

Abstract
Aims
Limitation of ischemia/reperfusion injury is a major therapeutic target after acute myocardial infarction (AMI). Toll‐like receptors are implicated in the inflammatory response that occurs during reperfusion. The triggering receptor expressed on myeloid cells (TREM)‐1 acts as an amplifier of the immune response triggered by toll‐like receptor engagement. We hypothesized that administration of a TREM‐1 inhibitory peptide (LR12) could limit reperfusion injury in a porcine model of AMI.
Methods and results
AMI was induced in 15 adult minipigs by a closed‐chest coronary artery occlusion‐reperfusion technique. Animals were randomized to receive LR12 or vehicle before reperfusion (LR12 n = 7, vehicle n = 8), and were monitored during 18 h.
AMI altered hemodynamics and cardiac function, as illustrated by a drop of mean arterial pressure, cardiac index, cardiac power index, ejection fraction, and real‐time pressure–volume loop‐derived parameters. TREM‐1 inhibition by LR12 significantly improved these dysfunctions (P < 0.03) and limited infarct size, as assessed by lower creatine phosphokinase and troponin I concentrations (P < 0.005).
Pulmonary, renal, and hepatic impairments occurred after AMI and were attenuated by LR12 administration as assessed by a better PaO2 to FiO2 ratio, a less positive fluid balance, and lower liver enzymes levels (P < 0.05).
Conclusion
Inhibition of the TREM‐1 pathway by a synthetic peptide limited myocardial reperfusion injury in a clinically relevant porcine model of AMI.
Keywords: Myocardial infarction, Reperfusion, Immune system, Inflammation
Introduction
Over the last 30 years, timely reperfusion strategies have dramatically improved survival rates in patients suffering from acute myocardial infarction (AMI).1 Despite this impressive success, mortality and onset of heart failure in survivors remain worrying. Novel therapeutic strategies are definitely needed to further improve myocardial salvage, as infarct size is closely linked to prognosis. Although reperfusion strategies are the cornerstone of myocardial salvage at the acute phase of AMI, they can worsen myocardial injury by themselves. This ‘double‐edge sword’ is termed ‘myocardial reperfusion injury’ phenomenon (RI). Indeed, RI could be responsible for up to half the final infarct size and could therefore be targeted to limit myocardial damage.2
Toll‐like receptors (TLRs) are highly conserved throughout species and act as sensors of danger signals. They recognize exogenous—pathogen‐associated molecular patterns—or endogenous—damage‐associated molecular patterns—molecules and induce innate immune response. During myocardial reperfusion, necrotic cells and damaged extracellular matrix components produce large amounts of damage‐associated molecular patterns, which, in turn, activate TLR signalling. It then promotes a pro‐inflammatory environment, especially through the activation of nuclear factor kappa B pathway, which has repeatedly been implicated in innate immunity activation and generation of lethal RI after cardiac ischemia.3, 4 As TLRs are implicated at the earliest stages of the inflammatory chain reaction, they appear ideal therapeutic targets for limitation of RI.5 Among TLRs present in the heart, TLR‐2 and TLR‐4 have the highest expression levels and have been extensively investigated in the context of RI.6
TREM‐1 is an immune receptor expressed by neutrophils, macrophages, and mature monocytes. It acts as an amplifier of the innate immune response triggered by TLRs engagement.7 It had regularly been shown that blockade of TREM‐1 activation protected from hyper‐responsiveness and death in various models of severe infection.8, 9, 10, 11 Beyond infectious diseases, TREM‐1 has also been found important during aseptic inflammatory disorders.12, 13, 14, 15, 16
The role of the immune system after cardiac ischemia is ambivalent as, on one hand, it appears crucial for debridement of the infarcted myocardium, but on the other hand, it leads to increased infarct size and detrimental ventricular remodelling. Considering this, a fine tuning of the immune response is required. Indeed, attempts to completely inhibit inflammation have failed to improve outcome.17, 18, 19, 20, 21, 22 Of note, TREM‐1 inhibition does not abrogate immune response but limits its self‐amplification and could therefore allow for a fine tuning between required and deleterious inflammatory signals.
We thus investigated LR12, an inhibitory TREM‐1 peptide that acts as a decoy receptor and binds to the yet unknown TREM‐1 ligand,11 during cardiac ischemia in a clinically relevant model of reperfused MI in adult minipigs. We show that LR12 improves hemodynamic parameters and cardiac function and limits remote organ dysfunction.
Methods
The experiments were performed in adherence with the National Institutes of Health Guidelines on the Use of Laboratory Animals and were approved by the ‘Comité d'éthique en expérimentation animale no. 66’ (Animal Experiments Ethical Committee no. 66) under the agreement number 00492.02. Our protocol was adapted from previously published ones.23, 24
Animal preparation
Adult male minipigs (Sus scrofa domestic, Vietnamese pot‐bellied minipigs, 40–68 kg) were purchased from Elevage Ferry (Vosges, France). Before surgery, animals were acclimated to the animal department for at least 5 days and fasted overnight with free access to water. Pre‐anaesthesia was performed through intramuscular injection of ketamine (10 mg/kg). Anaesthesia was induced and maintained with intravenously administered pentobarbital (initial bolus: 10 mg/kg, continuous infusion 10–12 mg/kg/h during the preparation period and 6–8 mg/kg/h after coronary occlusion), intermittent sufentanyl (10 µg every 2 h), and cisatracurium (Nimbex, GlaxoSmithKline, Marly‐le‐Roi, France) if necessary. Animals were mechanically ventilated (tidal volume 7 mL/kg, positive end‐expiratory pressure 4 cm H2O, FiO2 0.3, respiratory rate 16–18 breaths/min adjusted to maintain normocapnia).
Right jugular vein was exposed, and a multi‐lumen central line was inserted for perfusion and Swan–Ganz catheter insertion, allowing for the continuous recording of cardiac output, SvO2, and right atrial and pulmonary arterial pressures (14 Fr multi‐lumen access combining a 8 Fr sheath introducer and 2 conventional central lines, MAC, Teleflex Medical SAS, Le Faget, France). A 5 Fr introducer was placed in the right carotid artery, and a combined pressure‐conductance catheter (Ventri‐Cath 507, Millar Inc, Houston, Texas, USA) was positioned within the left ventricle (LV) under X‐ray guidance for cardiac function assessment. A right femoral arterial 7 Fr introducer was inserted for coronary catheter access and continuous measurement of arterial pressure. Right femoral vein was also catheterized (12 Fr introducer), and an 8–14 Fr Fogarty catheter (Edwards Lifesciences, Nyon, Switzerland) was placed for transient vena cava occlusion. A catheter in the bladder allowed urine collection. The animals received 2500 IU heparin, repeated every 4 h to avoid blood clotting of catheters, and 5 mg/kg amiodarone (Sanofi‐Aventis, Paris, France) 1 h before coronary occlusion to limit cardiac arrhythmias.
After surgery, animals were allowed to recover for 2 h before baseline measurements (defined as ‘H0’). Balanced saline solution (Isofundine, B Braun Medical SAS, Boulogne‐Billancourt, France) was continuously administered (10 mL/kg/h) throughout the study. Body temperature was kept constant (±1°C) using cold accumulators.
Experimental protocol
The timeline is presented in Figure 1. After baseline data collection (H0), myocardial infarction was induced by inflation of an angioplasty balloon in the proximal left anterior descending artery (LAD). An angiogram was performed after inflation of the balloon in order to verify the complete occlusion of LAD. Four animals died from refractory ventricular tachycardia within 15 min after LAD occlusion and were therefore excluded from further analyses. After 45 min, animals were randomized to receive LR12 (LR12 group, n = 7) or the vehicle (phosphate‐buffered saline) alone (control group, n = 8). LR12 consists of a 12 amino acid part of the extracellular domain of TLT‐1 (LQEEDAGEYGCM) and was chemically synthesized (Pepscan Presto BV, Lelystad, The Netherlands) as COOH terminally amidated peptide. The correct compound, free of endotoxin, was obtained with greater than 99% yield and was homogeneous after preparative purification, as confirmed by mass spectrometry and analytic reversed‐phase high‐performance liquid chromatography. This lyophilized peptide was extemporaneously solubilized in sterile phosphate‐buffered saline under aseptic conditions. A bolus of 5 mg/kg (in 150 mL) was intravenously delivered over 15 min, then a 1 mg/kg/h (30 mL/h) continuous infusion was started and lasted throughout the study period. This dosage was derived from previous experiments performed in minipigs and corresponded to the best effective dose.10 Control animals received the same amount of vehicle (sterile phosphate‐buffered saline). After a LAD occlusion period of 60 min, the balloon was deflated. An angiogram was performed in order to verify the restoration of the blood flow in the previously occluded artery. Sedation and mechanical ventilation were maintained throughout the 18 h study period.
Figure 1.
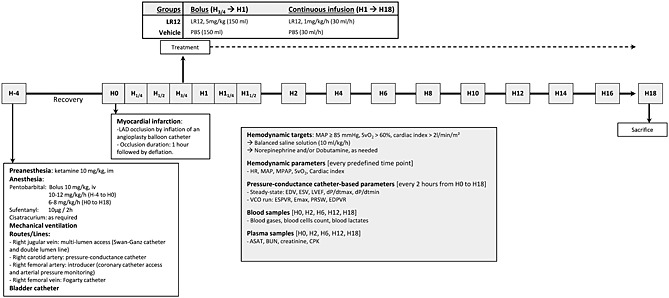
Experimental timeline. LAD, left anterior descending coronary artery; PBS, phosphate‐buffered saline; MAP, mean arterial pressure; SvO2, mixed venous oxygen saturation; HR, heart rate; MPAP, mean pulmonary arterial pressure; EDV, end‐diastolic volume; ESV, end‐systolic volume; LVEF, left ventricular ejection fraction; dP/dtmax, pressure development during isovolumic contraction; dP/dtmin, pressure development during isovolumic relaxation; VCO, vena cava occlusion; ESPVR and EDPVR, end‐systolic and end‐diastolic pressure volume relationship; Emax, maximal ventricular elastance; PRSW, preload recruitable stroke work; AST, aspartate aminotransferase; BUN, blood urea nitrogen; CK, creatine phosphokinase.
Experienced intensive care physicians, blinded for the given treatment, provided animal care. The main hemodynamic targets were to maintain mean arterial pressure (MAP) above 85 mmHg, cardiac index above 2.0 L/min/m2, and SvO2 above 60%. To achieve these goals, rapid fluid loading (Isofundine, B Braun Medical SAS, Boulogne‐Billancourt, France) was allowed provided that right atrial pressure and pulmonary capillary wedge pressure remained lower than 18 mmHg. In case of incomplete hemodynamic improvement, a continuous infusion of norepinephrine and/or dobutamine was started.
Animals were then killed under deep anaesthesia by KCl infusion 18 h after the induction of myocardial infarction.
Measurements
Hemodynamic parameters were continuously monitored throughout the study period and recorded at baseline, every 15 min from H0 to H1.5 and every 2 h from H2 to H18. The recorded parameters included MAP, mean pulmonary artery pressure, cardiac output, cardiac index, and SvO2. Systemic oxygen delivery (DO2) and systemic oxygen uptake (VO2), and pulmonary and systemic vascular resistance index were calculated by the Swan–Ganz monitor (Edwards Lifesciences, Irvine, CA, USA). Cardiac Power Index (W/m2) was calculated as MAP*cardiac index/451.25
Pressure–volume (PV) catheter was calibrated before data recordings. The arbitrary relative volume unit number was converted to true volume using pre‐warmed Rho calibration cuvette device (reference 910‐1060, Millar Inc, Houston, Texas, USA) to determine resistivity of body‐temperature heparinized blood. Parallel conductance (i.e. non‐LV blood conductance) was estimated by injection of 2.5 mL NaCl 10% into the pulmonary arterial line with simultaneous LV PV sampling. Pressure sensor was electronically calibrated with MPVSUltra Control Interface Software, according to manufacturer's recommendations. PV catheter‐based parameters were recorded every 2 h from H0 to H18. The ventilator was disconnected for 10 s, and steady state parameters were collected, including dP/dtmax, dP/dtmin (maximum and minimum values of the first derivative of ventricular pressure), end‐diastolic, and end‐systolic volumes of the left ventricle (EDV, ESV), ejection fraction, Tau (time constant of LV pressure decay by Glantz method), and stroke work (SW, i.e. the work performed by the left ventricle to eject the stroke volume into the aorta, calculated as the area within the PV loop). Immediately after, the Fogarty catheter was inflated in the inferior vena cava and pre‐load‐alternating PV data were collected during this vena cava occlusion run, including end‐systolic and end‐diastolic pressure volume relationships (ESPVR and EDPVR), maximum ventricular elastance (Emax), arterial elastance, ‘dP/dtmax to EDV’ relationship, pre‐load recruitable stroke work (PRSW), and pressure–volume area. ESPVR, dP/dtmax to EDV relationship, and PRSW are indexes of myocardial contractility that are relatively insensitive to pre‐load and afterload. Pressure–volume area is the specific area in the pressure–volume plane bounded by the ESPVR and EDPVR lines and the systolic segment of the PV loop. It is linearly correlated to left ventricular oxygen consumption.
Blood was sequentially drawn (H0, H2, H6, H12, and H18) for the determination of blood gases, arterial lactates, plasma concentration of alanine aminotransferase, blood urea nitrogen, creatinine, and creatine phosphokinase. Troponin I was determined by enzyme‐linked immuno assay according to manufacturer's instructions (porcine cardiac troponin I enzyme‐linked immuno assay kit, MBS024086, MyBioSource Inc, San Diego, CA, USA).
Statistical analysis
After testing for their normal distribution (Kolmogorov–Smirnov test), data are presented as means ± SD. Between group differences were tested by two‐way analysis of variance for repeated measures with Bonferroni correction or Student's t‐test when appropriate. Analyses were performed using GraphPad Prism software.
Results
LR12 improves hemodynamic parameters
After an immediate similar drop following LAD occlusion, MAP progressively increased until the reperfusion where it decreased again (Figure 2). By H6, MAP stabilized to baseline levels in the LR12 animals whereas it further decreased in the control group (94.9% vs. 71.3% of baseline value in LR12 vs. controls at the end of the study, P < 0.01). Two animals in each group required norepinephrine to reach hemodynamic targets, with a significantly lower cumulated dose in LR12 group (0.11 ± 0.07 vs. 2.85 ± 1.57 mg/kg, P < 0.01).
Figure 2.
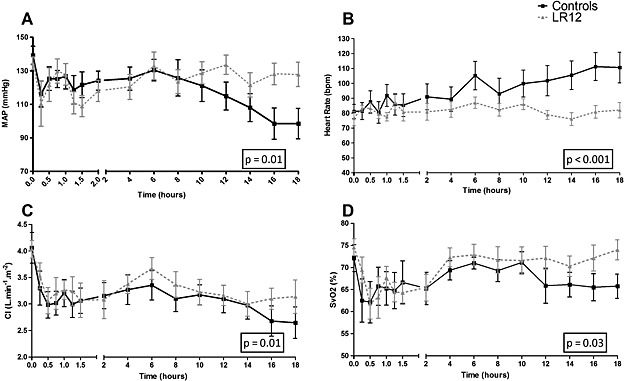
LR12 improves hemodynamics. Evolution of mean arterial pressure (A), heart rate (B), cardiac index (C), and SvO2 (D) during the study period. Hemodynamics parameters were improved in LR12‐treated animals as compared with control ones.
Heart rate progressively increased throughout the study in control pigs, whereas it remained stable in LR12 group (P < 0.0001).
Myocardial ischemia induced a rapid decline of cardiac index (83.1 and 79.6% of baseline value at H1 in LR12 and controls, respectively) that did not change significantly thereafter. Cardiac index was higher in LR12 than in control animals by H18 (78.9% vs. 65.9% of baseline, P < 0.01). This translated into lower SvO2 at H18 in the control group (P = 0.03). Two and three animals in, respectively, LR12 and control groups required dobutamine infusion to maintain adequate circulatory conditions, with similar cumulative dose (1.18 ± 0.33 vs. 0.93 ± 0.45 µg/kg in controls and LR12, respectively, P = 0.76).
LR12 improves cardiac function and limits infarct size
Cardiac power index, believed to better describe cardiac performance than conventional hemodynamic parameters, was significantly more depressed in controls than in LR12 pigs by H18 (47.2 ± 16.0% vs. 69.3 ± 11.5% of baseline value, P = 0.02) (Figure 3).
Figure 3.
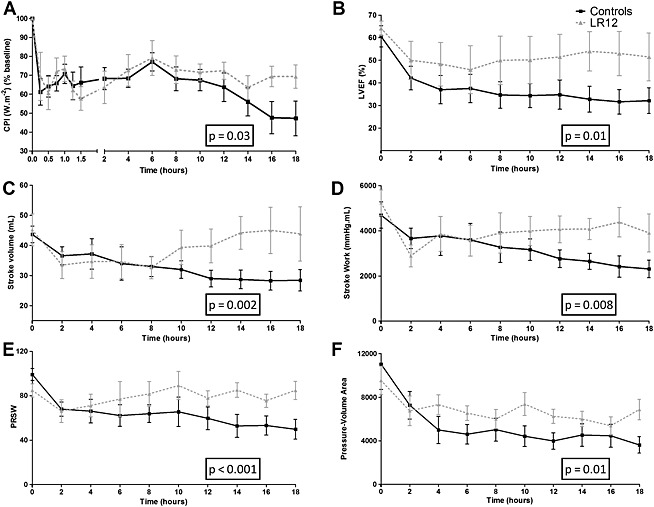
LR12 improves cardiac function. Evolution of cardiac power index (A) left ventricular ejection fraction (B), stroke volume (C), stroke work (D), preload recruitable stroke work (E), and pressure‐volume area (F) during the study period. Cardiac dysfunction was attenuated by LR12 treatment.
Pressure–volume loops studies at steady state revealed significantly lower stroke volumes (P = 0.002) and left ventricle ejection fraction in control group as compared with LR12‐treated pigs (P = 0.01). During vena cava occlusion, we noted an important decrease of PRSW in control group, corrected by LR12 administration (P < 0.001) (Figure 3). The detailed overview of the parameters obtained through PV loops studies is presented in Table 1.
Table 1.
Selected pressure‐volume loops derived parameters at baseline, 2 hours, and 18 hours after reperfusion
| H0 | H2 | H18 | ||
|---|---|---|---|---|
| Emax (mmHg/mL) | Controls | 7.2 ± 3.0 | 6.7 ± 4.1 | 6.8 ± 3.2 |
| LR12 | 7.1 ± 2.1 | 5.8 ± 1.0 | 6.6 ± 2.8 | |
| ESPVR (mmHg/mL) | Controls | 4.3 ± 2.7 | 3.6 ± 1.8 | 4.3 ± 0.9 |
| LR12 | 3.8 ± 1.9 | 3.1 ± 1.0 | 3.9 ± 1.2 | |
| PRSW (mmHg/mL) | Controls | 99.1 ± 16.0 | 68.0 ± 16.9 * | 49.8 ± 21.9 *, ** |
| LR12 | 84.8 ± 14.7 | 66.1 ± 27.1 | 84.8 ± 20.6 | |
| EDPVR (mmHg/mL) | Controls | 0.46 ± 0.39 | 0.61 ± 0.36 | 0.30 ± 0.06 |
| LR12 | 0.36 ± 0.20 | 0.48 ± 0.24 | 0.33 ± 0.07 | |
| Dp/dtmax (mmHg/s) | Controls | 2332 ± 652 | 2333 ± 468 | 1941 ± 402 |
| LR12 | 2286 ± 484 | 2008 ± 386 | 2553 ± 837 | |
| Dp/dtmin (mmHg/s) | Controls | −3207 ± 549 | −2548 ± 460 | −1767 ± 402 * |
| LR12 | −3389 ± 1186 | −2077 ± 715 | −2314 ± 499 | |
| EDV (mL) | Controls | 64.9 ± 17.7 | 80.6 ± 14.9 | 101.3 ± 41.8 * |
| LR12 | 66.8 ± 23.0 | 63.4 ± 17.1 | 84.0 ± 14.7 | |
| ESV (mL) | Controls | 28.6 ± 11.4 | 54.5 ± 26.2 | 67.2 ± 49.2 * |
| LR12 | 28.4 ± 15.6 | 34.5 ± 19.2 | 41.4 ± 23.3 | |
| LVEF (%) | Controls | 60.6 ± 14.8 | 42.1 ± 14.7 | 32.0 ± 16.0 *, ** |
| LR12 | 64.1 ± 8.5 | 50.0 ± 12.4 | 51.5 ± 10.6 | |
| EDP (mmHg) | Controls | 16.5 ± 10.5 | 29.9 ± 13.9 | 19.9 ± 9.6 |
| LR12 | 21.3 ± 6.0 | 24.0 ± 6.0 | 23.2 ± 9.5 | |
| ESP (mmHg) | Controls | 151.4 ± 24.7 | 133.8 ± 19.6 | 124.2 ± 22.2 |
| LR12 | 154.0 ± 22.0 | 124.2 ± 27.3 | 145.9 ± 22.7 | |
| Tau (ms) | Controls | 16.4 ± 2.7 | 17.4 ± 7.5 | 21.8 ± 7.0 |
| LR12 | 16.0 ± 4.8 | 23.5 ± 11.2 | 21.8 ± 5.6 | |
| SW (mL mmHg) | Controls | 4695 ± 1647 | 3663 ± 1285 | 2312 ± 862 * |
| LR12 | 5236 ± 1502 | 2890 ± 1278 | 3905 ± 1885 | |
| Ea (mmHg/mL) | Controls | 4.5 ± 2.0 | 3.9 ± 0.7 | 5.5 ± 2.3 |
| LR12 | 3.8 ± 1.3 | 4.3 ± 1.7 | 4.3 ± 1.8 | |
| PVA (mmHg/mL) | Controls | 11042 ± 6582 | 7270 ± 3340 | 3619 ± 1694 *, ** |
| LR12 | 9552 ± 3424 | 6749 ± 3613 | 6879 ± 2082 | |
| dP/dtmax‐EDV relationship | Controls | 13.5 ± 5.3 | 24.1 ± 23.2 | 30.9 ± 13.7 |
| LR12 | 14.9 ± 20.2 | 24.4 ± 15.9 | 40.2 ± 36.5 | |
| SVRi (dyn s/cm5/m2) | Controls | 2539 ± 447 | 3074 ± 779 | 2831 ± 614 |
| LR12 | 2453 ± 526 | 3015 ± 1170 | 3125 ± 1023 | |
| PVRi (dyn s/cm5/m2) | Controls | 199 ± 105 | 246 ± 70 | 400 ± 220 *, ** |
| LR12 | 174 ± 29 | 248 ± 56 | 220 ± 61 |
Emax, maximal ventricular elastance; ESPVR and EDPVR, end‐systolic and end‐diastolic pressure volume relationship; PRSW, preload recruitable stroke work; dP/dtmax, maximum pressure development during isovolumic contraction; dP/dtmin, minimum pressure development during isovolumic relaxation; EDV, end‐diastolic volume; ESV, end‐systolic volume; EDP, end‐diastolic pressure; ESP, end‐systolic pressure; LVEF, left ventricular ejection fraction; Tau (time constant of LV pressure decay by non‐zero asymptote method); SW, stroke work; Ea, effective arterial élastance; PVA, pressure‐volume area; SVRi and PVRi, systemic and pulmonary vascular resistance index (measured by pulmonary artery catheter).
P < 0.05 vs. H0.
P < 0.05 LR12 vs. controls.
Infarct size estimated by the area under the curve of plasma levels of creatine phosphokinase and troponin I was significantly reduced by LR12 (P < 0.005) (Figures 4 a and 4 b).
Figure 4.
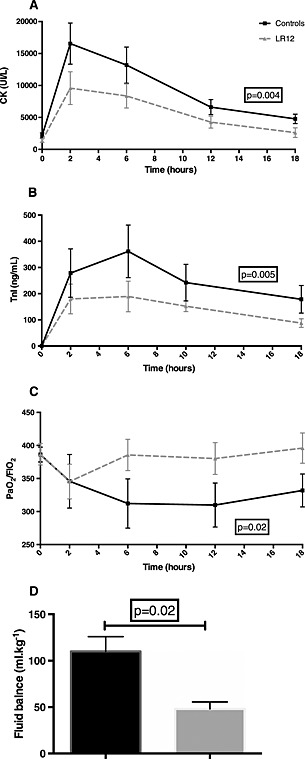
LR12 improves remote organ dysfunction and limit infarct size. Evolution of creatine phosphokinase (A), and troponin I plasma concentration (B), fluid balance (C), and PaO2 to FiO2 ratio (D) during the study period. Myocardial infarction was associated with impaired renal function, outlined by highly positive fluid balance, and altered gas exchange. These dysfunctions were corrected by LR12 administration. Moreover, LR12 limited infarct size, as assessed by lower creatine phosphokinase and troponin I levels throughout the study period.
LR12 reduces organ dysfunction
One hour after myocardial reperfusion, a drop of PaO2/FIO2 was observed in both groups. This phenomenon quickly reversed upon LR12 treatment while persisted throughout the study period in control animals (Figure 4 c).
We also observed renal function impairment: fluid balance was more positive in control pigs (P = 0.02) (Figure 4 d), and blood urea nitrogen progressively increased in controls and was higher than in the LR12 animals at the end of the study (Table 2).
Table 2.
Biological parameters
| H0 | H2 | H18 | ||
|---|---|---|---|---|
| BUN (g/L) | Controls | 0.16 ± 0.07 | 0.15 ± 0.07 | 0.22 ± 0.07*, ** |
| LR12 | 0.15 ± 0.03 | 0.12 ± 0.03 | 0.18 ± 0.06 | |
| Creatinine (mg/L) | Controls | 7.03 ± 1.60 | 7.82 ± 1.30 | 7.37 ± 1.78 |
| LR12 | 6.94 ± 1.08 | 5.57 ± 1.12 | 6.42 ± 0.92 | |
| ALT (IU/L) | Controls | 41 ± 24 | 96 ± 34* | 102 ± 44*, ** |
| LR12 | 44 ± 24 | 81 ± 41 | 65 ± 5 | |
| Platelets Count (G/L) | Controls | 330 ± 160 | 310 ± 160 | 200 ± 130* |
| LR12 | 360 ± 70 | 330 ± 70 | 240 ± 60* | |
| WBC (per μL) | Controls | 15640 ± 5026 | 17225 ± 4202 | 23671 ± 12676* |
| LR12 | 15786 ± 2251 | 20367 ± 5086 | 21383 ± 4672* | |
| CPK (IU/L) | Controls | 2175 ± 1076 | 16552 ± 8534 | 4770 ± 1980*, ** |
| LR12 | 1390 ± 782 | 9586 ± 6784 | 2620 ± 1845 | |
| Troponin (ng/mL) | Controls | 0.46 ± 0.27 | 278.80 ± 261.70* | 178.50 ± 149.30* |
| LR12 | 0.35 ± 0.25 | 179.70 ± 138.80*, ** | 87.70 ± 39.80*, ** |
BUN refers to blood urea nitrogen; ALT, alanine aminotransferase; WBC, white blood cells count; CPK, creatine phosphokinase.
P < 0.05 vs. H0.
P < 0.05 LR12 vs. controls.
Elevation of liver enzymes was observed as soon as 2 h after MI in the control pigs, whereas hepatic cytolysis did not occur upon LR12 treatment (Table 2).
Discussion
Failure of anti‐inflammatory therapies
Similarities exist between septic states and AMI, as innate immune response is quickly initiated by tissue injury, whether related to pathogens or other sterile aggressions such as ischemia. Attempts to inhibit inflammation in the setting of septic shock or AMI have failed to improve patients' outcome. Numerous clinical trials have investigated anti‐inflammatory strategies during these two acute pathological conditions, with sometimes the same evaluated drugs such as high‐dose corticosteroids17, 26 or interleukine‐1β receptor antagonist.27, 28 Despite encouraging pre‐clinical data, translation in the clinic has always been disappointing: failure of anti‐inflammatory approaches has been reviewed elsewhere, either during AMI29 or septic shock.30 However, experts in the field of cardiovascular diseases or sepsis still agree that targeting the inflammatory response could improve outcome but it would require (1) a paradigm shift taking into account the need for an adjusted balance between essential and detrimental facets of inflammation and (2) a more realistic and clinically relevant experimental approach.3, 31
TREM‐1 acts as an amplifier of the innate immune response. Its inhibition does not completely abrogate the inflammatory response but rather attenuates it.7 Conclusive data have been obtained by our group from several experimental animal models of septic shock, including clinically relevant species (i.e. pigs and non‐human primates).10, 32 These results fit with the new paradigm of limiting the magnitude of the initial inflammatory response rather than totally inhibiting it during septic shock.33 These features could also fit with the unmet need for therapies that allow a fine balance between required and deleterious immune response during AMI.
Hypothetical mechanisms
Inflammatory signals in the ischemic myocardium recruit neutrophils within the first hours, with a peak at 24 h, whereas monocytes infiltrate it later.34 Considering the length of our study period, neutrophils are most likely to be involved than other innate immune cells. A bulk of evidence has raised the role of neutrophil‐dependent myocardial injury during reperfusion through distinct mechanisms that could involve TREM‐1.35 First, the no reflow phenomenon has been linked to neutrophils entrapment into the microvasculature. Indeed, before infiltrating the myocardium, leucocytes interact closely with endothelial cells through complex molecular events involving integrins. This step enables leucocytes to roll along the vascular wall and then to stop, a pre‐requisite to transmigration. Recently, the critical role of TREM‐1 in transmigration has been shown in a different model of tissue injury (i.e. bacterial pneumonia) and could therefore explain some of the observed beneficial effects. In addition to microvascular obstruction, neutrophils can directly injure surrounding cardiomyocytes through the release of toxic product such as reactive oxygen species.36 Moreover, activation of the TLR‐4 pathway on circulating leucocytes, which is amplified by TREM‐1 signalling, results in decreased cardiomyocytes contractility.37
Reactive oxygen species release is a critical mediator of myocardial RI. Recruited leucocytes have the potential to produce large amounts of reactive oxygen species and therefore contribute to tissue damage within the first hours. 38 TREM‐1 inhibition reduces reactive oxygen species release in various inflammatory conditions and could thus limit RI through this mechanism.
Strengths
This work has several important strengths. First, we used adult minipigs. Although much more expensive than usual domestic pigs, these animals are physiologically close to adult humans and are well accepted by the scientific community for preclinical cardiovascular research. Second, we used a clinically relevant low‐invasive closed‐chest AMI model, and we tried to mimic what could be the use of this therapy if administered to humans (administration just before reperfusion). Third, we designed this study as a randomized and blinded one, therefore limiting experimental biases. Fourth, experienced intensive care physicians conducted resuscitation throughout the study period.
Limits
Nevertheless, several controversial points deserve discussion. Despite several consistent data sets obtained by different devices and techniques showing a beneficial effect of LR12 treatment, we could not find any significant change of ESPVR, whether between‐group or within‐group. This could be explained by an offset of the volume intercept V0 throughout the study period, a phenomenon previously reported in a similar model evaluating PV loop‐derived parameters during cardiogenic shock in pigs. 23 In the same way, we could not find any difference in dP/dtmax to EDV relationship, another load‐independent contractility index. This parameter has also been previously reported less reliable than PRSW.39 In our study, some issues could explain this result: an offset of volume intercept or a significant increase in heart rate in control animals. We collected many hemodynamic data, so this study is mostly descriptive. Further investigations are required to evaluate mechanistic insights raised in the previous paragraphs. Finally, the length of the study period was short in comparison with the whole process of leucocytes recruitment. Indeed, ischemic myocardium sequentially recruits various leucocytes subsets in a well‐compartmentalized reaction over the first week. However, whether a late therapeutic window to limit RI exists is not known and the current paradigm suggests that cardiomyocytes death mostly occurs quickly after blood flow restoration.2
Conclusion
The inhibition of the TREM‐1 pathway by the synthetic peptide LR12 was able to limit RI in a clinically relevant model of AMI. Although further studies are needed to decipher the precise mechanism of action of this treatment, this new promising strategy could be investigated in humans in the future.
Funding
This work was supported by the School of Surgery (Faculté de Médecine de Nancy, Université de Lorraine) and by INSERM UMR_S1116.
Conflict of interest
Marc Derive and Sébastien Gibot are co‐founders of INOTREM, a company developing TREM‐1 inhibitors
Lemarié, J. , Boufenzer, A. , Popovic, B. , Tran, N. , Groubatch, F. , Derive, M. , Labroca, P. , Barraud, D. , and Gibot, S. (2015), Pharmacological inhibition of the triggering receptor expressed on myeloid cells‐1 limits reperfusion injury in a porcine model of myocardial infarction. ESC Heart Failure, 2, 90–99. doi: 10.1002/ehf2.12029.
References
- 1. Puymirat E, Simon T, Steg PG, Schiele F, Guéret P, Blanchard D, Khalife K, Goldstein P, Cattan S, Vaur L, Cambou J‐P, Ferrières J, Danchin N. USIK USIC 2000 Investigators, FAST MI Investigators . Association of changes in clinical characteristics and management with improvement in survival among patients with ST‐elevation myocardial infarction. JAMA 2012; 308: 998–1006. [DOI] [PubMed] [Google Scholar]
- 2. Fröhlich GM, Meier P, White SK, Yellon DM, Hausenloy DJ. Myocardial reperfusion injury: looking beyond primary PCI. Eur Heart J 2013; 34: 1714–1722. [DOI] [PubMed] [Google Scholar]
- 3. Christia P, Frangogiannis NG. Targeting inflammatory pathways in myocardial infarction. Eur J Clin Invest 2013; 43: 986–995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Arslan F, de Kleijn DPV, Timmers L, Doevendans PA, Pasterkamp G. Bridging innate immunity and myocardial ischemia/reperfusion injury: the search for therapeutic targets. Curr Pharm Des 2008; 14: 1205–1216. [DOI] [PubMed] [Google Scholar]
- 5. Arslan F, de Kleijn DP, Pasterkamp G. Innate immune signaling in cardiac ischemia. Nat Rev Cardiol 2011; 8: 292–300. [DOI] [PubMed] [Google Scholar]
- 6. Feng Y, Chao W. Toll‐Like Receptors and Myocardial Inflammation. Int J Inflamm 2011; 2011: 170352, 21. doi: 10.4061/2011/170352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Bouchon A, Facchetti F, Weigand MA, Colonna M. TREM‐1 amplifies inflammation and is a crucial mediator of septic shock. Nature 2001; 410: 1103–1107. [DOI] [PubMed] [Google Scholar]
- 8. Gibot S, Kolopp‐Sarda M‐N, Béné M‐C, Bollaert P‐E, Lozniewski A, Mory F, Levy B, Faure GC. A soluble form of the triggering receptor expressed on myeloid cells‐1 modulates the inflammatory response in murine sepsis. J Exp Med 2004; 200: 1419–1426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Gibot S, Alauzet C, Massin F, Sennoune N, Faure GC, Béné M‐C, Lozniewski A, Bollaert P‐E, Lévy B. Modulation of the triggering receptor expressed on myeloid cells‐1 pathway during pneumonia in rats. J Infect Dis 2006; 194: 975–983. [DOI] [PubMed] [Google Scholar]
- 10. Derive M, Boufenzer A, Bouazza Y, Groubatch F, Alauzet C, Barraud D, Lozniewski A, Leroy P, Tran N, Gibot S. Effects of a TREM‐like transcript 1‐derived peptide during hypodynamic septic shock in pigs. Shock Augusta Ga 2013; 39: 176–182. [DOI] [PubMed] [Google Scholar]
- 11. Derive M, Bouazza Y, Sennoun N, Marchionni S, Quogley L, Washington V, Massin F, Max JP, Ford J, Alauzet C, Levy B, McVicar DW, Gibot S. Soluble TREM‐like transcript‐1 regulates leukocyte activation and controls microbial sepsis. J Immunol 2012; 188: 5585–5592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Kamei K, Yasuda T, Ueda T, Qiang F, Takeyama Y, Shiozaki H. Role of triggering receptor expressed on myeloid cells‐1 in experimental severe acute pancreatitis. J Hepato‐Biliary‐Pancreat Sci 2010; 17: 305–312. [DOI] [PubMed] [Google Scholar]
- 13. Gibot S, Massin F, Alauzet C, Derive M, Montemont C, Collin S, Fremont S, Levy B. Effects of the TREM 1 pathway modulation during hemorrhagic shock in rats. Shock Augusta Ga 2009; 32: 633–637. [DOI] [PubMed] [Google Scholar]
- 14. Schenk M, Bouchon A, Seibold F, Mueller C. TREM‐1–expressing intestinal macrophages crucially amplify chronic inflammation in experimental colitis and inflammatory bowel diseases. J Clin Invest 2007; 117: 3097–3106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Collins CE, La DT, Yang H‐T, Massin F, Gibot S, Faure G, Stohl W. Elevated synovial expression of triggering receptor expressed on myeloid cells 1 in patients with septic arthritis or rheumatoid arthritis. Ann Rheum Dis 2009; 68: 1768–1774. [DOI] [PubMed] [Google Scholar]
- 16. Bisson C, Massin F, Lefevre PA, Thilly N, Miller N, Gibot S. Increased gingival crevicular fluid levels of soluble triggering receptor expressed on myeloid cells (sTREM)‐1 in severe periodontitis. J Clin Periodontol 2012; 39: 1141–1148. [DOI] [PubMed] [Google Scholar]
- 17. Roberts R, DeMello V, Sobel BE. Deleterious effects of methylprednisolone in patients with myocardial infarction. Circulation 1976; 53: I204–I206. [PubMed] [Google Scholar]
- 18. Baran KW, Nguyen M, McKendall GR, Lambrew CT, Dykstra G, Palmeri ST, Gibbons RJ, Borzak S, Sobel BE, Gourlay SG, Rundle AC, Gibson CM, Barron HV. Limitation of myocardial infarction following thrombolysis in acute myocardial infarction (LIMIT AMI) study group. Double‐blind, randomized trial of an anti‐CD18 antibody in conjunction with recombinant tissue plasminogen activator for acute myocardial infarction: limitation of myocardial infarction following thrombolysis in acute myocardial infarction (LIMIT AMI) study. Circulation 2001; 104: 2778–2783. [DOI] [PubMed] [Google Scholar]
- 19. Faxon DP, Gibbons RJ, Chronos NAF, Gurbel PA, Sheehan F. HALT‐MI Investigators . The effect of blockade of the CD11/CD18 integrin receptor on infarct size in patients with acute myocardial infarction treated with direct angioplasty: the results of the HALT‐MI study. J Am Coll Cardiol 2002; 40: 1199–1204. [DOI] [PubMed] [Google Scholar]
- 20. Granger CB, Mahaffey KW, Weaver WD, Theroux P, Hochman JS, Filloon TG, Rollins S, Todaro TG, Nicolau JC, Ruzyllo W, Armstrong PW, COMMA Investigators . Pexelizumab, an anti‐C5 complement antibody, as adjunctive therapy to primary percutaneous coronary intervention in acute myocardial infarction: the COMplement inhibition in Myocardial infarction treated with Angioplasty (COMMA) trial. Circulation 2003; 108: 1184–1190. [DOI] [PubMed] [Google Scholar]
- 21. Mahaffey KW, Granger CB, Nicolau JC, Ruzyllo W, Weaver WD, Theroux P, Hochman JS, Filloon TG, Mojcik CF, Todaro TG, Armstrong PW. COMPLY Investigators . Effect of pexelizumab, an anti‐C5 complement antibody, as adjunctive therapy to fibrinolysis in acute myocardial infarction: the COMPlement inhibition in myocardial infarction treated with thromboLYtics (COMPLY) trial. Circulation 2003; 108: 1176–1183. [DOI] [PubMed] [Google Scholar]
- 22. APEX AMI Investigators , Armstrong PW, Granger CB, Adams PX, Hamm C, Holmes D, O'Neill WW, Todaro TG, Vahanian A, Van de Werf F. Pexelizumab for acute ST‐elevation myocardial infarction in patients undergoing primary percutaneous coronary intervention: a randomized controlled trial. JAMA 2007; 297: 43–51. [DOI] [PubMed] [Google Scholar]
- 23. How O‐J, Røsner A, Kildal AB, Stenberg TA, Gjessing PF, Hermansen SE, Myrmel T. Dobutamine‐norepinephrine, but not vasopressin, restores the ventriculoarterial matching in experimental cardiogenic shock. Transl Res J Lab Clin Med 2010; 156: 273–281. [DOI] [PubMed] [Google Scholar]
- 24. McCall FC, Telukuntla KS, Karantalis V, Suncion VY, Heldman AW, Mushtaq M, Williams AR, Hare JM. Myocardial infarction and intramyocardial injection models in swine. Nat Protoc 2012; 7: 1479–1496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Fincke R, Hochman JS, Lowe AM, Menon V, Slater JN, Webb JG, LeJemtel TH, Cotter G. SHOCK Investigators . Cardiac power is the strongest hemodynamic correlate of mortality in cardiogenic shock: a report from the SHOCK trial registry. J Am Coll Cardiol 2004; 44: 340–348. [DOI] [PubMed] [Google Scholar]
- 26. Lefering R, Neugebauer EA. Steroid controversy in sepsis and septic shock: a meta‐analysis. Crit Care Med 1995; 23: 1294–1303. [DOI] [PubMed] [Google Scholar]
- 27. Fisher CJ, Dhainaut JF, Opal SM, Pribble JP, Balk RA, Slotman GJ, Iberti TJ, Rackow EC, Shapiro MJ, Greenman RL. Recombinant human interleukin 1 receptor antagonist in the treatment of patients with sepsis syndrome. Results from a randomized, double‐blind, placebo‐controlled trial. Phase III rhIL‐1ra Sepsis Syndrome Study Group. JAMA 1994; 271: 1836–1843. [PubMed] [Google Scholar]
- 28. Abbate A, Van Tassell BW, Biondi‐Zoccai G, Kontos MC, Grizzard JD, Spillman DW, Oddi C, Roberts CS, Melchior RD, Mueller GH, Abouzaki NA, Rengel LR, Varma A, Gambill ML, Falcao RA, Voelkel NF, Dinarello CA, Vetrovec GW. Effects of interleukin‐1 blockade with anakinra on adverse cardiac remodeling and heart failure after acute myocardial infarction [from the Virginia Commonwealth University‐Anakinra Remodeling Trial (2) (VCU‐ART2) pilot study]. Am J Cardiol 2013; 111: 1394–1400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Dirksen MT, Laarman GJ, Simoons ML, Duncker DJGM. Reperfusion injury in humans: a review of clinical trials on reperfusion injury inhibitory strategies. Cardiovasc Res 2007; 74: 343–355. [DOI] [PubMed] [Google Scholar]
- 30. Webster NR, Galley HF. Immunomodulation in the critically ill. Br J Anaesth 2009; 103: 70–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Kempf T, Zarbock A, Vestweber D, Wollert KC. Anti‐inflammatory mechanisms and therapeutic opportunities in myocardial infarct healing. J Mol Med Berl Ger 2012; 90: 361–369. [DOI] [PubMed] [Google Scholar]
- 32. Derive M, Boufenzer A, Gibot S. Attenuation of responses to endotoxin by the triggering receptor expressed on myeloid cells‐1 inhibitor LR12 in nonhuman primate. Anesthesiology 2014; 120: 935–942. [DOI] [PubMed] [Google Scholar]
- 33. Xiao W, Mindrinos MN, Seok J, Cuschieri J, Cuenca AG, Gao H, Hayden DL, Hennessy L, Moore EE, Minei JP, Bankey PE, Johnson JL, Sperry J, Nathens AB, Billiar TR, West MA, Brownstein BH, Mason PH, Baker HV, Finnerty CC, Jeschke MG, López MC, Klein MB, Gamelli RL, Gibran NS, Arnoldo B, Xu W, Zhang Y, Calvano SE, McDonald‐Smith GP, Schoenfeld DA, Storey JD, Cobb JP, Warren HS, Moldawer LL, Herndon DN, Lowry SF, Maier RV, Davis RW, Tompkins RG. A genomic storm in critically injured humans. J Exp Med 2011; 208: 2581–2590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Nahrendorf M, Swirski FK, Aikawa E, Stangenberg L, Wurdinger T, Figueiredo J‐L, Libby P, Weissleder R, Pittet MJ. The healing myocardium sequentially mobilizes two monocyte subsets with divergent and complementary functions. J Exp Med 2007; 204: 3037–3047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Jordan JE, Zhao ZQ, Vinten‐Johansen J. The role of neutrophils in myocardial ischemia‐reperfusion injury. Cardiovasc Res 1999; 43: 860–878. [DOI] [PubMed] [Google Scholar]
- 36. Jaeschke H, Smith CW. Mechanisms of neutrophil‐induced parenchymal cell injury. J Leukoc Biol 1997; 61: 647–653. [DOI] [PubMed] [Google Scholar]
- 37. Tavener SA, Long EM, Robbins SM, McRae KM, Van Remmen H, Kubes P. Immune cell toll‐like receptor 4 is required for cardiac myocyte impairment during endotoxemia. Circ Res 2004; 95: 700–707. [DOI] [PubMed] [Google Scholar]
- 38. Braunersreuther V, Jaquet V. Reactive oxygen species in myocardial reperfusion injury: from physiopathology to therapeutic approaches. Curr Pharm Biotechnol 2012; 13: 97–114. [DOI] [PubMed] [Google Scholar]
- 39. Blaudszun G, Licker MJ, Morel DR. Preload‐adjusted left ventricular dP/dtmax: a sensitive, continuous, load‐independent contractility index. Exp Physiol 2013; 98: 1446–1456. [DOI] [PubMed] [Google Scholar]