

Abstract
Enzymes that catalyze DNA modifying activities including cytidine deamination and cytosine methylation play important biological roles and have been implicated pathologically in diseases such as cancer. Here, we report Direct Resolution of ONE dalton difference (DRONE), an UHPLC-based analytical method to track a single dalton change in the cytosine-to-uracil conversion catalyzed by the human apolipoprotein B m-RNA editing catalytic polypeptide-like 3 (APOBEC3) cytidine deaminases, implicated in cancer and antiviral defense. Additionally, we demonstrate broad applicability by tracking other important DNA modifications and assessing epigenetic enzyme inhibition. We have extended our methodology to obtain data on two distinct deamination events in the same oligonucleotide substrate designed from a putative APOBEC substrate, diversifying the utility of the described method. DRONE provides an important foundation for in-depth analysis of DNA-modifying enzymes and versatile detection of novel DNA modifications of interest.
Graphical Abstract
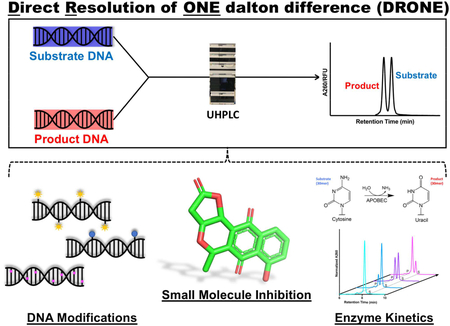
Enzymes involved in changes that modify DNA are important in virtually every aspect of human biology, from high fidelity DNA replication and repair to the maintenance of homeostasis. Many of these DNA modifying enzymes, which alter the sugar ring or nucleobase within a strand of DNA, are also intricately involved in pathological processes such as cancer and have therefore become important molecular targets for drug discovery efforts.1–4 In particular, Apolipoprotein B mRNA editing catalytic polypeptide like 3 (APOBEC3) enzymes, which catalyze a cytidine (C)-to-uridine (U) conversion in single stranded DNA (ssDNA), are recently discovered de novo mutators in cancer.5–8 APOBEC3-induced mutations can account for up to 60% of total somatic mutations in some cancers, underscoring the need to understand the mechanistic link between APOBEC3 enzyme catalysis and their role in human cancers.8
Biochemical investigation in the APOBEC cytidine deaminase field stems from early work on APOBEC1, the founding member of the APOBEC family in the editing of Apolipoprotein B mRNA. This posttranscriptional RNA editing was initially characterized biochemically using a primer extension based method developed by Driscoll et al. in which editing was detected by differential termination of polymerase extension via incorporation of specific dideoxynucleotides.9,10 Since this original work, many studies particularly by the laboratory of Harold Smith, have used the poisoned primer extension assay to characterize the APOBEC family of cytidine deaminase at large, including APOBEC1 and APOBEC3 family members.11–14 In parallel, a number of assays leveraging uracil DNA glycosylase (UDG) post-processing,15–18 PCR and restriction enzyme analysis,19,20 and E. Coli antibiotic resistance acquirement21,22 have also been utilized to assess cytidine deaminase activity. Those methods utilizing enzymatic processing have been adapted to a number of different formats (either gel-based densitometry readouts or 96 well plates equipping higher throughput capabilities23), allowing for efficient tailoring of the assay to better fit the needs of the researcher. These efforts converge in the goal of accurate biochemical characterization of APOBEC enzymes especially given their physiological and pathological relevance in vivo.
One challenge in the design of assays to detect APOBEC activity is that each involves a number of post-processing steps to efficiently detect the product of cytidine deamination. This analytical challenge stems from the fact that a cytidine deamination reaction results in the loss of a single dalton in a long strand of DNA (Figure 1A) which, in the absence of superior analytical methodologies, cannot be monitored directly given the miniscule change in molecular weight between the substrate and the product. In this study, we use a highly sensitive analytical chemistry methodology using ultra high performance liquid chromatography (UHPLC) to detect the small molecular weight difference between the substrate and the product. The use of HPLC and UHPLC MS/MS approaches to detect DNA modifications has long been established in the detection of DNA and RNA modifications, for instance, through the global measurement of DNA methylation and RNA adenosine methylation24–26 Diverse DNA and RNA modifications are able to be tracked using these next generation HPLCs, owing to the sensitive detection and efficient separation of DNA molecules. In particular, usage of volatile solvents allows for efficient coupling to MS/MS workflows, potentiating accurate identification of DNA modifications. We thus sought to develop a method leveraging the capability of UHPLC in the usage of sub-2 micron columns to perform unprecedented separations in detecting DNA modifications.
Figure 1. Design of DRONE, a UHPLC based assay to detect APOBEC activity.

A. Design of DRONE, a novel UHPLC based assay to detect the one Dalton difference between 30mer substrate and product oligonucleotides. B. Pilot baseline resolution separation of 30mer APOBEC3G substrate and product oligonucleotide.
Here, we report Direct Resolution of ONE dalton difference (DRONE), an analytical method using UHPLC to resolve the one dalton difference arising from the cytidine to uracil conversion catalyzed by APOBEC3 enzymes. As a first step in proof of concept, we sought to separate a 1:1 mixture of 30-mer reactant and product oligonucleotides of an APOBEC enzyme reaction (Figure 1A, Table S1). We modified multiple parameters including the use of a Poroshell 1.9 micron C18 column, a mobile phase consisting of the ion pairing HFIP-TEA buffer system (Buffer A: 400mM hexafluoroisopropanol (HFIP), 15mM trimethylamine (TEA); Buffer B: 50% Buffer A, 50% methanol), and optimized critical chromatographic conditions such as flow rate, temperature, and gradient for optimal oligonucleotide separation. In particular, selection of the ion pairing mobile phase was influenced by previously demonstrated success in oligonucleotide separations and unique properties of HFIP serving as a volatile, denaturing, and acidic solvent, allowing for buffering of the mobile phase and suitability for subsequent ionization and analysis by MS/MS.27–29
We determined that optimal separation is dependent on gradient steepness (40–45% buffer B) and on the length of the oligonucleotide. The developed method is capable of performing a one dalton baseline separation of a wide range of DNA molecules from 22–43 bases (Figure S1). Interestingly, we found that longer DNA strands containing more bases were more difficult to separate, but the usage of a tandem column setup utilizing two UHPLC columns restored baseline resolution. We also discovered that separations performed at the temperature range of 55–60°C yielded the best resolution (data not shown). Using these optimized conditions, we notably obtained baseline resolution of the substrate and product oligonucleotides monitoring A260 absorbance with a diode array detector. (Figure 1B).
In order to assess turnover with recombinant APOBEC3 enzyme, we developed a scheme to purify DNA for optimal chromatographic separation. Briefly, we optimized a two-step purification process, firstly removing proteins by phenol chloroform extraction, and secondly removing buffer components by dialysis in water (Figure 2). We then assessed the cytidine deamination kinetics of APOBEC3G, the most well studied APOBEC3 family member, by incubating it with a substrate oligonucleotide containing the preferred sequence of APOBEC3G deamination (5’-ACCCA, in which the deaminated cytosine is underlined) over the course of 2.5 hours (Figure 2). DRONE successfully tracked substrate to product conversion over time, and the rate of conversion calculated from the AUC of the substrate/product peaks were comparable to that observed with a radiolabeled UDG assay, a method widely used in the assaying of APOBEC cytidine deamination kinetics (Figure S2). DRONE can also be used for the detection of lower amounts of turnover, corresponding to 1.6%, 7.2%, 15%, 34%, successfully detection % product as low as 1% (Figure S3,S4). Furthermore, we were able to confirm that DRONE can be used to track the enzymatic reaction of a second APOBEC3 enzyme, APOBEC3B, which is closely associated with human cancers (Figure S5,S6), broadening the applicability of our method to more than one APOBEC3 enzyme.6–8
Figure 2. Applying DRONE to track APOBEC3G enzyme turnpover.

A. Development of a scheme to purify enzyme timepoint experiments by UHPLC analysis. B. A schematic depicting cytosine to uracil conversion catalyzed by recombinant APOBEC3G C. Conversion of substrate to product catalyzed by recombinant APOBEC3G over the course of 2.5 hours. Reaction was conducted under single turnover conditions at 5 μM recombinant APOBEC3G and 1 μM substrate oligonucleotide. Quantitation based on AUC analysis is shown on the bottom right representing mean +/− SEM of % product values based on three biological replicates per timepoint.
After successful utilization of DRONE to track APOBEC cytidine deamination, we expanded this method to detect other DNA modifications important in human biology. The DNA base cytosine is also a substrate for many epigenetic modifications caused by enzymes implicated in cancer including DNA methyltransferase (DNMT) catalyzed cytosine methylation and Ten-Eleven Translocation (TET) enzyme induced cytosine hydroxymethylation (Figure 3A).1,4 APOBEC activity can also act on methylated cytosine,18,30 so we validated whether the substrate and product of this particular conversion will be separated to reasonable resolution (Figure 3A). We tested the separation of substrate and product oligonucleotides of each enzyme-catalyzed modification in the context of a longer strand of DNA with DRONE. Remarkably, all modifications were baseline resolved using the DRONE methodology (Figure 3B).
Figure 3. Detection of epigenetic modifications and turnover by recombinant DNMT3B.

A. Schematic of epigenetic modifications on cytosine catalyzed by the respective enzymes. B. Separation of substrate and product oligonucleotides of several DNA modifying enzymes acting downstream of DNA cytosine. C. Turnover of the de novo DNA methyltransferase DNMT3B catalytic domain. Quantification is based on AUC analysis yielding mean +/− SEM from two biological replicates per timepoint.
To further explore epigenetic modifications such as DNA cytosine methylation, an alteration which is abundant in CpG islands present in promoter regions of many cancer genomes,1 we examined the reaction catalyzed by recombinantly expressed and purified DNMT3B, a de novo DNA cytosine methyltransferase implicated in several cancers including lung cancer and melanoma.1,31,32 While conversion of substrate to product occurred at a much slower rate as compared to APOBEC cytidine deamination, we achieved baseline separation of substrate and product peaks permitting accurate quantitation of the individual peaks (Figure 3C). Such built-in versatility extends DRONE into the monitoring of epigenetic DNA modifications implicated in human pathology and disease.
We next attempted to apply DRONE in the context of enzyme inhibitor evaluation. Given that inhibitor screens of DNA modifying enzymes often involve cumbersome method development including usage of radioactive material, we next attempted to evaluate inhibition of DNA modifying activity. As a pilot experiment, we decided to test the inhibitory activity of Nanaomycin A, which was found to be a potent and specific small molecule inhibitor of DNMT3B (Figure 4A).33 We performed dose response experiments and have found that Nanaomycin A inhibits DNMT3B activity is a dose- dependent manner. From this inhibition, we were able to extract out an IC50 value of 730 nM, consistent to the high nanomolar IC50 from a previously reported filter binding assay (Figure 4B).33 This preliminary data extends the use of DRONE in the evaluation of inhibitor efficacy in the context of in vitro enzymatic activity.
Figure 4. Applications of DRONE.

A. The structure of nanaomycin A. B. Inhibition of methyltransferase activity by the addition of DNMT3B inhibitory nanaomycin A. C. Adoption of fluorescent detection to boost sensitivity and signal of UHPLC traces. Inset represents the trace using A260 UV detection which has a lower signal to noise ratio. D. One hour incubation of an oligonucleotide tagged with 6-FAM fluorophore on the 5’ end with APOBEC3G in comparison to a substrate only control and a blank injection.
Detection methods other than monitoring absorbance, for instance fluorescence, would be beneficial in enhancing the sensitivity and signal of the analytical output.34 One immediate advantage of a fluorescent output would be the usage of DRONE in a cellular context, for instance in endogenously relevant cellular lysates, in which DNA modifying activity occurs at a much slower rate. We were curious to examine to what extent a fluorescent detection methodology will result in signal enhancement in comparison to detection of absorbance at 260 nm. In a proof-of-concept experiment, we incubated APOBEC3G with the 6-carboxyfluorescein (6-FAM) labeled substrate at the 5’ end for one hour to produce a near-1:1 ratio of substrate to product oligonucleotide. The oligonucleotide used for these experiments is a 5’ FAM labeled 27mer sequence containing a central APOBEC3G recognition site (5’-ACCCA-3’) (Table S1). Remarkably, fluorescence yielded a > 2-fold amplitude in peak height as compared to the data obtained using absorbance at 260nm. Furthermore, quantitation of the signal to noise ratio improved from 20:1 in A260 detection to 400:1 in fluorescent detection, potentiating detection of less abundant species (Figure 4C). Furthermore, we successfully validated efficient turnover of these modified oligonucleotides by APOBEC3G (Figure 4D). The use of such fluorescently labeled oligonucleotides could serve as a tool for increasing the sensitivity and engineering specificity in detection for challenging applications. This methodology could be easily adapted to detection by mass spectrometry as well.
Recent studies analyzing cancer genomes have suggested putative targets of the mutagenic APOBEC3 proteins, namely APOBEC3A and APOBEC3B.8,35–37 In a study conducted in 2014 by Henderson et al. focusing on head and neck squamous cell carcinomas38, two PIK3CA helical domain mutations (E542K and E545K) were identified as putative targets of APOBEC3. To validate this finding, we designed a 25mer single stranded oligonucleotide substrate containing these two sites (Figure 5), and we performed studies to observe the underlying deamination kinetics using our developed method. After assignment of each peak based on UHPLC runs of each product oligonucleotide individually and mixed together, we have found that both sites are efficiently deaminated (denoted P1 for the central C and P2 for the 5’ C) by APOBEC3B in a time-dependent manner over the course of 4 hours (Figure 5). Furthermore, we were able to detect sequential deamination events in the same oligonucleotide substrate shown as separately eluting peaks off of the UHPLC column. Quantification of the resulting peaks shows a time-dependent accumulation of a singly deaminated intermediate and its subsequent depletion to form the double deaminated product. We were able to establish sequential deamination of an endogenously relevant oligonucleotide substrate identified from bioinformatics studies.
Figure 5. Deamination Kinetics of a PIK3CA mimic substrate.

A. DNA sequence of the 25mer PIK3CA substrate with the two target cytosines denoted. B. Deamination by the recombinant APOBEC3B of the PIK3CA target substrate. Over the course of four hours, accumulation of P1 and P2 corresponding to the central and 5’ cytosine deamination event was observed, denoted on the timecourse.
In this study, we describe DRONE, an UHPLC-based analytical method to directly monitor reaction kinetics of several DNA modifying enzymes implicated in human cancers. The developed method is equipped with versatility, efficiently tracking APOBEC cytidine deamination at a one Dalton resolution, along with other DNA modifications including cytosine methylation and hydroxymethylation. This detection of enzymatic activity also allows for one to decipher more complex kinetic questions, an inherent challenge with traditional assays tracking DNA modifications. DRONE could thus be an important addition to existing method development workflows in the characterization of potent DNA mutators and DNA modifying enzymes implicated in human disease.
Supplementary Material
Acknowledgements
This material is based upon work supported by the Department of Defense Award W81XWH-15-1-0290 (KSA), National Science Foundation Graduate Research Fellowship Grant DGE-1122492 (TS), and NIH F31 fellowship support CA203254 granted by the National Cancer Institute (TS), We would also like to thank Agilent Technologies for technical advice and instrumental support which made this study possible. We also thank Dr. Xiaojiang Chen and Dr. Yang Fu at the University of Southern California for their kind gift of the MBP-APOBEC3B plasmid and very helpful advice on protein expression.
Footnotes
Supporting information
Associated content: Tables S1-S10 detailing descriptions of oligonucleotide sequences and chromatography conditions, Figures S1-S6 containing supplementary experimental figures, and an experimental materials and methods section.
References
- (1).Subramaniam D; Thombre R; Dhar A; Anant S Front Oncol 2014, 4, 80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Wyatt MD In Advances in Cancer Research, Vol 119, Tew KD; Fisher PB, Eds., 2013, pp 63–106. [DOI] [PubMed] [Google Scholar]
- (3).Kelley MR; Logsdon D; Fishel ML Future oncology (London, England) 2014, 10, 1215–1237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Rasmussen KD; Helin K Genes & Development 2016, 30, 733–750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Nik-Zainal S; Alexandrov LB; Wedge DC; Van Loo P; Greenman CD; Raine K; Jones D; Hinton J; Marshall J; Stebbings LA; Menzies A; Martin S; Leung K; Chen L; Leroy C; Ramakrishna M; Rance R; Lau KW; Mudie LJ; Varela I; McBride DJ; Bignell GR; Cooke SL; Shlien A; Gamble J; Whitmore I; Maddison M; Tarpey PS; Davies HR; Papaemmanuil E; Stephens PJ; McLaren S; Butler AP; Teague JW; Jonsson G; Garber JE; Silver D; Miron P; Fatima A; Boyault S; Langerod A; Tutt A; Martens JW; Aparicio SA; Borg A; Salomon AV; Thomas G; Borresen-Dale AL; Richardson AL; Neuberger MS; Futreal PA; Campbell PJ; Stratton MR Cell 2012, 149, 979–993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Burns MB; Lackey L; Carpenter MA; Rathore A; Land AM; Leonard B; Refsland EW; Kotandeniya D; Tretyakova N; Nikas JB; Yee D; Temiz NA; Donohue DE; McDougle RM; Brown WL; Law EK; Harris RS Nature 2013, 494, 366–370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Burns MB; Temiz NA; Harris RS Nat Genet 2013, 45, 977–983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Roberts SA; Lawrence MS; Klimczak LJ; Grimm SA; Fargo D; Stojanov P; Kiezun A; Kryukov GV; Carter SL; Saksena G; Harris S; Shah RR; Resnick MA; Getz G; Gordenin DA Nat Genet 2013, 45, 970–976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Driscoll DM; Wynne JK; Wallis SC; Scott J Cell 1989, 58, 519–525. [DOI] [PubMed] [Google Scholar]
- (10).Bransteitter R; Pham P; Scharff MD; Goodman MF Proc Natl Acad Sci U S A 2003, 100, 4102–4107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Smith HC Methods 1998, 15, 27–39. [DOI] [PubMed] [Google Scholar]
- (12).Smith HC Methods Enzymol 2007, 424, 389–416. [DOI] [PubMed] [Google Scholar]
- (13).Polevoda B; McDougall WM; Bennett RP; Salter JD; Smith HC Methods 2016, 107, 10–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).McDougall WM; Smith HC Biochem Biophys Res Commun 2011, 412, 612–617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Harris RS; Bishop KN; Sheehy AM; Craig HM; Petersen-Mahrt SK; Watt IN; Neuberger MS; Malim MH Cell 2003, 113, 803–809. [DOI] [PubMed] [Google Scholar]
- (16).Chelico L; Pham P; Calabrese P; Goodman MF Nat Struct Mol Biol 2006, 13, 392–399. [DOI] [PubMed] [Google Scholar]
- (17).Adolph MB; Love RP; Feng Y; Chelico L Nucleic Acids Res 2017, 45, 11925–11940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Fu Y; Ito F; Zhang G; Fernandez B; Yang H; Chen Xiaojiang S. Biochemical Journal 2015, 471, 25–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Nowarski R; Britan-Rosich E; Shiloach T; Kotler M Nat Struct Mol Biol 2008, 15, 1059–1066. [DOI] [PubMed] [Google Scholar]
- (20).Jaguva Vasudevan AA; Goering W; Haussinger D; Munk C Methods Mol Biol 2018, 1655, 97–107. [DOI] [PubMed] [Google Scholar]
- (21).Petersen-Mahrt SK; Harris RS; Neuberger MS Nature 2002, 418, 99–103. [PubMed] [Google Scholar]
- (22).Harris RS; Petersen-Mahrt SK; Neuberger MS Mol Cell 2002, 10, 1247–1253. [DOI] [PubMed] [Google Scholar]
- (23).Li M; Shandilya SM; Carpenter MA; Rathore A; Brown WL; Perkins AL; Harki DA; Solberg J; Hook DJ; Pandey KK; Parniak MA; Johnson JR; Krogan NJ; Somasundaran M; Ali A; Schiffer CA; Harris RS ACS Chem Biol 2012, 7, 506–517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Armstrong KM; Bermingham EN; Bassett SA; Treloar BP; Roy NC; Barnett MP Biotechnol J 2011, 6, 113–117. [DOI] [PubMed] [Google Scholar]
- (25).Lisanti S; Omar WA; Tomaszewski B; De Prins S; Jacobs G; Koppen G; Mathers JC; Langie SA PLoS One 2013, 8, e79044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Yuan BF Methods Mol Biol 2017, 1562, 33–42. [DOI] [PubMed] [Google Scholar]
- (27).Chen B; Mason SF; Bartlett MG J Am Soc Mass Spectrom 2013, 24, 257–264. [DOI] [PubMed] [Google Scholar]
- (28).Gong L; McCullagh JS Rapid Commun Mass Spectrom 2014, 28, 339–350. [DOI] [PubMed] [Google Scholar]
- (29).Studzinska S; Pietrzak L; Buszewski B Chromatographia 2014, 77, 1589–1596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Carpenter MA; Li M; Rathore A; Lackey L; Law EK; Land AM; Leonard B; Shandilya SMD; Bohn M-F; Schiffer CA; Brown WL; Harris RS The Journal of Biological Chemistry 2012, 287, 34801–34808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Lyko F; Brown R JNCI: Journal of the National Cancer Institute 2005, 97, 1498–1506. [DOI] [PubMed] [Google Scholar]
- (32).Micevic G; Muthusamy V; Damsky W; Theodosakis N; Liu X; Meeth K; Wingrove E; Santhanakrishnan M; Bosenberg M Cell reports 2016, 14, 2180–2192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Kuck D; Caulfield T; Lyko F; Medina-Franco JL Mol Cancer Ther 2010, 9, 3015–3023. [DOI] [PubMed] [Google Scholar]
- (34).An WF Methods Mol Biol 2009, 486, 97–107. [DOI] [PubMed] [Google Scholar]
- (35).Seplyarskiy VB; Soldatov RA; Popadin KY; Antonarakis SE; Bazykin GA; Nikolaev SI Genome Res 2016, 26, 174–182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Chan K; Roberts SA; Klimczak LJ; Sterling JF; Saini N; Malc EP; Kim J; Kwiatkowski DJ; Fargo DC; Mieczkowski PA; Getz G; Gordenin DA Nat Genet 2015, 47, 1067–1072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Saini N; Roberts SA; Sterling JF; Malc EP; Mieczkowski PA; Gordenin DA DNA Repair (Amst) 2017, 53, 4–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Henderson S; Chakravarthy A; Su X; Boshoff C; Fenton TR Cell Rep 2014, 7, 1833–1841. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.