

Abstract
The enantioselective synthesis of α-disubstituted N-heterocyclic carbonyl compounds has been accomplished using palladium-catalyzed allylic alkylation. These catalytic conditions enable access to various heterocycles, such as morpholinone, thiomorpholinone, oxazolidin-4-one, 1,2-oxazepan-3-one, 1,3-oxazinan-4-one and structurally related lactams, all bearing fully substituted α-positions. Broad functional group tolerance was explored at the α-position in the morpholinone series. We demonstrate the utility of this method by performing various transformations on our useful products to readily access a number of enantioenriched compounds.
Graphical Abstract
N,O-heterocycles such as morpholine, oxazolidine, and isoxazolidine are important pharmacophores in medicinal chemistry (figure 1).1–11 Notable morpholine-containing pharmaceuticals include edivoxetine2, an antidepressant and a treatment for ADHD; linezolid5, a synthetic antibiotic; and gefitinib4, an EGFR inhibitor used to treat certain breast, lung and other cancers. 5-Membered isoxazolidinone is the core structure of cycloserine8, an antibiotic for the treatment of tuberculosis. Quinocarcin7, possessing an oxazolidine ring in the 3,8-diazabicyclo[3.2.1]octane framework, has shown remarkable antiproliferative activity against lymphocytic leukemia. An antibiotic, FR-669799, isolated from Streptomyces sandaensis No. 6897, contains a 1,2-oxazinane moiety.
Figure 1.

Representative N,O-heterocyclic-containing pharmaceuticals and natural products
In addition to a wide variety of biological activity, N,O-heterocycles shown in Figure 2 are commonly used as synthetic intermediates, which provide various hydroxy acid moieties after acetal removal or N-O bond cleavage. Oxazolidin-4-ones have been reported as good platforms to α-hydroxyacids.12 For example, Ye et al.12a reported that the formal [3+2] cycloaddition between ketenes and oxaziridines could be applied in an enantioselective fashion to synthesize oxazolidin-4-one derivatives, which could be converted into the corresponding α-hydroxy acids. 1,2-Oxazinan-3-ones have proved to be excellent precursors to γ-hydroxy acid and γ-butyrolactone derivatives.13
Figure 2.

α-Tertiary and quaternary N,O-heterocycles
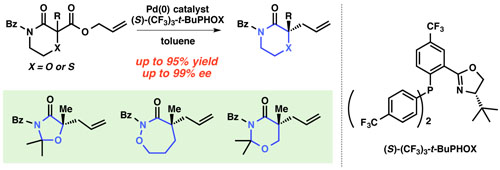
Our group has a sustained interest in the enantioselective synthesis of α-quaternary carbonyl compounds,14 – 16 which offers a novel solution for these challenging chiral centers in natural product synthesis. Influenced by our results, the Lupton group17a and the Shao group17b simultaneously reported that carbazolones are suitable substrates under our allylic alkylation conditions and applied the resulting chiral building blocks to total syntheses of indole alkaloids. Significant work in our laboratory has identified conditions for the enantioselective allylic alkylation to provide α-quaternary lactams in exceptional yields and enantioselectivities.14b As part of that endeavor, we reported that 2-allyl-2-methylmorpholin-3-one 2a was obtained in a similar manner in high yield (91% yield) and outstanding enantioselectivity (99% ee). We sought to extend the substrate scope to morpholine derivatives and postulated that a broadly expanded array of chiral N,O-heterocyles might be readily accessible using our palladium-catalyzed allylic alkylation. Herein, we describe enantioselective allylic alkylation of heterocycles, including morpholin-3-one, thiomorpholine-3-one, oxazolidin-4-one, 1,2-oxazepan-3-one and 1,3-oxazinan-4-one.18 Furthermore, the enantioenriched products obtained were successfully converted into useful asymmetric building blocks containing quaternary and tetrasubstituted tertiary chiral centers.
We prepared a collection of racemic morpholinone substrates 1a-e19 and performed palladium-catalyzed decarboxylative allylic alkylation with Pd2(dba)3 (5 mol %) and (S)-(CF3)3-t-BuPHOX ligand20 (12.5 mol %, PHOX = phosphinooxazo-line) in a 0.033 M solution of toluene (Figure 3). Simple α-benzyl substitution performed well in this chemistry; the desired 2-benzyl α-tetrasubstituted morpholinone 2b was obtained in 95% yield and 99% ee. Gratifyingly, other functionalized substrates (benzyl ether, methyl ester, nitrile) are well tolerated, affording α-functionalized morpholinones 2c, 2d and 2e in uniformly excellent enantioenrichment (99% ee), although the yield of 2d was moderate (60%). Having demonstrated a broad functional group tolerance within the side chain, we explored other ring sizes and frameworks. Replacement of oxygen with sulfur gave thiomorpholinone 2f in good yield, but slightly decreased enantioselectivity (79% yield, 86% ee). Like morpholinone, benzomorpholinone is also a good substrate class, delivering allylated product 2g in 76% yield and 95% ee. Additionally, α-tetrasubstituted oxazolidin-4-one 2h is produced in 82% yield and 96% ee with higher temperature applied (60 °C).21 Benzyloxazolinone 2i is also produced in good yield and enantioselectivity.
Figure 3. Substrate Scope of α-Tertiary Heterocyclesa.

aReaction performed with 0.1 mmol of 1, 5 mol % of Pd2(dba)3, 12.5 mol % of (S)-(CF3)3-t-BuPHOX at 0.033 M in toluene at 50 °C bDetermined by chiral SFC analysis. cReactions were performed on 1g, 1h and 1i at 60 °C. dThe ee of 2g was determined by chiral SFC analysis after Bz removal (see supporting information). ePd2(pmdba)3 (pmdba = bis(4-methoxybenzylidene)acetone) was used instead of Pd2(dba)3. fAbsolute configuration was assigned by vibrational circular dichroism (VCD) spectroscopy22 supported by theoretical calculations (see supporting information). gAbsolute stereochemistry assigned by conversion into (–)-methyl 2-hydroxy-2-methylpent-4-enoate.23
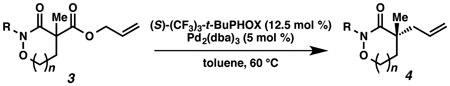
With excellent results on α,α-dialkyl 2-oxa- and thia-linked lactams in hand, we started to investigate allylic alkylation using cyclic hydroxamic acid derivatives to obtain a-quaternary N,O-heterocycles (Table 1). Isoxazolidin-3-one 3a (R = Bz), 3b (R = Boc) and 3c (R = CO2Ph) produced the desired alkylated compounds 4a-c in excellent yields (95–98%), but with modest enantioselectivities (72–73% ee) (entries 1–3). Benzoyl protected 1,2-oxazinan-3-one 3d underwent an unexpected side reaction, and produced only small amounts of 4d (entry 4).24 Despite of the low yield, the enantioselectivity of 4d is still satisfactory (88% ee), which encouraged us to identify an effective N-protecting group to circumvent the undesired reaction. A bulky pivaloyl group somehow suppresses the side reaction, but decreases the enantioselectivity (entry 5). An electron-rich N-benzylated 3f was a poor substrate for decarboxylative alkylation (entry 6).25 Finally, we discovered that carbamates 3g-i produced the desired products in good yields (67–89%) and acceptable enantioselectivities (84–87% ee) (entries 7–9), with little or none of the undesired side reactivity observed. We were delighted to find that 7-membered 3j is an excellent substrate in this class, furnishing 4j in a good yield and excellent enantioselectivity (entry 10, 81% yield, 93% ee).
Table 1.
Substrate Scope of α-Quaternary Cyclic Hydroxamic Acid Derivativesa
 | ||||
|---|---|---|---|---|
| entry | substrate | R | yield (%)b | ee (%)c |
| 1 | 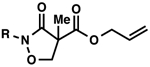 |
Bz (3a→4a)d | 98 | 73 |
| 2 | Boc (3b→4b) | 95 | 72 | |
| 3 | PhO(CO) (3c→4c) | 95 | 73 | |
| 4 | 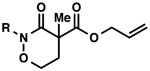 |
Bz (3d→4d) | 29 | 88 |
| 5 | Piv (3e→4e) | 48 | 73 | |
| 6 | Bn (3f→4f) | trace | ND | |
| 7 | Boc (3g→4g) | 67 | 85 | |
| 8 | Cbz (3h→4h) | 89 | 84 | |
| 9 | PhO(CO) (3i→4i) | 70 | 87 | |
| 10 | 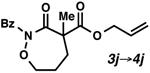 |
81 | 93 | |
Reaction performed under the conditions of Figure 3 at 60 °C.
All reported yields are for isolated products.
Enantiomeric excesses were determined by chiral SFC analysis.
Absolute configuration was assigned by VCD spectroscopy22 supported by theoretical calculations (see supporting information).
As shown in Scheme 1, we have also demonstrated allylic alkylation with 1,3-oxazinan-4-one 5 as an alternative β-hydroxy acid synthon of 3a. To our delight, 5 was successfully converted into 6 in 90% yield and 94% ee.
Scheme 1.

Synthesis of 1,3-oxazinan-4-one 6
We anticipate that our newly developed heterocycles could play important roles in medicinal agent discovery and also serve as useful chiral building blocks. To demonstrate the value and versatility of this new class of α-tetrasubstituted heterocycles, we implemented a number of product transformations (Scheme 2). For example, removal of the benzoyl group followed by reduction using LiAlH4 can readily convert morpholinone 2c into N-H morpholine 7. Acid treatment of 2h in methanol provided α-tertiary-hydroxy ester 8 in 71% yield without erosion of enantiopurity.26 a-Quaternary δ-lactone 9 was synthesized from 4j in a good yield by zinc mediated reduction of the N-O bond followed by acid catalyzed cyclization.
Scheme 2.

Derivatization of Allylic Alkylation Products
In conclusion, we have developed a variety of new classes of substrates for catalytic enantioselective allylic alkylation to generally form α,α-disubstituted 2-keto heterocycles, such as morpholinones, oxazolidinones, cyclic hydroxamic acid derivatives, and 1,3-oxazinanones. The asymmetric products formed in this communication are envisioned to be valuable pharmacophores in medicinal chemistry and their transformations afford a variety of important structures such as chiral hydroxy acid derivatives. Studies utilizing this method toward the synthesis of complex natural products and other bioactive small molecules are ongoing in our laboratory.
Supplementary Material
ACKNOWLEDGMENT
The authors wish to thank NIH-NIGMS (R01GM080269 to B.M.S. and R01GM075962 to K.N.H), Amgen, the Gordon and Betty Moore Foundation, the Caltech Center for Catalysis and Chemical Synthesis, and Caltech for financial support. Y.N. thanks Toray Industries Inc. for a postdoctoral fellowship. G.J.-O. and K.N.H. used the Extreme Science and Engineering Discovery Environment (XSEDE) supported by grant (OCI-1053575) along with the UCLA Institute of Digital Research and Education (IDRE). The authors are grateful to Dr. Rina Dukor (BioTools) for helpful discussions. The authors thank Scott Virgil (Caltech) for instrumentation assistance and Dr. Douglas C. Behenna (Caltech) for initial experimental results.
Footnotes
Supporting Information
Experimental procedures and compound characterization. This material is available free of charge via the Internet at http://pubs.acs.org.
Notes
The authors declare no competing financial interest.
REFERENCES
- (1).For reviews, see:Wijtmans R; Vink MKS; Schoemaker HE; van Delft FL; Blaauw RH; Rutjes FPJT Synthesis 2004, 641.Aroney M; Le Fevre RJW J. Chem. Soc. 1958, 3002.
- (2).(a) Dube S; Dellva MA; Jones M; Kielbasa W; Padich R; Saha A; Rao PJ Psychiatr. Res 2010, 44, 356. [DOI] [PubMed] [Google Scholar]; (b) Pangallo B; Dellva MA; D’Souza DN; Essink B; Russell J; Goldberger CJ Psychiatr. Res 2011, 45, 748. [DOI] [PubMed] [Google Scholar]
- (3).Hale JJ; Mills SG; MacCoss M; Finke PE; Cascieri MA; Sadowski S; Ber E; Chicchi GG; Kurtz M; Metzger J; Eiermann G; Tsou NN; Tattersall FD; Rupniak NMJ; Williams AR; Rycroft W; Hargreaves R; MacIntyre DE J. Med. Chem 1998, 41, 4607. [DOI] [PubMed] [Google Scholar]
- (4).(a) Morie T; Kato S; Harada H; Yoshida N; Matsumoto J Chem. Pharm. Bull 1994, 42, 877. [DOI] [PubMed] [Google Scholar]; (b) Mine Y; Yoshikawa T; Oku S; Nagai R; Yoshida N; Hosoki KJ Pharmacol. Exp. Ther 1997, 283, 1000. [PubMed] [Google Scholar]
- (5).(a) Barker AJ; Gibson KH; Grundy W; Godfrey AA; Barlow JJ; Healy MP; Woodburn JR; Ashton SE; Curry BJ; Scarlett L; Henthorn L; Richards L Bioorg. Med. Chem. Lett 2001, 11, 1911. [DOI] [PubMed] [Google Scholar]; (b) Sirotnak FM; Zakowski MF; Miller VA; Scher HI; Kris MG Clin. Cancer Res. 2000, 6, 4885. [PubMed] [Google Scholar]
- (6).Brickner SJ; Hutchinson DK; Barbachyn MR; Manninen PR; Ulanowicz DA; Garmon SA; Grega KC; Hendges SK; Toops DS; Ford CW; Zurenko GE J. Med. Chem 1996, 39, 673. [DOI] [PubMed] [Google Scholar]
- (7).Cang S; Ohta S; Chiba H; Johdo O; Nomura H; Nagamatsu Y; Yoshimoto AJ Antibiot. 2001, 54, 304. [DOI] [PubMed] [Google Scholar]
- (8).(a) Tomita F; Takahashi K; Shimizu K J. Antibiot 1983, 36, 463. [DOI] [PubMed] [Google Scholar]; (b) Takahashi K; Tomita F J. Antibiot 1983, 36, 468. [DOI] [PubMed] [Google Scholar]; (c) Tomita F; Takahashi K; Tamaoki T J. Antibiot 1984, 37, 1268. [DOI] [PubMed] [Google Scholar]
- (9).(a) Epstein IG; Nair KG; Boyd LJ Antibiot. Med. Clin. Ther 1955, 1, 80. [PubMed] [Google Scholar]; (b) Welch H; Putnam LE; Randall WA Antibiot. Med. Clin. Ther 1955, 1, 72. [PubMed] [Google Scholar]; (c) Hidy PH; Hodge EB; Young VV; Harned RL; Brewer GA; Phillips WF; Runge WF; Stavely HE; Pohland A; Boaz H; Sullivan HR J. Am. Chem. Soc 1955, 77, 2345. [Google Scholar]; (d) Ressler KJ; Rothbaum BO; Tannenbaum L; Anderson P; Graap K; Zimand E; Hodges L; Davis M Arch. Gen. Psychiatry 2004, 61, 1136. [DOI] [PubMed] [Google Scholar]
- (10).(a) Terano H; Takase S; Hosoda J; Kohsaka MJ Antibiot. 1989, 42, 145. [DOI] [PubMed] [Google Scholar]; (b) Rajski SR; Rollins SB; Williams RM J. Am. Chem. Soc 1998, 120, 2192. [Google Scholar]
- (11).(a) Kuehne ME; Konopka EA J. Med. Chem 1962, 5, 257. [DOI] [PubMed] [Google Scholar]; (b) Chylinska JB; Urbanski T; Mordarski M. J. Med. Chem 1963, 6, 484. [DOI] [PubMed] [Google Scholar]
- (12).(a) Shao P-L; Chen X-Y; Ye S Angew. Chem., Int. Ed 2010, 49, 8412. [DOI] [PubMed] [Google Scholar]; (b) Ooi T; Fukumoto K; Maruoka K Angew. Chem., Int. Ed 2006, 45, 3839. [DOI] [PubMed] [Google Scholar]; (c) Cardillo G; Hashem MA; Tomasini C J. Chem. Soc., Perkin Trans. 1 1990, 1487. [Google Scholar]; (d) Roush WR; Essenfeld AP; Warmus JS; Brown BB Tetrahedron Lett. 1989, 30, 7305. [Google Scholar]
- (13).(a) Ranganathan S; Ranganathan D; Mehrotra AK J. Am. Chem. Soc 1974, 96, 5261. [Google Scholar]; (b) Clarke PA; Cridland AP; Rolla GA; Iqbal M; Bainbridge NP; Whitwood AC; Wilson C J. Org. Chem 2009, 74, 7812. [DOI] [PubMed] [Google Scholar]
- (14).(a) Behenna DC; Mohr JT; Sherden NH; Marinescu SC; Harned AM; Tani K; Seto M; Ma S; Novak Z; Krout MR; McFadden RM; Roizen JL; Enquist JA Jr.; White DE; Levine SR; Petrova KV; Iwashita A; Virgil SC; Stoltz BM Chem. Eur. J 2011, 17, 14199. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Behenna DC; Liu Y; Yurino T; Kim J; White DE; Virgil SC; Stoltz BM Nature Chem. 2012, 4, 130. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Bennett NB; Duquette DC; Kim J; Liu W-B; Marziale AN; Behenna DC; Virgil SC; Stoltz BM Chem. Eur. J 2013, 19, 4414. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Reeves CM; Eidamshaus C; Kim J; Stoltz BM Angew. Chem., Int. Ed 2013, 52, 6718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).(a) Liu W-B; Reeves CM; Virgil SC; Stoltz BM J. Am. Chem. Soc 2013, 135, 10626. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Liu W-B; Reeves CM; Stoltz BM J. Am. Chem. Soc 2013, 135, 17298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Ma S; Han X; Krishnan S; Virgil SC; Stoltz BM Angew. Chem., Int. Ed 2009, 48, 8037. [DOI] [PubMed] [Google Scholar]
- (17).(a) Gartshore CJ; Lupton DW Angew. Chem., Int. Ed 2013, 52, 4113. [DOI] [PubMed] [Google Scholar]; (b) Li Z; Zhang S; Wu S; Shen X; Zou L; Wang F; Li X; Peng F; Zhang H; Shao Z Angew. Chem., Int. Ed 2013, 52, 4117. [DOI] [PubMed] [Google Scholar]
- (18).For examples of α,α-dialkylated ketopiperazine synthesis, see:Korch KM; Eidamshaus C; Behenna DC; Nam S; Horne D; Stoltz BM Angew. Chem. Int. Ed 2015, 54, 179.
- (19).For preparation of substrates, see ref 14b and supporting information.
- (20).McDougal NT; Streuff J; Mukherjee H; Virgil SC; Stoltz BM Tetrahedron Lett. 2010, 51, 5550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).The substrate 1h can be readily prepared from commercially available lactamide in three steps.19
- (22).Stephens PJ; Devlin FJ; Cheeseman JR VCD Spectroscopy for Organic Chemists; CRC Press: Boca Raton, FL, 2012. [Google Scholar]
- (23).Seto M; Roizen JL; Stoltz BM Angew. Chem., Int. Ed 2008, 47, 6873. [DOI] [PMC free article] [PubMed] [Google Scholar]
-
(24).We isolated 10 presumably due to retro [4+2] cycloaddition followed by palladium-catalyzed alkylation.
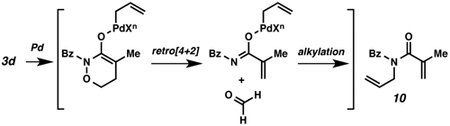
- (25).We also observed that electron-rich N-alkyl protection of amides decreased the reactivity under our conditions, see ref 14b.
- (26).For an alternative preparation of a-tertiary-hydroxy carbonyl compounds , see ref 23.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.