

Abstract
Recurrent panic attacks (PAs) are a common feature of panic disorder (PD) and post-traumatic stress disorder (PTSD). Several distinct brain regions are involved in the regulation of panic responses, such as perifornical hypothalamus (PeF), periaqueductal grey, amygdala and frontal cortex. We have previously shown that inhibition of GABA synthesis in the PeF produces panic-vulnerable rats. Here, we investigate the mechanisms by which a panic-vulnerable state could lead to persistent fear. We first show that optogenetic activation of glutamatergic terminals from the PeF to the basolateral amygdala (BLA) enhanced the acquisition, delayed the extinction and induced the persistence of fear responses 3 weeks later, confirming a functional PeF-amygdala pathway involved in fear learning. Similar to optogenetic activation of PeF, panic-prone rats also exhibited delayed extinction. Next, we demonstrate that panic-prone rats had altered inhibitory and enhanced excitatory synaptic transmission of the principal neurons, and reduced protein levels of metabotropic glutamate type 2 receptor (mGluR2) in the BLA. Application of an mGluR2 positive allosteric modulator (PAM) reduced glutamate neurotransmission in the BLA slices from panic-prone rats. Treating panic-prone rats with mGluR2 PAM blocked sodium lactate (NaLac)-induced panic responses and normalized fear extinction deficits. Finally, in a subset of patients with comorbid PD, treatment with mGluR2 PAM resulted in complete remission of panic symptoms. These data demonstrate that a panic-prone state leads to specific reduction in mGluR2 function within the amygdala network and facilitates fear, and mGluR2 PAMs could be a targeted treatment for panic symptoms in PD and PTSD patients.
Keywords: Fear extinction, Metabotropic glutamate 2 receptors, panic attacks, perifornical hypothalamus, amygdala, GABA
INTRODUCTION
Panic attack (PA) is a survival response1 that is normally evoked by imminent exteroceptive threats to survival (e.g., predators) or by certain interoceptive threats (e.g. carbon dioxide (CO2), sodium lactate (NaLac)) in vulnerable subjects. Panic attacks are characterized by cognitive symptoms of catastrophic fear accompanied by strong behavioral responses (i.e., fight or flight) as well as coordinated cardiorespiratory and metabolic responses to rapidly respond to these threats. Recurrent PAs, categorized as being either unexpected or expected, are the hallmark criteria for diagnosing panic disorder (PD), and are common symptoms of post-traumatic stress disorder (PTSD) and other fear disorders2.
The perifornical hypothalamic region (PeF) has long been recognized as a site that can elicit a ‘flight-or-fight’ response when activated, is critical for defense responses when confronted with predators, and is a putative node in generating coordinated responses characteristic of PAs. In humans, deep brain stimulation in the PeF produces self-reports of PA and fear of dying as well as somatic symptoms strongly associated with PAs in patients with PD (e.g., tachycardia, increased blood pressure, hyperventilation, thermal sensations and paresthesias)3–5. Similar panic associated responses are induced when disinhibiting the PeF region of rats, including escape behaviors, selective enhancement of fear, anxiety-like behavior, and increased blood pressure, heart rate, and respiration6–10. Importantly, chronic disinhibition of the PeF region produces rats that are prone to displaying panic-like responses to interoceptive stimuli, such as NaLac11–13 and CO2 [see review14]; stimuli that also reliably provoke PAs in subjects with PD15–17.
The mechanisms by which an initial PA contributes to persistent, disabling fear disorders such as PD or PTSD is not well understood. Mineka and Zinbarg have proposed that dysfunctional extinction of conditioned fear is important mechanism underlying the development and maintenance of PD18. There is evidence that persistent experience of fear and catastrophic cognitions following a PA is likely to lead to a higher incidence of PD and avoidance19. Similarly, while not always fully replicated, there is a general consensus that peri-traumatic PAs may worsen the likelihood of developing PTSD20, 21. Thus, one long-term consequence of PAs appears to be predisposing an individual to developing chronic fear disorders such as PD, PTSD and phobias. Our aim was to elucidate the potential mechanisms by which a panic-vulnerable state could lead to chronic fear disorders through a series of studies to determine the molecular basis of enhanced fear in panic vulnerable states. We hypothesized that panic response generated in the PeF region causes dysregulations in the amygdala, which leads to resistant extinction and higher vulnerability to phobia development. The amygdala is a critical brain region for the acquisition, consolidation, and extinction of fear memories22, and has reciprocal connections with the PA generating PeF region23–26. We investigated the acquisition and persistence of fear responses as well as molecular and network changes in the fear circuitry of panic-prone animals. We also conducted a preliminary translation of our hypotheses utilizing post-hoc analyses on a subset of data from a previous clinical study.
We show that optogenetic stimulation of PeF-amygdala projections enhances acquisition, delays the extinction of conditioned fear, and strengthens long-term fear memories. Using a standard fear conditioning paradigm, we first confirmed the delayed extinction of conditioned fear in chronic PeF disinhibited panic-vulnerable animals. Next, we conducted a series of studies to elucidate molecular determinants of panic vulnerability: 1) we evaluated changes in amygdala networks in panic-prone rats using whole-cell patch-clamp, which demonstrated a shift in excitatory/inhibitory ratio (E/I ratio) toward excitation and an increase in the excitability of BLA principal neurons; 2) we determined expression differences in the mGluR2 protein and mRNA levels in the amygdala; 3) we demonstrated that pre-treatment of amygdala slice preparations with selective mGluR2 PAM JNJ-42153605 reduced frequency of spontaneous excitatory postsynaptic potentials (sEPSPs); 4) we confirmed that application of mGluR2 PAM JNJ-42153605 significantly reduced glutamate release from optically stimulated PeF terminals within the BLA; 5) we tested the effects of treating panic-prone rats with a second mGluR2 PAM JNJ-40411813/ADX71149 that has been evaluated in phase 2 clinical studies and observed prevention of NaLac-induced panic responses and normalized conditioned fear extinction deficits; and finally, 6) we re-analyzed the results of a JNJ-40411813 clinical trial and uncovered remission of panic symptoms in PD patients following treatment with this mGluR2 PAM.
METHODS
Animals and housing conditions.
All experiments were conducted on adult male Sprague-Dawley rats (behavior: 250–300 g, electrophysiology: ~200 g, molecular analysis: 275–300 g) that were purchased from Envigo (Envigo, Indianapolis, IN) and were housed individually in plastic cages under standard environmental conditions (22ºC; 12/12 light/dark cycle) for 7–10 days prior to gathering data. For the optogenetic manipulation of fear behavior experiment, juvenile male Sprague-Dawley rats were ordered from Envigo and were group-housed upon arrival on postnatal day (PD) 22 (35–50 g). Food and water were provided ad libitum. All experiments were conducted in accordance with the NIH Guide for the Care and Use of Laboratory Animals, Eighth Edition (Institute for Laboratory Animal Research, The National Academies Press, Washington, DC, 2011) and the guidelines of the IUPUI Institutional Animal Care and Use Committee.
Surgical procedures.
Prior to and during surgery, rats were anesthetized with an isoflurane system (MGX Research Machine; Vetamic, Rossville IN). To produce panic-vulnerable and control rats, a 26 gauge T-shaped cannulae (#3260PG, Plastics One Inc., Ranoake VA) was directed at the PeF; cemented in place (Ortho-Jet; Lang, Wheeling, IL); connected to a subcutaneously implanted osmotic minipump (Alzet; Cupertino, CA) prefilled with the GABA synthesis inhibitor l-allyglycine (l-AG) solution or the inactive isomer (d-allylglycine, d-AG)13. The stereotaxic coordinates used for PeF transfection were −2.9 mm anterior-posterior (AP), ± 3.18 mm medial-lateral (ML), and −8.2 mm dorsal-ventral (DV), relative to bregma. We have previously reported significant reduction of activity of the GABA synthesis enzyme, Glutamic Acid Decarboxylase (GAD) and reduction of GABA content from 4 to 7 days after microinfusion of the GAD inhibitor l-AG11, 13, 27. All behavior, electrophysiological, and gene expression assessment experiments were performed between 5 to 7 days after the start of l-AG pump microinfusion. All animals were randomly selected for each treatment condition tested.
Optogenetic constructs.
AAV5-CaMKIIα-ChR2(H134R)-EYFP (ChR2) and AAV5-CaMKIIα-EYFP (Con) were obtained from the University of North Carolina Vector Core (Chapel Hill, NC). DNA constructs were provided by Dr. Karl Deisseroth, Stanford University. AAV5 vector titers were 6.2 × 1012 vg/ml (AAV5-CaMKIIα-ChR2(H134R)-EYFP) and 4.3 × 1012 vg/ml (AAV5-CaMKIIα-EYFP).
Optogenetic virus infusion and optic fiber implantation.
For bilateral viral transduction of the PeF (0.5 µl/side), juvenile male rats (P23-P25) were injected with stock concentrations of a virus expressing enhanced yellow fluorescent protein (eYFP) fused to the cation-pump channelrhodopsin-2 (ChR2) that is sensitive to blue light (AAV5-CaMKIIα-ChR2(H134R)-EYFP) or a control virus that mediates expression of eYFP alone (AAV5-CaMKIIα-EYFP). The stereotaxic coordinates used for PeF transfection were −2.1 mm AP, ± 1.1 mm ML, and −8.0 mm, relative to bregma. A 10 μl microsyringe was used to deliver the viral solutions into the PeF at a rate of 80 nl/min using a microsyringe pump (Harvard Apparatus, Holliston, MA) and injection needles were left in place for 10 min after infusion to allow for complete diffusion before removal. Animals were returned to their home cages for 4 weeks to permit high viral expression at the time of experiments, that was determined by electrophysiological and immunohistochemical methods. Then, optical fibers were bilaterally implanted above the BLA (optical fiber length: 9.0 mm; ferrule bore: 230 μm; Precision Fiber Products, Milpitas, CA). The stereotaxic coordinates used for bilateral fiber implants were −2.8 mm AP, ± 4.8 mm ML, and −8.0 mm DV, relative to bregma. Fiber implants were anchored in place with dental cement (Ortho-Jet; Lang, Wheeling, IL). Rats were subcutaneously injected with the analgesic buprenorphine (0.05 mg/kg) at the conclusion of each surgery and at 12 h increments (4x total) to mitigate post-surgical pain. All rats were allowed 7–10 days of recovery from surgery prior to behavioral testing. Final weight of animals at time of behavior experiments was ~250 g.
Assessment of fear behavior with optogenetic manipulation of PeF terminals in the BLA.
One week following bilateral fiber implants into the BLA (at least 5 weeks after viral injections into the PeF), animals were habituated to a sound attenuating fear conditioning chamber (Kinder Scientific, Poway, CA) for 10 min. Twenty four hours later, animals were placed in a rectangular testing chamber (18×10.5×15 inches) and optical fiber implants were connected via FC ceramic sleeves and 1.0 meter patch cables (#94024–100, Plexon Inc, Dallas, TX) to compact blue LED modules (465 nm, Plexon Inc). PeF terminals in the BLA were stimulated for 5 min (duration: 10 ms pulses, intensity: 10 mW; frequency: 10 Hz) using PlexBright system (Plexon Inc). Immediately following optical stimulation, animals were assessed for acquisition of fear using the same parameters outlined for the panic-prone l-AG or d-AG control rats described above. Animals underwent the same 5 min optical stimulation of PeF terminals in the BLA immediately prior to both consolidation and extinction testing of the fear conditioning protocol described in the previous section. Three weeks following activation of PeF terminals in the BLA, animals were assessed for spontaneous recovery of fear28, 29. This was tested by placing each animal that was randomly selected for its treatment condition in the same sound attenuating fear conditioning chamber that they had acquired fear in three weeks earlier. After a 120 s acclimation period, animals were given 10 trials of the CS (20 s, 80 dB) separated by 120 s intervals. The same experimenter (JLL) handled the rats during all sessions and was blinded to the treatment conditions. All trials were digitally video-recorded. The file names were re-coded by AIM. Freezing behavior (no visible signs of movement) was scored by a blind scorer (JLL) during the sound presentation and converted to percentage of total time. At the end of the experiments animals were sacrificed.
Assessment of conditioned fear behavior in panic-prone rats.
Panic-prone l-AG or d-AG control rats were habituated to the sound attenuating fear conditioning chamber on day 1 (Kinder Scientific, Poway, CA) for 10 min. For all experiments, the chamber was cleaned between animals. On day 2 acquisition of fear occurred where the rats were placed back into the chamber and after a 120 s acclimation period received 5 pairings (120 s inter-trial interval) of the conditioned stimulus (CS; 20 s, 80 dB) followed immediately by the unconditioned shock stimulus (US; 500 ms, 0.8 mA foot shock). On day 3, CS consolidation was assessed by placing the rats back into the chamber with a 120 s acclimation period followed by 5 presentations of the CS only (20 s, 80 dB) separated by 120 s. On day 4 extinction was assessed which consisted of the 120 s acclimation period followed by 20 trials of the CS (20 s, 80 dB) separated by 120 s intervals. The same experimenter (SDF) handled the rats during all sessions and was blinded to the phenotype. All trials were digitally video-recorded. The file names were modified by PLJ. Freezing behavior (no visible signs of movement) was scored by a blind scorer (SDF) during the sound presentation and converted to percentage of total time.
Electrophysiological studies of amygdala activity.
Five to seven days after initiation of l-AG or d-AG pump microinfusion, the Sprague-Dawley male rats (~200 g) were anesthetized with isoflurane and then trans-cardially perfused with 25–30 mL of protective artificial cerebrospinal fluid (aCSF) of the following composition (in mM): 93 N-methyl-D-glucamine (NMDG), 2.5 KCl, 1.2 NaH2PO4, 30 NaHCO3, 20 HEPES, 25 glucose, 2 thiourea, 5 Na-ascorbate, 3 Na-pyruvate, 0.5 CaCl2·4H2O, and 10 MgSO4·7H2O30. Rats were then decapitated, brains were quickly dissected and coronal slices (350 μm) were sectioned on either Leica VT1000S (Leica Biosystems, Buffalo Grove, IL) or Campden 7000smz-2 vibratomes (Lafayette Instrument Co, Lafayette, IN)31. For the initial recovery, slices were immersed in an oxygenated (mixture of 95% O2/5% CO2) NMDG-based aCSF at 30 °C for ≤15 min and then transferred to room temperature (RT) aCSF of the following composition (in mM): 130 NaCl, 3.5 KCl, 1.1 KH2PO4, 1.3 MgCl2, 2.5 CaCl2, 10 glucose, 30 NaHCO3. The osmolality of aCSF solutions was adjusted to ~315 mOsm. Once transferred to a submersion-type slice chamber and perfused at a rate of 2–3 ml/min with aCSF heated to 30°C, slices were visualized using either Nikon E600FN Eclipse (Nikon Instruments Inc., Melville, NY) or Scientifica SliceScope Pro 6000 (Scientifica, Uckfield, UK) upright microscopes. Whole-cell patch-clamp recordings were obtained using standard techniques with borosilicate glass electrodes (resistance 3–6 mΩ, WPI, Sarasota, FL) filled with a potassium gluconate-based recording solution with the following composition (in mM): 140 K-gluconate, 2 KCl, 1 MgCl2, 10 HEPES, 5 phosphocreatine, 2 K-ATP, 0.2 Na-GTP adjusted to pH 7.3 with KOH, and having an osmolality of 285–295 mOsm.
Individual neurons in the BLA and central nucleus of amygdala (CeA) were visualized using differential interference contrast microscopy with a 40x water immersion objective and displayed in real time on a monitor. Whole-cell recordings were made with a Multiclamp 700B amplifier using pClamp 10.3 software and a Digidata 1322A interface (Molecular Devices, Sunnyvale, CA). Experimenters (AIM and ETD) were blinded to the phenotype (l-AG versus d-AG) of the rats. Drugs were applied by adding them at the required concentration directly into the aCSF. To examine the stimulus-evoked postsynaptic currents, a concentric stimulating electrode (FHC, Bowdoinham, ME) was placed ~500 μm from the recorded neuron. A stimulus was repeated 3 times at a frequency of 0.2 Hz and then averaged for subsequent data analysis. The evoked excitatory and inhibitory (eEPSPs and eIPSPs, respectively), spontaneous excitatory (sEPSPs) and inhibitory (sIPSPs) postsynaptic potentials were recorded from different holding potentials ranging from −50 mV to −70 mV. For optogenetic experiments, animals expressing AAV5-CaMKIIα-hChR2(H134R)-eYFP within the PeF were utilized. To isolate light-evoked postsynaptic potentials, 2 mM QX314 was added to the recording solution and 100 µM picrotoxin, 1 µM CGP 53432, 100 µM AP4 were added to aCSF. The blue 490 nm light (10 ms duration) was delivered via 40x objective of Scientifica SliceScope Pro 6000 microscope outfitted with pE-2 four wavelength LED system (CoolLED, Andover, UK). The response was recorded at a membrane potential of −70 mV and was confirmed to be excitatory by loss of response following addition of 20 µM DNQX and 25 µM APV. All drugs were purchased from Sigma except for QX314, DNQX, APV, CGP 53432, SR 95531, which were obtained from Tocris Bioscience (Bio-Techne Co, Minneapolis, MN). Access resistance was continuously monitored; recordings in which resistance exceeded 20 MΩ or 15% change were excluded from analysis.
Gene Expression Assessments with TaqMan low-density array (TLDA).
Rats were euthanized by brief exposure to isoflurane and rapid decapitation 7 days after being implanted with l-AG or d-AG osmotic minipumps aimed at the PeF as described above. Locations of injectors were verified in the slices using a Leica dissecting microscope set to 4x magnification. The BLA and CeA were each dissected out of three adjacent 300 µm coronal brain slices [from approx. + 0.96 to + 2.76 mm from bregma32] using a 1.0 mm Harris Micro-punch (Electron Microscopy Sciences, Hatfield, PA). The tissue was processed and gene analysis was performed using the custom-designed TaqMan Low Density Array (TLDA) as previously described33. The glutamate and GABA-related gene expression panel was normalized using a geNorm34 approach as previously described in detail33. Relative gene expression was determined using delta delta Ct and data are relative to d-AG treated rats.
Western blot assessment of mGluR2 receptors in the amygdala.
Following 6 days of l-AG infusions into the PeF, animals were rapidly decapitated, brains flash-frozen in 2-methylbutane (Fisher Scientific, Waltham, MA), and then tissue was regionally dissected in the BLA and CeA from 300 μm coronal brain slices using a stainless steel microdissecting needle (1.0 mm (#18035–01) or 0.5 μm (#18035–50) diameter Neuropunch, Fine Science Tools, Foster City, CA). Tissue was then homogenized in RIPA buffer (Boston BioProducts, #BP-115DG) containing a protease inhibitor cocktail (Complete Mini Tablet; Roche, #11836153001). Protein was mixed with RIPA lysis buffer, centrifuged at 14,000 rpm for 20 min at 4ºC and supernatant was analyzed for protein concentration by the Bradford method (BioRad Laboratories, Hercules, CA). Then, 10 μg of protein incubated for 5 min at 95ºC prior to separation by 10% SDS-PAGE. Following electrophoresis, proteins were transferred to a nitrocellulose membrane (BioRad Laboratories). The membranes were blocked with Odyssey blocking buffer (LI-COR Biosciences, Lincoln, NE) in Tris-buffered saline (containing 0.1% Tween (TBS-T) for 60 min at RT and incubated with a rabbit primary polyclonal antibody to mGluR2 receptors (95 kDa; 1:20,000; Abcam, #ab150387) overnight 4°C. The membranes were rinsed for 5 min, 4 times, at RT in TBS-T. After the rinsing procedure, the membranes were incubated for 1 h at RT in IRDye 800-conjugated affinity purified anti-rabbit (1:15,000, #925–32211, LI-COR Biosciences) in Odyssey blocking buffer in 0.1% TBS-T. Control for protein loading was achieved by normalization to β-actin (43 kDa; 1:20,000, primary #sc47778, Santa Cruz, secondary IRDye 700-conjugated affinity purified anti-IgG H&L; 1:15,000, #925–6807, LI-COR Biosciences). Proteins were detected using the Odyssey infrared imaging system (excitation/emission filters at 780 nm/820 nm range; LI-COR Biosciences). For all Western analyses, data were normalized to d-AG.
Immunofluorescence of c-Fos in optogenetically stimulated animals.
Ninety minutes after the onset of PeF terminal optogenetic stimulation in the BLA, animals were deeply anesthetized with isoflurane (MGX Research Machine; Vetamic) and perfused trans-cardially with phosphate-buffered saline (PBS, pH 7.4), followed by 4% paraformaldehyde (PFA) in PBS (pH 7.4). Brains were removed, post-fixed overnight in 4% PFA at 4°C, then immersed in 30% sucrose in PBS and stored at 4°C until they were sectioned at 30 μm using a freezing microtome. Six alternate sets of slices were stored in cryoprotectant at –20 °C until they were processed for dual labeling immunohistochemistry. One set of alternate sections containing the rostrocaudal extent of the BLA were rinsed three times with PBS for 10 min at RT. Sections were then incubated for 20 min at RT in PBS containing 1% H2O2, rinsed two times in PBS for 10 min each, and then rinsed in PBS containing 0.3% Triton-X (PBS-T). Sections were then incubated in rabbit anti-c-Fos polyclonal antibody in PBS-T (1:1,000; sc-52; Santa Cruz Biotechnologies) overnight at RT and then rinsed 3 times for 10 min each in PBS. After rinsing with PBS, sections were incubated in donkey anti-rabbit Alexa Fluor 568 IgG (1:200; A10042, Life Technologies) for 90 min in PBST at RT. Tissue was then rinsed three times, for 10 min each, in PBS, incubated in mouse anti-GFP (1:100; A11120; Life Technologies) overnight, and rinsed again three times, 10 min each, in PBS. Sections were then placed in donkey anti-mouse Alexa Fluor 488 IgG (1:200; A11017; Life Technologies) for 90 min and then rinsed and stored in PBS until tissue was mounted onto slides and coverslipped using Vectashield mounting medium with DAPI (Vector; H-1200; Burlingame, CA). Sections were analyzed by an observer blind to the treatment conditions (JLL) and the number of c-Fos-immunoreactive (ir) cells in the BLA was counted.
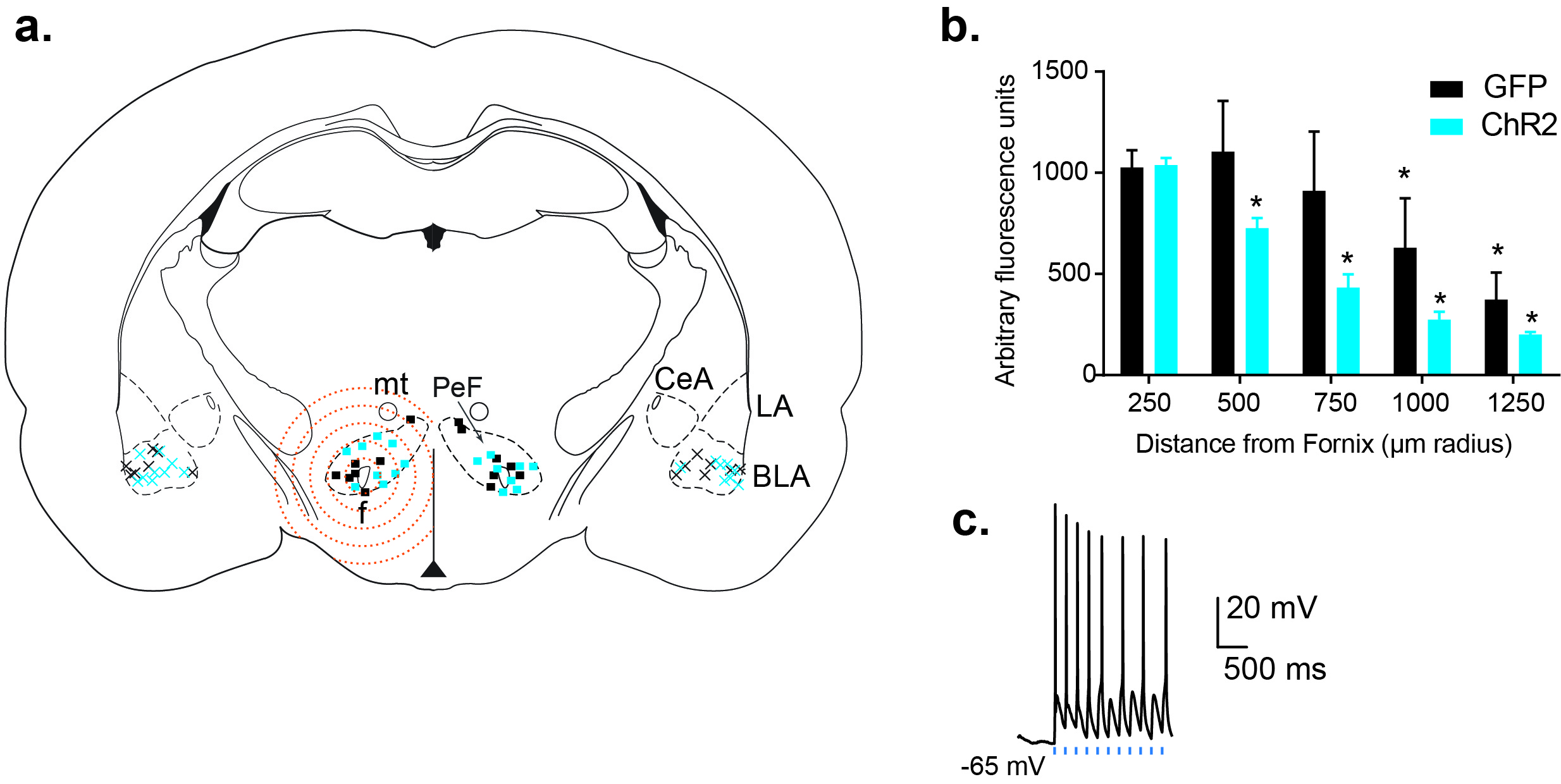
We used an additional set of slices to confirm the injection sites, viral infection, and spread of the virus in the PeF (Fig.1f,g, Suppl. Fig.1a). Only data from animals with injection localized to the PeF were included in the manuscript. The viral spread in the hypothalamic area containing PeF was analyzed by measuring fluorescence intensity in 250 μm radius concentric circles around the fornix. We observed a significant correlation between distance from fornix and fluorescence intensity in Chr2 and Control groups [2-way RM ANOVA, virus x distance interaction, F4,20 = 5.24, p = 0.005; distance effect, F4,20 = 53.86, p < 0.0001, Suppl. Fig.1a,b]. Tukey’s posthoc analysis confirmed significant reduction of fluorescence intensity by 500 μm from fornix in ChR2 group [p < 0.006, Suppl.Fig.1b].
Figure 1. Optogenetic activation in the amygdala of terminals from the panic-inducing PeF results in enhanced acquisition, consolidation, delayed extinction, and the persistence of conditioned fear.
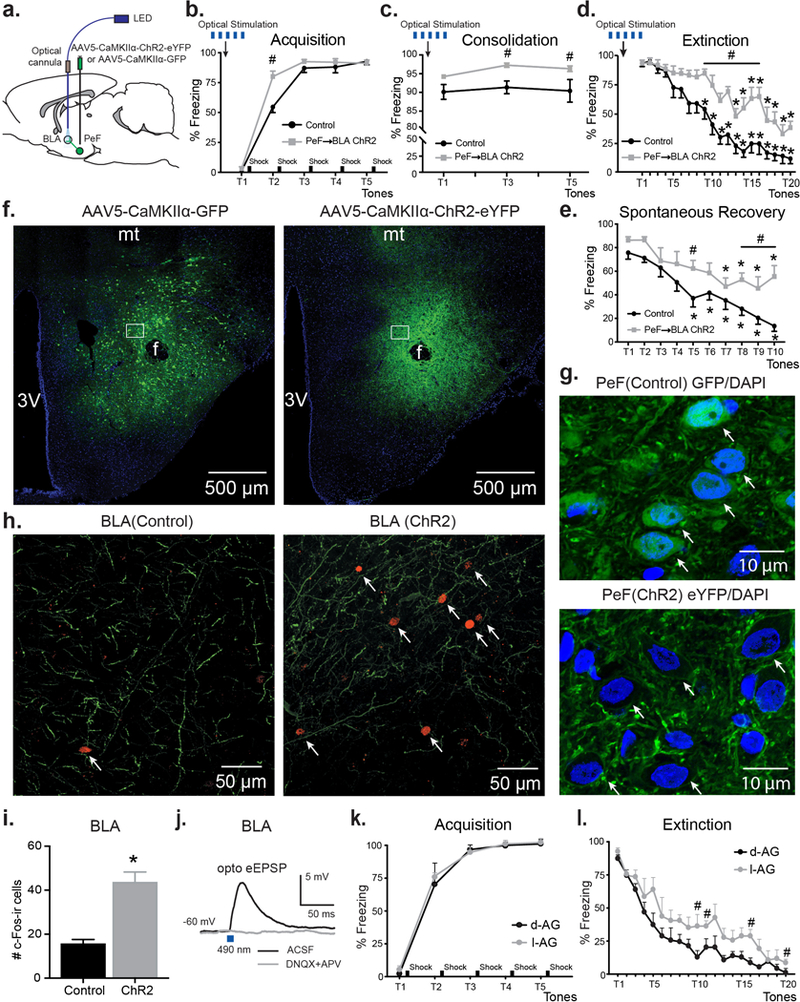
a) Schematic representation of the experimental setup. b-d) Optogenetic stimulation of PeF glutamatergic inputs in the BLA enhanced acquisition (b) as well as, consolidation (c), and delayed extinction (d) of fear memories compared to animals injected with control virus (n = 8–9/group). e) Three weeks after optical stimulation ChR2-expressing animals demonstrated significantly higher freezing during the spontaneous memory recovery test in the absence of additional optogenetic stimulation. f) Representative coronal hypothalamus containing sections from control (left) or ChR2 (right) animals showing GFP/eYFP (green) and DAPI (blue) double immunostaining. Scale bars, 500 μm. Bregma −2.92. g) Representative high magnification images showing regions in white boxes from (f) with dual GFP/DAPI immunostaining in PeF cells (arrows). green – GFP, blue – DAPI. Scale bars, 10 μm. Bregma −2.92. h) Representative fluorescent images showing GFP- and eYFP-positive PeF terminals (green) and c-fos (red) immunoreactivity (ir) in the BLA. Arrows represent c-fos-ir BLA neurons. Scale bars, 50 μm. Bregma −2.52. i) Group data showing an increased number of c-fos-ir neurons after stimulation of ChR2 PeF terminals in the BLA compared to control animals. *p < 0.05, unpaired t-test. j) Representative BLA traces depicting oEPSPs evoked by light pulses in the presence of aCSF and 10 min after perfusion with AMPA and NMDA antagonists DNQX (20 μm) and APV (25 μm). k) and l) Consistent with optogenetic results, rats with pharmacological disinhibition of PeF neurons, i.e., panic-prone rats (chronic l-AG infusions into the PeF to inhibit GABA synthesis) compared to control rats (chronic inactive d-AG infusions into the PeF), displayed delayed extinction of fear. Except where otherwise specified, *p < 0.05, ANOVA, Sidak’s within subject posthoc analysis, #p < 0.05, ANOVA, Sidak’s between subjects posthoc analysis. All data reported as mean + S.E.M.
Assessment of panic-associated responses in animals.
Social interaction (SI) test:
Anxiety-like behavior was measured utilizing the SI test35, which is a validated measure of anxiety-associated behaviors36 and is sensitive to current pharmacological treatments for anxiety disorders [acute benzodiazepine37 and chronic selective serotonin reuptake inhibitor (SSRI) treatments38]. The apparatus consisted of a solid box with an open roof approximately 0.9 m long x 0.9 m wide with walls 0.3 m high. A video camera was fixed above the box, and all behavioral tests were videotaped under low red-light conditions (approximately 10 lux) and in a familiar environment. The “experimental” rat and an unfamiliar “partner” rat were both placed individually in the center of the box and allowed to habituate to the environment for a 5 min period 24 h prior to each SI test. During the SI test, the two rats were placed together in the center of the box, and the total duration (sec) of non-aggressive physical contact (grooming, sniffing, crawling over and under, etc.) initiated by the “experimental” rat was quantified over the 5 min duration. Recorded sessions were re-coded by PLJ and were scored at a later time by an investigator (SDF) that was blind to any drug treatment.
Cardiovascular assessments:
Cardiovascular responses (i.e., mean arterial pressure, MAP, and heart rate, HR) were measured by a femoral arterial line connected to a telemetric probe which contained a pressure transducer [Cat. no. C50-PXT, Data Science International (DSI), St. Paul, MN]. DSI Dataquest software was used to monitor and record MAP and HR continuously in freely moving conscious rats for 20 min. The data reported are changes in HR and MAP from the average of the baseline (t −5 to t −1).
mGluR2 PAM compounds.
Two different mGluR2 PAM compounds have been used in the preclinical studies. They are both potent and selective mGluR2 PAMs [ED50 in vivo for JNJ-42153605 and JNJ-40411813 is 5.9 mg/kg and 21.0 mg/kg, respectively]39. Either compound is appropriate to perturb mGluR2 activity in preclinical panic models and advancement of JNJ-40411813 into phase 2 clinical studies40–42 aids translation between preclinical and clinical trials.
Testing the effects of mGluR2 PAM compounds on panic and phobia responses in panic-prone rats.
Five to seven days after initiation of minipump l-AG infusions into the PeF, panic-prone rats were pretreated i.p. with vehicle or 2.5 or 20 mg/kg doses of the mGluR2-PAM JNJ-4041181340-42 50–70 min prior to the i.v. infusion of 0.5M NaLac in a crossover design with at least 48 hrs between each crossover. Cardiovascular and general motor activity was assessed 5 min prior to and 15 min during the NaLac infusions. SI test was done immediately following offset of the NaLac infusions.
In a 2nd experiment, 5–7 days after initiation of minipump l-AG, panic-prone rats were habituated to the fear conditioning chamber on day 1. On day 2 panic-prone rats were systemically (i.p) pretreated with vehicle or the mGluR2-PAM JNJ-4215360543 50–70 min prior to tone + shock pairings. Rats were then exposed to the CS (5 tones) on consolidation day 3; and they were treated once again with JNJ-42153605 50–70 min prior to extinction on day 4 (20 tones) as described previously.
Testing of the efficacy of mGluR2 PAM on ameliorating the severity of panic symptoms in patients.
A phase 2, randomized, multicenter, double-blind, proof-of-concept study (ClinicalTrials.gov Identifier: NCT01582815) was conducted to evaluate the efficacy, safety, and tolerability of JNJ-40411813/ADX71149, a novel mGluR2 PAM as an adjunctive treatment for major depressive disorder (MDD) with significant anxiety symptoms (for details of the clinical study protocol, subject characteristics, and data analysis, see44). One hundred twenty-one patients (men and women, age between 18–64 years) were enrolled and had a DSM-IV-TR diagnosis of MDD, Hamilton Depression Rating Scale-17 (HDRS17) score of ≥18, HDRS17 anxiety/somatization factor score of ≥7, and an insufficient response to current treatment with a selective serotonin or serotonin-norepinephrine reuptake inhibitors. The study protocol was approved by an independent Ethics Committee and was conducted in accordance with ethical principles originating in the Declaration of Helsinki. This study was also in accordance with the International Conference on Harmonization Good Clinical Practice guidelines, applicable regulatory requirements, and in compliance with the study protocol. Furthermore, all patients provided written, informed consent to participate in study.
Exclusion criteria included a primary DSM-IV Axis I diagnosis other than MDD, >1 previously failed antidepressant treatment in the current episode of depression (excluding the current antidepressant), current major depressive episode length >6 months, and history of treatment resistance (≥3 lifetime treatment failures). During the study, patients were recommended not to take any over-the-counter or prescribed medications with moderate-to-strong modulation of cytochrome P450 3A4.
The double-randomized, 8-week double-blind treatment phase was comprised of two 4-week periods. We conducted a post-hoc analysis to examine the effects of JNJ-40411813 on panic anxiety symptoms, as measured by the Panic Disorder Severity Scale (PDSS) in five depressed subjects who met criteria at screening for comorbid panic disorder.
Data analysis.
The number of animals in each group was selected based on findings from our previous studies45, 46. Rats where cannulas or fiber implants were misplaced were removed from the analysis. Final group numbers are shown in figure legends.
First, a D’Agostino & Pearson test was used to assess the homogeneity of variance. All data passed the normality test and therefore we analyzed the data using parametric statistics. Social Interaction behavior was analyzed with an ANOVA with drug treatment as a main factor. In the presence of significance, between and within subjects posthoc analyses were assessed using Fisher’s LSD or Sidak’s tests. Cardiovascular activity was analyzed using an ANOVA with repeated measures with drug treatment as main factor and time as repeated measures. In the presence of significant main effects, between and within subjects posthoc tests were conducted using a parametric Fisher’s LSD or Dunnett’s posthoc tests, respectively. Fear conditioned freezing behavior was analyzed using an ANOVA with repeated measures (RM) with drug treatment as main factor. In the presence of significant main effect, posthoc analyses were assessed with a Fisher’s LSD posthoc test.
Electrophysiological eIPSPs data were analyzed using pClamp 10.3 (Molecular Devices, Sunnyvale, CA). Spontaneous sIPSPs were analyzed using the MiniAnalysis program (Synaptosoft, Decatur, GA). All events were identified visually to avoid errors in detection by automation. The threshold for detection of potentials was set at three times the root mean square baseline noise. For electrophysiological experiments n equals number of recorded cells. The electrophysiological and biochemistry data were collected from minimum of 3 animals for each condition. Electrophysiological experiments were evaluated either by Student’s two-sided t test or ANOVA when warranted. In the presence of significant main effect for ANOVAs, posthoc multiple comparisons were conducted using Sidak’s test. The amplitude of the hyperpolarization-activated current (Ih) was calculated as the difference between the amplitude of the steady-state current (ISS) measured at the end of the voltage step, and the amplitude of the instantaneous current (IINS) measured at the beginning of the voltage inward relaxation. The current/voltage (I-V) curves of the Ih were obtained by plotting the amplitude of the Ih against the membrane potential during the negative voltage step. Comparisons of gene expression between treatment groups were done using individual ANOVAs for each gene and posthoc comparisons were made using a Fisher’s LSD test33. For Western-blots, n equals number of separate experiments. Outliers were excluded if >2 standard deviations from the mean. Statistical significance throughout this manuscript was accepted with p < 0.05. All data are shown as means + s.e.m. All statistical analyses and graphs were produced with SPSS 22.0 (SPSS Inc, Chicago, IL, USA) or GraphPad Prism 7.0 (GraphPad Software, La Jolla, CA). The figure-plate illustrations were done using Adobe Illustrator CC 2018 (San Jose, CA, USA).
RESULTS
Optogenetic stimulation of PeF glutamatergic inputs to BLA enhanced conditioned fear.
To test the functional role of glutamatergic PeF terminals in fear conditioning, we injected rats with viral vectors encoding ChR2 under CaMKIIα promoter and identified PeF-derived eYFP-positive fibers in the BLA (Fig.1a,h). Using slices containing PeF, we also recorded PeF fluorescently labeled neurons (Fig.1f,g) and confirmed that they are responsive to brief blue (490 nm) light stimulation (Suppl.Fig.1c). In a separate set of experiments, we stimulated PeF terminals in the BLA (see Methods for details) of freely moving rats with blue light for 5 min periods before acquisition, consolidation and extinction of fear (Fig.1b-d). Optogenetic stimulation of PeF terminals in the BLA of ChR2 rats enhanced acquisition of fear compared to rats injected with control virus [tones x treatment interaction, F4,70 = 5.20, p = 0.001, Fig.1b]. Posthoc analysis of between subject effects at each tone demonstrated that ChR2 group had higher freezing at tone 2 compared to control group [Sidak’s test, p < 0.0001, Fig.1b]. Twenty-four hours after acquisition the consolidation of fear was confirmed by presenting rats with 5 tones in the same context. Animals from both groups demonstrated consolidation of fear memory but ChR2 group showed significantly higher freezing starting from tone 3, compared to control group [treatment effect, F1,14 = 22.93, p = 0.0003, Sidak’s test, p < 0.02, Fig.1c]. During extinction, both groups demonstrated extinction of fear memories [tones x treatment interaction F19,133 = 2.92, p = 0.0002, Fig.1d]. Sidak’s posthoc analysis within subjects showed significant reduction of freezing in the control group by tone 9, whereas ChR2 group did not demonstrate a reduction of freezing until tone 13. Therefore, we conclude that stimulation of PeF fibers in the BLA induces a significant delay of fear extinction [p < 0.05, Fig.1d]. Moreover, Sidak’s posthoc analysis of between subjects at each tone confirmed significantly higher freezing starting at tone 9 in the ChR2 group compared to controls group [p < 0.05, Fig.1d]. Three weeks following our fear conditioning protocol, all rats were returned to the same context for a single spontaneous recovery test (see Methods for details). Rats from both groups demonstrated significant reduction of freezing over time [tone effect F9,126 = 15.26, p < 0.0001, Fig. 1e], but ChR2 animals exhibited significantly higher freezing in response to tones, compared to controls [treatment effect F1,14 = 11.81, p = 0.004, and no tones x treatment interaction F9,126 = 1.363, p = 0.21, Fig.1e]. Sidak’s posthoc test, however, revealed differences in freezing over time within each group. Compared to the first tone for each respective group, freezing in the ChR2 group was not significantly reduced until tone 7, whereas freezing was significant lower starting at tone 5 in control group [p <0.03, Fig.1e].
Next, using immunochemistry we observed a significantly higher number of c-Fos positive BLA neurons in the ChR2 group, compared to the control group [t = 5.68, df = 12, p = 0.0001, Fig.1h,i]. DAPI nuclear staining co-labeled with diffuse cytosolic GFP virus expression and was enveloped by cell membrane ChR2-eYFP virus expression in the PeF of control and ChR2 virus injected animals, respectively (Fig. 1g). Additionally, using whole-cell patch-clamp recordings from a different set of rats, we also confirmed that eYFP-positive PeF terminals in the BLA are glutamatergic. All recorded neurons in the BLA showed optically-evoked EPSPs (oEPSPs), which were blocked in the presence of AMPA and NMDA antagonists, DNQX and APV, respectively (Fig.1j). Overall, this data confirmed that glutamatergic PeF projections to the BLA are involved in forming and maintaining cued fear associations.
Panic-prone rats show enhanced conditioned fear.
Rats made panic vulnerable following chronic inhibition of GABA synthesis with l-AG (n = 5), compared to control rats (d-AG, n = 5), displayed normal acquisition of fear conditioning on day 1 where freezing went from approximately 4–5% of time during tone prior to shock pairing to approximately 95% of time after last pairing [significant tone effect F4,32 = 210.1, p < 0.001; but no significant treatment x tones interaction F4,32 = 0.3, p = 0.875, or treatment effect F1,8 = 0.2, p = 0.665, Fig.1k]. On day 3, when only CS tone was presented 5 times both groups also had equivalent recall evidenced by both groups freezing for approximately 90% of time [data not shown: treatment x tones interaction F4,32 = 0.9, p = 0.483, treatment effect F1,8 = 0.001, p = 0.971]. However, during the extinction trial on day 3, the l-AG treated rats had delayed extinction compared to d-AG controls [treatment effect F1,8 = 9.5, p = 0.015, but no treatment x tones interaction F19,153 = 0.7, p = 0.793, Fig.1l]. Post-hoc analyses of between subjects at each time point revealed that l-AG treated rats had higher duration of freezing between tone 10 and tone 20 [Fisher’s LSD, p < 0.05, Fig.1l] indicating that disinhibition of the PeF alters fear learning.
Changes in intrinsic and synaptic properties of the amygdala neurons of panic-prone rats.
Basolateral Amygdala:
First, we assessed the chronic effect of l-AG on the intrinsic properties of BLA pyramidal neurons (Suppl. Table 1). Principal neurons were characterized as pyramidal shaped neurons that demonstrated spike-frequency adaptation and lack of spontaneous action potentials (APs) at resting membrane potential47. Seven-day minipump perfusion of l-AG into the PeF region induced a significant increase of the membrane input resistance of BLA pyramidal neurons [unpaired t-test, t = 4.84, df = 31, p < 0.0001, Fig.2a,e]. When tested with a series of hyperpolarizing current pulses (700 ms duration), all of the BLA principal neurons displayed time-dependent inward rectification “sags”, which become prominent in voltage responses more negative than −80 mV (Fig.2b). Furthermore, we found that the “sag” amplitude was lower in BLA neurons from l-AG animals than in those from d-AG controls [unpaired t-test, t = 5.5, df = 31, p < 0.0001, Fig.2b,f]. This hyperpolarizing “sag” results from deactivation of the channels mediating Ih current. Next, using voltage clamp mode, we further analyzed Ih current. Hyperpolarizing step pulses from −60 to −100 mV in 10 mV increments were used as activation voltage protocol (Fig.2c). In l-AG animals the amplitude of IINS and ISS were significantly reduced compared to d-AG [treatment x voltage interaction, F15,240 = 21.41, p < 0.0001, Fig.2c,d]. The amplitude of Ih current in BLA neurons, which was calculated as the difference between ISS and IINS currents, was significantly smaller in l-AG animals compared to d-AG controls [treatment x voltage interaction, F5,144 = 2.62, p = 0.027, Fig.2g]. Posthoc analysis confirmed significant differences from −90 mV to 110 mV [Sidak’s test, p < 0.04 Fig.2g]. Together, these findings indicate that the reduction of Ih current in l-AG animals could contribute to increased input resistance of the BLA neurons.
Figure 2. Disinhibition of panic network (with l-AG) was associated with an increase in the excitability of BLA pyramidal neurons.
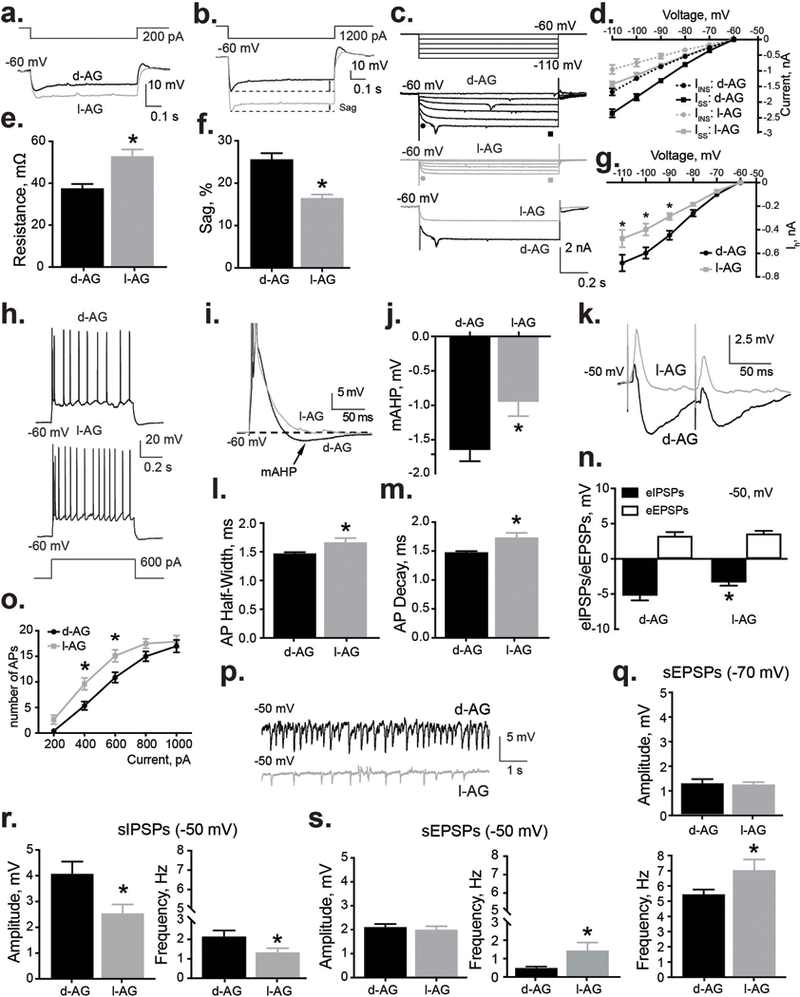
a,e) BLA neurons from I-AG animals (n = 14) displayed higher input resistance compared to d-AG controls (n = 19). a) Representative whole cell voltage responses to 600 pA current 700 ms pulses from the BLA of l-AG and d-AG animals. e) Group data indicating significantly higher input resistance in BLA neurons recorded from l-AG animals (l-AG, n = 9) compared to d-AG controls (d-AG, n = 14). b,f) BLA neurons from l-AG animals (n = 17) have smaller inward rectification (sag) compared to d-AG controls (n = 15). b) Whole cell voltage responses to 1200 pA current 700 ms pulses from the BLA neurons of l-AG and d-AG animals. f) Summary of the sag amplitude in d-AG and l-AG animals. c,d,g) Ih was suppressed in BLA neurons from l-AG animals (n = 14) compared to d-AG controls (n = 12). c) Representative current traces of Ih induced by applying hyperpolarizing voltage steps from −60 to 120 mV (step = −10 mV) in d-AG (top) and l-AG animals (middle). Representative examples of raw current traces (bottom) in response to voltage step to −110 mV from −60 mV. d) Plots of instantaneous current (Iins, ● and ●, as shown in (c) and steady state currents (Iss, ■ and ■, as shown in (c) against the membrane potential in the BLA neurons from l-AG (n = 14) and d-AG animals (n = 12). g) Plots of Ih against the membrane potential in the BLA neurons from l-AG (n = 14) and d-AG animals (n = 12). Note that the difference between Iss and Iins corresponds to Ih. *p < 0.05, ANOVA, Sidak’s between subjects posthoc analysis. h) and o) Depolarizing currents induced significantly more APs in neurons recorded from l-AG animals (n = 19), compared to d-AG animals (n = 14). *p < 0.05, ANOVA, Sidak’s between subjects posthoc analysis. i) Example traces showing the voltage response to current step used to induce a mAHP (APs are truncated). j) Group data indicating significant effect of l-AG treatment (l-AG, n = 9) on the amplitude of mAHP (d-AG, n = 14). l) Half-width and m) decay of APs of BLA neurons from l-AG animals (n = 16) were significantly different compared to d-AG rats (n = 16). k) Example traces of ePSPs in the BLA neurons from l-AG and d-AG animals. n) Group data showing that l-AG treatment significantly reduced amplitude of eIPSPs without affecting amplitude of eEPSPs (n = 18). p) Representative recordings of spontaneous activity in l-AG and d-AG animals. r) Group data showing that amplitude and frequency of sIPSPs were significantly reduced in l-AG animals (n = 16) compared to d-AG rats (n = 18) at holding potential of −50 mV. s,q) Group data showing that frequency, but not amplitude of sEPSPs were significantly increased in l-AG animals (n = 16) compared to d-AG control (n = 18) at holding potential of −50 mV (s) and −70 mV (q). Except where otherwise specified, *p < 0.05, t-test. All data reported as mean + S.E.M.
An increased input resistance is expected to increase neuronal excitability. We tested this prediction by analyzing action potential (AP) generation. Chronic treatment with l-AG significantly increased the number of APs generated in BLA principal neurons over a range of current injections [treatment effect, F1,145 = 18.46, p < 0.0001, but no current x treatment interaction F4,145 = 0.87, p = 0.48, Fig.2h,o]. Sidak’s posthoc test confirmed the significant increase of APs in l-AG group compared to d-AG group in response to 400 pA and 600 pA depolarizing current injections [p < 0.013, Fig.2o]. These results would suggest that an increase in the excitability of BLA projection neurons might contribute to the etiology of the anxiogenic behavior observed in panic-prone rats. Interestingly, in l-AG animals the half-width and decay of the APs were significantly different compared to d-AG animals [half-width: t = 2.18, df = 30, p = 0.037, decay: t = 2.74, df = 30, p = 0.01, Suppl. Table 1, Fig.2l,m]. Additionally, the amplitude of the medium afterhyperpolarization (mAHP) was also tested from traces with the same number of APs (current: 1000 pA), as the amplitude of these potentials is dependent on the number of spikes48. The mAHP amplitudes, measured as the peak negative potential after termination of the current step, were significantly blunted in the l-AG group [t = 2.64, df = 21, p = 0.015] compared to d-AG group indicative of reduced calcium-activated potassium channels (KCa) activity48 (Fig.2i,j).
Next, we investigated the effect of chronic infusion of l-AG into the PeF on inhibitory and excitatory neurotransmission in the BLA. First, we examined the response of BLA neurons to electrical stimulation of thalamic or cortical afferent pathways. The chloride reversal potential for GABAA receptor-mediated sIPSPs is approximately −70 mV. In order to fully appreciate the current, we shifted the holding potential to −50 mV. The amplitude of eIPSPs in l-AG animals at a holding potential of −50 mV was significantly lower, compared to d-AG-treated animals [t = 2, df = 33, p = 0.036, Fig.2k,n]. At a holding potential of −50 mV, no differences in BLA membrane input resistance between l-AG and d-AG animals were observed [t = 0.91, df = 31, p = 0.38, data not shown].
Further, chronic treatment with l-AG also induced significant reduction of sIPSPs amplitude [t = 2.51, df = 32, p = 0.017, Fig.2p,r] and frequency [t = 2.04, df = 32, p = 0.05, Fig.2p,r] compared to d-AG controls. Additionally, frequency, but not sEPSPs amplitude was significantly higher in l-AG group compared to d-AG group at the holding potential −50 mV [t = 2.18, df = 33, p = 0.036, Fig.2p,s] and −70 mV [t = 2.07, df = 32, p = 0.047, Fig.2q]. These data confirm that chronic treatment with l-AG in the PeF region results in reduced GABAergic inhibitory tone and increased presynaptic excitatory tone within the BLA.
Central nucleus of the amygdala:
Using whole-cell patch-clamp we also assessed the basic and synaptic properties of the neurons in the CeA. Overall, the CeA data suggest that chronic treatment with l-AG in the PeF region had no significant effects within the CeA area (Suppl. Fig.2).
Protein and gene expression changes in the amygdala of panic-prone rats.
Relative expression of GABA and glutamate related genes (delta delta Ct) in the BLA and CeA was compared between panic-prone (l-AG) and control (d-AG) rats using Taqman® low density array techniques33. Both GABA-related and glutamate-related genes were expressed differentially in the BLA [GABA-related gene x treatment interaction, F20, 84 = 2.022, p = 0.014, Fig.3a,d] of l-AG compared to d-AG rats. Of the twenty-one GABA related genes investigated, three had significantly altered expression. Here, expression of Slc32a1 (GABA vesicular transporter) and Gad2 (Glutamic acid decarboxylase 2 or Gad65) were significantly reduced in l-AG rats compared to d-AG controls [Fisher’s LSD, t = 2.48, p = 0.015 and t = 2.29, p = 0.025 respectively], while expression of Slc6a13 (GABA transporter, also known as Gat-2) was significantly increased in panic-prone rats compared to controls [Fisher’s LSD, t = 2.85, p = 0.005, Fig.3d].
Figure 3. Increased BLA excitability seen in panic-prone animals was associated with decreased mGluR2 protein and gene expression in the BLA and CeA and mGluR2 PAM reduced enhanced glutamatergic neurotransmission in the BLA.
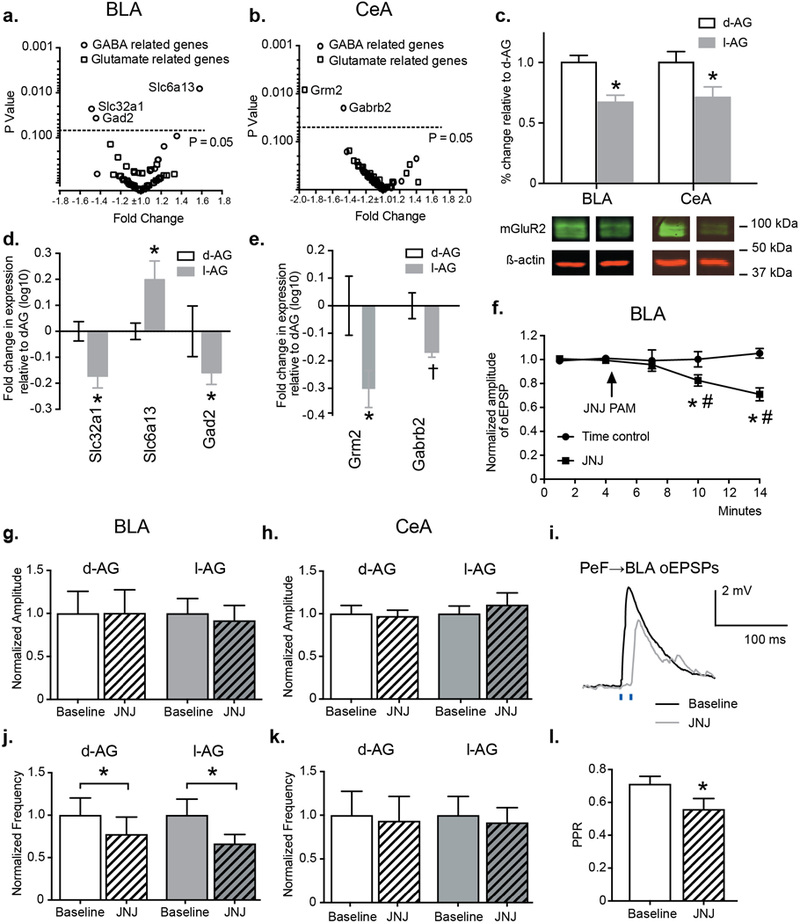
Pre-treatment with l-AG induced significant changes of a,d) GABA related genes in the BLA, and b,e) GABA/Glutamate related genes in the CeA. c) Group data and representative Western blots, normalized to β-actin, illustrating reduced mGluR2 protein levels in the BLA and CeA of l-AG rats (n = 8–9) compared to d-AG controls (n = 7). f,i) Bath application of JNJ-42153605 significantly reduced the amplitude of oEPSPs evoked by light stimulation of PeF→BLA terminals in ChR2-expressing animals (n = 8) compared to time control group (n = 4). Representative traces of oEPSPs evoked by light pulses before (black trace) and during (gray trace) bath application of JNJ-42153605 (i). *p ≤ 0.05, ANOVA. g,j) Bath application of JNJ-42153605 significantly reduced the frequency (j), but not the amplitude (g) of sEPSPs in recordings from the BLA of l-AG (n = 9) and d-AG animals (n = 9). h,k) Pre-incubation with JNJ-42153605 did not affect the amplitude (h) and the frequency (k) of sEPSPs in recordings from the CeA of l-AG (n = 7) and d-AG rats (n = 7). l) Bar graph indicating significant reduction of paired-pulse ratio (oEPSP2/oEPSP1) recorded 5 min before and 10 min after bath application of JNJ-42153605 (n = 8). Except where otherwise specified, *p < 0.05, t-test. All data represented as mean + S.E.M.
Overall, we noted different expression of GABA-related and glutamate-related genes in the CeA samples from l-AG rats compared to control rats [treatment effect GABA, F1,102 = 4.184, p = 0.043; glutamate F1,192 = 5.73, p = 0.018]. One GABA-related gene and one glutamatergic-related gene had significantly altered expression (Fig.3b,e). The expression of Gabrb2 (GABAA receptor beta2 subunit) and Grm2 (metabotropic glutamate receptor 2) were significantly reduced in l-AG rats compared to d-AG controls [Fisher’s LSD, t = 1.56, p = 0.023 and t = 3.13, p = 0.0086, respectively, Fig.3e].
Next, we measured mGluR2 protein levels using Western blot analysis and we confirmed significantly lower protein levels in the BLA [t = 4.69, df = 13, p = 0.0004] and CeA [t = 2.34, df = 14, p = 0.034] in l-AG animals compared to d-AG controls (Fig.3c).
Effects of the mGluR2 PAM JNJ-42153605 on glutamatergic neurotransmission in the BLA and CeA.
Next, we evaluated the effects of mGluR2 PAM JNJ-42153605 (2 μM) on sEPSPs in the BLA and CeA of d-AG and l-AG animals. We hypothesized that the mGluR2 PAM would reduce glutamate release. Indeed, pre-treatment with mGluR2 PAM JNJ-42153605 (2 μM) induced significant reduction of frequency [t = 2.18, df = 33, p = 0.036], but not amplitude [p = 0.62] of sEPSPs in the BLA of d-AG and l-AG animals (Fig.3g,j). Interestingly, bath application of JNJ-42153605 (2 μM) had no effect on amplitude and frequency of sEPSPs in the CeA (Fig.3h,k). Moreover, in a separate set of experiments, bath application of JNJ-42153605 (2 μM) significantly reduced the amplitude of blue light-evoked oEPSPs in acute brain slices [time effect, F4,40 = 3.89, p = 0.0092, treatment effect, F1,10 = 9.17, p < 0.013, time x treatment interaction: F4,40 = 7.19, p = 0.0002, Fig.3f,i]. Sidak’s posthoc within group analysis confirmed significant reduction of amplitude of oEPSPs at 10 min [p = 0.0009] and 14 min into recording in the presence of JNJ-42153605 [p < 0.0001] compared to baseline oEPSP (Fig.3f). Posthoc test also detected that the amplitudes of oEPSPs at 10 min [p = 0.0143] and 14 min [p < 0.0001] between JNJ-42153605-treated and time control groups were significantly different (Fig.3f). No changes in amplitude of oEPSPs were observed in time control group [0.1% DMSO in ACSF, p > 0.05]. The paired-pulse ratio (PPR) of oEPSPs was also significantly reduced [t = 2.56, df = 7, p = 0.037] supporting the presynaptic mechanism of action of JNJ-42153605 (Fig.3l).
Effects of the mGluR2 PAM JNJ-40411813 on NaLac-induced panic-like responses in panic-prone rats.
Anxiety-associated behavior:
To evaluate the effects of mGluR2 PAM JNJ-40411813 on anxiety-associated behavior, separate groups underwent SI testing before and after treatment with either NaLac and vehicle or NaLac and JNJ-40411813. An ANOVA with a Fisher’s posthoc test revealed significant differences between groups where panic-prone rats were systemically (i.p.) pretreated with 5 or 20 mg/kg JNJ-40411813 [F3,23 = 8.4, p = 0.001, n = 7,6,7,7, Fig.4a]. JNJ-40411813 attenuated NaLac-induced anxiety behavior (i.e., reductions in SI times, Fig.4a). Systemic treatment with either 5 or 20 mg/kg JNJ-40411813 had no effects on locomotor activity [p = 0.51, Fig.4b,c]. Additionally, we measured the number of “social avoidance” episodes during the SI test and observed a significantly higher number of episodes in NaLac-vehicle and a prevention of this effect in JNJ-40411813 treated groups [F3,20 = 11.87, p = 0.0001, Fig.4d].
Figure 4. Pretreating panic-prone rats (chronic l-AG) with mGluR2 PAM attenuated NaLac-induced panic symptoms and facilitated extinction of fear, without affecting general locomotor activity.
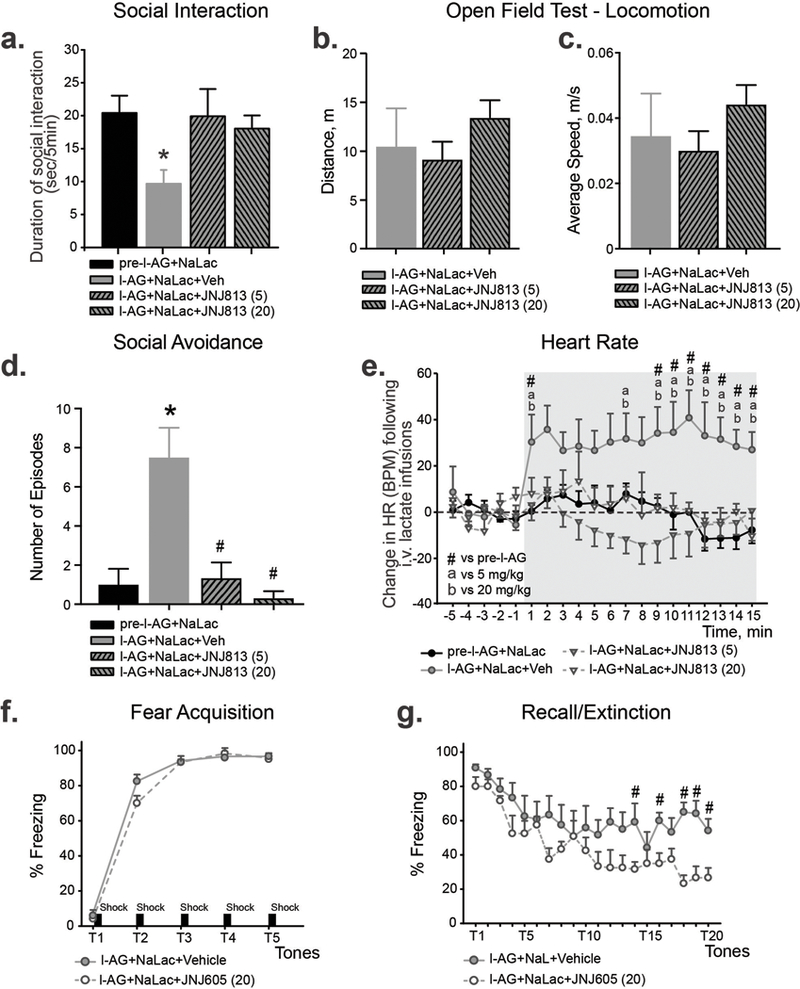
a) Anxiety-like behavior (as measured by reduced SI times) displayed by animals treated with 5 and 20 mg/kg i.p. of JNJ-40411813. b,c) JNJ-4041183 did not affect locomotor activity of panic-prone rats as measured by distance travelled (b) and average speed (c) during open field test. d) Bar graph demonstrating that panic-prone animals showed significant increase of social avoidance episodes. Pretreatment with 5 mg/kg or 20 mg/kg of JNJ-40411813 significantly reduced number of social avoidance episodes. *p < 0.05, compared to pre-l-AG group, #p < 0.05, compared to l-AG group. e) Heart rate response displayed by animals treated with 5 and 20 mg/kg doses of JNJ-40411813. n = 6–7 per group. Gray shading in line graphs indicates onset and duration of intravenous 0.5M NaLac infusions. #, a and bp < 0.05. f,g) Pretreating panic-prone rats with mGluR2 PAM JNJ-42153605 (20 mg/kg), did not alter acquisition of fear-induced freezing (f), but did attenuate the resistant extinction of fear-induced freezing on the recall/extinction (g). n = 5 per group. *p < 0.05, ANOVA. All data represented as mean + S.E.M.
Heart rate (HR) responses:
The same animals were examined for the effect of mGluR2 PAM on NaLac-induced cardiovascular responses. An overall ANOVA with drug treatment as main factor and time as a repeated measure detected a treatment by time interaction for the change in HR [Fig.4e for 5 and 20 mg/kg JNJ-40411813 effects on HR responses, F57,399 = 1.8, p < 0.001]. Between group analysis with a Fisher’s LSD posthoc determined that the NaLac induced marked increases in cardio-excitatory responses in l-AG rats (did not occur in the same rats prior to l-AG treatment, Fig.4e). Additionally, pre-treating panic-prone rats with JNJ-40411813 did not significantly affect baseline general motor or cardiovascular activity [baseline (5 min prior to intravenous infusion) HR [5 and 20 mg/kg, F3,16 = 0.8, p = 0.539, Fig.4e].
Effects of the mGluR2 PAM JNJ-42153605 on conditioned fear in panic-prone rats.
Here, panic-prone rats with repeated NaLac challenges showed low freezing (4–5%) during the tone prior to shock pairing and approached approximately 95% of time freezing after the 3rd pairing [significant time effect F4,40 = 626.3, p < 0.001, n = 5 per group, Fig.4f]. Although a significant drug x time interaction was detected [F4,40 = 3.1, p = 0.025], individual posthoc testing at each time point did not detect a difference between groups. On day 3, both groups also had equivalent recall evidenced by each group freezing for approximately 90% of time (data not shown). On day 4, there was no significant difference in the recall between groups (~ 90% vehicle, ~ 80% JNJ-42153605), but there was an overall treatment effect detected [F1,10 = 7.1, p = 0.023] with less overall freezing in JNJ-42153605 treated rats compared to vehicle treated rats, but no treatment x tones interaction [F19,190 = 1.4, p = 0.141, Fig.4g]. Post-hoc analyses also revealed that the mGluR2 PAM treated rats had enhanced extinction of fear-associated freezing indicating that modulation of mGluR2 can affect fear learning [Fig.4g].
Effects of an mGluR2-PAM on panic symptoms in patients.
Within a previously completed clinical trial reporting the effects of mGluR2 PAM compound JNJ-40411813, 5 subjects met criteria for a DSM-IV diagnosis of PD (comorbid with a diagnosis of major depressive disorder) at screening and reported mild to moderate panic symptoms at baseline. The study, which was double blinded and double randomized, aimed to dissect how addition of mGluR2 PAM to first-line antidepressants affected depressed patients with prominent anxiety symptoms. Each patient was randomized to either drug or placebo for the first 4 weeks (period 1), and then those patients randomized to drug were continued on drug, whereas those randomized to placebo who did not respond were re-randomized to either drug or placebo (1:1 ratio) for the last 4 weeks (period 2). All five patients that met criteria for our post-hoc analysis of this completed study had at least one 4 week period on the drug in dose ranges of 200 – 300 mg/day, with one patient being on placebo for the first 4 weeks and then getting randomized to 200 mg/day of the drug for the second 4 week period (the drug conditions are noted in parenthesis for each subject in Fig.5). The changes in PDSS show that all 5 subjects demonstrated complete remission of their panic symptoms within 2 – 4 weeks of mGluR2 PAM therapy. We also analyzed the individual panic relevant items of fear, cardiovascular and respiratory symptoms from Hamilton Anxiety scale (HAM-A) across all the subjects in the study (drug n = 61; placebo n = 58). Both groups showed significant reductions with no effect of treatment (See Suppl. Table 2).
Figure 5. Data demonstrating clinical effects of the mGluR2 PAM JNJ-40411813 on panic symptoms.
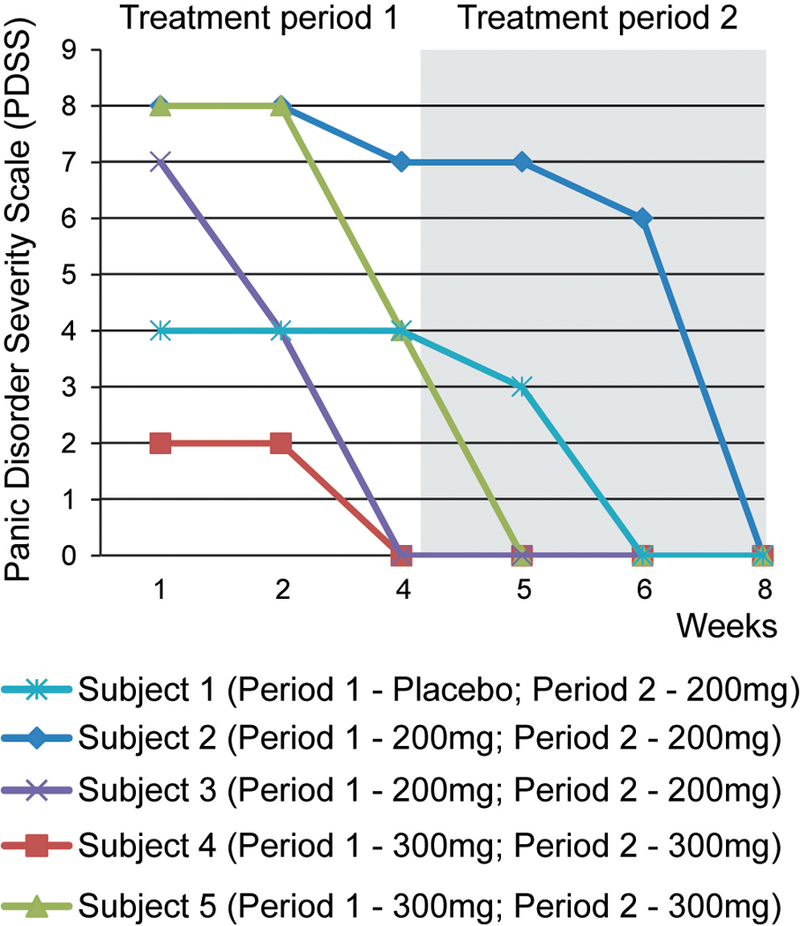
The changes in Panic Disorder Severity Scale (PDSS) show that all five subjects exhibited remission of their panic symptoms following 2 – 4 weeks of mGluR2 PAM therapy. NOTE: PDSS range = 0 – 28, Scores ≥ 8 consistent with DSM-IV Panic Disorder81.
DISCUSSION
In this study, we demonstrate that panic vulnerable rats show persistence of fear responses and delayed extinction of conditioned fear, similar to PD and PTSD patients who show greater resistance to extinguishing conditioned fear responses49–53. Furthermore, we provide mechanistic connections between the panic generating network and fear-regulating circuits by demonstrating that activation of panic pathways could substantially shift the fear network towards enhanced and persistent excitability. Disinhibition of the PeF, a panic-generating site, induces the long-term change of E/I balance in the amygdala characterized by reduced inhibition and enhanced excitation. Particularly, expression of mGluR2 was significantly reduced in the BLA and CeA in the panic-prone rats. Treating panic-prone rats with a selective mGluR2 PAM not only blocked the panic-responses following 0.5M NaLac, but also normalized fear extinction deficits. All of this pre-clinical evidence points to panic-prone state leading to reduced mGluR2 function in the amygdala fear network and facilitating the persistence of conditioned fear responses. This concept was further supported by preliminary human clinical data. We conducted a post-hoc analysis of a proof-of-concept clinical trial investigating the efficacy of the mGluR2 PAM JNJ-40411813 compound. In the subset of participants with comorbid panic disorder and depression which had not previously responded to SSRI/SNRI therapy (the typical first line therapy for PD and depression), treatment with the mGluR2 PAM resulted in complete remission of panic symptoms. These findings are also consistent with a previous study utilizing mGluR2 agonist that showed efficacy blocking CO2 provocation-induced anxiety symptoms in patients with PD54. Thus, mGluR2 PAMs may provide a uniquely targeted treatment for persistent fear disorders such as PD, PTSD, and phobias, especially in resistant groups that show limited improvements from first line SSRI therapies.
The panic-prone rats utilized in this study have been extensively characterized. The PeF panic model, since its inception in 199613, has consistently proven to be sensitive to interoceptive stimuli [see review14] that provoke PAs in subjects with PD15–17. The sensory pathways critical for NaLac response12, 55, anticipatory anxiety behaviors, and cardio-respiratory and sympatho-excitatory circuits have all been well mapped24. Glutamatergic/orexin neurons in the PeF play a critical role in generating panic responses in this model37 and enhance amygdalar-based fear conditioned behaviors56, 57 via their projections to both the BLA and CeA23. The amygdala (e.g., BLA and the CeA) is well known to play a critical role in acquisition and expression of conditioned fear responses22. Here, we have observed network, gene, and protein changes in the BLA that confirm a coherent pattern of synaptic physiology and result in diminished GABA synaptic transmission and enhanced glutamate neurotransmission. Our data is in agreement with previous reports that showed correlation between anxiety and fear and either reduced number of GABAergic interneurons58 or GABAergic inhibition in the BLA59–62. The network changes we have described in the BLA were associated with a significant reduction in local expression of the mGluR2 receptor protein, consistent with reduced presynaptic inhibition of glutamate release. However, we would like to point out that despite the significant reduction of mGlur2 protein and mRNA levels in the CeA, we didn’t detect any intrinsic membrane or synaptic changes in the CeA of l-AG animals. There are a number of differences between BLA and CeA neurons that could explain this finding. Firstly, the BLA projection neurons (those studied here) are glutamatergic, while the projection neurons of the CeA are predominantly GABAergic. Another difference is in the expression patterns of mGlur2. Unlike in the BLA, where mGlur2 is highly expressed in the cell bodies and the terminals, it has been shown that CeA mGlur2 is predominantly expressed in presynaptic terminals63. This organization could permit pathway and synapse specific control by mGlur2 within the amygdala. It is possible that glutamatergic terminals containing mGlur2 in the CeA are partly derived from projections that enter the CeA from the BLA64. In agreement with this, it has been previously reported that mGlur2 depressed BLA to CeA neurotransmission more potently than ventral amygdaloid CeA input65.
In addition to the observed reduction of presynaptic mGluR2 receptors in BLA neurons of l-AG animals, we also found alterations of basic intrinsic membrane properties. BLA neurons of l-AG animals had higher input resistance and increased number of APs. The increase in input resistance means that less ion channels are open. These intrinsic changes in BLA membrane properties could be the result of changes in voltage-gated potassium or calcium-dependent potassium channels66–68. Numerous studies have previously demonstrated that the HCN channels are involved in the control of BLA neuronal excitability69–71. In l-AG animals we have observed the reduction of HCN channel activity, which may also contribute to an overall increase of intrinsic BLA neuron excitability. Finally, closure of GABA-mediated chloride channels would also raise the input resistance and can increase the responsiveness of the neurons to other inputs72. In the BLA, we have observed a significant reduction of eIPSPs and amplitude and frequency of sIPSPs, which suggest a reduction in the amount of GABA released or an alteration of postsynaptic properties, which would likely modify chloride homeostasis. However, additional experiments using selective GABAA and GABAB antagonists are needed to fully investigate this possibility.
The above data suggest that panic leads to a greater persistence of conditioned fears, in part due changes in excitability and gene expression within the amygdala, thus providing a molecular mechanism for the clinical observation that patients with PAs are highly vulnerable to developing avoidance and disabling phobias73–75. Hyperexcitability of BLA neurons is also associated with an increase in most forms of anxiety disorders in humans76 and anxiety-like behaviors in rodents67, 68. Additionally, artificial activation of BLA principal neurons using optogenetics induced anxiety-like behaviors64. In light of this, there is increasing recognition that glutamatergic mechanisms may be a potential avenue to develop new treatments for anxiety problems such as PD77. Using the same panic-prone model, we have previously reported that pretreatment with either a specific mGluR2/3 allosteric agonist78 or a more selective mGluR2 agonist (i.e., CBiPES or THIIC)14 also attenuates panic responses without benzodiazepine-associated side effects such as sedation54. Treatment with mGluR2 PAM JNJ-40411813 significantly reduced NaLac-induced anxiety behaviors and cardio-excitation in panic-prone rats. Additionally, mGluR2 PAM facilitated fear extinction and attenuated the increased glutamatergic excitation within the BLA in panic-prone rats. In health human subjects, JNJ-40411813 is well tolerated, promotes deep sleep, and does not affect cognition or working memory79. Additionally, in distressed subjects, treatment with JNJ-40411813 shows several beneficial effects on behavioral symptoms80. Preliminary data presented here shows that patients with PD symptoms that were treated with the mGluR2 PAM JNJ-40411813 demonstrated remission of their panic symptom scores. This is consistent with previous studies showing that mGluR2 agonists reduce anxiety in PD subjects54. However, taking into account small number of subjects with PD in this trial, and the retrospective nature of this analysis, a randomized, prospective trial of JNJ-40411813 in patients with PD is needed to confirm its clinical efficacy. If such a study were positive, mGluR2 PAMs could be a safe and viable approach to patients with PD.
In conclusion, we provide a mechanistic framework for the interaction of panic and fear networks utilizing behavioral, molecular and electrophysiological paradigms. Data presented here suggests that mGluR2 PAMs could provide a novel, much needed, therapeutic approach to severe fear disorders such as PD, PTSD and other fear disorders, especially in those patients showing limited response to first line SSRI therapies.
Supplementary Material
{kind=link}
{kind=link}
Acknowledgements:
This work was supported by NIMH R01s MH052619, MH065702, and NCATS UL1 TR001108 to AS. The study was also supported by Janssen Pharmaceuticals Inc. and PLJ was supported by K01 AG044466. The authors would like to thank ADDEX Therapeutics for their involvement in the development of JNJ-40411813.
Footnotes
CONFLICT OF INTERESTS
Drs. L Ver Donck, M Ceusters and JM Kent are employees of Janssen. This work was supported by research grants to Indiana University with (Drs. Shekhar, Molosh and Johnson as PIs) from Janssen, but these authors have no other commercial conflicts.
REFERENCES CITED
- 1.Stein DJ, Bouwer C. A neuro-evolutionary approach to the anxiety disorders. JAnxietyDisord 1997; 11(4): 409–429. [DOI] [PubMed] [Google Scholar]
- 2.DSM- V. Diagnostic and Statistical Manual - Fifth Edn. (DSM - V) American Psychiatric Association: Washington, DC, 2013. [Google Scholar]
- 3.Rasche D, Foethke D, Gliemroth J, Tronnier VM. [Deep brain stimulation in the posterior hypothalamus for chronic cluster headache. Case report and review of the literature]. Schmerz 2006; 20(5): 439–444. [DOI] [PubMed] [Google Scholar]
- 4.Wilent WB, Oh MY, Buetefisch C, Bailes JE, Cantella D, Angle C et al. Mapping of microstimulation evoked responses and unit activity patterns in the lateral hypothalamic area recorded in awake humans. Technical note. Journal of neurosurgery 2011; 115(2): 295–300. [DOI] [PubMed] [Google Scholar]
- 5.Wilent WB, Oh MY, Buetefisch CM, Bailes JE, Cantella D, Angle C et al. Induction of panic attack by stimulation of the ventromedial hypothalamus. Journal of neurosurgery 2010; 112(6): 1295–1298. [DOI] [PubMed] [Google Scholar]
- 6.Anderson JJ, DiMicco JA. Effect of local inhibition of gamma-aminobutyric acid uptake in the dorsomedial hypothalamus on extracellular levels of gamma-aminobutyric acid and on stress-induced tachycardia: a study using microdialysis. JPharmacolExpTher 1990; 255(3): 1399–1407. [PubMed] [Google Scholar]
- 7.Samuels BC, Zaretsky DV, DiMicco JA. Tachycardia evoked by disinhibition of the dorsomedial hypothalamus in rats is mediated through medullary raphe. JPhysiol 2002; 538(Pt 3): 941–946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Shekhar A, DiMicco JA. Defense reaction elicited by injection of GABA antagonists and synthesis inhibitors into the posterior hypothalamus in rats. Neuropharmacology 1987; 26(5): 407–417. [DOI] [PubMed] [Google Scholar]
- 9.Shekhar A, Hingtgen JN, DiMicco JA. GABA receptors in the posterior hypothalamus regulate experimental anxiety in rats. Brain research 1990; 512(1): 81–88. [DOI] [PubMed] [Google Scholar]
- 10.Soltis RP, DiMicco JA. Hypothalamic excitatory amino acid receptors mediate stress-induced tachycardia in rats. AmJPhysiol 1992; 262(4 Pt 2): R689–R697. [DOI] [PubMed] [Google Scholar]
- 11.Johnson PL, Shekhar A. Panic-prone state induced in rats with GABA dysfunction in the dorsomedial hypothalamus is mediated by NMDA receptors. J Neurosci 2006; 26(26): 7093–7104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Shekhar A, Keim SR. The circumventricular organs form a potential neural pathway for lactate sensitivity: implications for panic disorder. J Neurosci 1997; 17(24): 9726–9735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Shekhar A, Keim SR, Simon JR, McBride WJ. Dorsomedial hypothalamic GABA dysfunction produces physiological arousal following sodium lactate infusions. Pharmacology, biochemistry, and behavior 1996; 55(2): 249–256. [DOI] [PubMed] [Google Scholar]
- 14.Johnson PL, Shekhar A. An animal model of panic vulnerability with chronic disinhibition of the dorsomedial/perifornical hypothalamus. Physiology & behavior 2012; 107(5): 686–698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gorman JM, Papp LA, Coplan JD, Martinez JM, Lennon S, Goetz RR et al. Anxiogenic effects of CO2 and hyperventilation in patients with panic disorder. AmJPsychiatry 1994; 151(4): 547–553. [DOI] [PubMed] [Google Scholar]
- 16.Pitts FN Jr., McClure JN Jr. Lactate metabolism in anxiety neurosis. NEnglJMed 1967; 277(25): 1329–1336. [DOI] [PubMed] [Google Scholar]
- 17.Woods SW, Charney DS, Goodman WK, Heninger GR. Carbon dioxide-induced anxiety. Behavioral, physiologic, and biochemical effects of carbon dioxide in patients with panic disorders and healthy subjects. Arch Gen Psychiatry 1988; 45(1): 43–52. [DOI] [PubMed] [Google Scholar]
- 18.Mineka S, Zinbarg R. A contemporary learning theory perspective on the etiology of anxiety disorders: it’s not what you thought it was. Am Psychol 2006; 61(1): 10–26. [DOI] [PubMed] [Google Scholar]
- 19.Asselmann E, Pane-Farre C, Isensee B, Wittchen HU, Lieb R, Hofler M et al. Characteristics of initial fearful spells and their associations with DSM-IV panic attacks and panic disorder in adolescents and young adults from the community. Journal of affective disorders 2014; 165: 95–102. [DOI] [PubMed] [Google Scholar]
- 20.Bryant RA, Brooks R, Silove D, Creamer M, O’Donnell M, McFarlane AC. Peritraumatic dissociation mediates the relationship between acute panic and chronic posttraumatic stress disorder. Behaviour research and therapy 2011; 49(5): 346–351. [DOI] [PubMed] [Google Scholar]
- 21.Fikretoglu D, Brunet A, Best SR, Metzler TJ, Delucchi K, Weiss DS et al. Peritraumatic fear, helplessness and horror and peritraumatic dissociation: do physical and cognitive symptoms of panic mediate the relationship between the two? Behaviour research and therapy 2007; 45(1): 39–47. [DOI] [PubMed] [Google Scholar]
- 22.Quirk GJ, Mueller D. Neural mechanisms of extinction learning and retrieval. Neuropsychopharmacology 2008; 33(1): 56–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Peyron C, Tighe DK, van den Pol AN, de Lecea L, Heller HC, Sutcliffe JG et al. Neurons containing hypocretin (orexin) project to multiple neuronal systems. JNeurosci 1998; 18(23): 9996–10015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Johnson PL, Truitt WA, Fitz SD, Lowry CA, Shekhar A. Neural pathways underlying lactate-induced panic. Neuropsychopharmacology 2008; 33(9): 2093–2107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Schmitt O, Usunoff KG, Lazarov NE, Itzev DE, Eipert P, Rolfs A et al. Orexinergic innervation of the extended amygdala and basal ganglia in the rat. Brain Struct Funct 2012; 217(2): 233–256. [DOI] [PubMed] [Google Scholar]
- 26.Myers B, Mark Dolgas C, Kasckow J, Cullinan WE, Herman JP. Central stress-integrative circuits: forebrain glutamatergic and GABAergic projections to the dorsomedial hypothalamus, medial preoptic area, and bed nucleus of the stria terminalis. Brain Struct Funct 2014; 219(4): 1287–1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Shekhar A, Johnson PL, Sajdyk TJ, Fitz SD, Keim SR, Kelley PE et al. Angiotensin-II is a putative neurotransmitter in lactate-induced panic-like responses in rats with disruption of GABAergic inhibition in the dorsomedial hypothalamus. J Neurosci 2006; 26(36): 9205–9215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Monfils MH, Cowansage KK, Klann E, LeDoux JE. Extinction-reconsolidation boundaries: key to persistent attenuation of fear memories. Science 2009; 324(5929): 951–955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Karpova NN, Pickenhagen A, Lindholm J, Tiraboschi E, Kulesskaya N, Agustsdottir A et al. Fear erasure in mice requires synergy between antidepressant drugs and extinction training. Science 2011; 334(6063): 1731–1734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhao S, Ting JT, Atallah HE, Qiu L, Tan J, Gloss B et al. Cell type-specific channelrhodopsin-2 transgenic mice for optogenetic dissection of neural circuitry function. Nature methods 2011; 8(9): 745–752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Molosh AI, Sajdyk TJ, Truitt WA, Zhu W, Oxford GS, Shekhar A. NPY Y1 receptors differentially modulate GABAA and NMDA receptors via divergent signal-transduction pathways to reduce excitability of amygdala neurons. Neuropsychopharmacology 2013; 38(7): 1352–1364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Paxinos G, Watson C. The rat brain in stereotaxic coordinates 5th Edition edn. Elsevier Academic Press: San Diego, 2005. [Google Scholar]
- 33.Truitt WA, Hauser SR, Deehan GA Jr., Toalston JE, Wilden JA, Bell RL et al. Ethanol and nicotine interaction within the posterior ventral tegmental area in male and female alcohol-preferring rats: evidence of synergy and differential gene activation in the nucleus accumbens shell. Psychopharmacology 2015; 232(3): 639–649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Vandesompele J, De Preter K, Pattyn F, Poppe B, Van Roy N, De Paepe A et al. Accurate normalization of real-time quantitative RT-PCR data by geometric averaging of multiple internal control genes. Genome Biol 2002; 3(7): RESEARCH0034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.File SE. The use of social interaction as a method for detecting anxiolytic activity of chlordiazepoxide-like drugs. JNeurosci Methods 1980; 2(3): 219–238. [DOI] [PubMed] [Google Scholar]
- 36.Sanders SK, Shekhar A. Regulation of anxiety by GABAA receptors in the rat amygdala. Pharmacology, biochemistry, and behavior 1995; 52(4): 701–706. [DOI] [PubMed] [Google Scholar]
- 37.Johnson PL, Truitt W, Fitz SD, Minick PE, Dietrich A, Sanghani S et al. A key role for orexin in panic anxiety. Nat Med 2010; 16(1): 111–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lightowler S, Kennett GA, Williamson IJ, Blackburn TP, Tulloch IF. Anxiolytic-like effect of paroxetine in a rat social interaction test. Pharmacology, biochemistry, and behavior 1994; 49(2): 281–285. [DOI] [PubMed] [Google Scholar]
- 39.Metcalf CS, Klein BD, Smith MD, Pruess T, Ceusters M, Lavreysen H et al. Efficacy of mGlu2 -positive allosteric modulators alone and in combination with levetiracetam in the mouse 6 Hz model of psychomotor seizures. Epilepsia 2017; 58(3): 484–493. [DOI] [PubMed] [Google Scholar]
- 40.Cid JM, Tresadern G, Duvey G, Lutjens R, Finn T, Rocher JP et al. Discovery of 1-butyl-3-chloro-4-(4-phenyl-1-piperidinyl)-(1H)-pyridone (JNJ-40411813): a novel positive allosteric modulator of the metabotropic glutamate 2 receptor. J Med Chem 2014; 57(15): 6495–6512. [DOI] [PubMed] [Google Scholar]
- 41.Lavreysen H, Langlois X, Donck LV, Nunez JM, Pype S, Lutjens R et al. Preclinical evaluation of the antipsychotic potential of the mGlu2-positive allosteric modulator JNJ-40411813. Pharmacol Res Perspect 2015; 3(2): e00097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lavreysen H, Ahnaou A, Drinkenburg W, Langlois X, Mackie C, Pype S et al. Pharmacological and pharmacokinetic properties of JNJ-40411813, a positive allosteric modulator of the mGlu2 receptor. Pharmacol Res Perspect 2015; 3(1): e00096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Cid JM, Tresadern G, Vega JA, de Lucas AI, Matesanz E, Iturrino L et al. Discovery of 3-cyclopropylmethyl-7-(4-phenylpiperidin-1-yl)-8-trifluoromethyl[1,2,4]triazolo[4 ,3-a]pyridine (JNJ-42153605): a positive allosteric modulator of the metabotropic glutamate 2 receptor. J Med Chem 2012; 55(20): 8770–8789. [DOI] [PubMed] [Google Scholar]
- 44.Kent JM, Daly E, Kezic I, Lane R, Lim P, De Smedt H et al. Efficacy and safety of an adjunctive mGlu2 receptor positive allosteric modulator to a SSRI/SNRI in anxious depression. Prog Neuropsychopharmacol Biol Psychiatry 2016; 67: 66–73. [DOI] [PubMed] [Google Scholar]
- 45.Johnson PL, Fitz SD, Engleman EA, Svensson KA, Schkeryantz JM, Shekhar A. Group II metabotropic glutamate receptor type 2 allosteric potentiators prevent sodium lactate-induced panic-like response in panic-vulnerable rats. J Psychopharmacol 2013; 27(2): 152–161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Johnson PL, Federici LM, Fitz SD, Renger JJ, Shireman B, Winrow CJ et al. Orexin 1 and 2 Receptor Involvement in Co2 -Induced Panic-Associated Behavior and Autonomic Responses. Depress Anxiety 2015; 32(9): 671–683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Washburn MS, Moises HC. Electrophysiological and morphological properties of rat basolateral amygdaloid neurons in vitro. J Neurosci 1992; 12(10): 4066–4079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Abel HJ, Lee JC, Callaway JC, Foehring RC. Relationships between intracellular calcium and afterhyperpolarizations in neocortical pyramidal neurons. J Neurophysiol 2004; 91(1): 324–335. [DOI] [PubMed] [Google Scholar]
- 49.Blechert J, Michael T, Vriends N, Margraf J, Wilhelm FH. Fear conditioning in posttraumatic stress disorder: evidence for delayed extinction of autonomic, experiential, and behavioural responses. Behaviour research and therapy 2007; 45(9): 2019–2033. [DOI] [PubMed] [Google Scholar]
- 50.Wessa M, Flor H. Failure of extinction of fear responses in posttraumatic stress disorder: evidence from second-order conditioning. Am J Psychiatry 2007; 164(11): 1684–1692. [DOI] [PubMed] [Google Scholar]
- 51.Garfinkel SN, Abelson JL, King AP, Sripada RK, Wang X, Gaines LM et al. Impaired contextual modulation of memories in PTSD: an fMRI and psychophysiological study of extinction retention and fear renewal. J Neurosci 2014; 34(40): 13435–13443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Michael T, Blechert J, Vriends N, Margraf J, Wilhelm FH. Fear conditioning in panic disorder: Enhanced resistance to extinction. J Abnorm Psychol 2007; 116(3): 612–617. [DOI] [PubMed] [Google Scholar]
- 53.Jovanovic T, Kazama A, Bachevalier J, Davis M. Impaired safety signal learning may be a biomarker of PTSD. Neuropharmacology 2012; 62(2): 695–704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Schoepp DD, Wright RA, Levine LR, Gaydos B, Potter WZ. LY354740, an mGlu2/3 receptor agonist as a novel approach to treat anxiety/stress. Stress (Amsterdam, Netherlands) 2003; 6(3): 189–197. [DOI] [PubMed] [Google Scholar]
- 55.Molosh AI, Johnson PL, Fitz SD, Dimicco JA, Herman JP, Shekhar A. Changes in central sodium and not osmolarity or lactate induce panic-like responses in a model of panic disorder. Neuropsychopharmacology 2010; 35(6): 1333–1347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Flores A, Valls-Comamala V, Costa G, Saravia R, Maldonado R, Berrendero F. The hypocretin/orexin system mediates the extinction of fear memories. Neuropsychopharmacology 2014; 39(12): 2732–2741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Sears RM, Fink AE, Wigestrand MB, Farb CR, de Lecea L, Ledoux JE. Orexin/hypocretin system modulates amygdala-dependent threat learning through the locus coeruleus. Proceedings of the National Academy of Sciences of the United States of America 2013. [DOI] [PMC free article] [PubMed]
- 58.Liu M, Fitzgibbon M, Wang Y, Reilly J, Qian X, O’Brien T et al. Ulk4 regulates GABAergic signaling and anxiety-related behavior. Transl Psychiatry 2018; 8(1): 43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Prager EM, Pidoplichko VI, Aroniadou-Anderjaska V, Apland JP, Braga MF. Pathophysiological mechanisms underlying increased anxiety after soman exposure: reduced GABAergic inhibition in the basolateral amygdala. Neurotoxicology 2014; 44: 335–343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Almeida-Suhett CP, Prager EM, Pidoplichko V, Figueiredo TH, Marini AM, Li Z et al. Reduced GABAergic inhibition in the basolateral amygdala and the development of anxiety-like behaviors after mild traumatic brain injury. PLoS One 2014; 9(7): e102627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Prager EM, Bergstrom HC, Wynn GH, Braga MF. The basolateral amygdala gamma-aminobutyric acidergic system in health and disease. J Neurosci Res 2016; 94(6): 548–567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Liu ZP, Song C, Wang M, He Y, Xu XB, Pan HQ et al. Chronic stress impairs GABAergic control of amygdala through suppressing the tonic GABAA receptor currents. Mol Brain 2014; 7: 32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Gu G, Lorrain DS, Wei H, Cole RL, Zhang X, Daggett LP et al. Distribution of metabotropic glutamate 2 and 3 receptors in the rat forebrain: Implication in emotional responses and central disinhibition. Brain research 2008; 1197: 47–62. [DOI] [PubMed] [Google Scholar]
- 64.Felix-Ortiz AC, Burgos-Robles A, Bhagat ND, Leppla CA, Tye KM. Bidirectional modulation of anxiety-related and social behaviors by amygdala projections to the medial prefrontal cortex. Neuroscience 2016; 321: 197–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Neugebauer V, Zinebi F, Russell R, Gallagher JP, Shinnick-Gallagher P. Cocaine and kindling alter the sensitivity of group II and III metabotropic glutamate receptors in the central amygdala. J Neurophysiol 2000; 84(2): 759–770. [DOI] [PubMed] [Google Scholar]
- 66.Hetzel A, Rosenkranz JA. Distinct effects of repeated restraint stress on basolateral amygdala neuronal membrane properties in resilient adolescent and adult rats. Neuropsychopharmacology 2014; 39(9): 2114–2130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Rosenkranz JA, Venheim ER, Padival M. Chronic stress causes amygdala hyperexcitability in rodents. Biol Psychiatry 2010; 67(12): 1128–1136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Rau AR, Chappell AM, Butler TR, Ariwodola OJ, Weiner JL. Increased Basolateral Amygdala Pyramidal Cell Excitability May Contribute to the Anxiogenic Phenotype Induced by Chronic Early-Life Stress. J Neurosci 2015; 35(26): 9730–9740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Park K, Lee S, Kang SJ, Choi S, Shin KS. Hyperpolarization-activated currents control the excitability of principal neurons in the basolateral amygdala. Biochem Biophys Res Commun 2007; 361(3): 718–724. [DOI] [PubMed] [Google Scholar]
- 70.Park K, Yi JH, Kim H, Choi K, Kang SJ, Shin KS. HCN channel activity-dependent modulation of inhibitory synaptic transmission in the rat basolateral amygdala. Biochem Biophys Res Commun 2011; 404(4): 952–957. [DOI] [PubMed] [Google Scholar]
- 71.Zhang S, You Z, Wang S, Yang J, Yang L, Sun Y et al. Neuropeptide S modulates the amygdaloidal HCN activities (Ih) in rats: Implication in chronic pain. Neuropharmacology 2016; 105: 420–433. [DOI] [PubMed] [Google Scholar]
- 72.Duvarci S, Pare D. Glucocorticoids enhance the excitability of principal basolateral amygdala neurons. J Neurosci 2007; 27(16): 4482–4491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Noyes R Jr., Crowe RR, Harris EL, Hamra BJ, McChesney CM, Chaudhry DR. Relationship between panic disorder and agoraphobia. A family study. Arch Gen Psychiatry 1986; 43(3): 227–232. [DOI] [PubMed] [Google Scholar]
- 74.Yates WR. Phenomenology and epidemiology of panic disorder. Annals of clinical psychiatry : official journal of the American Academy of Clinical Psychiatrists 2009; 21(2): 95–102. [PubMed] [Google Scholar]
- 75.Kessler RC, Chiu WT, Jin R, Ruscio AM, Shear K, Walters EE. The epidemiology of panic attacks, panic disorder, and agoraphobia in the National Comorbidity Survey Replication. Arch Gen Psychiatry 2006; 63(4): 415–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Rauch SL, Shin LM, Wright CI. Neuroimaging studies of amygdala function in anxiety disorders. Ann N Y Acad Sci 2003; 985: 389–410. [DOI] [PubMed] [Google Scholar]
- 77.Gorman JM. New molecular targets for antianxiety interventions. JClinPsychiatry 2003; 64 Suppl 3: 28–35. [PubMed] [Google Scholar]
- 78.Shekhar A, Keim SR. LY354740, a potent group II metabotropic glutamate receptor agonist prevents lactate-induced panic-like response in panic-prone rats. Neuropharmacology 2000; 39(7): 1139–1146. [DOI] [PubMed] [Google Scholar]
- 79.Ahnaou A, de Boer P, Lavreysen H, Huysmans H, Sinha V, Raeymaekers L et al. Translational neurophysiological markers for activity of the metabotropic glutamate receptor (mGluR2) modulator JNJ-40411813: Sleep EEG correlates in rodents and healthy men. Neuropharmacology 2016; 103: 290–305. [DOI] [PubMed] [Google Scholar]
- 80.Salih H, Anghelescu I, Kezic I, Sinha V, Hoeben E, Van Nueten L et al. Pharmacokinetic and pharmacodynamic characterisation of JNJ-40411813, a positive allosteric modulator of mGluR2, in two randomised, double-blind phase-I studies. J Psychopharmacol 2015; 29(4): 414–425. [DOI] [PubMed] [Google Scholar]
- 81.Shear MK, Rucci P, Williams J, Frank E, Grochocinski V, Vander Bilt J et al. Reliability and validity of the Panic Disorder Severity Scale: replication and extension. J Psychiatr Res 2001; 35(5): 293–296. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.