

Abstract
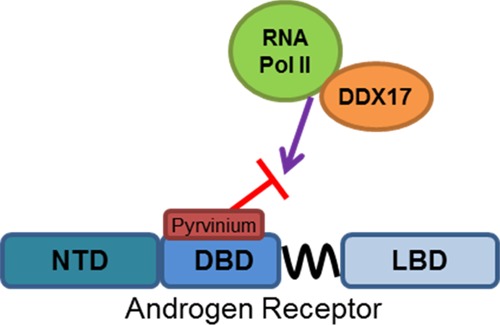
Pyrvinium was identified as the first small molecule inhibitor of the androgen receptor (AR) DNA-binding domain (DBD). It was also among the first small molecules shown to directly inhibit the activity of AR splice variants (ARVs), which has important clinical implications in the treatment of castration-resistant prostate cancer. Important questions about pyrvinium’s mechanism of action remain. Here, we demonstrate through mutational analysis that amino acids 609 and 612 are important for pyrvinium action. Nuclear magnetic resonance demonstrates a specific interaction between a soluble pyrvinium derivative and the AR DBD homodimer–DNA complex. Chromatin immunoprecipitation and electrophoretic mobility shift assay experiments demonstrate that, despite an interaction with this complex, pyrvinium does not alter the DNA-binding kinetics in either assay. AR immunoprecipitation followed by mass spectrometry was used to identify proteins whose interaction with AR is altered by pyrvinium. Several splicing factors, including DDX17, had reduced interactions with AR in the presence of pyrvinium. RNA sequencing of prostate cancer cells treated with pyrvinium demonstrated changes in splicing, as well as in several other pathways. However, pyrvinium did not alter the levels of ARVs in several prostate cancer cell lines. Taken together, our new data pinpoint the direct interaction between pyrvinium and AR DBD and shed light on the mechanism by which it inhibits AR transcriptional activity.
Introduction
Despite the approval of several new agents to treat metastatic prostate cancer following the development of castration resistance, the disease remains incurable, and prostate cancer is still the second leading cause of cancer death in men in the United States.1 It is now well-established that sustained androgen receptor (AR) activity is a key mechanism driving resistance in castration-resistant prostate cancer, despite the castrate levels of serum androgens.2 To address this resistance, several novel compounds have been developed that target the AR signaling pathway, including the FDA-approved drugs abiraterone3 and enzalutamide,4 as well as others in clinical development, including galeterone5 and ARN-509.6 Despite promising responses to these agents in many men, none appear to be curative, and both de novo and acquired resistance to these drugs are widespread. Although there is evidence of an increase of truly AR-independent cancers that arise following multiple lines of hormonal therapy,6 there is also strong evidence that a significant fraction of prostate tumors treated with next-generation androgen/AR-directed therapies continue to demonstrate a molecular signature consistent with continued AR signaling.7 Furthermore, the majority of men who progress on abiraterone and enzalutamide have rising prostate-specific antigen (PSA) levels, strongly suggesting that these tumors remain AR-driven.4,8
Several mechanisms have been proposed to account for continued AR signaling in the setting of advanced AR targeting. Point mutations in the AR ligand-binding domain (LBD) have been identified that confer resistance to abiraterone9 and enzalutamide.10 Likewise, the expression of AR splice variants (ARVs) has been documented to mediate resistance to abiraterone and enzalutamide.11 ARVs are truncated AR isoforms that lack LBD but retain the N-terminal domain (NTD) and DNA-binding domain (DBD) and are thus constitutively active even in the absence of ligands. Many ARV species have been found in clinical samples, and the presence of ARVs, ARV-7 in particular, has been correlated with a poor response to abiraterone and enzalutamide in several clinical studies (for review, see ref (11)). Although not as well-studied in a clinical setting, several signaling pathways have been shown to activate AR signaling in the absence of ligands in prostate cancer models, including HER2, IL-6, and others (for review, see ref (12)). The majority of these pathways are proposed to activate AR through its NTD, either by direct interactions or by post-translational modifications. It has also been proposed that in some cancers, the glucocorticoid receptor (GR) can replace AR and drive the expression of AR target genes.13 GR and AR have highly homologous DBDs and have very similar preferences for DNA-binding sites; so, it is very plausible that GR could bind to and activate AR target genes. These mechanisms are not necessarily mutually exclusive, and each could play a role in different subsets of cancers to contribute to the “AR-active” molecular signature observed in many cancers resistant to next-generation hormonal therapies. Regardless of the mechanism at play, it is clear that the continued expression of AR target genes is driving much of the resistance, and new therapies are necessary to treat these cancers.
We identified pyrvinium pamoate (PP) in a screen for noncompetitive AR inhibitors14 and subsequently found it to be the first bona fide AR inhibitor that functions via the AR DBD.15 We have previously demonstrated that (1) pyrvinium is the active component of the compound, (2) it functions synergistically with competitive antagonists, (3) it does not degrade AR, (4) it does not prevent nuclear translocation, (5) it does not prevent AR DNA binding, but (6) it does prevent RNA pol II recruitment to the transcription start sites.16 We have also demonstrated that PP interacts with AR and prevents its proteolysis.15 The AR mutants lacking the NTD or LBD are sensitive to PP, but those lacking the DBD are insensitive to PP, strongly suggesting that PP functions via the DBD. This is important as PP is able to inhibit the constitutive activity of clinically relevant ARVs.15 Interestingly, AR inhibition is cell type and tissue selective, and PP can inhibit the activity of GR and several other nuclear receptors with varying potency in prostate cancer cells.15 In this study, we confirm a biophysical interaction between a soluble pyrvinium derivative and the AR DBD–DNA complex using nuclear magnetic resonance (NMR) and pinpoint the AR DBD residues that mediate the interaction with pyrvinium by mutational analysis. We further explore the mechanism of PP inhibition of AR activity using unbiased proteomic and transcriptomic approaches and find that the reduced interaction with splicing proteins may contribute to PP’s activity.
Results and Discussion
Pyrvinium Binds the AR DBD–DNA Complex
Our previous work demonstrated that pyrvinium functioned to inhibit AR activity via the DBD, and our computer modeling predicted interactions with amino acids K609, N610, and P612.15 We therefore created alanine mutations of these three residues and tested the ability of pyrvinium to inhibit the transcriptional activity of these AR mutants in a luciferase reporter assay (Figure 1A). PP inhibited the activity of the N610 AR mutant nearly as potently as it inhibited the wild-type AR, but the other two mutants were resistant to PP inhibition, suggesting that K609 and P612 are critical contacts for pyrvinium action. We next sought biophysical evidence of pyrvinium interaction with the AR DBD–DNA complex using both crystallography and NMR approaches. As both techniques are more feasible with compounds that are soluble in aqueous solutions and pyrvinium is not, we endeavored on a small medicinal chemistry undertaking (to be described in a later publication) to develop a soluble pyrvinium derivative. We arrived at one such compound, P24, which had nearly an equivalent potency to pyrvinium (Figure S1A) but was readily soluble in aqueous solutions. Using a standard crystallography screen, the reddish P24 compound was found to be present in the crystals of purified AR DBD in complex with the ADR3 DNA element,17 as evidenced by the reddish color of the treated crystals (Figure S1B); however, repeated attempts to obtain interpretable spectra from various crystal preparations, even those without P24, failed. NMR, however, provided strong evidence of a physical interaction between P24 and the AR DBD–ADR3 DNA complex. When AR DBD was added to DNA duplex, the heteronuclear single quantum coherence (HSQC) spectrum of AR DBD changed dramatically accompanied with line broadening (Figure 1B,C). This confirmed the formation of a complex between AR DBD and the DNA duplex. Some cross-peaks of free AR DBD disappeared completely, which indicated that no free AR DBD was detectable with the existence of 30% excess of DNA. When P24 was added to the AR DBD and DNA complex, chemical shifts of most AR DBD peaks were not affected, except that more than 20 peaks disappeared (Figure 1D). These changes are shown more clearly in Figure 1E, which is the zoomed-in overlay of Figure 1C,D. This suggested that some residues of AR DBD interact with P24 when AR DBD is bound to DNA. The interaction between P24 and AR DBD bound to DNA did not break the complex between the protein and DNA duplex as the HSQC spectra in Figure 1C,D are very similar but different from the HSQC spectrum of free AR DBD, as shown in Figure 1B. When P24 was added to the AR DBD protein without DNA, no chemical shift was observed, even up to a 4:1 molar ratio of P24:AR DBD (Figure S1C). This result suggested that P24 does not interact with AR DBD in the absence of DNA. On the basis of the dynamics of the band shifts, the affinity constant of P24 of the DBD–DNA complex is predicted to be 30–300 nM, which is in line with the IC50 of P24 in the luciferase reporter assay (Figure S1A). The HSQC spectra suggest that P24 neither blocks the formation of the protein–DNA complex, nor does it break or reverse the formed complex. To further investigate this, we added P24 to the protein and DNA separately before adding the protein to DNA to form the protein–DNA complex. The HSQC spectrum acquired on the protein–DNA complex prepared this way is identical to that with the addition of P24 after the formation of the protein–DNA complex. This is in line with our computer modeling showing that the pyrvinium pharmacophore interacts with the DBD–DNA complex.15
Figure 1.

Interaction with the DBD. (A) Alanine mutations of AR at K609, N610, and P612 were transfected into PC3 prostate cancer cells, along with PSA-luciferase and SV40-renilla luciferase control reporter plasmids. Following overnight treatment with the indicated drugs, the luciferase activity was quantified in quadruplicate samples. PP inhibits the activity of wild type and N610A AR but not K609A or P612A AR. (B–E) 1H–15N HSQC spectra of 15N-labeled AR DBD and its complex with the DNA duplex in the absence and presence of P24. (B) HSQC spectrum of 15N-labeled AR DBD. (C) HSQC spectrum of 15N-labeled AR DBD in complex with DNA. (D) HSQC of the 15N-labeled AR DBD and DNA complex in presence of P24. (E) Zoomed-in region of overlay from the spectra of C (in red) and D (in blue) to show the difference caused by the addition of P24 to the protein–DNA complex.
Effect of Pyrvinium on DNA Binding
Because pyrvinium interacts with the AR DBD–DNA complex, we investigated if pyrvinium alters the DNA-binding parameters using chromatin immunoprecipitation (ChIP) and electrophoretic mobility shift assay (EMSA) approaches. We had previously observed that PP did not prevent the dihydrotestosterone (DHT)-induced binding of AR to a panel of AR-binding sites (ARBSs) after 4 h of treatment.16 As computer modeling suggests that PP interacts with both the protein and DNA, we hypothesized that PP might “lock” the protein on the DNA, altering the on/off rate which could lead to a reduced transcriptional activity. Therefore, we treated LNCaP cells with DHT, PP, and/or enzalutamide for 4 h. To perform a washout experiment, we also treated cells as above but removed the media containing drugs and replaced with drug-free media for 90 min prior to the ChIP assay. In a preliminary experiment, we found that removing DHT causes a decrease of AR binding at most sites between 0.5 and 2 h. We therefore examined AR binding at two previously identified ARBSs18 after a 1.5 h washout. We confirmed our previous finding that PP does not reduce AR binding to DNA as detected by ChIP16 and further found that PP treatment does not prevent the loss of AR binding upon withdrawal of DHT (Figure 2A). To more directly quantify the effect on the affinity of AR for DNA, we performed EMSAs using the soluble derivative P24. We used a minimal fragment of AR comprising the DBD (residues 557–647) and a 23 bp DNA probe containing a high affinity site (GTACGGAACAAAATGTACTGTAC). We then incubated twofold dilutions of AR DBD (starting at 5 μM) with DNA (at 5 nM) in the absence and presence of two P24 concentrations (300 nM and 100 μM). Under these conditions, we found that the affinity of AR DBD for the site was Kd = 29.6 and that the addition of P24 did not significantly lower the affinity (Kd = 34.7 and Kd = 35.3). Thus, we conclude that the addition of P24 does not have a significant effect on the affinity of AR DBD for specific DNA binding.
Figure 2.

Effect of pyrvinium on DNA binding. (A) LNCaP cells were treated as indicated for 4 h with or without a 1.5 h drug washout, at which point cells were fixed and AR ChIP was performed. DHT causes enrichment of AR at two previously identified ARBSs, which are not blocked by PP. Drug washout reduces AR occupancy at these binding sites and PP does not retain AR at the ARBSs. (B,C) EMSAs were used to measure the affinity of AR DBD for a specific AR-binding sequence in the presence of the control vehicle BCD or P24. The shifted band represents an AR DBD dimer bound to DNA. Although the affinity of AR DBD for DNA is slightly reduced in the presence of two different concentrations of P24, the effect is not significant (p = 0.24 or 0.16).
IP–Mass Spectrometry To Identify AR-Binding Proteins Affected by Pyrvinium
Although pyrvinium interacts with AR DBD, it does not alter the AR interaction with DNA, leading to the question as to how it prevents the transcriptional activity of AR. We previously demonstrated that pyrvinium reduces the interaction between RNA pol II and AR by western blot and that it reduces the amount of RNA pol II at the KLK3 transcription start site, as determined by ChIP.16 To identify the additional factors that could contribute to the loss of AR transcriptional activity, LNCaP cells were treated with DHT ± PP. The cells were lysed, AR was immunoprecipitated, and the proteins were resolved by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE). Sections from a Coomassie-stained gel were isolated and prepared for mass spectrometry (MS) analysis (Figure 3A). Western blotting demonstrated the specificity of the AR pull-down and confirmed that PP reduced the AR interaction with RNA pol II (Figure 3A). A semiquantitative analysis of protein levels was accomplished by comparison to a known amount of standard spiked into each sample. Among the proteins that were identified in the cells treated with DHT only but not in the DHT + PP-treated cells were two DEAD box proteins, DDX5 and DDX17, also known as p68 and p72. These proteins are RNA helicases known to be involved in splicing19−21 and have been identified as coregulators for AR and other nuclear receptors.22−25 We first confirmed the altered association between AR and these proteins by IP followed by western blot (Figure 3B). We found that the presence of PP reduced the interaction between DDX17 and AR in a co-IP more so than it did in the interaction between DDX5 and AR. We tested whether the overexpression of DDX17 could rescue the AR inhibition caused by PP treatment. Indeed, when DDX17 was overexpressed by ∼twofold [as measured by reverse transcription (RT)–quantitative polymerase chain reaction (qPCR)], we found that the expression of the androgen-responsive gene FKBP5 was not as potently inhibited by PP in LNCaP or LAPC4 cells (Figure 3C,D). This suggests that DDX17, at least in part, mediates the response to PP in these two prostate cancer cell lines.
Figure 3.

IP–MS to identify AR-binding proteins affected by pyrvinium. (A) LNCaP cells were treated with 1 nM DHT ± 100 nM PP (indicated by a “–“ or “+” adjacent to “PP”. Cells were lysed, AR was immunoprecipitated and proteins resolved by SDS-PAGE. Sections from a Coomassie-stained gel were isolated and prepared for MS analysis. A western blot was also performed to demonstrate the specificity for AR pull-down and PP activity, as it is known to block the interaction with RNA pol II. The table indicates the arbitrary score from each lane for the detection of AR and DDX proteins, with approximate mass. (B) To confirm the loss of DDX protein binding, LNCaP cells were treated with vehicle, DHT, or DHT + PP for 24 h, at which point the cells were lysed and AR was immunoprecipitated. Western blot for AR and DDX17 demonstrates a loss of coprecipitation of DDX17 with AR in the presence of PP. (C) LNCaP or (D) LAPC4 cells were transfected with a DDX17 expression vector or control vector. The following day, the indicated drugs were added, and 24 h later, RNA was harvested. qPCR demonstrated a decrease in the efficacy of PP to inhibit the transcription of the AR target gene FKBP5 upon DDX17 overexpression. * significantly different compared to DHT alone (p < 0.05).
Transcriptomic Analysis of Pyrvinium Treatment
We performed RNA-seq on LAPC4 cells treated with vehicle, DHT, or DHT + PP. We found that biological duplicate samples clustered together by principal component analysis (Figure 4A) and by hierarchical clustering (Figure 4B), as expected. Several hundred genes were found to be significantly differentially expressed when comparing any two groups. Gene ontology analysis using the IPA upstream regulator module demonstrated that DHT treatment increased androgen/AR signaling, as evidenced by the “activated” signatures for AR, DHT, and the AR agonist metribolone (Figure 4C). The addition of PP caused decreases not only in androgen/AR signaling (“inhibited” DHT signature), but also in estrogen and glucocorticoid signaling (Figure 4D). As we have shown previously, PP has some activity against other hormone nuclear receptors in prostate cancer cells;16 so, this is expected as well. However, PP also appears to inhibit the p53 and HER2 signaling signatures while increasing the signaling pathways mediated by EIF2AK3 and ATF4, as well as the signatures of tosedostat and tunicamycin treatment. Pathway analysis suggested that this upregulation was associated with increased ER stress, response to unfolded protein, and apoptosis. These additional points of regulation might contribute to the profound antiprostate cancer effect of PP.
Figure 4.

Transcriptomic analysis of pyrvinium treatment. LAPC4 cells were treated overnight with vehicle, 0.3 nM DHT, or DHT +100 nM PP. RNA was harvested and sequenced. Principal component analysis (A) demonstrates clustering of biological duplicates, as does hierarchical clustering (B). Gene ontology demonstrates appropriate response to DHT (C). (D) PP treatment caused decreased nuclear hormone receptor signaling as well as P53 and ERBB2 signaling; it also caused activation of pathways unrelated to nuclear hormone signaling.
Because several of the proteins identified in the IP–MS experiment were known to be splicing factors, we interrogated the RNA-seq data to determine if there were changes in the splicing patterns with PP treatment. Although we found few if any significant differences with PP treatment in terms of mutually exclusive exon splice forms, alternative 5′ or 3′ splice site usage, or retained introns, we did observe an increase in the transcripts with differentially skipped exons with PP treatment. There were 31 unique transcripts that lacked an exon in the PP-treated cells compared to the cells treated with DHT alone, and there were 37 unique transcripts that lacked an exon in the DHT-treated cells compared to the PP-treated cells. Gene ontology suggested that the majority of these genes were involved in alternative splicing or RNA processing (Figure 5A). Several arginine-/serine-rich splicing factors (SRSFs), including SRSFs 3, 6, and 7, were on this list. These factors are known to contain a “poison cassette” exon that, when included in the transcript, causes a premature termination codon (PTC) to be expressed which targets the transcript for nonsense-mediated mRNA decay.26 Using the approach of Lareau, et al.26 we used qPCR to quantify the changes in the inclusion of the poison cassettes in SRSFs 3 and 7 (Figure 5B,C). We confirmed the RNA-seq data which demonstrated that PP caused a decrease in the expression of both SRSF3 and SRSF7 total transcript levels (constitutive transcription) and that it caused a decreased inclusion of the PTC exon. Interestingly, DHT treatment itself decreased the total and PTC-containing transcript levels, which further decreased in a dose-dependent fashion by PP but was not blocked by enzalutamide treatment. The SRSF proteins have been shown to control intron inclusion in the transcripts; therefore, we queried our RNA-seq data to determine if PP was causing intron inclusion in the transcripts. However, we found no evidence of this in the data. Although we have shown that PP causes there to be fewer SRSF transcripts containing a poison cassette, the functional significance of this finding is unknown.
Figure 5.

Effect of pyrvinium on alternative splicing. (A) Gene ontology analysis of transcripts with differential exon inclusion with PP treatment demonstrated that most are involved in alternative splicing and RNA processing. qRT-PCR analysis of (B) SRSF3 or (C) SRSF7 transcript levels. DHT treatment reduces the total levels of mature transcripts (constitutive) as well as the levels of PTC-containing transcripts, which are further reduced in a dose-dependent fashion by PP treatment. (D) qRT-PCR analysis of AR exons 1 and 7 (with the ratio of exon1/exon7 shown in the inset) and ARV-7. * significantly different compared to DHT treatment with (p < 0.05).
Finally, we examined the expression of ARV-7 in LAPC4 cells. We saw a low-level expression of ARV-7 compared to full-length AR but no change with DHT or PP treatment (Figure 5D). As ARVs other than ARV-7 may be present, we also quantified the expression of AR exon 1 and AR exon 7, with the idea that an increased ARe1:ARe7 ratio would indicate an increased ARV expression. We found that DHT treatment decreased the levels of both exon 7 and exon 1, with the decrease in exon 1 being more pronounced. The addition of PP to DHT further decreased the expression of both exons. Furthermore, we found that DHT decreased the ARe1/ARe7 ratio and that the addition of PP did not change the DHT-associated decrease of the ARe1/ARe7 ratio (Figure 5D), suggesting PP does not alter the levels of ARVs in these prostate cancer cells.
Pyrvinium was previously identified as the first AR DBD inhibitor. Such an inhibitor could contribute significantly to the treatment of prostate cancers driven by ARVs and other forms of nonligand AR activation. Another series of compounds proposed to act through AR DBD was recently reported.27 These compounds appear to interact with the amino acids Q592 and Y594 on a slightly different surface of AR DBD than that of pyrvinium. The compounds are not as potent as PP but do appear to have increased selectivity for AR versus other nuclear receptors. This may or may not be advantageous, as limiting off-target effects might reduce the clinical adverse effects, but targeting multiple nuclear receptors could prevent the functional replacement of AR as the driver of cancer cell growth. An AR NTD inhibitor is also in development,28 and although there is some question about whether or not it acts directly on AR NTD,29 it is able to inhibit the growth of prostate cancers in several different models. Although an AR NTD inhibitor should inhibit prostate cancers driven by ARVs and other forms of nonligand AR activation as would DBD inhibitors, there is a legitimate concern that resistance could more readily arise in patients treated with NTD inhibitors. The NTD, especially the AF-1 region to which the EPI series of compounds is purported to bind, is a hotspot for mutation, even in the absence of drug selection.30 Conversely, very few mutations have been found in the highly conserved DBD, which is absolutely essential for canonical AR transcriptional activity. In fact, attempts to derive PP-resistant cell lines over the last several years have been mostly fruitless. We have failed to develop resistant LNCaP and LAPC4 cell lines, and the only 22Rv1 cell line we developed that was resistant to PP appears to grow in the presence of PP simply by exporting the drug, as the fluorescent PP is observed at much lower levels in the resistant cells (Figure S2). In contrast, it is remarkably easy to develop LNCaP and LAPC4 cell lines that are resistant to bicalutamide and enzalutamide, as we and others have done.10,31 This might suggest that the development of resistance to PP or other AR DBD inhibitors would be less likely in humans as well.
In this manuscript, we confirm a biophysical interaction between a soluble pyrvinium derivative, P24, and the AR DBD–DNA complex using NMR. Although the strong peaks in the central region of the proton dimension spectrum may indicate partial protein denaturization, most of the changes in the spectrum associated with P24 do not occur in this central region; so, the results still support P24 binding. Although crystallographic analysis failed to provide detailed spectra, the color of the crystal strongly suggests pyrvinium derivative binding as well. The synthesis and subsequent characterization of P24 will be described elsewhere as it is being developed as a clinical lead. We also identified important AR DBD residues that mediate PP sensitivity, confirming our previous computational modeling.15 As we had previously determined that PP did not prevent AR interaction with DNA, we hypothesized that it might inhibit the transcriptional activity by altering the normal DNA-binding kinetics, which has been shown to be important for AR target gene transcription.32 Although EMSA and ChIP washout experiments were carried out with two slightly different molecules, making direct comparisons difficult, neither assay detected any differences in AR binding to DNA, and thus the results suggest that PP and P24 do not disrupt the normal DNA-binding kinetics. Other techniques such as fluorescence recovery after photobleaching will be necessary to confirm this. Although different binding sites/DNA molecules were used for these and NMR studies, making direct comparisons among the techniques difficult, the combined results demonstrating the inability of P24 to disrupt AR DNA binding further suggest that pyrvinium and its derivatives do not affect AR DNA binding. Instead, we propose that pyrvinium and its derivatives prevent AR transcriptional activity by altering the conformation of AR (as shown in our fluorescent resonance energy transfer assay14) in such a way that it prevents the association of necessary cofactors. We previously demonstrated that RNA pol II has reduced interaction with AR and was absent at the KLK3 transcription start site,14 and here, using a proteomics approach, we identified other important factors whose interactions with AR are diminished in the presence of PP. PP appears to affect several proteins involved in the splicing process, including DDX17, which, when overexpressed, reduced the potency of PP. Although the transcriptomic approach did not detect widespread changes in splicing, including no difference in the expression of ARVs, it did identify changes in several splicing proteins, specifically the arginine-/serine-rich splicing factors. Although exactly how the inhibition of the interaction between AR and the splicing factors relates to these changes in splicing remains to be determined, it is likely that the altered association of AR and the splicing factors contributes to PP’s activity. Although pyrvinium has been shown to affect mitochondrial activity and have nonspecific toxicity,27 these effects occur at concentrations well above those that affect the AR activity and that were used in our studies. In summary, the mechanism of action of pyrvinium has been further defined, and the derivatives of pyrvinium have real promise in the treatment of prostate cancers resistant to advanced hormonal therapies.
Methods
Cells, Culture Conditions, and Reagents
LNCaP and PC3 cells were purchased from ATCC, whereas LAPC4 cells were a gift from Charles Sawyers. Cells lines have undergone cell line authentication by ATCC in the last 2 years. The LNCaP and LAPC4 cells were maintained in phenol red-free RPMI 1640 supplemented with 10% FBS and antibiotics, whereas the PC3 cells were maintained in Dulbecco’s modified Eagle’s medium with 10% fetal bovine serum (FBS) and antibiotics. The cells were transferred to charcoal-stripped (C/S) media prior to luciferase and qPCR assays. DHT was purchased from steraloids and PP from Sigma. The synthesis of the soluble pyrvinium derivative P24 will be described elsewhere.
Plasmids, Transfections, and Luciferase Reporter Assays
Alanine mutations of the AR DBD residues were created by site-directed mutagenesis. An AR expression plasmid was amplified with mutant primers using KAPA high-fidelity polymerase (Kapa Biosystems) using the manufacturer’s protocol. The parent plasmid was digested with DpnI (Agilent) for 1 h. The mutant plasmids were transformed into NEB 5-alpha competent cells (NEB). The mutations were screened by Sanger sequencing. The DDX17 expression plasmid was obtained from the Harvard plasmid repository (clone HsCD00458034). The cells were transfected using Lipofectamine LTX & Plus (Thermo Fisher). For luciferase studies, the cells were transfected with PSA-luciferase18 and pRL-SV40 (Promega) as a control. The cells were transferred to quadruplicate wells of a 96-well plate in C/S media 24 h after transfection and treated with drugs. The luciferase activity was assayed 24 h after treatment using the dual-luciferase reporter assay system (Promega). Student’s t test (two-sided and equal variance) was performed, and the association was considered significant when p < 0.05 and indicated by an asterisk.
Expression and Purification of AR DBD and15 N-Labeled AR DBD
The DNA sequences encoding the DBD of the human AR (AR-B: 557-647) were cloned into an N-terminal his6-tagged vector (pET28a, Novagen). The vector was transformed into BL21DE3 Gold Escherichia coli (Agilent) cells and grown to an OD600 of between 0.2 and 0.4. The temperature was reduced to 27 °C, ZnCl2 was added to the final concentration of 10 μM, and the expression was induced with 0.5 mM IPTG for 4 h when OD600 reached 0.8. The cells were spun down at 6000g for 15 min, resuspended in loading buffer (25 mM TrisHCl, pH 7.5, 500 mM NaCl, 15 mM imidazole, 1 mM DTT, 1 mM PMSF), and lysed with an EmulsiFlex C3 homogenizer. The cell debris was spun down at 40 000 rpm in a Beckman Ti-75 rotor for 1 h at 4 °C. The supernatant was loaded onto a HisTrap FF column (GE Life Sciences) in loading buffer. The unbound protein was washed off in low imidazole buffer (25 mM TrisHCl, pH 7.5, 500 mM NaCl, 30 mM imidazole), and his6-AR DBD was eluted with a linear gradient from 30 to 375 mM imidazole. The cleanest fractions were pooled and dialyzed overnight at 4 °C into 20 mM TrisHCl, pH 7.5, 50 mM NaCl, 2.5 mM CaCl2, and 1 mM DTT containing ∼10 U/mg thrombin (Sigma) to cleave the his6-tag. The precipitate was pelleted (40 000 rpm, 1 h, 4 °C), and the supernatant was loaded onto a cation-exchange column (HiTrap SP HP, GE Life Sciences), pre-equilibrated with 20 mM TrisHCl, pH 7.5, 50 mM NaCl, and 1 mM DTT, and eluted in a linear gradient of NaCl from 50 to 350 mM. The fractions containing DBD were pooled, concentrated (Amicon Ultra—3K, Millipore), filtered (Ultrafree-CL), run over a gel filtration column (16/600 Superdex 200 PG, GE Life Sciences) in 20 mM HEPES, pH 7.7, 100 mM NaCl, and 1 mM DTT. The purified protein was collected, run on a 12% SDS-PAGE to measure purity, and quantified by A280 (ε = 5095 M–1 cm–1). The 15N-labeled version was expressed from a single colony of BL21-DE3 transformed with pET28a-AR DBD and was grown in a 5 mL LB with 100 μg/mL AMP overnight. The culture was spun down in the morning, inoculated into 3 L of fresh LB with 100 μg/mL AMP, and grown to an OD600 ∼0.8. These cultures were then spun down (4000g, 10 min at room temperature) and resuspended in M9 minimal medium, supplemented with vitamins (5 mL of Centrum dissolved in 50 mL water) and 1 g 15NH4Cl, and grown to an OD600 of 1 at 18 °C and then induced overnight with 0.5 mM IPTG. The culture was then spun down, resuspended in Ni2+ loading buffer, and purified as described above.
Nuclear Magnetic Resonance
A 1.5 mg 15N-labeled AR DBD protein in 7 mL 20 mM HEPES buffer was exchanged to 50 mM phosphate buffer (pH 6.5) containing 1 mM DTT and 0.02% NaN3 and concentrated to 0.12 mM. The DNA duplex was prepared by annealing the complementary ssDNA oligos (CCAGAACATCAAGAACAC and GTGTTCTTGATGTTCTGG) at 95 °C for 5 min in annealing buffer (10 mM Tris, pH 7.5–8.0, 50 mM NaCl, 1 mM ethylenediaminetetraacetic acid (EDTA)) and then cooling to room temperature. Stock 120 μM DNA sample was prepared in 50 mM phosphate buffer (pH 6.5) containing 1 mM EDTA. The AR DBD and DNA complex was prepared by adding 270 μL of 24 μM AR DBD gradually to 600 μL of 7.2 μM DNA duplex on ice. The complex was then concentrated using a 3 kDa cutoff amicon cell to about 180 μL. Stock P24 solution was prepared in 50 mM phosphate buffer with pH 6.5 and 10% D2O. P24 (molar ratio of 5.5:1 of P24/AR DBD) was added to the protein–DNA complex. Alternatively, P24 was added to the protein and DNA samples before the protein was added to the DNA. In such a preparation, the final molar ratio of 2:1 of P24/AR DBD was used. NMR experiments were carried out on a Bruker 700 MHz equipped with a TXI-triple resonance inverse cryoprobe at 25 °C. The spectrum width used for 1H–15N HSQC is 13 and 27.5 ppm with carrier frequency on water and 116.75 ppm for 1H and 15N, respectively. The acquisition points are 2048 and 88 for the 1H and 15N dimensions, respectively. The number of scans is 760. The data were processed using a Bruker TopSpin 3.5.
Electrophoretic Mobility Shift Assay
A DNA duplex containing an AR-binding site (GTACGGAACAAAATGTACTGTAC) was first created by labeling the N-terminus with Cy5 and annealing with the unlabeled complement oligo in annealing buffer (10 mM TrisHCl, pH 8.0, 100 mM NaCl, 5 mM MgCl2) at 95 °C for 5 min, followed by slow cooling to 4 °C over 2.5 h. The annealed duplex were then stored in a concentrated form at 4 °C until use. To perform the EMSAs, we first diluted our DNA to 20 nM (a 4× stock) in binding buffer (20 mM TrisHCl, pH 8.0, 5 mM MgCl2, 5% glycerol, 1 mM EDTA, 300 mM KCl, 200 ng/μL bovine serum albumin (BSA), 1 mM DTT). The protein was diluted from a 426 μM frozen stock of AR DBD to 10 μM (a 2× stock) in binding buffer. The 10 μM AR DBD stock was then serially diluted 2× in binding buffer 13 or 14 times in 96-well plates, with the lowest concentration then being 2.4 or 1.2 nM. We then made two 200× stocks of P24 in 100 mg/mL (2-hydroxypropyl)-β cyclodextran (BCD). For the low concentration at 200×, we diluted 20 mg/mL (38 mM) P24 to 60 μM in 100 mg/mL BCD, and for the high concentration stock we diluted to 20 mM in 100 mg/mL BCD. We then diluted each stock to 1:50 in binding buffer (to be used as 4× stock). We then added 10 μL of DNA to 10 μL of P24 in 96-well plates, then added 20 μL of the protein dilutions, and equilibrated at room temperature for 1 h. While these were being equilibrated, we pre-ran 8% (19:1 acrylamide/bis-acrylamide) native acrylamide gels in 1× Tris-glycine at 200 V. While still running, we loaded 5 μL of the P24/AR DBD/DNA mixture onto the gel and ran for 25 min. At the end of the run, the gel was removed from the plates and placed in water and then imaged immediately (BioRad ChemiDoc MP Imaging System-Image Lab 4.1 Software) using the preprogrammed Cy5 channel. Affinities were calculated by the fraction bound using the disappearance of the free DNA band. The bands were then quantified from gel images using GE ImageQuant TL 7.0 software, using an unbound area for background correction. The fraction of DNA bound was plotted against the log of protein concentration and fit to the following equation (GraphPad Prism 7.00): y = 100 × “X”h/(Kdh + “X”h). An average Kd and the standard error were determined from four repeats performed on different days.
RT and qPCR
The total RNA was isolated from the cells using the GeneJET RNA purification kit (Thermo Scientific). The isolated RNA was then reverse-transcribed with Moloney murine leukemia virus reverse transcriptase (Invitrogen). The relative target gene expression was then assessed by qPCR with an SYBR green detection dye (Invitrogen) and Rox reference dye (Invitrogen) on the StepOne Real-Time PCR System (Applied Biosystems). Using the ΔΔCt relative quantification method, the target gene readouts were normalized to RPL19 and GADPH transcript levels. The experiments are the average of biological triplicates; p values were calculated using a two-tailed Student’s t- test.
AR ChIP
The LNCaP cells in a medium containing C/S FBS were treated with 0.1% DMSO vehicle, 1 nM DHT, DHT + 100 nM PP, or DHT + 100 nM enzalutamide for 4 h at 37 °C. For some cells, the media were removed, washed once with media, and replaced with media without drugs for 1.5 h prior to fixation. Fixation was performed in 1% formaldehyde for 3 min as the dishes cooled from 37 to 22 °C and 125 mM glycine for 10 min as the dishes cooled from 22 to 4 °C. The cells were lysed in immunoprecipitation lysis buffer (50 mM Hepes-KOH, pH 7.4, 1 mM EDTA, 150 mM NaCl, 10% glycerol, 0.5% Triton X-100, supplemented with protease inhibitors) and harvested by scraping. The nuclei were collected by centrifugation (500g for 5 min at 4 °C), resuspended in 2 mL of immunoprecipitation buffer (10 mM Tris-HCl, pH 8.0, 1 mM EDTA, 150 mM NaCl, 5% glycerol, 0.1% sodium deoxycholate, 0.1% SDS, 1% Triton X-100, supplemented with protease inhibitors), and sonicated until an average DNA fragment size of 100–500 bp was achieved (assessed by agarose gel electrophoresis). A 10 μg of anti-AR antibody (PG-21; Millipore) or normal rabbit IgG was used for immunoprecipitation. The immunoprecipitated material was washed with immunoprecipitation buffer containing 300 mM NaCl + 100 μg/mL yeast tRNA and resuspended in 80 μL of proteinase K solution (pH 8.0, 0.7% SDS, 200 μg/mL proteinase K). After reversing the cross-links, the DNA fragments were purified using the QIAquick PCR Purification kit (Qiagen). Real-time PCR was carried out as described above using primers for the AR-occupied regions previously described,18 and ChIP data were normalized to a region 140 bp upstream of the HSPA1A gene, which is not occupied by AR.
RNA-Seq
RNA sequencing was performed by the City of Hope Integrative Genomics core facility. cDNA synthesis and library preparation were performed using the TruSeq RNA Library prep kit in accordance with the manufacturer-supplied protocols. Libraries were sequenced on the Illumina Hiseq 2500 with a single read of 40 bp reads. The 40 bp long single-ended sequence reads were mapped to the human genome (hg19) using TopHat, and the frequency of the RefSeq genes was counted with customized R scripts. The raw counts were then normalized using the trimmed mean of M values method and compared using Bioconductor package “edgeR”. The average coverage for each gene was calculated using the normalized read counts from “edgeR”. Differentially regulated genes were identified using one-way analysis of variance with linear contrasts to calculate p values, and genes were only considered if the false discovery rate was <0.25 and the absolute value of the fold change was >2. There were over 40.2 million reads on average, with greater than 90% aligned to the human genome. Gene ontology analyses were performed using GSEA,33 DAVID,34 and Ingenuity Pathway Analysis (Qiagen).
IP–MS Analysis
The LNCaP cells were treated with drugs for 24 h and lysed in TBS, 0.1% Triton X-100, protease, and phosphatase inhibitors (Roche). Immunoprecipitation was performed using anti-AR (AR441, Santa Cruz). Western blot was used to detect AR (PG-21, Millipore), RNA polymerase II (clone 8WG16; Covance), DDX5 (Millipore #05-850), or DDX17 (Millipore). The immunoprecipitated AR was separated by SDS-PAGE and stained with SimplyBlue (Themo Fisher). Gel bands were excised and destained in ammonium bicarbonate. After disulfide bond reduction with 10 mM tris(carboxyethyl)phosphine and thiol alkylation with 50 mM iodoacetamide, the gel bands were incubated with trypsin (Promega) overnight. Peptides were extracted with 0.1% trifluoroacetic acid/70% acetonitrile and lyophilized. The samples were prepared and loaded along with a protein standard (yeast alcohol dehydrogenase) for analysis on a MALDI Q-TOF instrument (Waters) following separation by liquid chromatography using a C18 column (Waters). Peptide analysis was performed using Scaffold (Proteome Software, Inc.), with the relative quantities determined only on proteins with >95% probability of identity and a minimum of five peptides per sample.
Acknowledgments
We would like to thank Richard Jaramillo for advice on protein purification and crystallization; Lu Yang and the Integrative Genomics core facility staff for bioinformatic analyses; and Roger Moore, Teresa Hong, and Markus Kalkum with assistance and advice on MS analysis.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsomega.8b03205.
Supplementary methods on crystallization efforts and drug-resistant cell line creation; activity of P24, AR DBD-P24 crystals, and NMR spectra; and P24-resistant cell line (PDF)
Author Present Address
∥ Department of Translational Genomics, University of Southern California. 1975 Zonal Ave, Los Angeles, CA 90033.
Author Present Address
⊥ Fulgent Genetics 4978 Santa Anita Ave Suite 205, Temple City, CA 91780.
This work was supported by grants from the World Cancer Research Fund and the Margaret E. Early Research Trust, as well as a donation from Beverly and Steve Dorfman to J.O.J. Research reported in this publication also included work performed in the Integrative Genomics, NMR, and Mass Spectrometry Core Facilities supported by the National Cancer Institute of the National Institutes of Health under award number P30CA033572. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
The authors declare no competing financial interest.
Supplementary Material
References
- Siegel R. L.; Miller K. D.; Jemal A. Cancer statistics, 2015. Ca-Cancer J. Clin. 2015, 65, 5–29. 10.3322/caac.21254. [DOI] [PubMed] [Google Scholar]
- Bluemn E. G.; Nelson P. S. The androgen/androgen receptor axis in prostate cancer. Curr. Opin. Oncol. 2012, 24, 251–257. 10.1097/cco.0b013e32835105b3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blumentals W.; Foulis P.; Schwartz S.; Mason T. Does warfarin therapy influence the risk of bladder cancer?. Thromb. Haemostasis 2004, 91, 801–805. 10.1160/TH03-08-0553. [DOI] [PubMed] [Google Scholar]
- Scher H. I.; Fizazi K.; Saad F.; Taplin M.-E.; Sternberg C. N.; Miller K.; de Wit R.; Mulders P.; Chi K. N.; Shore N. D.; Armstrong A. J.; Flaig T. W.; Fléchon A.; Mainwaring P.; Fleming M.; Hainsworth J. D.; Hirmand M.; Selby B.; Seely L.; de Bono J. S. Increased survival with enzalutamide in prostate cancer after chemotherapy. N. Engl. J. Med. 2012, 367, 1187–1197. 10.1056/nejmoa1207506. [DOI] [PubMed] [Google Scholar]
- Purushottamachar P.; Godbole A. M.; Gediya L. K.; Martin M. S.; Vasaitis T. S.; Kwegyir-Afful A. K.; Ramalingam S.; Ates-Alagoz Z.; Njar V. C. O. Systematic structure modifications of multitarget prostate cancer drug candidate galeterone to produce novel androgen receptor down-regulating agents as an approach to treatment of advanced prostate cancer. J. Med. Chem. 2013, 56, 4880–4898. 10.1021/jm400048v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aggarwal R. R.; Small E. J. Small-cell/neuroendocrine prostate cancer: a growing threat?. Oncology 2014, 28, 838. [PubMed] [Google Scholar]
- Antonarakis E. S.; Boudadi K. Resistance to Novel Antiandrogen Therapies in Metastatic Castration-Resistant Prostate Cancer. Clin. Med. Insights: Oncol. 2016, 10, 1–9. 10.4137/CMO.S34534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Bono J. S.; Logothetis C. J.; Molina A.; Fizazi K.; North S.; Chu L.; Chi K. N.; Jones R. J.; Goodman O. B. Jr.; Saad F.; Staffurth J. N.; Mainwaring P.; Harland S.; Flaig T. W.; Hutson T. E.; Cheng T.; Patterson H.; Hainsworth J. D.; Ryan C. J.; Sternberg C. N.; Ellard S. L.; Fléchon A.; Saleh M.; Scholz M.; Efstathiou E.; Zivi A.; Bianchini D.; Loriot Y.; Chieffo N.; Kheoh T.; Haqq C. M.; Scher H. I. Abiraterone and increased survival in metastatic prostate cancer. N. Engl. J. Med. 2011, 364, 1995–2005. 10.1056/nejmoa1014618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen E. J.; Sowalsky A. G.; Gao S.; Cai C.; Voznesensky O.; Schaefer R.; Loda M.; True L. D.; Ye H.; Troncoso P.; Lis R. L.; Kantoff P. W.; Montgomery R. B.; Nelson P. S.; Bubley G. J.; Balk S. P.; Taplin M.-E. Abiraterone treatment in castration-resistant prostate cancer selects for progesterone responsive mutant androgen receptors. Clin. Cancer Res. 2015, 21, 1273–1280. 10.1158/1078-0432.ccr-14-1220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joseph J. D.; Lu N.; Qian J.; Sensintaffar J.; Shao G.; Brigham D.; Moon M.; Maneval E. C.; Chen I.; Darimont B.; Hager J. H. A clinically relevant androgen receptor mutation confers resistance to second-generation antiandrogens enzalutamide and ARN-509. Cancer Discov. 2013, 3, 1020–1029. 10.1158/2159-8290.cd-13-0226. [DOI] [PubMed] [Google Scholar]
- Antonarakis E. S.; Armstrong A. J.; Dehm S. M.; Luo J. Androgen receptor variant-driven prostate cancer: clinical implications and therapeutic targeting. Prostate Cancer Prostatic Dis. 2016, 19, 231–241. 10.1038/pcan.2016.17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schweizer M. T.; Yu E. Y. Persistent androgen receptor addiction in castration-resistant prostate cancer. J. Hematol. Oncol. 2015, 8, 128. 10.1186/s13045-015-0225-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arora V. K.; Schenkein E.; Murali R.; Subudhi S. K.; Wongvipat J.; Balbas M. D.; Shah N.; Cai L.; Efstathiou E.; Logothetis C.; Zheng D.; Sawyers C. L. Glucocorticoid receptor confers resistance to antiandrogens by bypassing androgen receptor blockade. Cell 2013, 155, 1309–1322. 10.1016/j.cell.2013.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones J. O.; Diamond M. I. A cellular conformation-based screen for androgen receptor inhibitors. ACS Chem. Biol. 2008, 3, 412–418. 10.1021/cb800054w. [DOI] [PubMed] [Google Scholar]
- Lim M.; Otto-Duessel M.; He M.; Su L.; Nguyen D.; Chin E.; Alliston T.; Jones J. O. Ligand-independent and tissue-selective androgen receptor inhibition by pyrvinium. ACS Chem. Biol. 2014, 9, 692–702. 10.1021/cb400759d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones J. O.; Bolton E. C.; Huang Y.; Feau C.; Guy R. K.; Yamamoto K. R.; Hann B.; Diamond M. I. Non-competitive androgen receptor inhibition in vitro and in vivo. Proc. Natl. Acad. Sci. U.S.A. 2009, 106, 7233–7238. 10.1073/pnas.0807282106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarraf P.; Mueller E.; Jones D.; King F. J.; DeAngelo D. J.; Partridge J. B.; Holden S. A.; Chen L. B.; Singer S.; Fletcher C.; Spiegelman B. M. Differentiation and reversal of malignant changes in colon cancer through PPARγ. Nat. Med. 1998, 4, 1046–1052. 10.1038/2030. [DOI] [PubMed] [Google Scholar]
- Bolton E. C.; So A. Y.; Chaivorapol C.; Haqq C. M.; Li H.; Yamamoto K. R. Cell- and gene-specific regulation of primary target genes by the androgen receptor. Genes Dev. 2007, 21, 2005–2017. 10.1101/gad.1564207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark E. L.; Fuller-Pace F. V.; Elliott D. J.; Robson C. N. Coupling transcription to RNA processing via the p68 DEAD box RNA helicase androgen receptor co-activator in prostate cancer. Biochem. Soc. Trans. 2008, 36, 546–547. 10.1042/bst0360546. [DOI] [PubMed] [Google Scholar]
- Mooney S. M.; Goel A.; D’Assoro A. B.; Salisbury J. L.; Janknecht R. Pleiotropic effects of p300-mediated acetylation on p68 and p72 RNA helicase. J. Biol. Chem. 2010, 285, 30443–30452. 10.1074/jbc.m110.143792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicol S. M.; Fuller-Pace F. V. Analysis of the RNA helicase p68 (Ddx5) as a transcriptional regulator. Methods Mol. Biol. 2010, 587, 265–279. 10.1007/978-1-60327-355-8_19. [DOI] [PubMed] [Google Scholar]
- Clark E. L.; Coulson A.; Dalgliesh C.; Rajan P.; Nicol S. M.; Fleming S.; Heer R.; Gaughan L.; Leung H. Y.; Elliott D. J.; Fuller-Pace F. V.; Robson C. N. The RNA helicase p68 is a novel androgen receptor coactivator involved in splicing and is overexpressed in prostate cancer. Cancer Res. 2008, 68, 7938–7946. 10.1158/0008-5472.can-08-0932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Endoh H.; Maruyama K.; Masuhiro Y.; Kobayashi Y.; Goto M.; Tai H.; Yanagisawa J.; Metzger D.; Hashimoto S.; Kato S. Purification and Identification of p68 RNA Helicase Acting as a Transcriptional Coactivator Specific for the Activation Function 1 of Human Estrogen Receptor α. Mol. Cell. Biol. 1999, 19, 5363–5372. 10.1128/mcb.19.8.5363. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Fuller-Pace F. V.; Ali S. The DEAD box RNA helicases p68 (Ddx5) and p72 (Ddx17): novel transcriptional co-regulators. Biochem. Soc. Trans. 2008, 36, 609–612. 10.1042/bst0360609. [DOI] [PubMed] [Google Scholar]
- Wortham N. C.; Ahamed E.; Nicol S. M.; Thomas R. S.; Periyasamy M.; Jiang J.; Ochocka A. M.; Shousha S.; Huson L.; Bray S. E.; Coombes R. C.; Ali S.; Fuller-Pace F. V. The DEAD-box protein p72 regulates ERα-/oestrogen-dependent transcription and cell growth and is associated with improved survival in ERα-positive breast cancer. Oncogene 2009, 28, 4053–4064. 10.1038/onc.2009.261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lareau L. F.; Inada M.; Green R. E.; Wengrod J. C.; Brenner S. E. Unproductive splicing of SR genes associated with highly conserved and ultraconserved DNA elements. Nature 2007, 446, 926–929. 10.1038/nature05676. [DOI] [PubMed] [Google Scholar]
- Li H.; Ban F.; Dalal K.; Leblanc E.; Frewin K.; Ma D.; Adomat H.; Rennie P. S.; Cherkasov A. Discovery of small-molecule inhibitors selectively targeting the DNA-binding domain of the human androgen receptor. J. Med. Chem. 2014, 57, 6458–6467. 10.1021/jm500802j. [DOI] [PubMed] [Google Scholar]
- Flavin R.; Peluso S.; Nguyen P. L.; Loda M. Fatty acid synthase as a potential therapeutic target in cancer. Future Oncol. 2010, 6, 551–562. 10.2217/fon.10.11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brand L. J.; Olson M. E.; Ravindranathan P.; Guo H.; Kempema A. M.; Andrews T. E.; Chen X.; Raj G. V.; Harki D. A.; Dehm S. M. EPI-001 is a selective peroxisome proliferator-activated receptor-gamma modulator with inhibitory effects on androgen receptor expression and activity in prostate cancer. Oncotarget 2015, 6, 3811–3824. 10.18632/oncotarget.2924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu D.; Sunkel B.; Chen Z.; Liu X.; Ye Z.; Li Q.; Grenade C.; Ke J.; Zhang C.; Chen H.; Nephew K. P.; Huang T. H.-M.; Liu Z.; Jin V. X.; Wang Q. Three-tiered role of the pioneer factor GATA2 in promoting androgen-dependent gene expression in prostate cancer. Nucleic Acids Res. 2014, 42, 3607–3622. 10.1093/nar/gkt1382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Copeland B. T.; Bowman M. J.; Boucheix C.; Ashman L. K. Knockout of the tetraspaninCd9in the TRAMP model ofde novoprostate cancer increases spontaneous metastases in an organ-specific manner. Int. J. Cancer 2013, 133, 1803–1812. 10.1002/ijc.28204. [DOI] [PubMed] [Google Scholar]
- Nenseth H. Z.; Dezitter X.; Tesikova M.; Mueller F.; Klokk T. I.; Hager G. L.; Saatcioglu F. Distinctly different dynamics and kinetics of two steroid receptors at the same response elements in living cells. PLoS One 2014, 9, e105204. 10.1371/journal.pone.0105204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramanian A.; Tamayo P.; Mootha V. K.; Mukherjee S.; Ebert B. L.; Gillette M. A.; Paulovich A.; Pomeroy S. L.; Golub T. R.; Lander E. S.; Mesirov J. P. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. U.S.A. 2005, 102, 15545–15550. 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang D. W.; Sherman B. T.; Lempicki R. A. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 2009, 4, 44–57. 10.1038/nprot.2008.211. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.