

Abstract
It has been well established that mutations in K-Ras and N-Ras proto-oncogenes can convert them into active oncogenes. Current molecular cancer research has been focused on determining the key steps by which cellular genes become oncogenes and not on the underlying and fundamental chemical damage mechanism and susceptibility to damage. In this study, we investigate the damage hot spots present in the N-Ras and K-Ras genes upon exposure to UVC radiation. Detection of damage is accomplished by a simple, sensitive, mix-and-read assay using an EvaGreen probe in a 96-well microtiter plate. Our results show that, although there is high degree of sequential similarities among K-Ras and N-Ras genes, they show different degrees of UV damage in different portions of their genomes. Our experiments demonstrate that overall, the K-Ras genome is more prone to UVC damage than the N-Ras genome. We observe that the extent of damage increases with increasing number of TTs in a sequence, consistent with previous results that show that thymine cyclobutyl photodimers are the primary DNA damage photoproducts upon UVC irradiation. This understanding of the effect of UVC radiation on various codons of K-Ras and N-Ras genes will help to increase our understanding about hot spots of DNA damage and the chemical damage mechanism.
Introduction
Mutational activation of the Ras family of genes has been found to be one of the principal oncogenic events in cancer.1,2 The Ras family encodes small GTPase proteins involved in cellular signal transduction and consist of three proto-oncogenes, K-Ras, N-Ras, and H-Ras.1,3,4 Although there is a high degree of homogeneity among the different Ras gene sequences, preliminary studies have demonstrated that many different tumors are identified with different Ras genes.5 However, no correlation has been established between an activated Ras oncogene and the tumor type present. Apparently, mutation of Ras proto-oncogenes is not essential for tumorigenesis, but it can still be a contributing factor to human carcinogenesis.6
Much effort has been made to investigate how mutation leads to the activation of the Ras genes. Past research has shown that mutation at any of the codons 12, 13, and 61 in any of the three Ras genes is capable of activating their oncogenic functions.3,7,8 Interestingly, it has been found that mutation preferentially occurs at codon 12 of K-Ras rather than at codon 13 or 61 of K-Ras or at any N-Ras or H-Ras condon.7,9,10 Furthermore, on investigating the repair mechanism between codon 12 and the other condons, it was observed that there was no substantial difference in their repair rates. These findings raise a pivotal question: What factors determine the oncogenic properties of Ras proto-oncogenes? In this paper, we explore one aspect of this question, the relationship between DNA damage and mutational hot spots of Ras genes.
It is well known that UV radiation is one of the major causes of skin cancer.11 Animal model studies12,13 have inferred that UV irradiation can introduce point mutations in the Ras proto-oncogene. These point mutations lead to the production of Ras protein with diminished or no GTPase activity. Loss of GTPase activity shifts the equilibrium between active GTP-bound Ras protein and inactive GDP-bound Ras protein to produce a more active GTP. The active GTP-bound Ras protein can now stimulate a plethora of downstream processes causing unregulated signal transduction and facilitating the development of skin cancer.8,14−16 The major photoproducts induced in DNA by UVC light are the cyclobutyl pyrimidine photodimers (CPDs) and (6–4) pyrimidine-pyrimidinone photoproducts.13,17 In vitro UVC irradiation experimental studies have already shown that UV light forms a point mutation at codons 12 and 618 in the N-Ras gene. Another site-directed mutagenesis experiment performed by introducing CPDs at predetermined sites showed that the photodimer could induce a point mutation at the modified positions.18 However, it is equally important to determine if the Ras genes are activated by the production of CPD in the gene. Recent in vitro experiments have demonstrated that proto-oncogene expression in human epidermis increases as a result of UV radiation, leading to a high rate of cell proliferation and differentiation.19
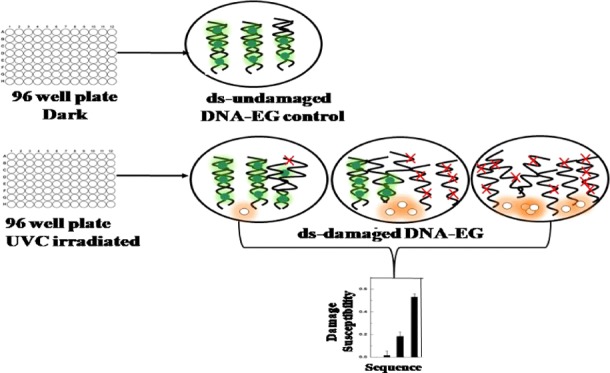
Current molecular cancer research has been focused on determining the key steps by which cellular genes become oncogenes, not on the underlying and fundamental chemical damage mechanisms and susceptibility to damage. To address this, there is a need to develop a new assay that can screen the entire genome of DNA in a high-throughput fashion with nucleotide resolution. Microarray technology is being extensively used for the study of gene expression and its changes in different cell states for a large population of genes simultaneously.20,21 However, the wider acceptance of the microarray is limited due to the use of time-consuming and potentially biased image processing software22 for sample analysis. A DNA hybridization assay combined with fluorescence detection by label-free probes serves to be a simple, fast, inexpensive and sensitive method to study DNA damage.23,24 Unlike the microarray, hybridization assays neither use laborious software for data analysis nor expensive fluorescently labeled DNA probes for damage quantification. In this work, we use a 96-well microplate platform coupled with a fluorescent intercalating dye, EvaGreen (EG),25,26 to develop a multiplexed solution-phase hybridization assay for investigating the mutational hot spots of damage present in Ras genes. Earlier studies have already proven that K-Ras is more susceptible to mutation when compared to N-Ras.3,15,27 Thus, in this study, we chose to investigate the effect of UVC radiation on the entire genomes of both K-Ras and N-Ras.
Results and Discussion
K-Ras and N-Ras Damage by UVC Radiation
The oligonucleotide sequences used in this study that make up the K-Ras and N-Ras genes are listed in Table 1. To enable better localization of damage and to attempt to correlate damage susceptibility with sequence, each Ras gene was split into 32 oligonucleotide sequences of 7 codons each, with the first and last codons in each sequence overlapping in the previous and following sequences, respectively. All oligonucleotides for both Ras genes gave a positive damage response to UVC radiation, i.e., all oligonucleotides showed a decrease in fluorescence from the EG dye upon increasing UVC irradiation. EG dye is an intercalating dye constructed of two monomeric units joined through a flexible linker. This dimeric dye is inactive in the absence of dsDNA and assumes a closed-looped conformation in a solution with two chromophores in close proximity to one another. In this conformation, the fluorescence is quenched. This looped conformation of EG opens upon binding and intercalation to dsDNA when present, separating the two chromophores. In this open, intercalated form, the EG exhibits maximum fluorescence. The equilibrium between the inactive and active form of EG in the presence of DNA is regulated by the DNA double helical structure. Upon damage of dsDNA, a change in its helical structure occurs due to the disruptions to the normal base paring, leading to a release of bound EG and a consequent reduction in EG fluorescence.25 Thus, we see a decrease in the fluorescence intensity with increasing damage (damage susceptibility). Damage susceptibility increases from 0 to 83% for K-Ras and 0–69% for N-Ras, depending on the nucleobases present in different sequences. The results of UVC-induced damage are listed in Table 2 for K-Ras and Table 3 for N-Ras oligonucleotide sequences.
Table 1. K-Ras and N-Ras Sequences Used in This Studya.
| K-Ras |
N-Ras |
||
|---|---|---|---|
| name | sequence | name | sequence |
| S1-7 | 5′-ATG ACT GAA TAT AAA CTT GTG-3′ | S1-7 | 5′-ATG ACT GAG TAC AAA CTG GTG-3′ |
| S7-13 | 5′-GTG GTA GTT GGA GCT GGT GGC-3′ | S7-13 | 5′-GTG GTG GTT GGA GCA GGT GGT-3′ |
| S13-19 | 5′-GGC GTA GGC AAG AGT GCC TTG-3′ | S13-19 | 5′-GGT GTT GGG AAA AGC GCA CTG-3′ |
| S19-25 | 5′-TTG ACG ATA CAG CTA ATT CAG-3′ | S19-25 | 5′-CTG ACA ATC CAG CTA ATC CAG-3′ |
| S25-31 | 5′-CAG AAT CAT TTT GTG GAC GAA-3′ | S25-31 | 5′-CAG AAC CAC TTT GTA GAT GAA-3′ |
| S31-37 | 5′-GAA TAT GAT CCA ACA ATA GAG-3′ | S31-37 | 5′-GAA TAT GAT CCC ACC ATA GAG-3′ |
| S37-43 | 5′-GAG GAT TCC TAC AGG AAG CAA-3′ | S37-43 | 5′-GAG GAT TCT TAC AGA AAA CAA-3′ |
| S43-49 | 5′-CAA GTA GTA ATT GAT GGA GAA-3′ | S43-49 | 5′-CAA GTG GTT ATA GAT GGT GAA-3′ |
| S49-55 | 5′-GAA ACC TGT CTC TTG GAT ATT-3′ | S49-55 | 5′-GAA ACC TGT TTG TTG GAC ATA-3′ |
| S55-61 | 5′-ATT CTC GAC ACA GCA GGT CAA-3′ | S55-61 | 5′-ATA CTG GAT ACA GCT GGA CAA-3′ |
| S61-67 | 5′-CAA GAG GAG TAC AGT GCA ATG-3′ | S61-67 | 5′-CAA GAA GAG TAC AGT GCC ATG-3′ |
| S67-73 | 5′-ATG AGG GAC CAG TAC ATG AGG-3′ | S67-73 | 5′-ATG AGA GAC CAA TAC ATG AGG-3′ |
| S73-79 | 5′-AGG ACT GGG GAG GGC TTT CTT-3′ | S73-79 | 5′-AGG ACA GGC GAA GGC TTC CTC-3′ |
| S79-85 | 5′-CTT TGT GTA TTT GCC ATA AAT-3′ | S79-85 | 5′-CTC TGT GTA TTT GCC ATC AAT-3′ |
| S85-91 | 5′-AAT AAT ACT AAA TCA TTT GAA-3′ | S85-91 | 5′-AAT AAT AGC AAG TCA TTT GCG-3′ |
| S91-97 | 5′-GAA GAT ATT CAC CAT TAT AGA-3′ | S91-97 | 5′-GCG GAT ATT AAC CTC TAC AGG-3′ |
| S97-103 | 5′-AGA GAA CAA ATT AAA AGA GTT-3′ | S97-103 | 5′-AGG GAG CAG ATT AAG CGA GTA-3′ |
| S103-109 | 5′-GTT AAG GAC TCT GAA GAT GTA-3′ | S103-109 | 5′-GTA AAA GAC TCG GAT GAT GTA-3′ |
| S109-115 | 5′-GTA CCT ATG GTC CTA GTA GGA-3′ | S109-115 | 5′-GTA CCT ATG GTG CTA GTG GGA-3′ |
| S115-121 | 5′-GGA AAT AAA TGT GAT TTG CCT-3′ | S115-121 | 5′-GGA AAC AAG TGT GAT TTG CCA-3′ |
| S121-127 | 5′-CCT TCT AGA ACA GTA GAC ACA-3′ | S121-127 | 5′-CCA ACA AGG ACA GTT GAT ACA-3′ |
| S127-133 | 5′-ACA AAA CAG GCT CAG GAC TTA3′ | S127-133 | 5′-ACA AAA CAA GCC CAC GAA CTG-3′ |
| S133-139 | 5′-TTA GCA AGA AGT TAT GGA ATT-3′ | S133-139 | 5′-CTG GCC AAG AGT TAC GGG ATT-3′ |
| S139-145 | 5′-ATT CCT TTT ATT GAA ACA TCA-3′ | S139-145 | 5′-ATT CCA TTC ATT GAA ACC TCA-3′ |
| S145-151 | 5′-TCA GCA AAG ACA AGA CAG GGT-3′ | S145-151 | 5′-TCA GCC AAG ACC AGA CAG GGT-3′ |
| S151-157 | 5′-GGT GTT GAT GAT GCC TTC TAT-3′ | S151-157 | 5′-GGT GTT GAA GAT GCT TTT TAC-3′ |
| S157-163 | 5′-TAT ACA TTA GTT CGA GAA ATT-3′ | S157-163 | 5′-TAC ACA CTG GTA AGA GAA ATA-3′ |
| S163-169 | 5′-ATT CGA AAA CAT AAA GAA AAG-3′ | S163-169 | 5′-ATA CGC CAG TAC CGA ATG AAA-3′ |
| S169-175 | 5′-AAG ATG AGC AAA GAT GGT AAA-3′ | S169-175 | 5′-AAA AAA CTC AAC AGC AGT GAT-3′ |
| S175-181 | 5′-AAA AAG AAG AAA AAG AAG TCA-3′ | S175-181 | 5′-GAT GAT GGG ACT CAG GGT TGT-3′ |
| S181-187 | 5′-TCA AAG ACA AAG TGT GTA ATT-3′ | S181-187 | 5′-TGT ATG GGA TTG CCA TGT GTG-3′ |
| S183-189 | 5′-ACA AAG TGT GTA ATT ATG TAA-3′ | S184-190 | 5′-TTG CCA TGT GTG GTG ATG TAA-3′ |
Oligonucleotide sequences for K-Ras and N-Ras genes are listed. The subscripts in the “Name” column denote the start and end codon numbers. Note that the first and last codons in each sequence overlap those on the previous and following strands, respectively. However, the last sequence for K-Ras has 5 and N-Ras has 4 overlapping codons from the previous strands. This is done to maintain a constant sequence length of 21 nucleobases. All the sequences listed above were annealed with their complementary sequences to form their respective dsDNA oligonucleotide.
Table 2. UVC-Induced Damage Susceptibility and Number of Bipyrimidine Sites of K-Ras Oligonucleotidesa.
| sequence number | damage susceptibility | TT | CC | CT |
|---|---|---|---|---|
| S175-181 | 0.83 ± 0.01 | 10 | 0 | 7 |
| S163-169 | 0.73 ± 0.02 | 9 | 0 | 4 |
| S97-103 | 0.80 ± 0.01 | 8 | 0 | 6 |
| S85-91 | 0.75 ± 0.01 | 7 | 0 | 3 |
| S139-145 | 0.72 ± 0.01 | 7 | 1 | 4 |
| S181-187 | 0.38 ± 0.02 | 7 | 0 | 4 |
| S133-139 | 0.62 ± 0.01 | 6 | 1 | 5 |
| S79-85 | 0.53 ± 0.02 | 6 | 1 | 1 |
| S115-121 | 0.48 ± 0.01 | 6 | 2 | 3 |
| S183-189 | 0.33 ± 0.02 | 6 | 0 | 1 |
| S157-163 | 0.46 ± 0.01 | 5 | 0 | 4 |
| S169-175 | 0.42 ± 0.02 | 5 | 1 | 4 |
| S25-31 | 0.43 ± 0.01 | 5 | 1 | 4 |
| S1-7 | 0.62 ± 0.01 | 4 | 0 | 4 |
| S127-133 | 0.40 ± 0.01 | 4 | 2 | 6 |
| S49-55 | 0.36 ± 0.02 | 4 | 1 | 7 |
| S43-49 | 0.20 ± 0.03 | 4 | 1 | 6 |
| S103-109 | 0.40 ± 0.01 | 3 | 1 | 8 |
| S91-97 | 0.32 ± 0.02 | 3 | 1 | 6 |
| S73-79 | 0.31 ± 0.05 | 3 | 6 | 7 |
| S37-43 | 0.24 ± 0.02 | 3 | 3 | 8 |
| S145-151 | 0.15 ± 0.02 | 3 | 2 | 7 |
| S31-37 | 0.12 ± 0.02 | 3 | 1 | 6 |
| S13-19 | 0.19 ± 0.03 | 2 | 3 | 5 |
| S151-157 | 0.16 ± 0.03 | 2 | 2 | 5 |
| S55-61 | 0.14 ± 0.02 | 2 | 1 | 7 |
| S61-67 | 0.14 ± 0.03 | 2 | 1 | 6 |
| S121-127 | 0.11 ± 0.01 | 2 | 1 | 7 |
| S19-25 | 0.00 ± 0.03 | 2 | 0 | 6 |
| S7-13 | 0.18 ± 0.02 | 1 | 4 | 4 |
| S67-73 | 0.01 ± 0.03 | 0 | 4 | 6 |
| S109-115 | 0.00 ± 0.01 | 0 | 4 | 6 |
This table is ordered by the number of TT bipyrimidine sites in the K-Ras oligonucleotides used in this study. The damage susceptibility is calculated as (1 – F), where F = Fi/Fc, Fi is the fluorescence intensity of EG-dsDNA upon damage and Fc is the fluorescence intensity of the undamaged DNA-EG control sample. The columns headed as “TT”, “CC”, and “CT” list the number of respective bipyrimidine sites in the oligonucleotides.
Table 3. UVC-Induced Damage Susceptibility and Number of Bipyrimidine Sites of N-Ras Oligonucleotidesa.
| sequence number | damage susceptibility | TT | CC | CT |
|---|---|---|---|---|
| S37-43 | 0.69 ± 0.02 | 6 | 1 | 4 |
| S151-157 | 0.53 ± 0.03 | 6 | 1 | 3 |
| S169-175 | 0.33 ± 0.02 | 6 | 0 | 5 |
| S139-145 | 0.69 ± 0.01 | 5 | 2 | 4 |
| S115-121 | 0.32 ± 0.01 | 5 | 2 | 3 |
| S85-91 | 0.26 ± 0.02 | 5 | 0 | 3 |
| S127-133 | 0.19 ± 0.00 | 5 | 2 | 3 |
| S49-55 | 0.18 ± 0.02 | 5 | 2 | 3 |
| S13-19 | 0.31 ± 0.01 | 4 | 3 | 1 |
| S25-31 | 0.23 ± 0.03 | 4 | 1 | 5 |
| S103-109 | 0.45 ± 0.02 | 3 | 1 | 5 |
| S133-139 | 0.30 ± 0.02 | 3 | 4 | 4 |
| S163-169 | 0.26 ± 0.01 | 3 | 2 | 3 |
| S157-163 | 0.23 ± 0.02 | 3 | 1 | 5 |
| S121-127 | 0.22 ± 0.01 | 3 | 2 | 4 |
| S43-49 | 0.21 ± 0.01 | 3 | 2 | 3 |
| S79-85 | 0.16 ± 0.03 | 3 | 1 | 2 |
| S97-103 | 0.41 ± 0.02 | 2 | 2 | 6 |
| S91-97 | 0.26 ± 0.00 | 2 | 3 | 4 |
| S19-25 | 0.19 ± 0.01 | 2 | 2 | 7 |
| S73-79 | 0.18 ± 0.03 | 2 | 4 | 8 |
| S184-190 | 0.13 ± 0.01 | 2 | 2 | 0 |
| S61-67 | 0.09 ± 0.01 | 2 | 1 | 5 |
| S1-7 | 0.12 ± 0.03 | 2 | 1 | 4 |
| S175-181 | 0.34 ± 0.02 | 1 | 4 | 6 |
| S55-61 | 0.10 ± 0.03 | 1 | 2 | 5 |
| S31-37 | 0.01 ± 0.02 | 1 | 3 | 5 |
| S7-13 | 0.00 ± 0.02 | 1 | 5 | 2 |
| S67-73 | 0.00 ± 0.03 | 1 | 2 | 8 |
| S145-151 | 0.00 ± 0.02 | 1 | 4 | 5 |
| S181-187 | 0.00 ± 0.02 | 1 | 3 | 1 |
| S109-115 | 0.00 ± 0.03 | 0 | 4 | 4 |
This table is ordered by the number of TT bipyrimidine sites in the N-Ras oligonucleotides used in this study. The damage susceptibility is calculated as (1 – F), where F = Fi/Fc, Fi is the fluorescence intensity of EG-dsDNA upon damage and Fc is the fluorescence intensity of the undamaged DNA-EG control sample. The columns headed as TT, CC, and CT list the number of respective bipyrimidine sites in the oligonucleotides.
To understand the relationship between UVC damage and the sequence composition, we plotted the correlation diagrams for all possible nucleobase pairs. Here it is worth mentioning that only the correlation depicted in Figure 1 shows a significant positive correlation of all the correlations between sequence and damage susceptibility attempted; all other plots are included in the Supporting Information (Figures S1 and S2) for completeness. Figure 1 indicates the plot with damage susceptibility as a function of number of TT sites for both K-Ras and N-Ras oligonucleotide sequences. It is already known that DNA damage by UVC radiation primarily occurs due to the formation of pyrimidine dimers such as cyclobutane pyrimidine dimers (CPDs), [6–4] pyrimidine pyrimidinone photoproducts ([6–4] PPs), and photohydrates.28,29 The CPDs and [6–4] PPs can potentially be formed between TT, CC, CT, and TC30 nucleotide pairs. From Figure 1, it can be clearly seen that the damage shows a strong positive linear correlation with increasing numbers of TT sites; no significant correlation is seen between damage and other bipyrimidine sites (see the Supporting Information). This result is expected for UVC-induced dsDNA damage, since the yield of T<>T photodimers is the highest (∼77%) compared to any other bipyrimidine lesion.31−33 It is interesting that the UVC-induced dsDNA damage gives a better R2 value for K-Ras (0.90) than for N-Ras (0.23) oligonucleotides. This could be because of the presence of a higher total number (134) and larger range (0–10) of TT sites in K-Ras when compared to the number (93) and range (0–6) of TT sites in N-Ras oligonucleotides. Another interesting result obtained by the study of damage susceptibility as a function of sequence composition is a small, negative correlation between damage and GG (R2 = 0.44) or GT (R2 = 0.35) bipyrimidine sites in K-Ras (Figures S1 and S2), suggesting that GG and GT bipyrimidine sites may provide a mild protective effect to UVC irradiation. No such effect is observed in the N-Ras sequences.
Figure 1.

UVC damage susceptibility (1 – F) as a function of the number of TT dinucleotide sites for K-Ras and N-Ras. The damage susceptibility on the y-axis is 1 – F, where F = Fi/Fc, Fi is the fluorescence intensity of EG-dsDNA upon UVC damage and Fc is the fluorescence intensity of the undamaged DNA-EG control sample. The EG dye fluorescence intensity at 530 nm is measured in the presence of 1 μM dsDNA (10 mM Tris, 1 mM ethylenediaminetetraacetic acid (EDTA), 10 mM NaCl, pH 7.4). The solid line through the filled square points is a linear fit for the K-Ras sequences with its R2 value indicated in the upper portion of the plot. The dashed line through the empty circle points is the linear fit for the N-Ras sequences with its R2 value indicated in the lower portion of the plot.
The correlation between damage susceptibility and increasing number of TT sites in the K-Ras oligonucleotides deserves further comment. Although a statistically significant correlation is observed, there are some deviations from this behavior when sequences with the same number of TT sites are compared. For example, comparing oligonucleotide sequences S85-91, S139-145, and S181-187 with 7 TT sites shows that sequence S181-187 exhibits a much lower damage susceptibility than the other two and much lower than expected from the correlation. In fact, the damage susceptibility for S181-187 is more closely consistent with the damage susceptibility for the closely related sequence S183-189, which shares the last 5 condons with S181-187. Furthermore, investigating sequences with 7 TT sites, we found that sequences that include a guanine adjacent to TT sites give lower damage susceptibility. In sequence-dependent studies on T<>T photodimer rates, Sarasin et al.31 and Kohler et al.33 have established that in tetrads of the type XTTY, the rate of dimer formation is higher for CTTC, TTTC, TTTA, TTTT, ATTC, CTTA, and ATTA and lower for GTTG, GTTC, GTTT, and GTTA. This model predicts, then, that all the tetrads with Gs adjacent to a TT site lower the rate of T<>T photodimer formation and that sequence S181-187 with 2 GTTT tetrads would have lower damage susceptibility, which is what is observed.
Having established that the damage of K-Ras sequences by UVC irradiation appears to be related to T<>T photodimer formation, modulated by adjacent guanines, we now similarly analyze the N-Ras gene sequences. Table 3 shows the UVC-induced damage susceptibilities for the N-Ras sequences arranged in the order of decreasing number of TT sites. Here, we also see a general decrease in damage susceptibility with decreasing number of TT sites. However, we see a few more exceptions for N-Ras than for K-Ras, which will be discussed in detail.
On comparing sequences S37-43, S151-157, and S169-175 with 6 TT sites, the damage susceptibility for sequence S169-175 is much lower than that of the other two sequences with 6 TT sites. On correlating the damage to the neighboring nucleotides, no obvious justification could be established for this deviation. In addition, we investigated the position of TT sites for all the above sequences. We found that if the TT sites are distributed throughout the dsDNA, the T<>T photodimer formation will effectively destabilize the dsDNA more when compared to sequences with TT sites clustered more closely together. This effect of the distribution of TT site positions on the change in damage susceptibility is confirmed for sequence S169-175, where all the 6 TT sites are located at the terminal and semiterminal positions24 between nucleobase number 1 and 11. Thus, hydrogen bonding between nucleobase number 12 to 21 of the dsDNA remains unperturbed, and this provides the required hydrophobic environment for EG to intercalate between the nucleobases of the second half of dsDNA, contributing to the higher fluorescence intensity. On the contrary, the 6 TT sites in oligonucleotide sequence S37-43 are randomly located at terminal, semiterminal, and central position of both the strands of the dsDNA. This distribution will destabilize the dsDNA structure throughout the entire 21 nucleobase sequence and give a higher damage susceptibility due to the presence of more nonfluorescent EG in solution. This result is in agreement with the earlier findings of the significant effect of mismatch position on DNA duplex stability.24
Furthermore, comparing the N-Ras sequences with 5 TT sites (S139-145, S115-121, S85-91, S127-133, and S49-55), only sequence S139-145 gives a high damage susceptibility (∼60%) and all the other sequences exhibit a low damage susceptibility (20–30%). In these oligonucleotide sequences, the presence of neighboring Gs to the TT sites does account for the discrepancy in damage susceptibility. A detailed analysis shows that oligonucleotide sequences S115-121 (0.32 ± 0.01) and S85-91 (0.26 ± 0.02) have GTTC tetrads and oligonucleotide sequence S127-133 (0.19 ± 0.00) contains GTTT, GTTC, and CTTG tetrads. Similarly, oligonucleotide sequence S49-55 gives a low damage susceptibility of 0.18 ± 0.02 and contains GTTT and GTTG tetrads.
The above predictive model is further verified by comparing the olignucleotide sequence S85-91 of both K-Ras and N-Ras. These two sequences differ only in the neighboring nucleobases at 1 TT site out of the 4 TT sites present (excluding terminal TT sites with the least effect on the apparent damage susceptibility24). The tetrad ATTT in K-Ras sequence is replaced by GTTC in N-Ras. Basically, replacing the adenosine of the K-Ras tetrad with guanosine in N-Ras tetrad drops the damage susceptibility of N-Ras by ∼3 times. Thus, the quenching effect of a neighboring G nucleobase can play a vital role in the T<>T photodimer formation rate and the subsequent damage susceptibility.34,35
These results for UVC-induced DNA damage in K-Ras and N-Ras sequences establishes an excellent agreement between damage and the effect of neighboring gaunine nucleobase on T<>T photodimer formation rate. This result is consistent with the sequence dependence of T<>T damage formation observed by Sarasin et al.31 and Kohler et al.33 In addition, the position of TT sites has been shown to account for the different damage susceptibilities of different K-Ras and N-Ras oligonucleotide sequences, as well as of the genes themselves.
A potential discrepancy in the trend of damage susceptibility is observed for sequences with 1–4 TT sites. With more than 4 TT sites, the formation of T<>T photodimers seems to dominate the damage susceptibility, as discussed above. However, greater deviations from the correlation are seen for oligonucleotide sequences with 4 or fewer TT sites. For example, oligonucleotide sequences S1-7, S49-55, and S127-133 for K-Ras and S97-103, S103-109, and S175-181 for N-Ras have 1–4 TT sites and show a damage susceptibility ranging between 30 and 65%. This could be due to the fact that UVC radiation damages other bipyrimidine sites, such as TC, CT, and CC, to some extent. The bipyrimidine dimer formation yields upon UVC radiation is reported to occur in the order TT > TC > CT > CC.33 Thus, in the absence or presence of a low number of T<>T photodimer, there is a probability of forming other bipyrimidine photoproducts. Sequence S1-7 of K-Ras with only 4 TT sites exhibits a damage susceptibility of 0.62 ± 0.01. This high damage susceptibility might be attributed to the presence of 4 TC/CT sites.
From Tables 2 and 3, it is clear that the damage susceptibility of most K-Ras oligonucleotide sequences is larger than their analogous N-Ras oligonucleotide sequence. Several biological studies performed on different tumor types have confirmed K-Ras genes to be more prone to mutation than N-Ras genes.27 The COSMIC (the Catalog of Somatic Mutations in Cancer) data set confirms that 22% of K-Ras genes are mutated in comparison to 8% of N-Ras genes in all tumors analyzed.3 Consistent with the biological results, our data give a larger damage susceptibility (∼83%) for K-Ras than for N-Ras (∼69%). This is mainly due to the presence of the higher number of TT sites in the K-Ras gene, making them more prone to DNA damage. However, the oligonucleotide sequences that correspond to the biologically active codons 12, 13, and 61 did not show any remarkable UVC damage susceptibility in either of the Ras genes. This result is not too surprising, since UVC irradiation may not be the primary insult to lead to oncogenic Ras genes. To understand the role of the biologically active codons of Ras genes in further detail, more research on the biological insults responsible for transforming the proto-oncogenes into oncogenes is required. However, we can use our method to further explore other types of DNA damage, such as oxidative and chemical damage, to gain further insight into the proto-oncogene formation mechanism.
Conclusions
The different mutagenic susceptibility of K-Ras and N-Ras genes has been shown here to correlate with molecular-level differences in UVC-induced damage susceptibility. Parallelizing a simple and rapid mix-and-read assay using EG dye as a fluorescence reporter and splitting the genes into shorter oligonucleotide sequences allow us to easily specify the cause of damage due to thymine–thymine cyclobutyl photodimers (CPDs), modulated by gaunosine residues on either side of the TT pairs within each codon.
Experimental Section
Materials
Oligonucleotide targets were obtained from Integrated DNA Technologies, Inc. (Coralville, IA). Hydrochloric acid was obtained from Anachemia (Montreal, QC, Canada), sodium chloride was obtained from ACP Chemical Inc. (Montreal, Quebec), Tris was obtained from ICN biomedicals (Aurora, OH), and ethylenediaminetetraacetic acid (EDTA) was obtained from BDH Inc. (Toronto, ON, Canada). The complete sequences of K-Ras and N-Ras genes are listed in Tables S1 and S2, respectively (see the Supporting Information). These genes are further subdivided into 32 overlapping sequences, with each sequence having a length of 21 nucleobases (Table 1).
DNA Damage Induced by UV Radiation
For DNA damage experiments, 147 μL of 1.36 μM nitrogen-purged dsDNA samples of each target sequence mentioned in Table 1 was placed in a separate well of a 96-well plate (Corning Special Optics, NY). UV light from UVC lamps emitting at 254 nm was chosen for irradiation. The UVC lamps in a Luzchem (Ottawa, ON, Canada) DEV photoreactor were turned on for 20 min prior to the experiment to ensure a stabilized light source. The photoreactor was purged continuously with nitrogen to remove oxygen and minimize ozone generation from the lamp. Finally, the 96-well microplate was placed in the photoreactor. Each well of the 96-well microplate was exposed to UVC light continuously for 2 h. Control samples were handled under identical condition, but were not exposed to UVC light and kept in the dark. After irradiation, the 96-well microplates were removed from the photoreactor and EG dye was added to each well. The final concentrations of the dsDNA and EG dye were a fixed ratio of 1:1.33.
Fluorescence Measurements
Room-temperature EvaGreen fluorescence intensities for UV-exposed dsDNA solutions were measured using a Safire fluorescence plate reader (Tecan, Mannendorf, Switzerland) after the addition of EG dye followed by incubation at 37 °C for 20 min in the dark. Fluorescence emission spectra were recorded using an excitation wavelength of 490 nm and an emission wavelength of 530 nm. The fluorescence spectra of dsDNA alone gave a minimum or zero background at the 530 nm emission wavelength.
Acknowledgments
The authors acknowledge the financial support for this project from the Canadian Natural Sciences and Engineering Research Council (NSERC) Discovery Grants and the Faculty of Science, University of Alberta.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsomega.8b03017.
The authors declare no competing financial interest.
Supplementary Material
References
- Jin Y.; Shima Y.; Furu M.; Aoyama T.; Nakamata T.; Nakayama T.; Nakamura T.; Toguchida J. Absence of oncogenic mutations of RAS family genes in soft tissue sarcomas of 100 Japanese patients. Anticancer Res. 2010, 30, 245–251. [PubMed] [Google Scholar]
- Ramdzan Z. M.; Pal R.; Vadnais C.; Vandal G.; Cadieux C.; Leduy L.; Davoudi S.; Yao L.; Karnezis A. K.; Paquet M.; Dankort D.; Nepveu A.; et al. RAS-Transformation in cancer cells require CUX1-dependent repair of oxidative DNA damage. PLoS Biol. 2014, 12, e1001807 10.1371/journal.pbio.1001807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prior I. A.; Lewis P. D.; Mattos C. A comprehensive survey of Ras mutations in cancer. Cancer Res. 2012, 72, 2457–2467. 10.1158/0008-5472.CAN-11-2612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macaluso M.; Russo G.; Cinti C.; Basan V.; Gebbia N.; Russo A. Ras family genes: An interesting link between cell cycle and cancer. J. Cell Physiol. 2002, 192, 125–130. 10.1002/jcp.10109. [DOI] [PubMed] [Google Scholar]
- To M. D.; Rosario R.; Westcott P. M.; Banta K. L.; Balmain A. Interactions between wild-type and mutant Ras genes in lung and skin carcinogenesis. Oncogene 2013, 32, 4028–4033. 10.1038/onc.2012.404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernández-Medarde A.; Santos E. Ras in cancer and developmental diseases. Genes Cancer 2011, 2, 344–358. 10.1177/1947601911411084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng Z.; Hu W.; Chen J. X.; Pao A.; Li H.; Rom W.; Hung M. C.; Tang M. S. Preferential DNA damage and poor repair determine ras gene mutational hotspot in human cancer. J. Natl. Cancer Inst. 2002, 94, 1527–1536. 10.1093/jnci/94.20.1527. [DOI] [PubMed] [Google Scholar]
- Pierceall W. E.; Kripke M. L.; Ananthaswamy H. N. N-ras mutation in ultraviolet radiation-induced murine skin cancers. Cancer Res. 1992, 52, 3946–3951. [PubMed] [Google Scholar]
- Graziano S. L.; Gamble G. P.; Newman N. B.; Abbott L. Z.; Rooney M.; Mookherjee S.; Lamb M. L.; Kohman L. J.; Poiesz B. J. Prognostic significance of K-ras codon 12 mutations in patients with resected stage I and II non-small-cell lung cancer. J. Clin. Oncol. 1999, 17, 668–675. 10.1200/JCO.1999.17.2.668. [DOI] [PubMed] [Google Scholar]
- Slebos R. J.; Hruban R. H.; Dalesio O.; Mooi W. J.; Offerhaus G. J.; Rodenhuis S. Relationship between K-ras oncogene activation and smoking in adenocarcinoma of the human lung. J. Natl. Cancer Inst. 1991, 83, 1024–1027. 10.1093/jnci/83.14.1024. [DOI] [PubMed] [Google Scholar]
- Glass A. G.; Hoover R. N. The emerging epidemic of melanoma and squamous cell skin cancer. JAMA, J. Am. Med. Assoc. 1989, 262, 2097–2100. 10.1001/jama.1989.03430150065027. [DOI] [PubMed] [Google Scholar]
- Su F.; Viros A.; Milagre C.; Trunzer K.; Bollag G.; Spleiss O.; Reis-Filho J. S.; Kong X.; Koya R. C.; Flaherty K. T.; et al. RAS mutations in cutaneous squamous-cell carcinomas in patients treated with BRAF inhibitors. N. Engl. J. Med. 2012, 366, 207–215. 10.1056/NEJMoa1105358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamiya H.; Murata N.; Murata T.; Iwai S.; Matsukage A.; Masutani C.; Hanaoka F.; Ohtsuka E. Cyclobutane thymine dimers in a ras proto-oncogene hot spot activate the gene by point mutation. Nucleic Acids Res. 1993, 21, 2355–2361. 10.1093/nar/21.10.2355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roddey P. K.; Garmyn M.; Park H.; Bhawan J.; Gilchrest B. A. Ultraviolet irradiation induces c-fos but not c-Ha-ras proto-oncogene expression in human epidermis. J. Invest. Dermatol. 1994, 102, 296–299. 10.1111/1523-1747.ep12371785. [DOI] [PubMed] [Google Scholar]
- Baines A. T.; Xu D.; Der C. J. Inhibition of Ras for cancer treatment: the search continues. Future Med. Chem. 2011, 3, 1787–1808. 10.4155/fmc.11.121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schubbert S.; Zenker M.; Rowe S. L.; Böll S.; Klein C.; Bollag G.; van der Burgt I.; Musante L.; Kalscheuer V.; Wehner L.; et al. Germline KRAS mutations cause Noonan syndrome. Nat. Genet. 2006, 38, 331–336. 10.1038/ng1748. [DOI] [PubMed] [Google Scholar]
- Climent T.; González-Ramírez I.; González-Luque R.; Merchán M.; Serrano-Andrés L. Cyclobutane Pyrimidine Photodimerization of DNA/RNA Nucleobases in the Triplet State. J. Phys. Chem. Lett. 2010, 1, 2072–2076. 10.1021/jz100601p. [DOI] [Google Scholar]
- Banerjee S. K.; Borden A.; Christensen R. B.; LeClerc J. E.; Lawrence C. W. SOS-dependent replication past a single trans-syn T-T cyclobutane dimer gives a different mutation spectrum and increased error rate compared with replication past this lesion in uninduced cells. J. Bacteriol. 1990, 172, 2105–2112. 10.1128/jb.172.4.2105-2112.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brenner M.; Coelho S. G.; Beer J. Z.; Miller S. A.; Wolber R.; Smuda C.; Hearing V. J. Long-lasting molecular changes in human skin after repetitive in situ UV irradiation. J. Invest. Dermatol. 2009, 129, 1002–1011. 10.1038/jid.2008.325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei H.; Kuan P. F.; Tian S.; Yang C.; Nie J.; Sengupta S.; Ruotti V.; Jonsdottir G. A.; Keles S.; Thomson J. A.; Stewart R. A study of the relationships between oligonucleotide properties and hybridization signal intensities from NimbleGen microarray datasets. Nucleic Acids Res. 2008, 36, 2926–2938. 10.1093/nar/gkn133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Powell J.; Bennett M.; Waters R.; Reed S. A novel global genome method to measure and map DNA damage: application for chemotherapy treatment stratification. Lancet 2014, 383, S83 10.1016/S0140-6736(14)60346-9. [DOI] [Google Scholar]
- Russo G.; Zegar C.; Giordano A. Advantages and limitations of microarray technology in human cancer. Oncogene 2003, 22, 6497–6507. 10.1038/sj.onc.1206865. [DOI] [PubMed] [Google Scholar]
- El-Yazbi A. F.; Loppnow G. R. Fluorescence of terbium ion-DNA complexes: a sensitive inexpensive probe for UV-induced DNA damage. Anal. Chim. Acta 2013, 786, 116–123. 10.1016/j.aca.2013.04.068. [DOI] [PubMed] [Google Scholar]
- El-Yazbi A. F.; Wong A.; Loppnow G. R. A luminescent probe of mismatched DNA hybridization: Location and number of mismatches. Anal. Chim. Acta 2017, 994, 92–99. 10.1016/j.aca.2017.09.036. [DOI] [PubMed] [Google Scholar]
- El-Yazbi A. F.; Loppnow G. R. Probing DNA damage induced by common antiviral agents using multiple analytical techniques. J. Pharm. Biomed. Anal. 2018, 157, 226–234. 10.1016/j.jpba.2018.05.019. [DOI] [PubMed] [Google Scholar]
- Shoute L. C. T.; Loppnow G. R. Characterization of the binding interactions between EvaGreen dye and dsDNA. Phys. Chem. Chem. Phys. 2018, 20, 4772–4780. 10.1039/C7CP06058K. [DOI] [PubMed] [Google Scholar]
- Johnson L.; Mercer K.; Greenbaum D.; Bronson R. T.; Crowley D.; Tuveson D. A.; Jacks T. Somatic activation of the K-ras oncogene causes early onset lung cancer in mice. Nature 2001, 410, 1111–1116. 10.1038/35074129. [DOI] [PubMed] [Google Scholar]
- Francesco L.; Horspool W.. CRC handbook of Organic Photochemistry and Photobiology, 2nd ed.; CRC Press, 2003; pp 140.1–140.8. [Google Scholar]
- Ravanat J. L.; Douki T.; Cadet J. Direct and indirect effects of UV radiation on DNA and its components. J. Photochem. Photobiol., B 2001, 63, 88–102. 10.1016/S1011-1344(01)00206-8. [DOI] [PubMed] [Google Scholar]
- Zavala A. G.; Morris R. T.; Wyrick J. J.; Smerdon M. J. High-resolution characterization of CPD hotspot formation in human. Nucleic Acid Res. 2014, 42, 893–905. 10.1093/nar/gkt912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bourre F.; Renault G.; Seawell P. C.; Sarasin A. Distribution of ultraviolet-induced lesions in Simian Virus 40 DNA. Biochimie 1985, 67, 293–299. 10.1016/S0300-9084(85)80071-7. [DOI] [PubMed] [Google Scholar]
- Douki T. Low ionic strength reduces cytosine photoreactivity in UVC-irradiated isolated DNA. Photochem. Photobiol. Sci. 2006, 5, 1045–1051. 10.1039/b604517k. [DOI] [PubMed] [Google Scholar]
- Law Y. K.; Forties R. A.; Liu X.; Poirier M. G.; Kohler B. Sequence-dependent thymine dimer formation and photoreversal rates in double-stranded DNA. Photochem. Photobiol. Sci. 2013, 12, 1431–1439. 10.1039/c3pp50078k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng K. W.; Chen Z.; Hao Y. H.; Tan Z. Molecular crowding creates an essential environment for the formation of stable G-quadruplexes in long double-stranded DNA. Nucleic Acids Res. 2010, 38, 327–338. 10.1093/nar/gkp898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawanishi S.; Hiraku Y.; Oikawa S. Mechanism of guanine-specific DNA damage by oxidative stress and its role in carcinogenesis and aging. Mutat. Res., Rev. Mutat. Res. 2001, 488, 65–76. 10.1016/S1383-5742(00)00059-4. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.