopathologic examination and molecular analysis after applying decalcifying biopsy material.5 Until recently,6 the lack of therapies for metastatic castration-resistant prostate cancer (mCRPC) limited the value of investigating individual metastatic molecular profiles. However, as a result of therapeutic advances, therapeutic targeting of the androgen receptor signaling pathway, and the emergence of novel drug combinations, molecular profiling of metastases, despite the challenges of interrogating skeletal metastases, has become increasingly relevant. Advances in molecular techniques now allow the potential for interrogating nucleic acid expressions in formalin-fixed, paraffin-embedded tissue7 as long as adequate tissue is obtained.
Because the usefulness of concomitant evaluation of histopathologic and aberrant nucleic acid expression patterns from skeletal metastases in mCRPC is unclear, we evaluated in a metastatic biopsy-based prospective cohort study the results of a uniformly applied protocol for obtaining metastatic tissue for molecular and histopathologic studies. The success of obtaining adequate yield likely impacts the future clinical application of targeted therapies that are also based on genomic aberrations unique to the state of disease progression after androgen deprivation therapy.
Patients and Methods
Study Population
Patients with advanced metastatic prostate cancer who experienced progression while receiving androgen deprivation therapy and those with progression to a castrate-resistant state were invited to participate in a prospectively conducted biopsy and molecular profiling trial. Details of the PROMOTE (Prostate Cancer Medically Optimized Genome-Enhanced Therapy) (clinicaltrials.gov Identifier: NCT #01953640) study have been published previously.2 Briefly, the study was activated on May 17, 2013, to determine associations between tumor exome and transcriptome alterations obtained from biopsy specimens from metastatic sites before and after starting treatment for mCRPC state with a novel drug, abiraterone acetate. Two serial metastatic tissue biopsies were performed, with the first one before initiation of abiraterone acetate–prednisone drug therapy and a second one after 12 weeks of treatment. All biopsies of metastases were performed after obtaining written consent from patients enrolled in this institutional review board–approved study.
Lesion Selection and Biopsy Technique
Computed tomography and bone scans performed as part of the study protocol were reviewed by 1 of 3 dedicated study interventional radiologists (T.D.A., P.W.E., B.P.M.) to select the biopsy target at study entry and 12 weeks after treatment. For the initial biopsy (visit 1), soft tissue lesions were targeted preferentially whenever available. If no appropriate soft tissue target was present, an osseous metastatic lesion was selected. If multiple skeletal lesions were present, lesions with a lytic component or new or rapidly growing lesions were given preference for biopsy, as long as their sampling was considered safe. For the second biopsy (visit 2), a new target was preferred when present on the 12-week repeated imaging over the original target lesion biopsied; if a new target was not available, the same lesion was targeted again. All biopsies were performed using standard technique, including computed tomography or ultrasound guidance, sterile technique, and local anesthesia (Figure 1). Moderate intravenous sedation was administered at the discretion of the performing interventional radiologist. For soft tissue lesions, 4 to 6 18-gauge core samples were obtained with an automated biopsy instrument (Bard Monopty, Bard Biopsy Systems). For sclerotic osseous lesions, 11- and/or 13-gauge cores were obtained using either a manual trephinated bone needle (Osteo-Site and Ackerman biopsy needles, Cook Medical) or a powered device (OnControl, Vidacare Corp). Four to 6 passes were made into the lesion to yield sufficient material, with each biopsy core yielding at least a 0.5-cm length of tissue. Following the biopsy, patients were observed for up to 2 hours in the radiology department recovery area. Date and time of the biopsy were recorded by the research assistant or determined retrospectively from time data embedded in the stored images. Follow-up with all patients was performed within 24 hours for assessing safety and adverse effects of the procedure.
Figure 1.
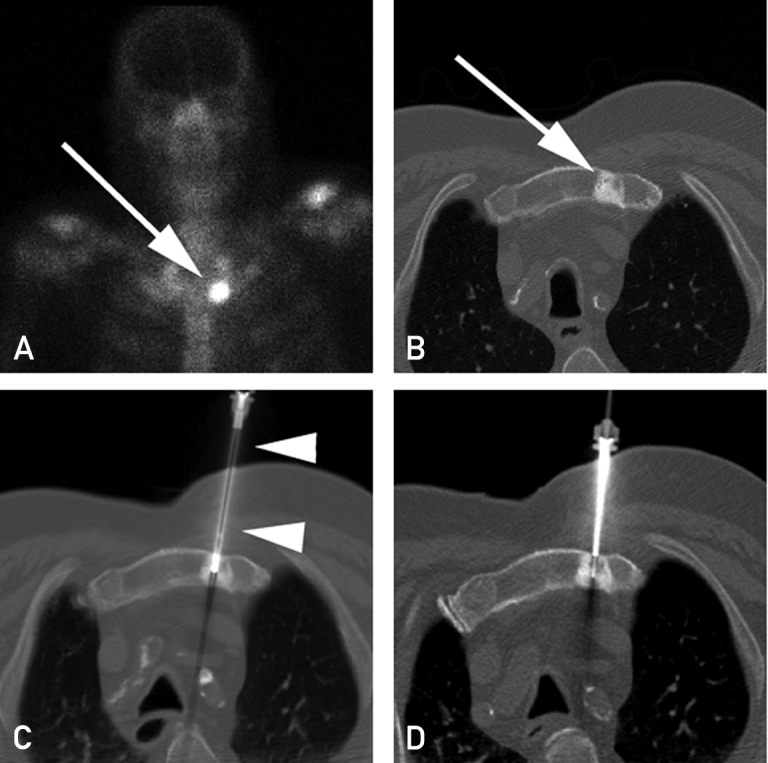
A, Technetium Tc 99m bone scan shows focal activity in the left side of the manubrium (arrow) consistent with a metastasis. B, Computed tomographic image shows focal sclerosis in the left side of the manubrium (arrow) corresponding to the bone scan and consistent with a metastasis. C, Computed tomographic image obtained during biopsy shows an 11-gauge bone biopsy device (arrowheads) sampling the metastasis. D, Computed tomographic image obtained during second biopsy event 4 months later, showing 13-gauge needle through an 11-gauge introducer.
Sample Handling
The number of cores attempted at each visit ranged from 1 to 6. The first core (designated S1) was submitted for DNA sequencing, while the second (designated S2) was submitted for RNA sequencing. The third and fourth cores (designated X3 and X4, respectively) were submitted for xenograft implantation.8 At the radiology suite, and before submission of these cores for sequencing and engraftment, 1- to 2-mm segments from both the tip ends of each biopsy core were separated and fixed in formalin for assessing histopathologic characteristics. In a minority of cases, a fifth and sixth core (designated F5 and F6, respectively) were obtained and entirely submitted for either additional xenograft implantation or formalin fixation and histopathologic analysis. Heavily calcified bone samples were placed in decalcifying solution (formic acid 20%) after fixation for about 30 minutes. Less calcified bone samples underwent surface decalcification with 5% hydrochloric acid.
Histopathologic Analysis
Hematoxylin and eosin–stained sections of samples submitted for histopathologic analysis were reviewed by a single pathologist (R.E.J.) . The presence of any amount of recognizable tumor was sufficient to determine a sample as “positive,” independent of the size of the tumor deposit or cellularity. No immunohistochemical stains were used to corroborate the hematoxylin-eosin impression. Cases with equivocal or only atypical findings were not considered positive. Tumors with any recognizable gland formation were labeled as adenocarcinomas, while tumors lacking gland formation were labeled as poorly differentiated carcinomas. Small cell differentiation was excluded by the absence of characteristic morphologic features.
Nucleic Acid Purity Evaluation
DNA quantity was measured using the Invitrogen Qubit dsDNA BR (broad range) assay kit on the Qubit fluorometer. Whole-exome sequencing of DNA was performed as previously described.2, 8 In brief, exome sequencing was performed using the Agilent Technologies Inc SureSelect Human All ExonV4+UTR capture kit (plus additional baits covering the AR gene), followed by alignment with Novoalign, somatic mutation calling using 3 somatic callers, and copy number (CN) estimation using PatternCNV.9 Purity (percent of tumor cells) was estimated 2 different ways. For tumors with significant somatic CN alterations, plots of CN log2 ratio and B-allele frequency were examined to determine the log ratio levels corresponding to +1 gain and −1 deletion. For diploid tumors, if X represents the purity (defined as the percent of tumors cells), the log2 ratio of +1 gains is log2 (Gain) = [x*3+(1−x)*2]/2α and the log2 ratio of a single deletion is log2 (Deletion) = [x*1+(1−x)*2]/2α, with α being a normalization factor. For tetraploid tumors, we used the tetraploid level as the baseline and computed gains and deletions relative to the 4-copy state giving these alternate equations log2 (Gain) = [x*5+(1−x)*2]/2α and the log2 ratio of deletion is log2 (Deletion) = [x*3+(1−x)*2]/2α. We then compute differences between different levels (gains and/or deletions), which gives us equations for purity that do not include the α factor. Inverting these equations, we solved for the gain purity and deletion purity and reported the average. For samples without major CN alterations, we used the 2 × the median of the mutant allele frequency (MAF), defined as reads supporting mutant allele divided by total reads at mutation site (Supplemental Figure 1, available online at http://mcpiqojournal.org). Samples with 10% or greater tumoral DNA purity were considered positive.
The RNA purity was estimated when a matching DNA sample passing DNA quality control was available. Estimation was based on computation of the MAF (eg, the fraction of reads with the mutant allele) in both the RNA and DNA from somatic mutations discovered in the DNA. The RNA purity was calculated by multiplying the copy number variant–derived DNA cellularity (purity) with a robust mean of the ratio of the MAF in RNA over that of the DNA MAF for each variant with 30 reads in both RNA and DNA. At least 10 variants were required to determine the mean to avoid bias from allelic-specific expression (RNA) or from CN regions (DNA). The DNA MAF was required to be consistent with the copy number variant–based purity to be usable. Quality of RNA was assessed by determining an RNA integrity number (RIN), which is based on the ratio between the 18s and 28s peaks of ribosomal RNA as determined using the Agilent Bioanalyzer. For samples with a RIN of less than 6, a DV200 (the percentage of RNA fragments >200 nucleotides as measured on the Agilent Bioanalyzer) score was determined and interpreted as high quality (DV200 >70%), medium (50%-70%), low (30%-50%), and very low (<30%). Quantity of RNA was determined using a Thermo Scientific NanoDrop spectrophotometer. Samples with 10% or greater tumoral RNA purity were considered positive.
Statistical Analyses
Descriptive statistics for pathologic and nucleic acid yields at visits 1 and 2 are provided with ranges and means and medians wherever applicable. Comparison between groups for a number of pathologic and nucleic acid variables was performed using the χ2 test. For comparison of means and medians, a Mann-Whitney test calculator was used wherever applicable.10 P<.05 was considered statistically significant.
Results
A total of 89 patients enrolled in the study between May 30, 2013,and September 10, 2015, underwent 172 core needle biopsy procedures (89 at visit 1 and 83 at visit 2), rendering a total of 508 cores (mean number of cores per biopsy procedure, 2.9). The distribution of cores included 171 S1 cores, 151 S2 cores, 124 X3 cores, 44 X4 cores, 17 F5 cores, and 1 F6 core. The distribution of metastatic sites biopsied included bone (131), lymph nodes (31), liver (5), lung (3), and penile (1) and prostate bed (1) soft tissues (Figure 2).
Figure 2.

A, Distribution of sites biopsied per visit (colors correspond to visit 2 biopsy). B, Patient and sample flow diagram for both visits. HP = histopathology; V1 = visit 1; V2 = visit 2.
Histopathologic Findings
Overall, in 85 of 172 biopsy procedures (49%) at least 1 of the obtained cores was positive for metastatic tumor on histopathologic examination of tissue obtained from the tip of the biopsy specimen (Supplemental Figure 2, available online at http://mcpiqojournal.org). A higher number of cores were positive in visit 1 than visit 2, with 53 of 88 (60%) visit 1 biopsies and 32 of 83 (39%) visit 2 biopsies positive for histopathologic tumor content (P=.006). The positive biopsies resulted in a total of 152 cores (of the 508 obtained [30%]) positive for tumor; their distribution per visit and core type are summarized in Table 1. There was a significant difference in tumor yield between S1 and S2 cores compared to X3 and X4 cores (P<.001). Of the 152 positive cores, 85 (56%) had greater than 50% tumor cellularity. Tumors were classified as metastatic adenocarcinoma in 70 of 85 biopsies and poorly differentiated carcinoma in 15 of 85. No small cell carcinoma differentiation was identified. Gleason grade applied to the metastatic tumor ranged from 3+4 to 5+5. Characteristics of the positive cores are summarized in Table 2. Metastatic carcinoma was observed in 50 of 130 bone lesion samples (38%), compared to 35 of 41 nonbone sites (85%) (P<.001) (Table 3).
Table 1.
Core Positivity by Histopathology According to Visit and Type of Corea
| Variable | No. of cores obtained | No. of positive cores by HP | % Positive | P valueb | P valuec |
|---|---|---|---|---|---|
| Visit 1 biopsies | |||||
| Core S1 | 88 | 42 | 48 | ||
| Core S2 | 79 | 32 | 41 | .35 | <.001 |
| Core X3 | 57 | 12 | 21 | .02 | |
| Core X4 | 17 | 4 | 24 | .83 | |
| Core F5 | 7 | 5 | 71 | NA | |
| Core F6 | 1 | 1 | 100 | NA | |
| Visit 2 biopsies | |||||
| Core S1 | 83 | 23 | 28 | ||
| Core S2 | 72 | 18 | 25 | .70 | .01 |
| Core X3 | 67 | 9 | 13 | .09 | |
| Core X4 | 27 | 3 | 11 | .76 | |
| Core F5 | 10 | 3 | 30 | NA | |
| Core F6 | 0 | 0 | 0 | NA |
HP = histopathologic examination; NA = not analyzed. See text for definition of core categories.
Compared preceding row (ie, S2 vs S1, X3 vs S2, X4 vs X3).
Value when S1+S2 cores are compared to X3+X4 cores for each visit. F5 and F6 cores not included in analysis because of small numbers and different handling.
Table 2.
Tumor Quantitation by Histopathology According to Visit and Type of Core
| Variable | Visit 1 |
Visit 2 |
|||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| S1 | S2 | X3 | X4 | F5 | F6 | S1 | S2 | X3 | X4 | F5 | |
| Positive cores with cellularity >50% | 67% (28/42) | 56% (18/32) | 42% (5/12) | 50% (2/4) | 100% (5/5) | 100% (1/1) | 52% (12/23) | 39% (7/18) | 44% (4/9) | 33% (1/3) | 67% (2/3) |
| Tumor size (mm) | |||||||||||
| <1 | 12% (5/42) | 13% (4/32) | 25% (3/12) | 0 | 0 | 0 | 13% (3/23) | 11% (2/18) | 11% (1/9) | 0 | 33% (1/3) |
| 1-5 | 81% (34/42) | 81% (26/32) | 75% (9/12) | 100% (4/4) | 80% (4/5) | 100% (1/1) | 78% (18/23) | 83% (15/18) | 89% (8/9) | 100% (3/3) | 67% (2/3) |
| >5 | 7% (3/42) | 6% (2/32) | 0 | 0 | 20% (1/5) | 0 | 9% (2/23) | 6% (1/18) | 0 | 0 | 0 |
Table 3.
Tumor Detection by Histopathology and Nucleic Acid Analysis
| Variable | Visit 1 | Visit 2 | P-value | Nonbone | Bone | P-value |
|---|---|---|---|---|---|---|
| Histopathology | 60% (53/88a) | 39% (32/83) | .005 | 85% (35/41) | 38% (50/130a) | <.001 |
| DNA | ||||||
| Median DNA concentration, ng/μL (range) | 39 (1-302) | 35 (1-256) | .18 | 39 (1-245) | 37 (1-302) | .71 |
| Cases with ≥10% tumoral DNA purity | 89% (64/72) | 80% (45/56) | .18 | 94% (30/32) | 82% (79/96) | .11 |
| RNA | ||||||
| Median RNA concentration, ng/μL (range) | 57 (2-2098) | 30 (1-924) | .01 | 43 (1-924) | 41 (2-2098) | .33 |
| Median RNA integrity number | 4.9 | 2.6 | .001 | 7.3 | 2.6 | <.001 |
| Cases with ≥10% tumoral RNA purity | 79% (55/70) | 59% (41/69) | .01 | 85% (29/34) | 64% (67/105) | .02 |
| Tumor detection by any method (histopathology, DNA purity, RNA purity) | 83% (74/89) | 72%(60/83) | .03 | 90% (37/41) | 74% (97/131) | .09 |
One bone biopsy at Visit 1 did not render a histopathologically examinable sample.
Nucleic Acid Yield and Purity
A total of 161of the 172 biopsies (82 at visit 1 and 79 at visit 2) yielded specimens with DNA material; purity could be calculated in 128 (72 samples at visit 1 and 56 at visit 2; 32 nonbone and 96 bone samples) (80%). Similarly, 158 of 172 biopsies yielded specimens with RNA material (83 at visit 1 and 75 at visit 2), in 139 of which purity could be calculated (70 samples at visit 1 and 69 at visit 2; 34 nonbone and 105 bone samples) (88%). Mean and median concentrations were 56.8 and 38.7 ng/μL for DNA and 112.0 and 41.2 ng/μL for RNA, respectively (further breakdown of details based on visit provided in Table 3). For RNA samples, mean and median RIN was 4.4 and 3, respectively. DV200 was calculated for 91 of 112 cases with a RIN of 6 or less. Mean and median DV200 values were 48.1 and 46, respectively. In 78 of the 91 cases (86%), the DV200 value was 30 or greater. Overall, by RIN and DV200 parameters, RNA was considered of acceptable quality in 123 of 158 specimens (78%). Mean and median tumoral nucleic acid purity was 35% and 31% for DNA and 35% and 28% for RNA, respectively. In all, 134 of 172 procedures (78%) yielded tumor material either by histopathologic or nucleic acid purity analysis. Forty-eight biopsy specimens with negative histopathologic results had 10% or greater tumoral DNA or RNA purity, whereas in 10 cases with positive histopathologic results, 10% or greater tumoral DNA or RNA purity could not be demonstrated. More than 10% tumoral DNA purity was observed in 89% and 80% of visit 1 and visit 2 biopsy specimens, respectively. Similarly, more than 10% tumor RNA purity was observed in 79% of visit 1 vs 59% for visit 2 (P=.008). Table 3 shows the difference in histopathologic positivity, nucleic acid concentration, purity, RIN, and tumor detection by any method among samples from visit 1 vs visit 2 and bone vs nonbone samples.
Discussion
The implementation of precision medicine in advanced cancer treatments is dependent on the identification of actionable biological signatures in metastases.11 This issue has become clinically relevant because genomic signatures in metastases can be used for identifying primary and acquired resistance pathways, prognostic signals, and prediction of treatment outcomes at the genome level.12 Harvesting of mCRPC, however, is cumbersome, especially from skeletal metastases, and there is limited data available on concomitant histopathologic and genomic material yield from biopsy specimens from patients with increasing prostate-specific antigen levels during ongoing androgen deprivation therapy. In our prospectively conducted study, which was performed in the context of identifying associations between tumor genome and transcriptome alterations and clinical outcomes, we systematically determined the histopathologic and nucleic acid material yield because it may have implications for clinical practice in the future. Patients underwent an initial biopsy on enrollment, and a second metastatic site biopsy was obtained after 12 weeks of treatment with abiraterone acetate–prednisone. Our results suggest that biopsy of mCRPC is feasible and provides adequate tissue for pursuing histopathologic and sequencing studies in most, but not all, cases. Overall, 78% of procedures yielded tumoral tissue either by histopathologic or nucleic acid analysis. This figure is lower than the 93% yield recently reported by Robinson et al3 in a somewhat similar population of patients, with the difference being that in their study, bone biopsies corresponded to only 40% of all biopsies compared to 76% of all metastatic sites biopsied in our cohort study. The sampling in the Robinson et al cohort was also done in the context of standard-of-care approaches or through a cohort of prospective clinical trials and consisted of biopsies of bone or soft tissue metastases obtained under radiographic guidance. Tissue was snap frozen, and further procurement was guided by frozen section of the tissue. Of 189 patients who underwent biopsy, tumor presence was pathologically confirmed in 175 (93%), and greater than 20% tumor purity was obtained in 150. In the Robinson et al series, 3.6% had a neuroendocrine phenotype, ranging from usual adenocarcinoma with neuroendocrine differentiation to small cell carcinoma. The lower yield associated with biopsy of skeletal metastases was observed in our cohort as well, with only 74% of skeletal biopsies yielding tumoral material (by histopathologic and nucleic acid analysis) vs 90% in nonskeletal metastases. Thus, despite the recognition that in advanced prostate cancer the dominant metastatic site is bone, selection of skeletal metastasis for biopsy runs the risk of a 1 in 4 chance for obtaining no tumoral material for downstream successful genomic sequencing and analyses. Also expectedly, tumor yield was lower in second biopsies after treatment exposure compared to first biopsies, presumably the result of therapy effect following first biopsy. Interestingly, we also observed that each subsequent serial pass at the time of biopsy resulted in a lower chance for obtaining a positive core, and this was statistically significant when the first 2 passes were compared to the third and fourth passes (P<.001). This finding suggests that first and second passes are the best specimens for sequencing studies. Other factors, such as distance from the skin to the lesion edge, may also affect tumor yield.13
Our study does not allow a comparison between nucleic acid and histopathologic analysis. Although there was a substantially lower yield in histopathologic analysis compared to extracted DNA and RNA tumor purity, the study design was heavily weighted toward maximum submission of tumor for nucleic acid extraction, while only a 1- to 2-mm section of the end from each core was submitted for formalin fixation. This is underscored by the higher yield obtained in the few F5 and F6 cores, which were entirely submitted for histopathologic analysis. The current study also found that histopathologic analysis is useful not only in determining presence of tumor but also in identifying tumor type and other morphologic characteristics. Further, immunohistochemical analysis of archived tissue may complement genomic and proteomic studies.
Other than the 2 aforementioned prospective cohort studies, previous attempts to harvest mCRPC have been based on rapid autopsy procurement or retrospective series.14, 15 In 2014, Hong et al16 reported successful tissue procurement from mCRPC in 7 patients, using image-guided biopsies in 5 while 2 underwent a therapeutic surgical procedure. Of 9 sites sampled, 8 yielded samples suitable for genomic analysis. Van Allen et al17 also reported successful whole-exome sequencing from a prostate cancer bone metastasis biopsy. The authors were able to identify genomic alterations in the phosphoinostide 3–kinase pathway as well as germline variants in the BRCA2 gene, both abnormalities potentially actionable. Efstathiou et al18 were able to successfully analyze mCRPC by immunohistochemistry for androgen receptor expression and other markers from tissue harvested from bone marrow biopsies. However, they did not target specific metastatic lesions. Despite this, they were able to harvest tumoral tissue in 47% and 53% of biopsy specimens obtained before and after 8-week enzalutamide therapy, respectively. More recently, Sailer et al5 described a bone biopsy protocol for mCRPC that yielded adequate tissue for DNA and RNA procurement. They described an 86% tumor detection rate, with an 82% success rate in whole-exome sequencing and a 33% success rate in RNA sequencing. The protocol included a combination of frozen-section and formalin-fixed tissue for histopathologic analysis and a combination of tissue and blood clot from the biopsy site for sequencing. Contrary to our study, all tissue was submitted for histopathologic analysis if the initial assessment was negative for tumor.
The relevance of these findings should be analyzed against the background of the emergence of new noninvasive techniques of interrogating prostate cancer cells, such as cell-free DNA,19 circulating tumor cells,20, 21 extracellular vesicles,22 or other components of liquid biopsies. While promising, these techniques are in early stages of development and will require validation by comparison with robust data obtained from traditional biopsy sampling.23 Some clinically relevant questions, such as the significance of a heterogeneous molecular landscape in mCRPC,24, 25 may only be answered by location-specific sampling. Genomic analysis of obtained tumoral DNA is currently in progress, and results of this analysis will be the topic of future publications.
Conclusion
Image-guided core needle biopsy of mCRPC is feasible and provides adequate tissue for pursuing histopathologic and sequencing studies. Histopathologic analysis of extracted material mostly, but not completely, correlates with DNA/RNA sequencing data. Bone lesions yield considerably less tumoral material than nonosseous sites, but a proper biopsy technique and adequate sample handling may provide acceptable rates of tumor yield.
Acknowledgments
The funding organizations had no role in the design and execution of the study, in the collection, analyses, and interpretation of the data, or in the preparation, review, or approval of the submitted manuscript.
Footnotes
Dr Kohli is now with H. Lee Moffitt Cancer Center, Tampa, FL.
Grant Support: The PROMOTE study is funded in part by the Mayo Clinic Center for Individualized Medicine, A.T. Suharya and D.H. Ghan, Joseph and Gail Gassner, and the Mayo Clinic Schulze Center for Novel Therapeutics in Cancer Research. Other groups contributing to the study include the Mayo Clinic Cancer Center and the Pharmacogenomics Research Network. Funding support for this work was provided in part by grant R01 CA21209 from the National Institutes of Health, National Cancer Institute and award W81XWH-15-1-0634 from the US Department of Defense (M.K.).
Potential Competing Interests: Drs Jimenez, Atwell, Sicotte, Wang, Eiken, McMenomy, Tan, Wang, and Kohli have received royalties from Mayo Clinic Ventures/Champion Oncology (waived and paid to their institution).
Contributor Information
Rafael E. Jimenez, Email: jimenez.rafael@mayo.edu.
Manish Kohli, Email: Manish.kohli@moffitt.org.
Supplemental Online Material
Supplemental Figure 1.

Supplemental Figure 2.
Supplemental material can be found online at http://mcpiqojournal.org. Supplemental material attached to journal articles has not been edited, and the authors take responsibility for the accuracy of all data.
References
- 1.Siegel R.L., Miller K.D., Jemal A. Cancer statistics, 2018. CA Cancer J Clin. 2018;68(1):7–30. doi: 10.3322/caac.21442. [DOI] [PubMed] [Google Scholar]
- 2.Wang L., Dehm S.M., Hillman D.W., et al. A prospective genome-wide study of prostate cancer metastases reveals association of Wnt pathway activation and increased cell cycle proliferation with primary resistance to abiraterone acetate-prednisone. Ann Oncol. 2018;29(2):352–360. doi: 10.1093/annonc/mdx689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Robinson D., Van Allen E.M., Wu Y.M., et al. Integrative clinical genomics of advanced prostate cancer. Cell. 2015;161(5):1215–1228. doi: 10.1016/j.cell.2015.05.001. [published correction appears in Cell. 2015;162(2):454] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Grasso C.S., Wu Y.M., Robinson D.R., et al. The mutational landscape of lethal castration-resistant prostate cancer. Nature. 2012;487(7406):239–243. doi: 10.1038/nature11125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sailer V., Schiffman M.H., Kossai M., et al. Bone biopsy protocol for advanced prostate cancer in the era of precision medicine. Cancer. 2018;124(5):1008–1015. doi: 10.1002/cncr.31173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mateo J., Carreira S., Sandhu S., et al. DNA-repair defects and olaparib in metastatic prostate cancer. N Engl J Med. 2015;373(18):1697–1708. doi: 10.1056/NEJMoa1506859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Van Allen E.M., Wagle N., Stojanov P., et al. Whole-exome sequencing and clinical interpretation of formalin-fixed, paraffin-embedded tumor samples to guide precision cancer medicine. Nat Med. 2014;20(6):682–688. doi: 10.1038/nm.3559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kohli M., Wang L., Xie F., Sicotte H., Yin P., Dehm S.M., et al. Mutational landscapes of sequential prostate metastases and matched patient derived xenografts during enzalutamide therapy. PLoS One. 2015;10(12):e0145176. doi: 10.1371/journal.pone.0145176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wang C., Evans J.M., Bhagwate A.V., et al. PatternCNV: a versatile tool for detecting copy number changes from exome sequencing data. Bioinformatics. 2014;30(18):2678–2680. doi: 10.1093/bioinformatics/btu363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Social Science Statistics Mann-Whitney U test calculator. www.socscistatistics.com/tests/mannwhitney/
- 11.Mullane S.A., Van Allen E.M. Precision medicine for advanced prostate cancer. Curr Opin Urol. 2016;26(3):231–239. doi: 10.1097/MOU.0000000000000278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Beltran H., Eng K., Mosquera J.M., et al. Whole-exome sequencing of metastatic cancer and biomarkers of treatment response. JAMA Oncol. 2015;1(4):466–474. doi: 10.1001/jamaoncol.2015.1313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.McKay R.R., Zukotynski K.A., Werner L., et al. Imaging, procedural and clinical variables associated with tumor yield on bone biopsy in metastatic castration-resistant prostate cancer. Prostate Cancer Prostatic Dis. 2014;17(4):325–331. doi: 10.1038/pcan.2014.28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mehra R., Kumar-Sinha C., Shankar S., et al. Characterization of bone metastases from rapid autopsies of prostate cancer patients. Clin Cancer Res. 2011;17(12):3924–3932. doi: 10.1158/1078-0432.CCR-10-3120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rubin M.A., Putzi M., Mucci N., et al. Rapid ("warm") autopsy study for procurement of metastatic prostate cancer. Clin Cancer Res. 2000;6(3):1038–1045. [PubMed] [Google Scholar]
- 16.Hong M.K., Sapre N., Phal P.M., Macintyre G., Chin X., Pedersen J.S., et al. Percutaneous image-guided biopsy of prostate cancer metastases yields samples suitable for genomics and personalised oncology. Clin Exp Metastasis. 2014;31(2):159–167. doi: 10.1007/s10585-013-9617-2. [DOI] [PubMed] [Google Scholar]
- 17.Van Allen E.M., Foye A., Wagle N., et al. Successful whole-exome sequencing from a prostate cancer bone metastasis biopsy. Prostate Cancer Prostatic Dis. 2014;17(1):23–27. doi: 10.1038/pcan.2013.37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Efstathiou E., Titus M., Wen S., et al. Molecular characterization of enzalutamide-treated bone metastatic castration-resistant prostate cancer. Eur Urol. 2015;67(1):53–60. doi: 10.1016/j.eururo.2014.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yin C., Luo C., Hu W., Ding X., Yuan C., Wang F. Quantitative and qualitative analysis of circulating cell-free DNA can be used as an adjuvant tool for prostate cancer screening: a meta-analysis. Dis Markers. 2016;2016:3825819. doi: 10.1155/2016/3825819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.McDaniel A.S., Ferraldeschi R., Krupa R., et al. Phenotypic diversity of circulating tumour cells in patients with metastatic castration-resistant prostate cancer. BJU Int. 2017;120(5B):E30–E44. doi: 10.1111/bju.13631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.León-Mateos L., Vieito M., Anido U., López López R., Muinelo Romay L. Clinical application of circulating tumour cells in prostate cancer: from bench to bedside and back. Int J Mol Sci. 2016;17(9):1580. doi: 10.3390/ijms17091580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Biggs C.N., Siddiqui K.M., Al-Zahrani A.A., et al. Prostate extracellular vesicles in patient plasma as a liquid biopsy platform for prostate cancer using nanoscale flow cytometry. Oncotarget. 2016;7(8):8839–8849. doi: 10.18632/oncotarget.6983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jiang R., Lu Y.T., Ho H., et al. A comparison of isolated circulating tumor cells and tissue biopsies using whole-genome sequencing in prostate cancer. Oncotarget. 2015;6(42):44781–44793. doi: 10.18632/oncotarget.6330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Haffner M.C., Mosbruger T., Esopi D.M., et al. Tracking the clonal origin of lethal prostate cancer. J Clin Invest. 2013;123(11):4918–4922. doi: 10.1172/JCI70354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Taylor B.S., Schultz N., Hieronymus H., et al. Integrative genomic profiling of human prostate cancer. Cancer Cell. 2010;18(1):11–22. doi: 10.1016/j.ccr.2010.05.026. [DOI] [PMC free article] [PubMed] [Google Scholar]