

Abstract
The atomistic change of C(sp3)—H to C(sp3)—O can have a profound impact on the physical and biological properties of small molecules. Traditionally, chemical synthesis has relied on pre-existing functionality to install new functionality, and directed approaches to C—H oxidation are an extension of this logic. The impact of developing undirected C—H oxidation reactions with controlled site-selectivity is that scientists gain the ability to diversify complex structures at sites remote from existing functionality, without having to carry out individual de novo syntheses. This perspective offers a historical view of why as recently as 2007, it was thought that the differences between aliphatic C—H bonds of the same bond type (for example, 2° aliphatic) were not large enough to distinguish them preparatively with small molecule catalysis in the absence of directing groups or molecular recognition elements. We give an account of the discovery of Fe(PDP)-catalyzed non-directed aliphatic C—H hydroxylations and how the electronic, steric, and stereoelectronic rules for predicting site-selectivity that emerged have affected a shift in how the chemical community views the reactivity among these bonds. The discovery that site-selectivity could be altered by tuning the catalyst [i.e. Fe(CF3-PDP)] with no changes to the substrate or reaction now gives scientists the ability to exert control on the site of oxidation on a range of functionally and topologically diverse compounds. Collectively, these findings have made possible the emerging area of late-stage C—H functionalizations for streamlining synthesis and derivatizing complex molecules.
Graphical Abstract
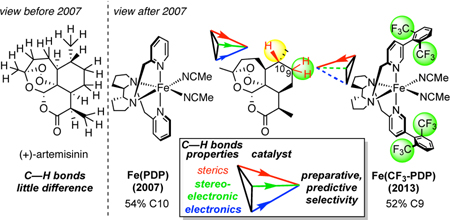
Introduction
The impact of developing non-directed aliphatic C—H oxidation reactions analogous to those found in nature is substantial. Chemists gain the ability to diversify complex structures at sites remote from existing functionality for the purpose of changing physical and/or biological properties, without carrying out individual de novo syntheses. Prior to 20071, 2, the chemical community mindset was that the physical differences between aliphatic C—H bonds of the same bond type (for example, 2° C—H bonds) was too small to distinguish between them preparatively without a directing group or molecular recognition approach, vide infra. In 2018, there has been a paradigm shift and it is widely accepted that the electronic, steric, and stereoelectronic differences between aliphatic C—H bonds in complex molecule settings makes them susceptible to site-selective oxidations. The power of such late-stage functionalizations to rapidly change the properties of drugs is being recognized by programs in the pharmaceutical industry. This perspective will provide an accurate historical account on how the literature on C—H oxidations prior to 2007 shaped the mindset that such bonds were indistinguishable and our view on what led to the paradigm shift in thinking by 2018.
Adding hydroxyl groups is a fast track to function.
The atomistic change of transforming aliphatic C(sp3)—H bonds to C(sp3)—O can have a profound impact on the properties of molecules. The introduction of oxygen functionality into molecules alters their physical properties (solubility, polarity) and can significantly impact on their interactions with biomolecular targets. The precise positioning of oxidized functionality acting as hydrogen bond donors and acceptors in three-dimensional space enables small molecules to interact with specific protein sites and effect varying physiological responses. Several examples are shown in Figure 1 where installation of a ketone or hydroxyl moiety can change the smell or taste of a small molecule.3 In the pharmaceutical realm, oxygen moieties in drugs can be essential for the physiological response. For example, in the drugs paclitaxel (Taxol®), erythromycin A, cilofungin, and ritonavir, removal of the benzoate or key hydroxyl moieties results in either complete or partial loss of the desired biological activity.4 Introduction of hydroxyl moieties is also critical in metabolic processes that clear drugs from our bodies.5
Figure 1.
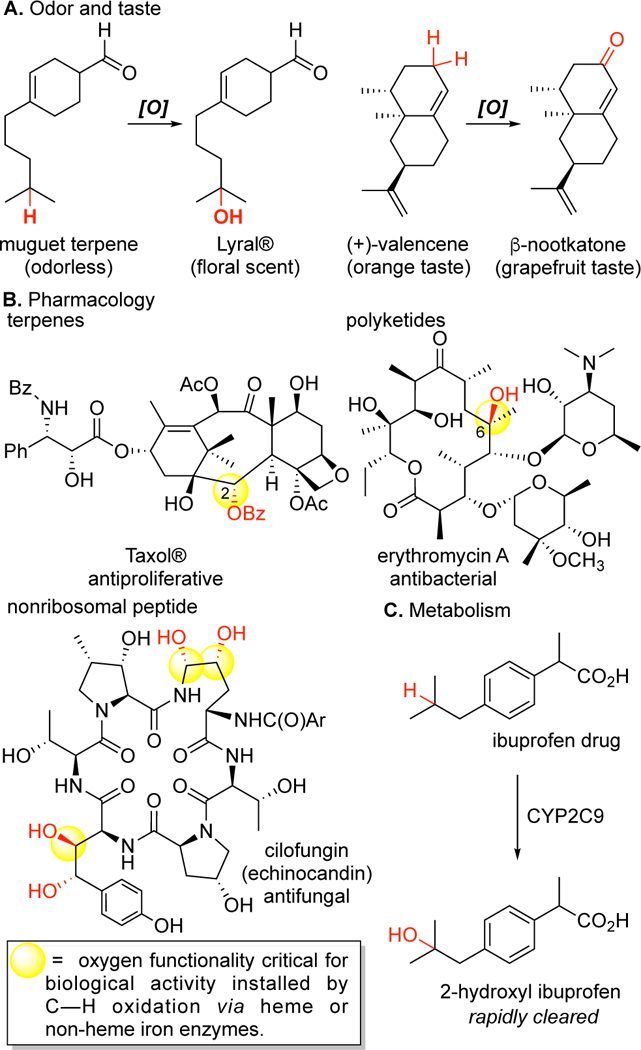
Atomistic change of C(sp3)–H to C(sp3)–O impacts a molecule’s properties. Introduction of a hydroxyl or ketone functionality can change (A) the smell and taste, (B) the pharmacological properties, and (C) the physical properties of small molecules (for example, solubility).
Aliphatic C—H hydroxylation: nature’s way.
Carbon-oxygen is a versatile functionality: it may be elaborated into other common functional groups (N, S, C) or further derivatized (e.g. sugars, phosphates). Traditional synthetic approaches rely on pre-existing functional groups on molecules to install new functionality. What strategic advantages exist in installing oxygen via C—H hydroxylation? Nature has evolved C—H hydroxylation reactions using heme and non-heme iron enzymes [e.g. cytochrome P-450 (CYP), α-ketoglutarate dependent oxygenases] that install oxygen functionality independently and remotely from existing functionality. In the biosynthesis of the natural product drugs, such enzymes are used to install key hydroxyl groups for biological activity in the final structure (Figure 1B). In echinocandin B, non-heme and heme iron enzymes (Ecd-G, Ecd-H) perform aliphatic C—H oxidations in both free L-homotyrosine and the assembled macrocyclic peptide (at orni-thine).6 In the biosynthesis of erythromycin, the C-6 hydroxyl moi-ety, key for medicinal potency, is installed onto the 6- deoxyerythronolide B (6-dEB) core via a designated P-450 (EryF) at late stages of the synthetic sequence.7 In the metabolism of ibuprofen, a liver CYP2C9 mediates C—H hydroxylation at C2 that leads to its rapid clearance from the body.5
Non-directed aliphatic C—H oxidation.
The strength and ubiquity of aliphatic C—H bonds (pKa ~ 50, BDE ~96–101 kcal/mol8) in complex molecules renders them among the most challenging bonds to selectively oxidize. Despite sporadic use prior to 2007, C— H oxidation reactions and their strategic use in chemical synthesis was largely overlooked by the synthetic community. Although we and others had begun to delineate the strategy of late-stage oxidation for streamlining and diversifying chemical synthesis,9 preparative and selective aliphatic C—H oxidation reactions did not exist to fully demonstrate the power of this strategy. In support of this, C— H oxidation and the associated streamlining logic does not appear in prominent textbooks10 on total synthesis prior to 2007. The majority of 20th century examples were of TFDO oxidations at tertiary sites on steroids (vide infra, Figures 4). The lack of substrate structural diversity, the operational difficulty of the oxidant, and paucity of ra-tionale for site-selectivities in part explains why prominent reviews in 2007 and 2006 stated that differences between aliphatic C—H bonds was not large enough to achieve useful levels of site-selectiv-ity: “…there is very little difference in reactivity between the various C—H bonds in alkanes, so targeting a specific C—H bond is diffi-cult”1 and “Selective targeting of internal isolated sp3 C—H bonds in complex targets has hitherto been limited to enzymatic or biomimetic systems equipped with a recognition element capable of arranging the C—H bond of interest and the reactive center in proximal and favorable orientation. At present, it seems that the reactivity differences between isolated alkyl C—H bonds (e.g., two adjacent methylenes in the middle of a long alkyl group) are very small to tackle this problem in the absence of a molecular recognition element.”2
A report came in 2007 from our group of [Fe(PDP)] catalysis capable of predictable, site-selective aliphatic C(sp3 )—H oxidations among 3° C—H bonds remote from existing functionality in complex natural products.11 This was followed by a report in 2009/2010 demonstrating that even the most challenging secondary C(sp3 )— H bonds can similarly be site-selectively oxidized.12 A third report in 2013 showed that the site of oxidation can be rationally altered by modifying the catalyst.13 Through these studies, we elucidated that C—H bonds can be predictably differentiated by catalysts based on their electronic, steric, and stereoelectronic properties. This fundamental insight led to a quantitative model that enables the site of oxidation to be predicted in complex settings as a function of the catalyst.13 Fe(PDP) catalysis has now been demonstrated to effect preparative, predictable late-stage oxidations in a range of complex mol-ecules derived from nature- terpenes, steroids, alkaloids,14 peptides,15 lactams16 - at strong aliphatic C—H bonds remote from functionality (Figure 2).
Figure 2.
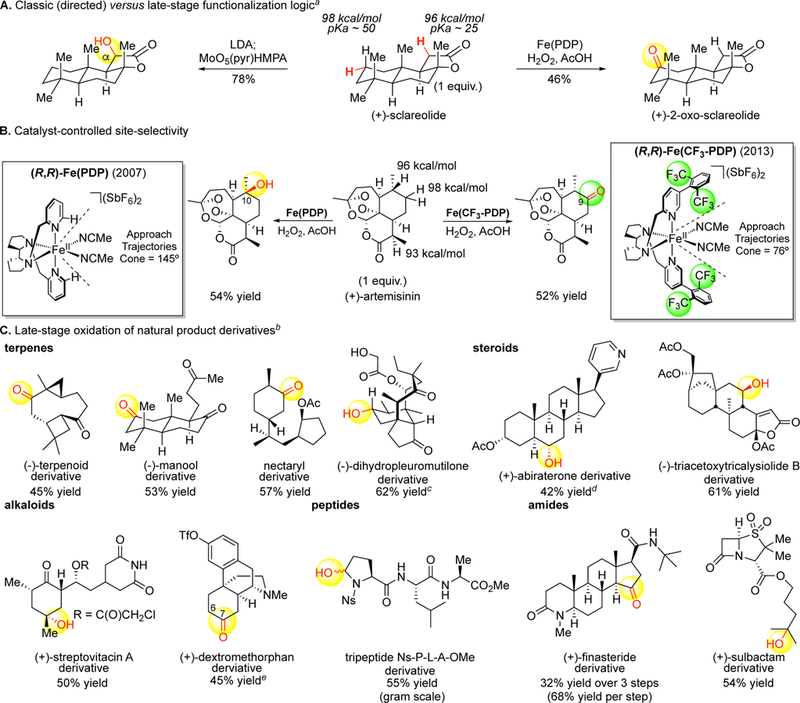
Late-stage functionalization via Fe(PDP)- and Fe(CF3-PDP)-catalyzed aliphatic C–H oxidation. (A) Traditional synthetic logic using existing functionality to introduce new functionality versus late-stage functionalization logic where new functionality is introduced remote from existing functionality. (B) The invention of Fe(PDP) and Fe(CF3-PDP) catalysts for remote, late-stage aliphatic C–H oxidations. Site-selectivity operates under catalyst control: using the same molecule (artemisinin) and changing only the catalysts, preparative oxidation occurs at two distinct sites. (C) Fe(PDP) and Fe(CF3-PDP) catalysis has been developed to enable late-stage oxidation of a wide range of natural products (terpenes, steroids, alkaloids, peptides, amides). aDirected sclareolide α-oxidation, ref 53c. bAll yields are isolated yields based on 1 equiv of substrate. c2:1 alcohol:ketone ratio. d6:1 alcohol:ketone ratio. e2.5:1 C7ketone:C6alcohol ratio.
Whereas it was known that site-selectivity could be achieved between different C—H bond types (for example, 3° versus 2°, 1° versus 2° /3°, vide infra, Figures 4, 5, 21), the Fe(PDP) work caused a shift in the community’s thinking on the ability to achieve selectivity among the same type of aliphatic C—H bond.17 Additionally, Fe(PDP)-oxidations first demonstrated that such site-selectivities can furnish preparative amounts of mono-oxidized products and follow easy rules that predict the oxidation site in complex molecules. These aspects of Fe(PDP) catalysis contrasted prior reactions run with large excesses of substrate, furnishing <1% oxidized products, and proceeding with variable levels of predictability (vide infra, Figures 4, 5, 21). Following these advances, it is now widely recognized that undirected C(sp3 )—H functionalizations can follow predictable site-selectivity rules first delineated with Fe(PDP) catalysis based on subtle interactions of the catalyst/oxidant with the electronic, steric, and stereoelectronic properties of C—H bonds.17 Accordingly, there has been an explosion of research in the flourishing area of late-stage functionalization to derivatize drugs and natural products and streamline synthesis.18, 19, 20, 21, 22, 23 Reviews in 2018 now state: “…natural products contain C—H bonds in a variety of steric and electronic environments. …electron-rich C—H bonds are cleaved faster than electron-poor C—H bonds… the steric properties of the reagent reacting with the C—H bonds can affect the regioselectivity of C—H bond functionalization.”24
Historic Approaches to Aliphatic C—H Hydroxylations
Since the late 1800’s, non-directed aliphatic C—H hydroxylations were considered to be the least selective C—H functionalizations, lacking the selectivity, preparative utility, and generality required for routine utilization in synthesis.18 Two general approaches towards C—H hydroxylations, stoichiometric reagents (hy-droxyl/alkoxyl radical and organic oxidants) and metal-catalyzed processes, are discussed below. A significant distinction between these approaches is that organic oxidants often rely on substrate controlled selectivity whereas metal mediated processes may exert catalyst control, particularly in cases where C—H cleavage and functionalization occur closely associated with the metal center.
Hydroxyl radicals and strained dioxiranes.
In 1894, Fenton reported that the combination of hydrogen peroxide with acidic ferrous sulfate (Fe2+SO4) leads to a powerful oxidant that can oxidize aliphatic compounds.25 Forty years later, Haber and Weiss provided evidence that the Fenton reaction proceeds via HO· radical,26 the most reactive of all radicals and second only to fluorine with respect to its oxidation potential. Accordingly, Fenton chemistry is used in environmental remediation of ground or water chemical spills.27 Because of the highly exothermic nature of the reaction that generates a strong bond (H—OH, BDE ~ 119 kcal/mol), the early transition state renders HO· among the least selective reagent forming substantial amounts of secondary and even primary radicals in addition to tertiary.28 Because secondary and primary radicals are not capable of strong stabilization of a positive charge in the subsequent electron transfer steps proposed for hydroxylations, they often lead to dimerized products.29
In the 1980’s, iron salts with monodentate pyridine ligands (often in solvent quantities) were evaluated for aliphatic C—H oxidations. Collectively known as “Gif”, these systems were proposed to proceed via iron oxo species primarily due to the fact that secondary to tertiary ratios of hydroxylated products on adamantane contrasted with the 1:1 ratios observed for Fenton reactions. Fenton chemistry in the presence of molecular oxygen (O2) leads to a radical autoxidation pathway with hydrocarbons that forms alkoxide radicals (Figure 3A). Later studies revealed that the site-selectivities were due to both the generation of alkoxide radicals via autoxidation and adamantylation of pyridine (Figure 3B).30
Figure 3.

Aliphatic C–H oxidation reactivity via hydroxyl radicals promoted with (A) iron salts (Fenton reaction) and (B) iron salts in the presence of pyridine and carboxylic acid additives (Gif reaction).
Stoichiometric electrophilic organic heterocycles (e.g. dioxiranes, oxaziridines) are weak oxidants used primarily for tertiary C—H hydroxylation. Dioxirane oxidants like TFDO31 derive from an expensive, volatile reagent (l, l, l-trifluoro- 2- propanone, boiling point 22 ºC) and must be formed and run under conditions that limit reproducibility, that is glassware free of trace metal contaminants, −20 °C temperatures, and no visible light. If these conditions are not met, TFDO decomposes to carbon centered radicals that may react at diffusion controlled rates with molecular oxygen to generate alkoxide radicals, sharing similar reactivity profiles and limitations to those discussed above with Fenton chemistry (Figure 4A).32 In molecules prone to radical decomposition pathways, the conversion is limited to maintain high selectivities (Figure 4B).33 Because of TFDO’s limited reactivity, methylene oxidation does not effectively compete with tertiary oxidation. In the 1990’s, intermolecular reactions performed with steroids gave unexplained preferences for 3° C—H hydroxylations depending on the steroid (Figure 4C).34, 35 Stoichiometric reactions with organic oxidants are limited by preparative challenges to tune the oxidant for reactivity and selectivity. Consistent with this, site-selectivities have only been altered by changing the substrate (Figure 4C).
Figure 4.
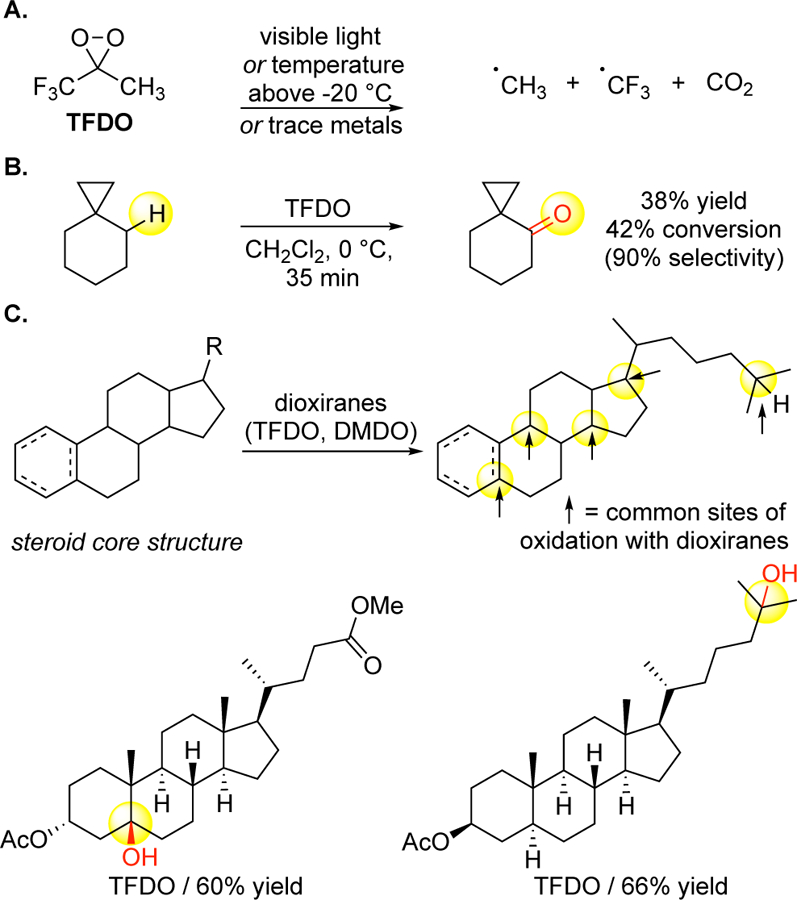
Aliphatic C–H oxidations via stoichiometric dioxirane oxidants. (A) TFDO [methyl(trifluoromethyl)dioxirane] must be made and used at cryogenic temperatures (−20 °C) under conditions that exclude visible light and trace metals to avoid decomposition into free radicals. (B) Compounds housing functionality prone to decomposition in the presence of free radicals are not run under conditions that afford high conversion with TFDO (often requiring elevated temperatures). (C) Aliphatic C–H oxidations of steroids with TFDO and DMDO (dimethyldioxirane) occur at tertiary sites. Changes in site-selectivities happen only by changing the substrate and are not explained.
Organometallic reactions.
In the early 1970’s, platinum salts (K2[PtCl4]) were shown to catalyze hydroxylation and chlorination of alkanes in water via high-valent Pt(IV) intermediates (Figure 5A).36 Remarkable site-selectivities, distinct from those of the free radicals or dioxiranes, favored oxidation at very strong primary C— H bonds. A bipyrimidine Pt catalyst was later reported that uses sulfuric acid (H2SO4) as an oxidant and functionalization reagent to furnish methyl bisulfate from methane with impressive yields (70% based on methane) and catalyst turnover numbers (ca. 500 tn) (Figure 5B).37 Extensive mechanistic studies on this38 and related reactions of rhodium and iridium with higher order alkanes39 elucidated that the site-selectivity among different bond types to favor functionalization at 1º C—H bonds is due to a kinetic and thermodynamic preference to form the least sterically hindered C—M bond (Figure 5C). Extensive work has been done on such organometallic systems, including the discovery of new oxidants,40, 41 alternate reactivity modes (e.g. dehydrogenations42), alternative functionalization reagents not requiring oxidants (i.e. carbonylations43, borylations44). Despite this, reactions suffer from reactivity issues requiring large excesses of substrate and falling short of furnishing useful yields of mono- functionalized products (Figure 5D).45 The development of preparative (1 equiv. substrate), non-directed, catalytic aliphatic primary C—H oxidations are a significant unmet need in the area of aliphatic C—H oxidations.46, 47
Figure 5.
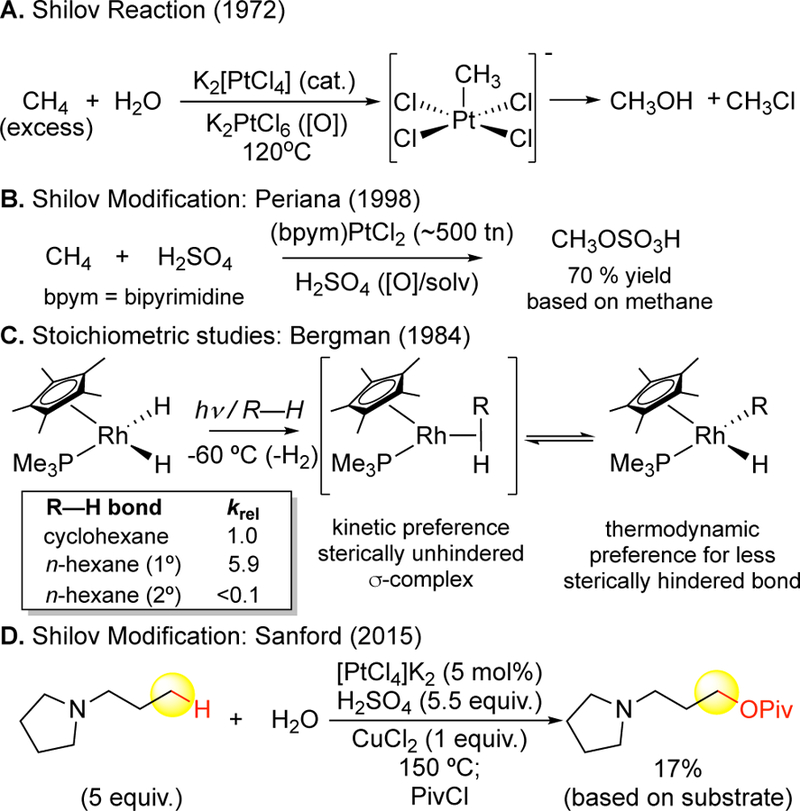
Aliphatic C–H oxidation reactivity via organometallic intermediates. (A) The Shilov reaction converts methane to methanol and chloromethane using platinum salts. (B) A modification of the Shilov reaction that converts methane to methyl bisulfate features the ability to use platinum in catalytic amounts, with sulfuric acid acting as both the functionalization reagent and the stoichiometric oxidant. (C) Stoichiometric studies elucidated that the primary site-selectivities of reactions that proceed via organometallic intermediates are primarily due to a kinetic preference to proceed via an unhindered σ-complex and a thermodynamic preference to form the less sterically hindered C–M bond. (D) Reactivity challenges persist with the Shilov reaction, requiring suprastoichiometric amounts of substrate.
Cytochrome P-450 Mimics: Iron and Manganese Porphyrins.
The selective, non-directed oxidations of unactivated hydrocarbons by cytochrome P-450 enzymes stimulated interest in developing chemical model systems.48 Well-defined iron49 and manganese50 catalysts with heme ligands that can be tuned were extensively explored as models for cytochrome P450 to catalyze aliphatic C—H hydrox-ylations (Figure 6). Spectroscopic evidence, KIE studies, as well as H2O18 exchange experiments support that the active oxidant is a high valent metal oxo species.51 Unfortunately, aliphatic C—H oxidation reactions with these catalysts were very low yielding requiring solvent quantities of hydrocarbon substrate. Site-selectivities for hydroxylation were investigated with electronically biased substrates and shown to be minimal (Figure 6A).49 In both the iron and manganese porphyrin systems, strong evidence exists that free radical processes operate contemporaneously with more selective metal(oxo) chemistry.51b Aliphatic C—H hydroxylations and epoxidations are not stereoretentive, leading to long-lived carbon-centered radicals that can open cyclopropane rings (Figure 6B-C).49b, 52 Moreover, in the presence of halogens (catalyst counterion) or oxygen (air), large amounts of byproducts are generated from radical halogenation and/or autoxidation processes.50a, 50c Mn(IV)(TMP)oxo’s have been isolated and used in stoichiometric oxygen transfer reactions (Figure 6D). Interestingly, under anaerobic conditions the yields of oxidized products drop significantly and autoxidation products are still observed, signifying that an active oxidant has significant oxygen centered radical character.52
Figure 6.
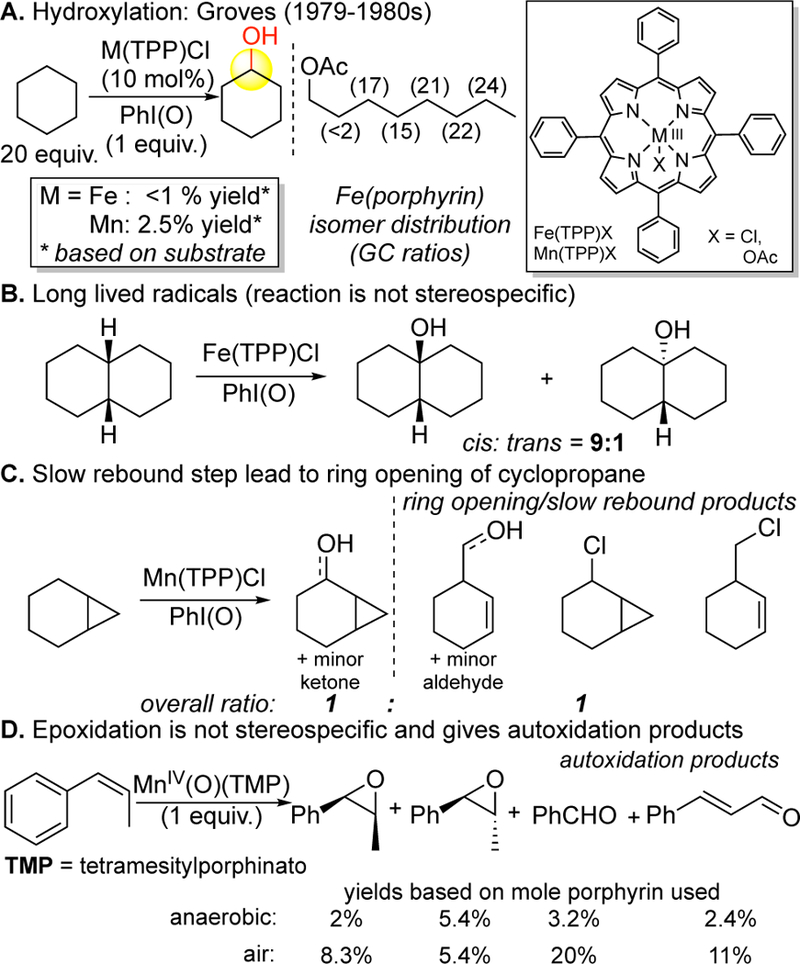
Biomimetic aliphatic C–H oxidation and epoxidation reactivity with iron and manganese heme catalysts. (A) Reactivity and site-selectivity challenges. (B,C) The reactions are not stereospecific, leading to long-lived carbon-centered radical species that scramble stereochemistry and open cyclopropane rings. (D) Epoxidation experiments performed with stoichiometric manganese(oxo) porphyrin complexes show the presence of autoxidation products under both aerobic and anaerobic conditions, signifying an oxidant is formed with significant oxygen-centered radical character.
The Site-Selectivity Challenge
Directing Groups.
Many viewed, directing groups as the most promising approach for achieving site-selectivity in unactivated aliphatic C—H oxidations. Averting underlying reactivity and selectivity challenges, the directed approach to C—H oxidation relies on traditional synthetic logic by using existing functionality on a molecule to introduce new functionality (Figure 7).53 A pre-existing functional group on the substrate is transformed into functionality that can support a species that effects C—H cleavage and subsequent functionalization, generally beta or gamma to the directing group. Such approaches for Fenton-type reactions install functionality poised to generate oxygen radicals on pre-existing alcohols. Because such reactions have strict geometric requirements they are most effective in rigid substrates like steroids54 (Figure 7A). In organometallic reactions, the directing group acts as a ligand for the metal, directing the C—H metallation and functionalization step and enabling reactivity in transformations that intermolecularly do not occur at any appreciable rate (Figure 7B). A very early catalytic demonstration by Murahashi in the 1950’s was on arenes (C(sp2)—H bonds) in the context of carbonylations55 (Figure 7C). Despite numerous stoichiometric reports of “tail-biting” reactions observed with ligands of metals,56 the next catalytic system appeared from Murai in the 1990’s for aromatic C—H alkylation.57 In the 21st century, this approach has been extensively developed to include a wide range of metals, directing groups and has been extended to C(sp3)— H bond oxidations (Figure 7E, 7F).40b, 58
Figure 7.
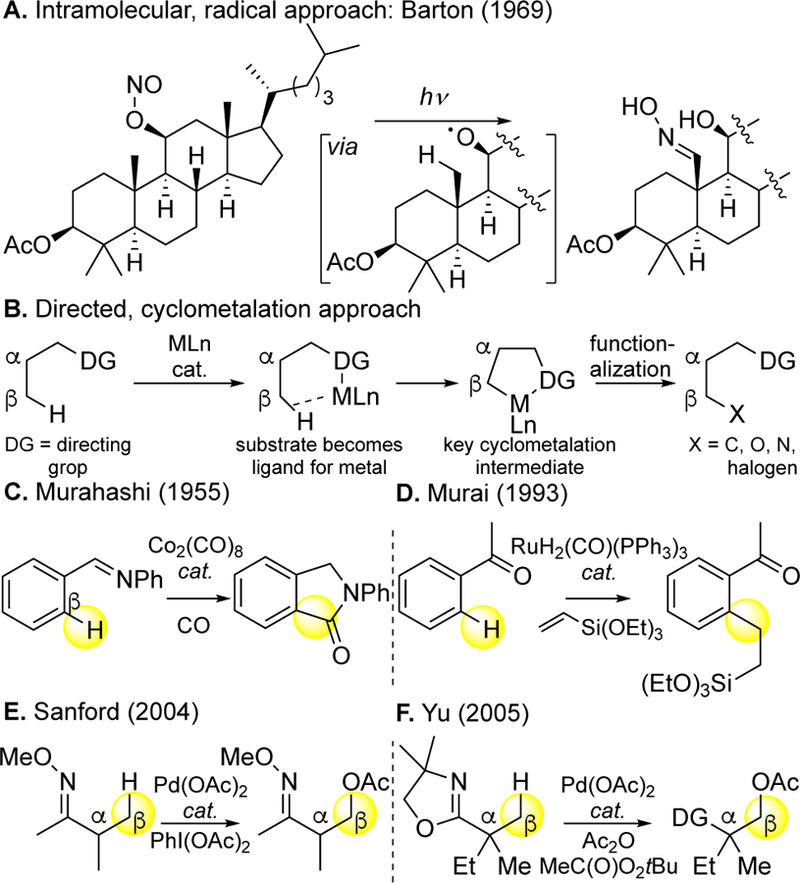
Substrate directing group approaches for improving reactivity and controlling site-selectivity. (A) Intramolecular hydroxyl radical reactions require rigid substrates like steroids. (B) General design principles for directed organometallic C−H functionalizations. (C,D) Directed organometallic aromatic C−H functionalizations. (E,F) Directed organometallic aliphatic C−H oxidations.
Substrate directing group approaches have significant practical limitations for late-stage functionalizations. In the directing group approach, a substrate must have a pre-existing functionality that is:
Amenable to being chemically transformed into a directing group,
Located at a site that can direct and is desirable for oxidation, and
Compete effectively with other functionality on the substrate for binding to the metal. Despite these limitations, directed C—H oxidations can achieve functionalization at sites that are currently inaccessible to non-directed systems, specifically those sites proximal to and deactivated by electron withdrawing functionality (vide infra,Figure 15).
Molecular Recognition.
Metal porphyrin catalysis explored bioinspired approaches for site-selectivity with elaborate binding pockets to exploit shape (Figure 8) and functional group (Figure 9) recognition. Shape-recognition strategies that create cylinders encapsulating the reactive site showed a slight preference for hydroxylating linear topologies;59, 60 however, the site-selectivities and yields of oxidized products were not synthetically useful. Moreover, serious issues ensued with catalyst turnover due to strong binding of the products, rendering most reactions stoichiometric. Functional group recognition strategies generally require covalent modifications of pre-existing functionality on the substrate and the catalyst to furnish a host-guest interaction.61, 62 This approach has yielded modest yields and site-selectivities of hydroxylated products in steroids. More recently a site-selective hydroxylation of ibuprofen was demonstrated using hydrogen bonding recognition between the carboxylic acid native to the substrate and an acid on the catalyst’s lig-and.63 Molecular recognition approaches to site-selectivity have seen a renaissance in organometallic C—H functionalization.64 The work with steroids supports the views expressed in the literature prior to 20071,2 that methylene C—H bonds could not be preparatively distinguished without molecular recognition elements. Analogous to enzyme engineering, such approaches struggle to provide general solutions across a range of topologically and functionally diverse substrates (Figure 9A, B). Significantly, the same site-selectivities are now demonstrated on steroid core using Fe(CF3-PDP) catalysis without molecular recognition elements (vide infra, Figure 17C, 26D).
Figure 8.
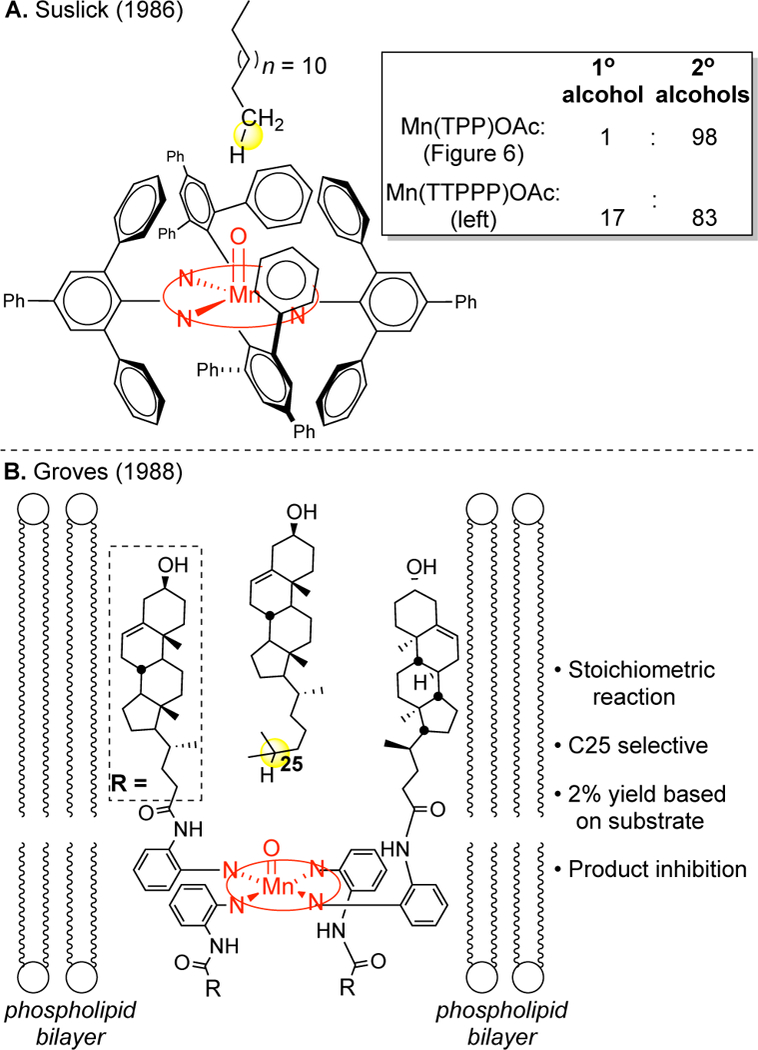
Shape–molecular recognition strategies for controlling site-selectivity in aliphatic C–H oxidations with manganese porphyrins. Panel B: Reproduced with permission from ref 60. (Stereochemistry at C20 of cholesterol corrected.) Copyright 1988 ACS.
Figure 9.
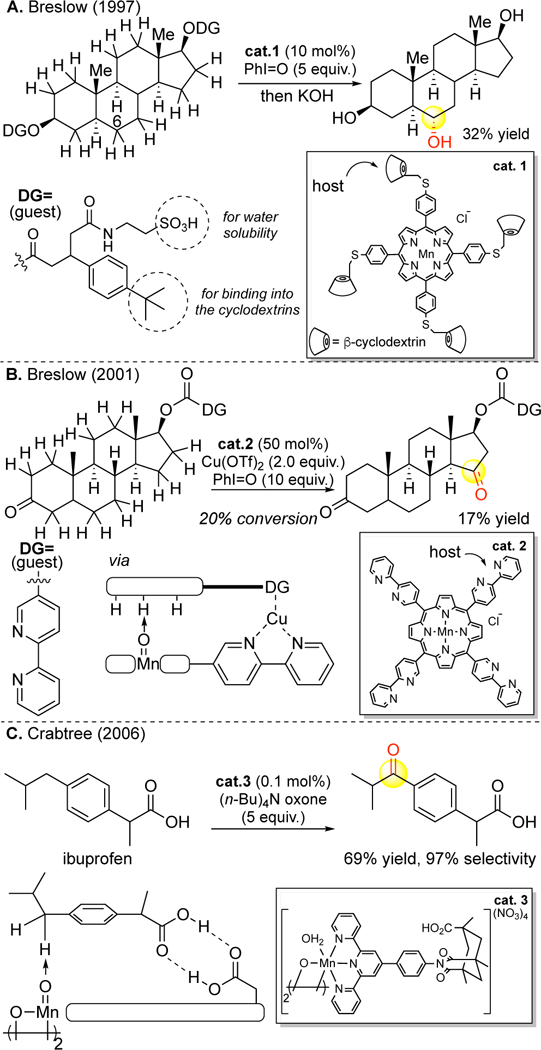
Functional group–molecular recognition strategies for controlling site-selectivity in aliphatic and benzylic C–H oxidations with manganese catalysts. Panel A: Reproduced with permission from ref 61a. Copyright 1997 ACS. Panel B: Reproduced from ref 62. Copyright 2001, with permission from Elsevier. Panel C: From ref 63. Reproduced with permission from AAAS.
The Advent of Fe(PDP) Catalysis
Endothermic C—H Cleavage.
Development of a non-directed system that distinguishes between the small energetic differences needed to effect selectivity in isomeric product ratios (e.g. enanti-oselection between two prochiral faces, 2.7 kcal/mol difference leads to 99:1 e.r.)65 , while providing the driving force to overcome the high energetic barriers needed to effect aliphatic C—H bond cleavage, is a challenging goal. We started with some general ideas of where to find the kind of reactivity and selectivity that would render undirected aliphatic C—H hydroxylations into reactions suitable for chemical synthesis. We endeavored to discover a catalyst where hydrogen abstraction via a metal oxidant is endothermic and proceeds via a late, product-like transition state. One approach is to identify a catalyst that generates a metal(hydroxide) intermediate that forms a weak O—H bond, significantly weaker than water (119 kcal/mol) or alkoxide (104 kcal/mol) formed via Fenton-type reactions. In some aspects this is analogous to the increased site-selectivities achieved with radical bromination28, 66 versus chlorination. The challenge lies in finding an oxygen-based oxidant (typically the most reactive) that renders the C—H cleavage step endothermic. Doing this would enhance the catalyst’s ability to discriminate between subtle electronic, steric, and stereoelectronic differences that we thought would distinguish C—H bonds in complex molecule settings (Figure 10).
Figure 10.
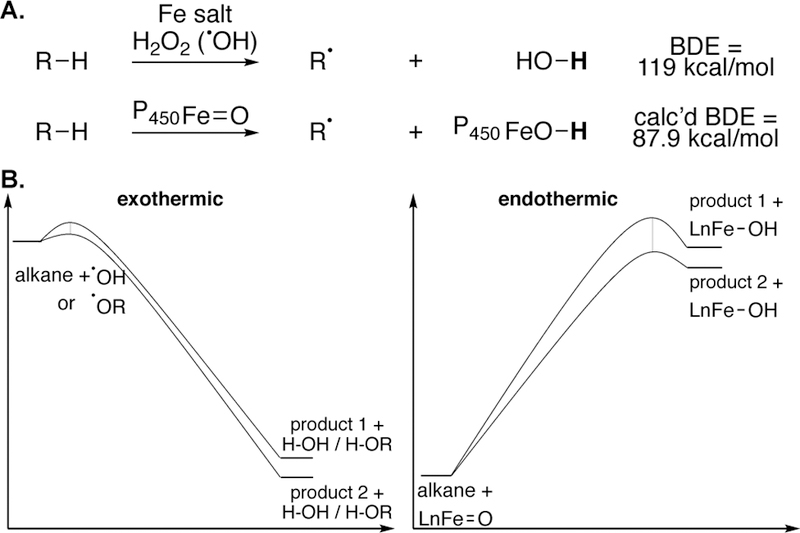
Our guiding hypothesis for developing catalysts for site-selective C–H hydroxylations: the discovery of metal(oxo) intermediates that abstract hydrogen to form weak metal O–H bonds and will proceed via endothermic pathways with late, product-like transition states that may afford sensitivity to differences in the C–H bonds’ chemical environment within complex molecules.
Whereas the Fe(O—H) bond strength of P450 is unknown, theoretical calculations predicted that the C—H cleavage step of an aliphatic C—H bond by P450 is endothermic.67 Strong σ-donating thiolate axial ligation is thought to be an important factor in the selectivity observed with P450 enzymes.68 The failure in P450 mimics to achieve this selectivity (Figure 6) may be due to the inability to tolerate thiolate axial ligation: the reversible nature of monodentate ligand binding to the axial site of a metal(heme) in solution would lead to rapid oxidation of the sulfur.
We hypothesized that irreversible, chelating σ-donating ligation may be more readily achieved using a non-heme ligand framework. Additionally, unlike flat heme ligands, non-heme ligands may adopt coordination to the metal that places steric elements closer to the reactive metal oxidant. We pursued a non-heme ligand design that was modular, easy to modify electronically and sterically. Such general guiding principles ultimately led us to the first synthetically useful system for aliphatic C—H hydroxylations.
Non-Heme Iron Functional Mimic.
The non-heme iron oxidase enzymes (e.g. methane monooxygenase, prolyl-4-hydroxylase) effect highly selective aliphatic C—H oxidations via high valent iron oxo intermediates. However, perhaps because of their flexible, less well-defined, histidine and carboxylate-rich ligand environments, their chemical model systems have been scantily explored. The development and study of structural mimics have led to a more detailed understanding of non-heme iron enzymes such as methane-monooxygenase, however, these systems do not act as functional mimics for aliphatic C—H hydroxylation chemistry.69 In the 1990’s well-defined pyridine/amine ligated non-heme iron (II) complexes began to appear as small molecule functional mimics for the non-heme iron oxidases. The polydentate amine ligands tris(2-pyridylmethyl)amine (TPA)70 and N,N’-dimethyl-N,N’-bis(2-pyridylmethyl)ethylene-1,2-diamine (MEP)71 and others72 were introduced and complexed with iron salts to furnish both monomeric and dimeric iron complexes that showed C—H hydroxylation activity with cyclohexane (Figure 11). Preparatively, these systems did not distinguish themselves from the previous state-of-the-art: reactions were run with large excesses of substrate and afforded trace yields of oxidized products.73 However, mechanistically these systems appeared to us distinct. Most significantly, hydroxylations of cis-1,2-dimethylcyclohexane were stereospecific with no observed trans-alco-hol.73 Labeling experiments with H2O18 showed that the iron-bound oxidant generated from H2O2 was able to exchange with water, suggestive of an iron(oxo) intermediate (Figure 11B).73b
Figure 11.
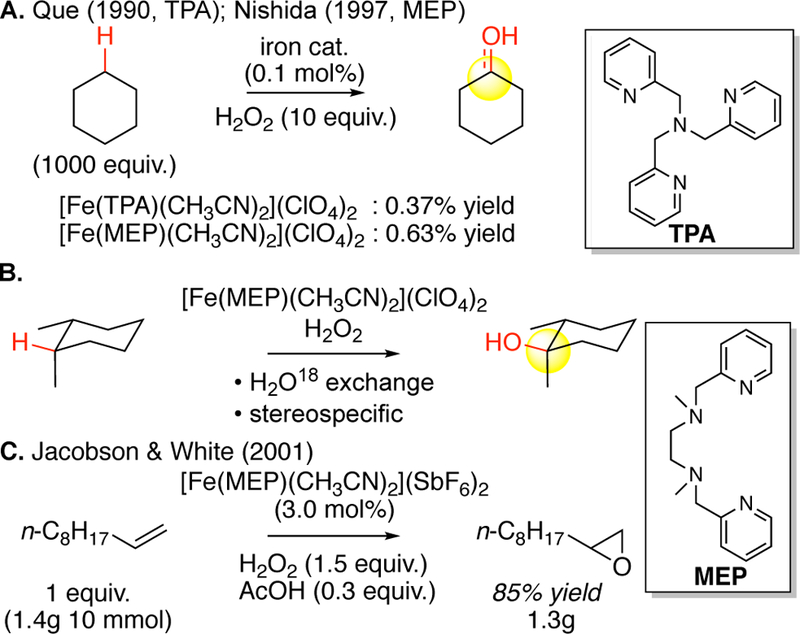
(A) Biomimetic aliphatic C–H hydroxylation reactivity using non-heme iron catalysts Fe(TPA) and Fe(MEP). (B) Aliphatic C–H oxidations catalyzed by Fe(MEP) are stereospecific, and H2O18 labeling studies indicate that they proceed via iron carbonyls. (C) The first example of preparative oxidation catalyzed by a non-heme iron complex is in the epoxidation of terminal olefins.
In the oxidations evaluated, the Fe(MEP) complexes exhibited higher catalytic activity than those of Fe(TPA).71, 73 Additionally, the MEP ligand has strong s-donating character via the bidentate tertiary amine ligation, and a symmetrical, modular structure that al-lows electronic and steric structure-activity relationships to be evaluated in a synthetically straightforward way. In 2001, one of us discovered that [Fe(II)(MEP)(CH3CN)2](SbF6)2 is a preparatively useful catalyst with H2O2 for epoxidations of terminal olefins.74 Albeit a much lower energy process than aliphatic C—H oxidation, this discovery represented the first time a non-heme iron oxidation system had been rendered preparatively useful. Collectively, these considerations led us to pursue the Fe(MEP) system for aliphatic C—H oxidations.
A Breakthrough: Fe(PDP).
A breakthrough was made by our group in 2007 when the MEP system was strategically altered to enable the first preparative, undirected C—H hydroxylation of aliphatic compounds (Figure 12).11 The MEP iron complex itself provided modest selectivity for tertiary C—H hydroxylations (entry 1, 56%, selectivity is defined as yield of desired product/conversion of starting material). Studies on the MEP ligand had shown that in-creased flexibility led to an increased lability of its iron complexes,75 suggesting to us that MEP iron complexes decompose during the reaction to species that catalyze unselective oxidations. We hypothe-sized that increasing the σ-donation of the amine ligands and simultaneously rigidifying the ligand would result in improvements in selectivity. After significant experimentation, exchanging the eth-ylene diamine backbone with pyrrolidine rings76 ultimately furnished Fe(PDP) catalyst ([Fe(II)(PDP)(CH3CN)2](SbF6)2) that afforded a significant improvement in selectivity for C—H hydrox-ylation (entry 2, 92%). A key to the reactivity was the use of acetic acid as an additive. We were inspired to try this first in the epoxidation system and later in the aliphatic C—H oxidation because of the carboxylate rich frameworks of non-heme iron enzymes (Figure 11).69, 74 The acetic acid additive resulted in an increase in catalytic activity without significantly altering selectivity (entries 3,4). Rudimentary mechanistic studies involving varying concentrations of reaction components suggested that catalyst decomposition was occurring via bimolecular pathways. This led to the development of the “iterative addition protocol” where catalyst, H2O2, and acetic acid are added in a portionwise manner three times. Now widely adopted in other C—H functionalizations, this and other.slow addition12c protocols maintain low concentrations of the active oxidant in solution (entry 5). Collectively, this gave a further improvement in yield to afford the first preparatively useful aliphatic C—H hydroxylation reaction for both tertiary (3°) and secondary (2°) aliphatic C—H bonds (vide infra).11, 12 In contrast to the low yields observed with Fe(MEP), Fe(PDP) was now capable of cleaving the strong, nonpolar C—H bonds of cyclohexane (~98 kcal/mol) to furnish high yields of cyclohexanone (92%) based on only 1 equivalent substrate.11
Figure 12.
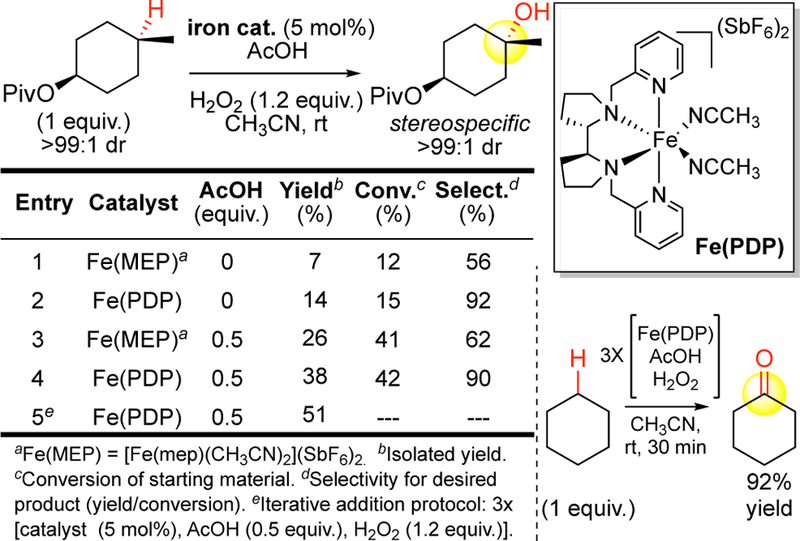
Discovery of Fe(PDP) catalysis for preparative aliphatic C–H oxidations. Table from ref 11. Reproduced with permission from AAAS.
Mechanistic Considerations with Fe(PDP) Hydroxylations.
A biomimetic mechanistic scenario for Fe(PDP)-mediated C—H hydroxylations has emerged where an iron (oxo)carboxylate abstracts a hydrogen to generate an FeO—H intermediate and a very short-lived carbon-centered radical. The Fe—OH rebound to the carbon-centered radical furnishes hydroxylated product and regenerates the Fe(PDP) catalyst. Several lines of evidence support that Fe(PDP)-catalyzed aliphatic C—H hydroxylation proceeds via a metal bound oxidant (likely an oxenoid) rather than a free radical pathway. Site-selectivities are not solely dependent on bond dissociation energies (BDE); numerous examples have emerged of stronger C—H bonds being selectively oxidized in the presence of weaker ones (Figure 13B, vide infra 19C). In the presence of olefins only epoxides are formed with no allylic oxidized products that are typical in oxygen-centered radical reactions (Figure 6D).77 Additionally, the functionalization step is tightly regulated at the metal center. The reaction is stereospecific in substrates with stereochemically defined tertiary stereogenic centers (Figure 13C). Moreover, C—H hydroxylation alpha to cyclopropanes does not lead to ring opened products, even in cyclopropane radical probe experiments (Figure 13C). This indicates that rebound of the Fe—OH to the carbon centered radical must be very rapid, with a very short-lived carbon centered radical (lifetime < 1 × 10 −11 s).78 The profile of oxidation products does not change depending on the reaction atmosphere (i.e. Ar versus air), supporting either no intermediate or a very short-lived one.
Figure 13.
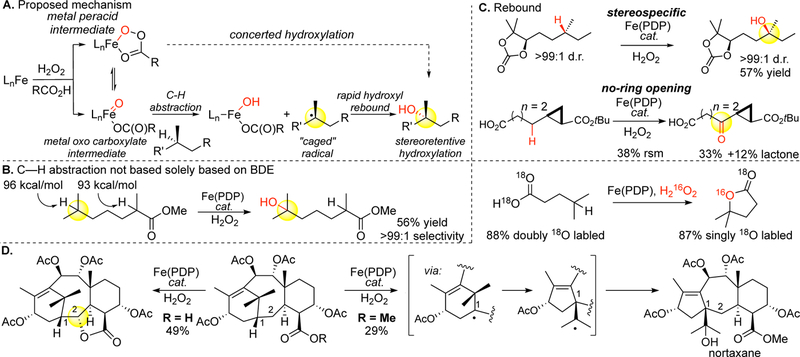
Mechanistic considerations with Fe(PDP) hydroxylations. (A) Proposed mechanism for Fe(PDP)-mediated C(sp3)–H hydroxylation. (B) The site-selectivity in C–H cleavage with the Fe(PDP) oxidant is not solely based on BDE. (C) Oxidations with Fe(PDP) do not scramble stereocenters or open cyclopropane rings, indicating that no long-lived carbon-centered radicals are formed. (D) Evidence for a stepwise mechanism is seen in Fe(PDP) oxidation of a taxane derivative where a ring-contracted nortaxane product is formed. Carboxylic acids on the substrate can direct oxidation away from the site that is intermolecularly favored with Fe(PDP). Collectively this suggests that Fe(PDP) oxidations proceed via a carboxylate-bound oxidant through a late, product-like C–H cleavage step and a rapid hydroxyl rebound step.
Physical organic studies done in our laboratories78 and spectroscopic studies done by others79 on the mechanism of the acetic acid effect suggest that carboxylic acid binding promotes oxo formation by providing a critical proton to the H2O2 to eliminate water. Exposure of 18O-labeled carboxylic acid substrate to the reaction conditions leads to predominantly singly labeled lactone, suggestive that lactonization proceeds predominantly via hydroxyl, not carboxylate, rebound.80 Recently, others have suggested an iron peracid, that could effect a concerted C—H insertion as an intermediate to an iron carboxylate oxo species81 (Figure 13A). Evidence against a concerted mechanism is seen in our oxidation of the taxane skeleton. Oxidation with Fe(PDP) of a taxane derivative at C1 furnishes a nortaxane product, i.e. A-ring contracted oxidized product, likely via a radical rearrangement induced by relief of ring strain.80 Analogous products have been reported in Barton deoxygenation reactions at C1 of taxanes proceeding via carbon centered radical intermediates (Figure 13D).82 When carboxylic acid moieties are incorporated into the substrate, with no additional acid, they can divert C—H abstraction from electronically and/or sterically favored sites. For example, when a carboxylic acid is revealed on the taxane, it diverts intermolecular C—H oxidation at C1 in favor of intramolecular oxidation at C2 to form a diastereomerically pure butyrolactone product (Figure 13D).80 Collectively this supports carboxylic acid binding to the catalyst prior to the active oxidant formation and that a step-wise mechanism is viable.
Selectivity Rules: Electronics, Sterics, and Stereoelectronics
Once preparative utility had been achieved with Fe(PDP), factors influencing site-selectivities for aliphatic C—H oxidations could be systematically delineated for the first time. Mechanistic studies indicated that the iron oxidant (vide supra) is devoid of free radical character, significantly contrasting what was observed with porphyrin catalysts. We hypothesized that such an iron-bound oxidant will achieve a late product-like transition state that can discriminate the electronic differences between hydrogens in complex molecule settings (Figure 14). The pyridyl rings provide the opportunity to install substituents in the approach cone to the reactive iron oxidant providing a second element of steric differentiation. The proposed mechanism of the reaction invokes slight sp2 hybridization in the transition state for oxidation, suggesting alleviation of ring strain and other stereoelectronic considerations may lead to further discrimination in C—H oxidation. Systematic studies were performed on simple substrates designed to test individually the effects of electronics, sterics, and stereoelectronics on the site-selectivity of Fe(PDP) oxidation among C—H bonds of the same relative bond strengths (i.e. 2° or 3°).11, 12 These seminal studies illustrated for the first time that a C—H hydroxylations could proceed with predictable site-selectivities based on electronics, sterics, and stereoelectronics factors and that these selectivities could be large enough to furnish useful yields of mono-oxidized products: 1 equivalent of substrate afforded average isolated yields of 50% for the major mono-oxidized product.
Figure 14.
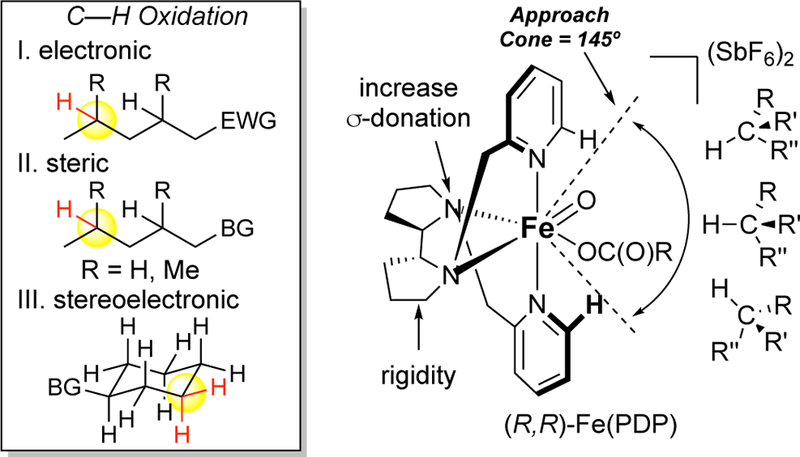
Fe(PDP) is proposed to discriminate C(sp3)–H bonds on the basis of their different electronic, steric, and stereoelectronic environments.
Electronics.
Fe(PDP) is thought to generate a highly electrophilic iron oxidant that proceeds via a late, product-like transition state. We systematically evaluated the hypothesis that such an oxidant will be able to discriminate between C—H bonds based on their electronic properties. In substrates having two tertiary sites, it was delineated that C—H hydroxylation occurred preferentially at the most electron rich tertiary site, most remote from the electron-withdrawing groups (EWG).11 EWG’s could effectively deter oxidation even at sites up to three carbons away. The protection of tertiary amines with HBF4 generated an EWG stronger than halogens14 or acetate11 (Figure 15B). Despite their higher bond dissociation energies and more ubiquitous nature, the same preparative site-selectivities based on electronic effects were observed with Fe(PDP) on methylene (2°) sites (Figure 15C).12
Figure 15.

Fe(PDP) and Fe(CF3-PDP) catalysts discriminate between aliphatic C–H bonds on the basis of their electronic properties. (A) General trends for electronically driven site-selectivities are established. (B) Fe(PDP) affords preparative yields of mono-oxidized products at the more electron-rich site in substrates housing two electronically distinct tertiary C–H sites. (C) Fe(PDP) and Fe(CF3-PDP) afford preparative yields of mono-oxidized product at the site most remote from an electron-withdrawing group in substrates housing multiple electronically distinct secondary C–H sites. aCombined isolated yields for regioisomeric oxidation products.
Since 2007, numerous examples of catalysts/oxidants operating on these now archetypical substrates according to the site-selectivity rules delineated with Fe(PDP) have been shown to effect similar site-selective C—H oxidations,83, 84 chlorinations,85 brominations,85a, fluorinations,87 azidations,88, 89 xanthylations90, and alkylations91 (Figure 16). Many of these reactions proceed via radical C—H abstraction with suprastoichiometric N-or I-centered electrophilic radical species that may not be practical to modify to alter site-selectivity and reactivity. Site-selectivities based on electronic factors have been demonstrated with such species to favor the most electron rich C— H site, generally with modest site-selectivity and reactivity for methylene sites (Figure 16).
Figure 16.
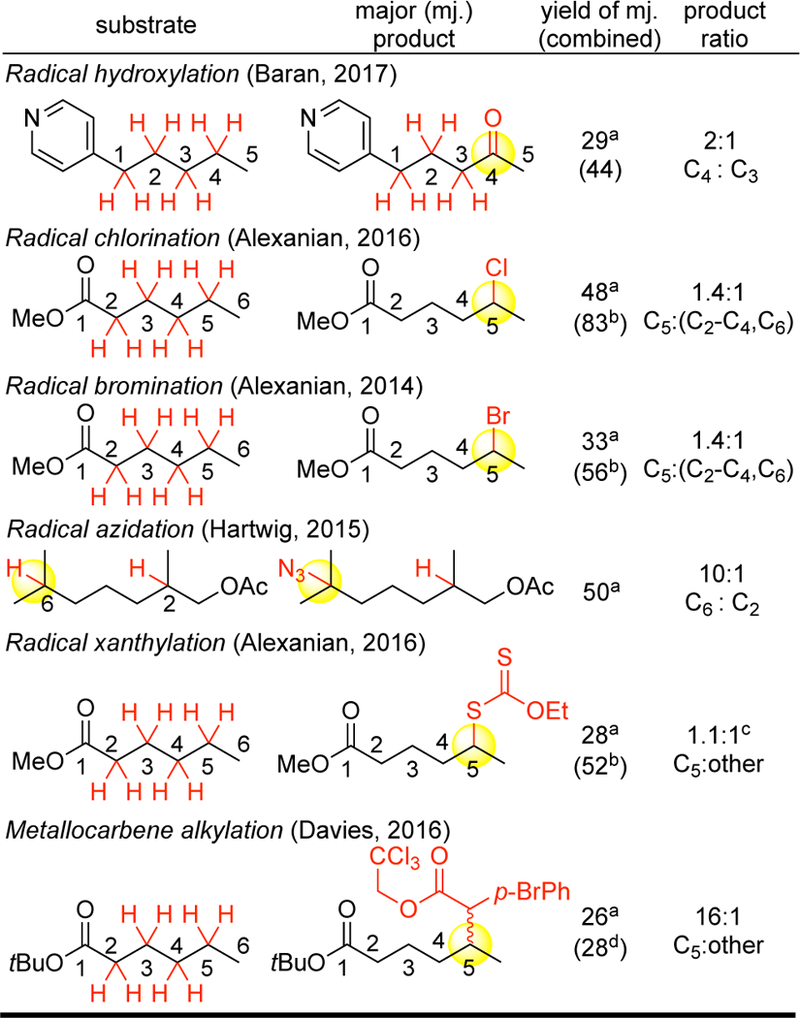
Generality of electronic rules for site-selectivity established with Fe(PDP) for radical and metallocarbene-mediated C–H functionalizations. aCalculated yield of major product. bGC yields. c53% selectivity. d3 equiv of substrate; yield based on substrate.
Sterics.
X-ray crystallographic analysis of the [Fe(II)(PDP)(CH3CN)2](SbF6)2 catalyst revealed a restricted cone of possible approach trajectories of the substrate to the iron oxidant defined by the pyridyls of the PDP ligand (approach cone = 145º, Figure 14). This feature of the PDP ligand offers additional im-portant advantages over porphyrin systems, stoichiometric dioxirane systems, and free radical-based approaches. With the PDP lig-and framework, the iron complex bears steric elements that can easily be tuned in the vicinity of the oxidant, enabling nonbonding substrate/catalyst interactions to provide additional discrimination in site-selectivity. Oxidation with Fe(PDP) was shown to be favored at unhindered sites, with steric bulk on the substrate influencing site-selectivity by shielding sites proximal to it from oxidation (Figure 17A).11 Significantly, the studies on analogous substrates with TFDO92 or a radical promoted azidation reaction89a showed either no selectivity or alternate selectivities based primarily on electronic factors (Figure 17A). In trans-1,2-dimethylcyclohexane, TFDO preferentially oxidizes the sterically hindered axial 3° C—H bond over sterically more accessible methylene sites, likely because it is the most electron rich bond in the molecule with the lowest BDE (Figure 17B).31 In contrast, Fe(PDP), favors oxidation at the sterically more accessible but stronger methylene sites. Steric modifications of Fe(PDP) leading to Fe(CF3-PDP) (vide infra, Figure 22) can en-hance this site-selectivity and furnish even higher levels of discrimination during oxidation between C—H bonds based on their steric environment (3º:2º = 1:2 to 1:10, Figure 17B).13 Comparison of Fe(CF3-PDP) with TFDO oxidation of the androsterone steroid core shows an analogous trend where TFDO oxidization occurs at an axial 3º C—H bond,93 whereas Fe(CF3-PDP) predictably oxidizes at a sterically accessible methylene site (HBF4 is used to mask the basic pyridine nitrogen).14 Underscoring that TFDO site-selectivities are substrate dependent whereas Fe(PDP) site-selectivities are an interplay of catalyst-substrate interactions, no rationale is given as to why TFDO preferentially oxidizes the C14 3º site over other tertiary sites, whereas the site of Fe(CF3-PDP) oxidation can be predicted using a quantitative model for that catalyst.13
Figure 17.
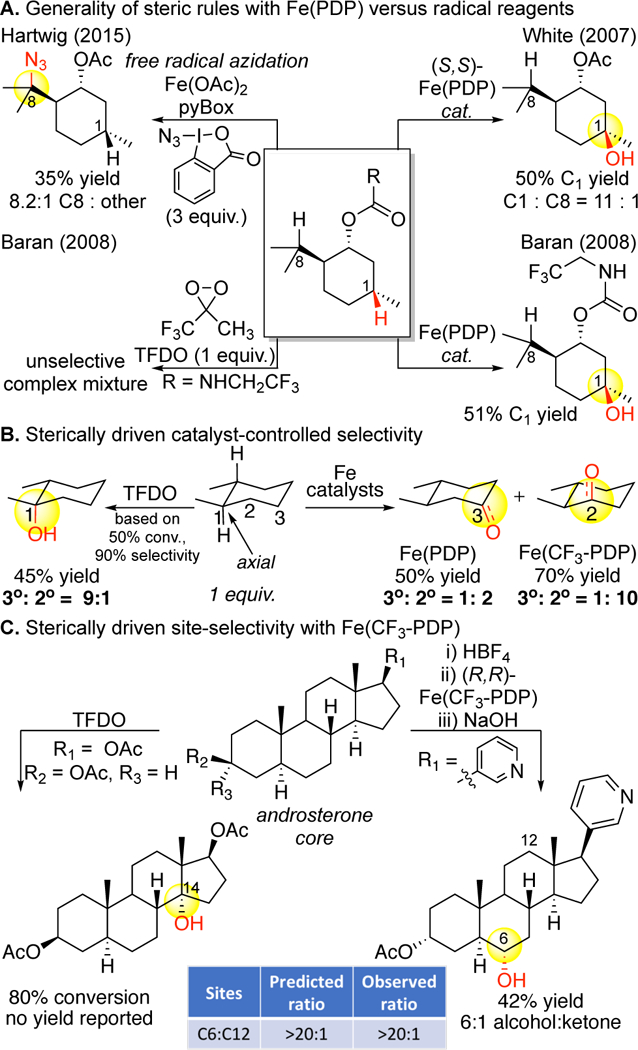
Fe(PDP) and Fe(CF3-PDP) catalysts discriminate between aliphatic C–H bonds based on their steric properties. Preparative yields of mono-oxidized products are seen in Fe(PDP)- and Fe(CF3-PDP)-catalyzed oxidations of (A) a menthol derivative, (B) trans-1,2-dimethylcyclohexane, (C) and an androsterone derivative. Small radical and dioxirane reagents show either no or alternate selectivities.
Stereoelectronics.
Sites that are activated through stereoelectronic effects (e.g. hyperconjugative activation, relief of 1,3-diaxial interactions and torsional strain) additionally undergo facile oxidation with Fe(PDP) and other oxidants (Figure 18A).9c, 12, 90, 94 Although quaternary carbons generally divert oxidations remote from a-hydrogens on a cyclohexane ring, a cyclopropane, having appreciable p-character in the C—C bonding orbitals, can activate these methylenes via hyperconjugation (Figure 18B).12 In radical mediated processes the functionalization step is not linked to C—H abstraction, resulting in long-lived free radical carbon intermediates that scramble stereogenic centers, undergo skeletal rearrangements, and often afford low product yields (possibly because of homodimerization). Cyclopropane-containing substrates were examined in manganese porphyrin hydroxylations, halogenation and azidation reactions that proceed via free radicals and shown to afford large quantities of the ring-opened products (Figure 6C, 18C).50a, 85a, 87, 88 In contrast, with Fe(PDP) catalysis no products from ring opening are observed. Stereoelectronic activation is effective in complex settings with other possible sites of oxidation: a terpenoid substrate afforded as the major product oxidation adjacent to the cyclopropane ring (Figure 18B).12
Figure 18.
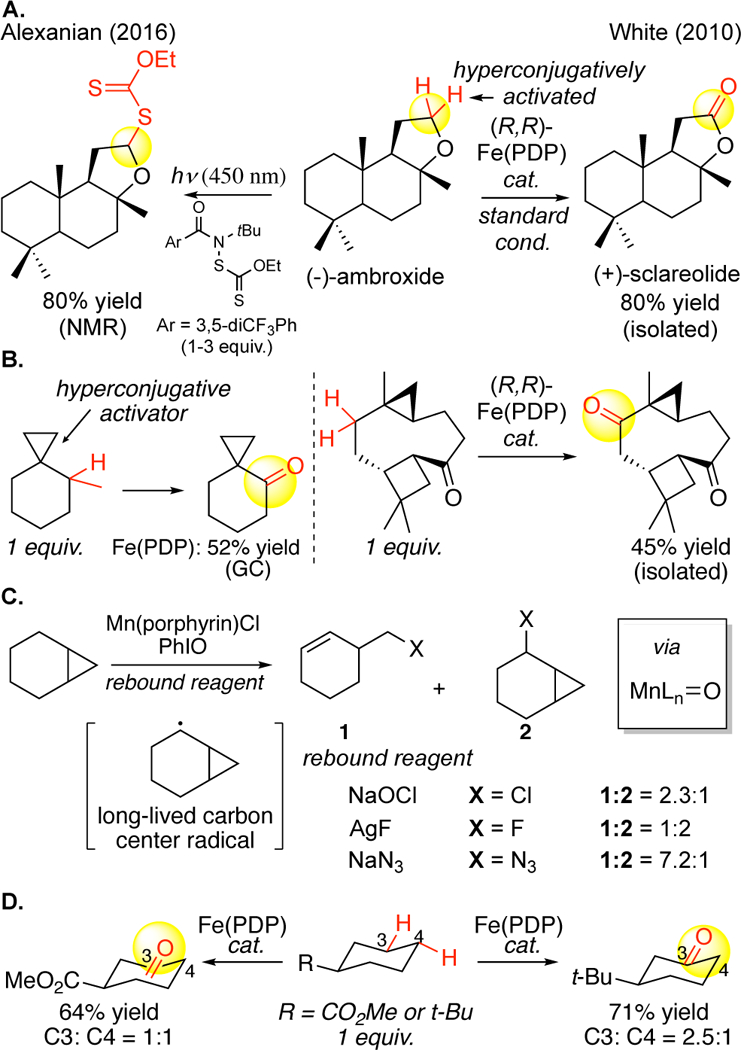
Fe(PDP) discriminates between aliphatic C–H bonds on the basis of their stereoelectronic properties. (A) Oxygen functionality hyperconjugatively activates adjacent methylene sites to furnish preparative yields of mono-oxidized products with Fe(PDP) oxidations and light-promoted radical xanthylation. (B,C) Cyclopropane rings that act as hyperconjugative activators in Fe(PDP) catalysis are opened with radical reagents. (D) Ring-strain release (release of torsional strain) promotes oxidation at C3 of substituted cyclohexane rings with Fe(PDP) catalysis.
Examination of a series of 6-membered rings with electron withdrawing functionality showed no preference for the electronically favored remote (C4) site over the more proximal C3 site (C3:C4 = 1:1, statistically corrected) (Figure 18D). When a more sterically demanding tert-butyl substituent that was no longer electronically deactivating was examined, the site-selectivity favoring C3 increased (C3: C4 = 2.5:1, statistically corrected).12 This trend with simple molecules first suggested that stereoelectronic parameters based on conformational effects (e.g. relief of torsional strain, 1,3-diaxial interactions) will be a contributing factor to the product distributions with Fe(PDP), particularly in cyclohexane oxidations within complex molecules.
Complex Molecules.
The site-selectivity rules that emerged rendered aliphatic C—H hydroxylation predictable for the first time, even in complex molecule settings and enabled the first examples of site-selective late-stage oxidation to appear in the literature that were not on steroidal structures (Figure 19 A, B, C).11,12 When Fe(MEP) was examined in complex molecule settings, significantly lower yields were observed due to unselective oxidations.
Figure 19.
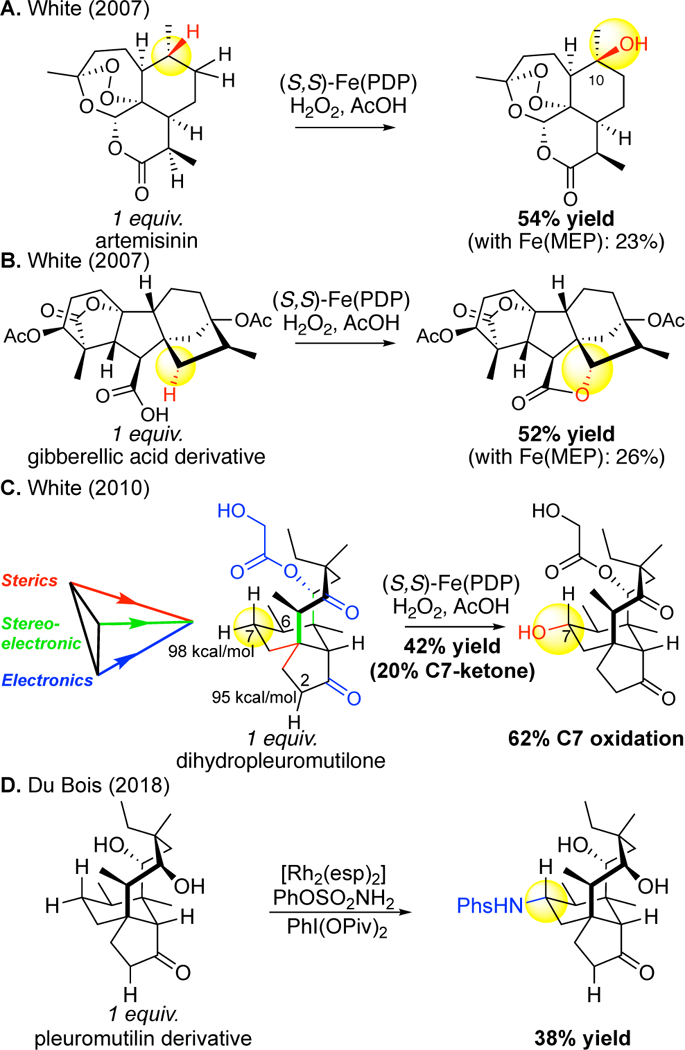
First examples of predictable, site-selective late-stage aliphatic C–H oxidations on complex molecules that are not steroids: (A) artemisinin (2007), (B) gibberellic acid derivative (2007), and (C) dihydropleuromutilone (2010). (D) Analogous site-selectivities are seen in 2018 with rhodium nitrene-based C–H aminations.
Examination of C—H oxidations in complex molecules suggested to us that Fe(PDP) relies on an interplay of interactions with the substrate based on electronics, sterics, stereoelectronics in controlling the site of C—H hydroxylation. When these factors combine constructively, one site is favored towards oxidation. An illustrative example is seen in the site-selective methylene oxidation of a pleuromutilin derivative having a total of 11 possible sites of oxidation (6 methylene, 4 tertiary, and an unprotected hydroxyl moiety, primary C—H bonds are not oxidized with this catalyst).12 The four tertiary sites are electronically (alpha to ketones or ester) and sterically deactivated towards oxidation (C6 3° C—H is in a sterically disfavored axial orientation) (Figure 19C). Regarding methylene oxidation the ketones electronically deactivate the 8- and 5-membered rings, leaving the 6-membered ring’s methylene sites as the most likely for oxidation. The most distal site from the sterically bulky quaternary center is C7; moreover repulsive 1,3-diaxial interactions with the eight-membered ring may be alleviated by C—H bond oxidation at C7. Consistent with this analysis, C7 is the major site of oxidation (62% yield based on 1 equiv. substrate) with the major product being the result of a diastereoselective hydroxylation at the sterically more accessible equatorial C—H bond. It is significant to note that a methylene site at C2 alpha to a carbonyl with a bond dissociation energy ca. 3 kcal/mol lower than the C7 methylene is not oxidized with this catalyst.8 Analogous site-selectivities have now been reported for methylene C—H aminations with rhodium nitrene-based oxidants, albeit with modest yields (Figure 19D).95
Hydroxyls are generally rapidly oxidized to ketones under these oxidation conditions due to hyperconjugative activation of the C— H bond by the newly installed hydroxyl oxygen. If the C—H bond is sterically difficult to access due to a combination of catalyst and sub-strate sterics, a significant amount of the hydroxylated product, often as one diastereomer, is formed (Figure 17C). The development of catalyst systems that stop at methylene hydroxylation to furnish alcohols across a broader scope of substrates is a significant future goal.
The only instances where Fe(PDP) weighs one of these physical organic properties of C—H bonds (electronics, sterics, stereoelectronics) over the others to dictate site-selectivity is in the case of hyperconjugative activation. For example, despite many other sites being electronically and sterically accessible in ambroxide, the methylene next to the ethereal oxygen is oxidized selectively to furnish sclareolide in 80% yield based on 1 equivalent of substrate (Figure 18A, 20).12 Similar electronic, steric, stereoelectronic considerations delineated above can be used to predict sclareolide’s major site of methylene oxidation (C2). Sclareolide has proven to be a “privileged substrate” for a wide range of electrophilic radical C—H functionalizations.85a, 86–88, 90, 95
Figure 20.
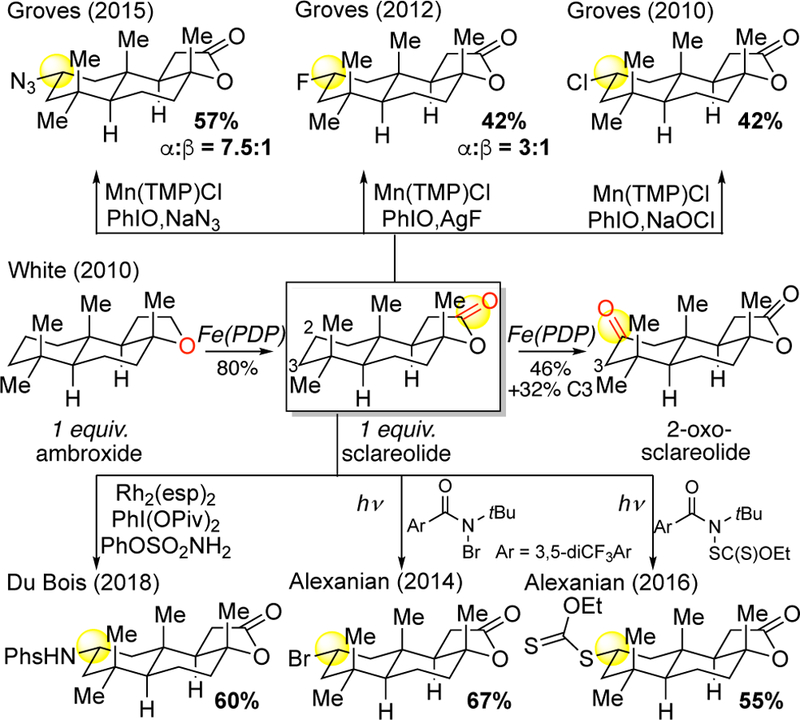
Sclareolide sequential C–H oxidation was first demonstrated with Fe(PDP) catalysis and later as a “privileged substrate” for electrophilic metallonitrene and radical C(sp3)–H functionalizations.
Hindsight.
Our findings and the work that directly followed stands in stark contrast to the mindset prior to 2007 that reactivity differences between unactivated, aliphatic C–H bonds of the same bond type were too small affect preparative levels of site-selectivity.1,2 In 2018 it is widely accepted that this is possible, and others have demonstrated the “Fe(PDP) Rules” in the context of C(sp3)–H azidations, halogenations, aminations, alkylations, etc…17, 24, 84–91, 95 Reviews retrospectively extrapolating examples of electronic, steric, and stereoelectronic effects impacting site-selec-tivities in C—H hydroxylations, aminations, alkylations, and halogenations have also appeared.96, 97 Given this, why were these exam-ples entirely overlooked prior to 2007 both in reviews and in synthetic applications?1, 2, 10 Our view is that most examples were not preparative and the site-selectivities failed to furnish useful amounts of oxidized products (Figure 21).98, 99, 100, 44b A reasonable assumption made by the community is that if the reactivity of the oxidizing species is increased, the site-selectivities disappear. Additionally, the majority of C—H oxidations in complex molecules that were preparative were performed with TFDO on steroid substrates. The privileged nature of steroids (rigid and functionally sparse), the paucity of rationale given for the observed site-selectivities, as well as the inability to alter site-selectivities without changing the molecule contributed to the perception that the selectivities were substrate-specific and not general (Figure 4).33, 34, 35
Figure 21.

Examples of site-selective aliphatic C–H functionalization reactivity prior to 2007 lacked preparative utility.
Altering Site-Selectivities via Catalyst Modifications: Fe(CF3-PDP)
It has been noted that one of the greatest unmet challenges is the development of catalysts that are capable of undirected C—H functionalization of complex molecules that override the “inherent reactivity” of C—H bonds.97 We recognized that the site-selectivities with Fe(PDP) were governed by subtle interactions between the catalyst and the physical organic properties (electronic, steric and stereoelectronic properties) of C—H bonds, i.e. “inherent reactivity”. This can be gleaned from the examples where site-selectivities deviate from those of more simple electrophilic oxidants like TFDO or N- and I-based radicals (Figure 17). Consequently, site–selectivity should be amenable to being altered in a predictable way by designing catalysts that weigh one of the physical organic properties of C—H bonds more heavily than the others. In 2013 our group first demonstrated this with the discovery of catalyst Fe(CF3-PDP).13 Site-selectivities could be predictably altered in aliphatic C—H oxi-dations with no changes made to the substrate or to the reaction. Fe(PDP) was structurally modified to make it more responsive to the steric environment of C–H bonds than to their electronic/stereoelectronic environment. Whereas Fe(PDP) relies on an interplay of interactions, Fe(CF3-PDP) relies primarily on steric interactions with the substrate to control site-selectivities.
In evaluating the 3D-structure of Fe(PDP), a relatively wide cone angle (145°) of possible approach trajectories of the substrate to the ironoxo can be seen so that a combination of electronic and steric/stereoelectronic factors may influence site selectivity. Catalyst modifications that incorporated steric block elements were evaluated with the aim of restricting approach trajectories to render catalyst/substrate steric interactions paramount. Ultimately, Fe(CF3-PDP) was discovered with pendent diCF3aryl rings that narrow the cone of possible approach trajectories to 76°.13 An important consequence of these modifications is that the electron withdrawing CF3 groups render Fe(CF3-PDP) a stronger oxidant, well-suited for oxidizing strong methylene C—H bonds (Figure 22).
Figure 22.
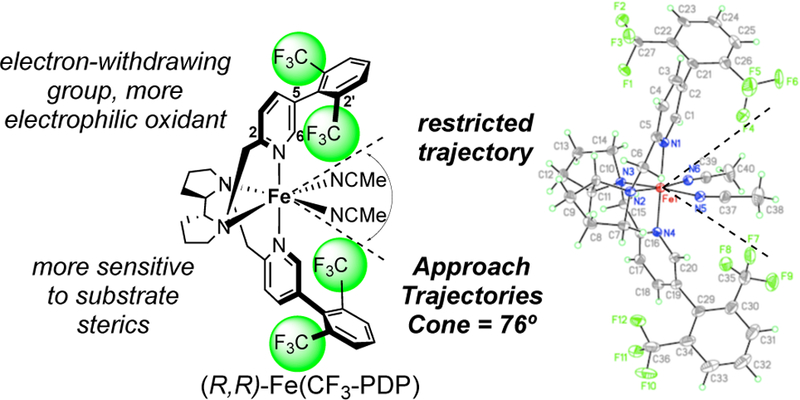
Designing the Fe(CF3-PDP) catalyst to predictably alter site-selectivities. By narrowing the approach trajectory to the active oxidant, Fe(CF3-PDP) weighs the steric environment of C–H bonds more heavily than their electronic/stereoelectronic environment. Reproduced with permission from ref 13. Copyright 2013 ACS.
In contrast to Fe(PDP) whose selectivities are dictated by its interplay of electronic and steric/stereoelectronic effects with the substrate, Fe(CF3-PDP) relies primarily on non-bonding interactions with the substrate to control site-selectivities. Substrates where steric and electronic factors strongly diverge to favor distinct sites are poorly selective with Fe(PDP). In these substrates Fe(CF3-PDP) furnished useful site-selectivities based on its ability to override electronic influences in the substrate in favor of steric ones (Figure 23A). Additionally for substrates that achieve site-selectivity with Fe(PDP) based on a combination of favorable electronics and moderate sterics, alternate site-selectivities can be achieved with Fe(CF3-PDP) based primarily on sterics (Figure 23B).13
Figure 23.
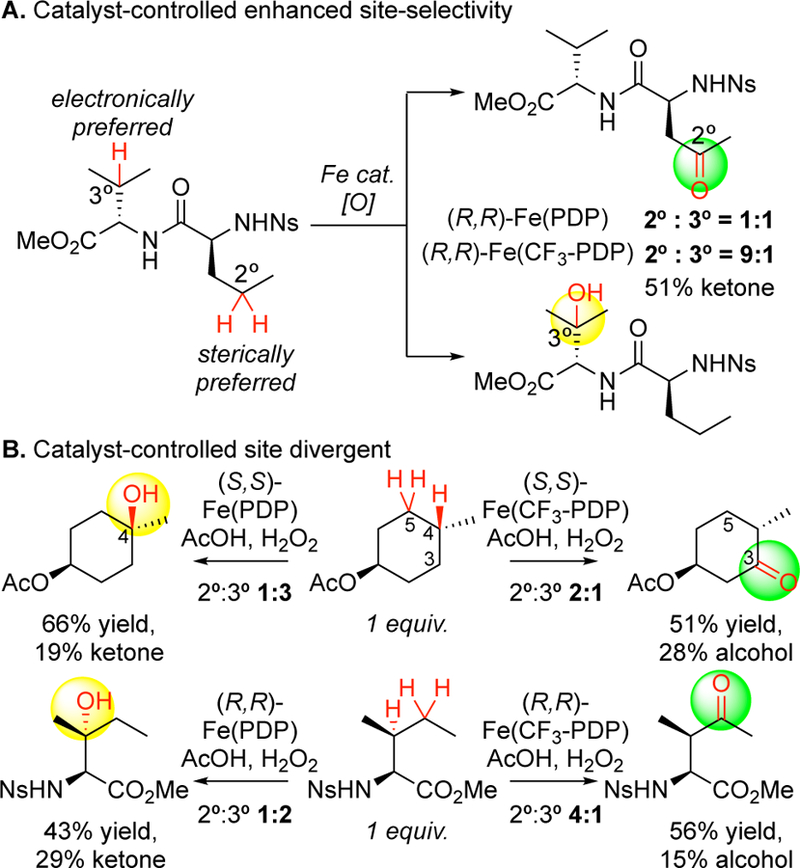
Catalyst-controlled site-selectivities with Fe(CF3-PDP). (A) When electronic and steric preferences at sites on the substrate diverge, Fe(CF3-PDP) overrides electronic preferences in favor of sterics and is selective. (B) In cases where Fe(PDP) selects for a site based on favorable electronics and moderate sterics, Fe(CF3-PDP) overturns that selectivity in favor of the more sterically accessible site.
The discovery of Fe(CF3-PDP) demonstrated that simple catalysts can be designed to predictably alter site-selectivities on complex molecules - with no changes to the molecule or the reaction (Figure 24). In antimalarial compound artemisinin, C10 and C9 that are remote from existing functionality and activated towards oxidation. Fe(PDP) favors aliphatic C—H hydroxylation at the electronically and stereoelectronically preferred C10 site with 2:1 selectivity (C10:C9) that, because all the other sites are deactivated, furnishes a synthetically useful isolated yield of mono-oxidized product (54%). The Fe(CF3-PDP) catalyst overrides this bias in favor of oxidation at the less hindered C9 secondary site-with preparatively useful selectivity (1:11 C10:C9) and isolated yield 52% based on 1 equivalent of substrate.13 The calculated selectivities obtained using the quantitative model (vide infra) are consistent with what is empirically observed. Previously, oxidation at the C9 position had only been accomplished using a P-450 enzyme specifically engineered for artemisinin.12, 101 In contrast, Fe(CF3-PDP) has displayed such catalyst control in numerous substrates of diverse topologies and functionalities. For example, in a recent report on the diversification of the cyanthiwigin natural product class, Fe(PDP) and Fe(CF3-PDP) demonstrated divergent site-selectivities on the same tricyclic core. 102It is significant to note that Fe(PDP) and Fe(CF3-PDP) are chiral catalysts and both the (R,R) and (S,S) versions should be evaluated, particularly in oxidations of rigid, chiral molecules. The diminished yields seen with (S,S)-Fe(PDP) tertiary oxidation relative to (R,R)-Fe(CF3-PDP) methylene oxidation of the cyanthiwigin core may be the result of a mismatch of the chirality of the Fe(PDP) catalyst with the chirality of the substrate.
Figure 24.
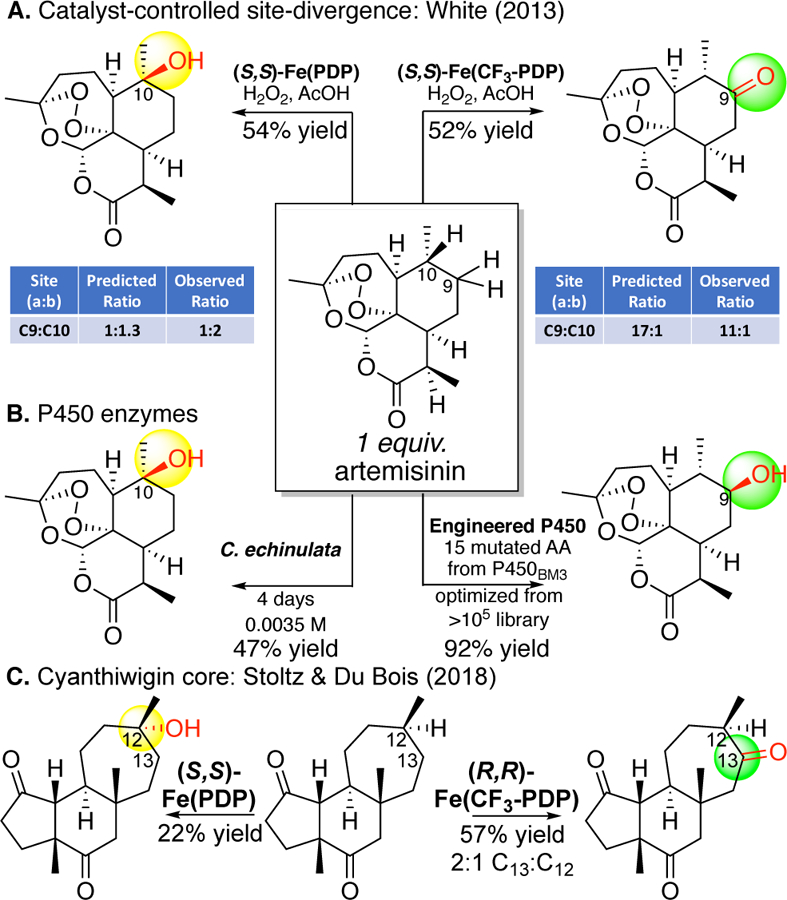
Catalyst controlled site-divergence. (A) Predictable site-divergent selectivities in natural product artemisinin using Fe(PDP) or Fe(CF3-PDP). (B) Prior to Fe(PDP), site-selective oxidations on artemisinin had only been reported with enzymes. (C) Site-divergence in the cyanthiwigin core. The modest yields for tertiary hydroxylation underscores the importance of finding the “matched” version of the chiral Fe(PDP) catalyst with chiral substrates. Panel A: Reproduced with permission from ref 13. Copyright 2013 ACS.
These constitute the first examples where changes made only to a small molecule catalyst preparatively alter site-selectivities- with no change made to the substrate or reaction. Our approach to site-selective catalysis is demonstrating itself to be predictable and highly general as it applies to a range of different reactions and to all molecules that have C—H bonds in different electronic, steric and stereoelectronic environments. Recently, radical azidations,88, 89 chlorinations85b and metallonitrene aminations95 have collectively shown the same site-selectivities on analogous substrates (Figure 25).102 Recently, stoichiometric N-centered radicals reagents have been sterically tuned to alter site-selectivity in C—H chlorinations reactions (Figure 25A).85c Current limitations of these reactions are that they proceed with low yields of C—H functionalized products, particularly at methylene sites, and free radical-based reactions ablate stereocenters at tertiary sites (Figure 25). These limitations may be addressed in the future by the discovery of catalysts that tightly regulate both the C—H cleavage and functionalization steps at the metal center.
Figure 25.
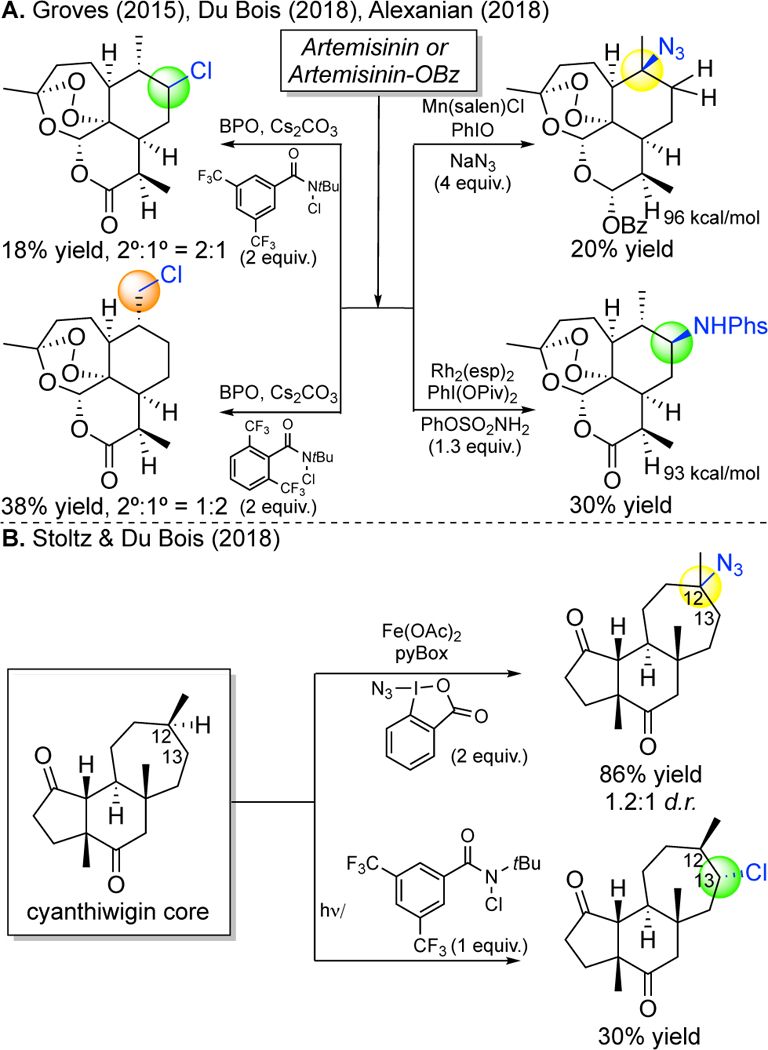
Reactivities and selectivities with radical chlorinations and azidations and metallonitrene C–H aminations in (A) artemisinin and (B) the cyanthiwigin core.
A Predictive, Quantitative Model for the Selectivity Rules
Fe(PDP) and Fe(CF3-PDP) can be viewed as decoders, illuminating the most electron rich/stereoelectronically activated and/or sterically accessible sites in complex molecules. With this fundamental insight, we developed a quantitative model that enables the site of oxidation to be predicted in complex molecules.13 For many complex molecules, electronic, steric and stereoelectronic factors cannot be readily determined by the simple “back of the envelope” analysis prior to performing the oxidation reaction. Such knowledge prior to C—H oxidation is beneficial when choosing which catalyst to use, particularly when the substrate availability is limited. Although 13C NMR spectroscopic methods claim to be predictive for the site of oxidation with small electrophilic oxidants, they cannot be predictive for catalysts that additionally operate based on steric and stereoelectronic factors. 96, 103
The catalyst reactivity models quantitatively correlate the physical organic properties of C—H bonds to the empirical site-selectivities as a function of catalyst (Figure 26).13 A site-filter first identifies likely sites of oxidation that are remote from existing functionality and sterically accessible in a quantitative manner that is based on parameterization of electronic [E = NPA charges] and steric/stereoelectronics [S, assigned based in Winstein-Holness values (A-values)]. These values are systematically categorized across all substrates according to a predetermined algorithm. Only sites that have two highly reactive (red) or one highly reactive and one moderately reactive (red and purple) parameter(s) are considered susceptible to oxidation with either catalyst (Figure 26A). Importantly, because these categorizations are done using the same algorithm, irrespective of substrate, knowledge of the empirical results for the oxidation of a molecule will not bias which sites pass through the filter.
One site is held constant and compared to the other sites that passed through the filter based on electronic (ΔE) or steric (ΔS) differences. This data is inputted into the mathematical models developed for each catalyst based on a correlation of these parametrized physical properties with the empirically observed site-selectivities (Figure 26B). These mathematical models express quantitatively how each catalyst is weighing the differences in electronics, sterics, and stereoelectronics that exist between the C—H bonds of the molecule. The models predict the major site of oxidation and calculate the magnitude and direction of the site-selectivity in complex substrates (Figure 26C). The model for Fe(PDP) supports the empirical observations that Fe(PDP) weighs electronic and steric/stereoelectronic factors equally; whereas Fe(CF3-PDP) weighs sterics more heavily than electronics. The model is predictive for substrates that were not part of the training set, and remarkably even accommodates functionality not incorporated, e.g. basic nitrogen protected via a protonation strategy and amides protected as imidate salts (Figure 17C, 26D).14, 16
Figure 26.

Development of a predictive, quantitative model for site-selective aliphatic C-H oxidations with Fe(PDP) and Fe(CF3-PDP). (A) Parameterization of electronic, steric, and stereoelectronics properties of C-H bonds enables development of a site-filter that identifies likely sites for oxidation with PDP catalysts that are remote from existing functionality, relatively unhindered, and stereoelectronically activated. (B) Development of a predictive model entails mathematically correlating the substrate properties with the empirically observed site-selectivities as a function of catalyst. (C) Predicted site-selectivity on a nectaryl derivative. (D) Predicted site-selectivity on a finasteride derivative. (E) A linear correlation was found between the predicted and observed site-selectivities as a function of catalyst. Panels B/E: Reproduced with permission from ref 13. Copyright 2013 ACS.
Comparing the calculated and empirical ΔΔG for Fe(PDP) and Fe(CF3-PDP) for all substrates used to create the models provides a good linear correlation (Figure 26E). This study represents the first quantitative evidence that the physical organic parameters of electronics, sterics, and stereoelectronics of C—H bonds correlate to preparative site-selectivities in C—H oxidation and that this relationship can be varied as a function of the catalyst.13 Since 2007, these site-selectivity rules have been routinely applied to analyze the observed site-selectivities in undirected C—H oxidation reactions.17, 24, 84–91, 94–97
The Advent of Late-Stage Functionalization
Natural Product Diversification.
Historically, examples of late-stage natural product C–H functionalizations were limited to stoichiometric dioxiranes reactions involving hydroxylation of tertiary sites in steroidal substrates (Figure 4C).34, 35, 93, 104 Oxidation of 2° C—H sites on steroids appeared to require a molecular engineering approach for small molecule catalysts (Figure 9).61, 62 Fe(CF3-PDP) catalysis has demonstrated the ability to introduce oxygen on analogous steroids with the same site-selectivities without the requirement for elaborate catalyst designs and substrate modifications: the 2° C6a position in an abiraterone acetate analog and the 2° C15 positions in a finasteride analog can be predictably site selectively oxi-dized with Fe(CF3-PDP) catalysis (Figure 17C, 26D).14, 16 Fe(PDP) or Fe(CF3-PDP) catalysis has additionally demonstrated the ability to effect site-selective and -divergent aliphatic C—H oxidations in other medicinally important classes of natural products: terpenes, peptides, alkaloids. In all cases, mono-oxidized products can be furnished predictably and in preparative yields (Figure 2).11–16
Nature’s iron oxidation enzymes are critical components in the biosynthesis of structurally and functionally diverse secondary metabolites that have impacted human health (Figure 1). Fe(PDP) and Fe(CF3-PDP) catalysis has enabled for the first time a nonribosomal peptide synthesis (NRPS) inspired strategy for diversification of amino acids (preassembly modifications) and peptides (post-assembly modifications) to be implemented in an in vitro setting.15 The targeted C—H oxidative modifications of amino acids with preservation of acenter chirality enabled four chiral pool amino acids to be transformed into twenty-one unnatural chiral amino acids (Figure 27A). Late-stage C—H functionalization of a single tripeptide with Fe(PDP) catalysis furnished eight tripeptides, each having distinct unnatural amino acids. In addition to generating diversity in the chiral pool, such a late-stage functionalization strategy streamlines synthetic sequences. For example, traditional chemical routes to make all eight peptides would require eight de novo syntheses, including separate syntheses of the unnatural, chiral amino acid building blocks (Figure 27B). Because the iron catalysts are able to oxidize proline selectively at any position in a peptide, a proline residue in a pentapeptide macrocycle used as a turn element to promote cyclizations, can be transformed into a linear ornithine residue, inducing a major change in the peptide structure (Figure 28).
Figure 27.
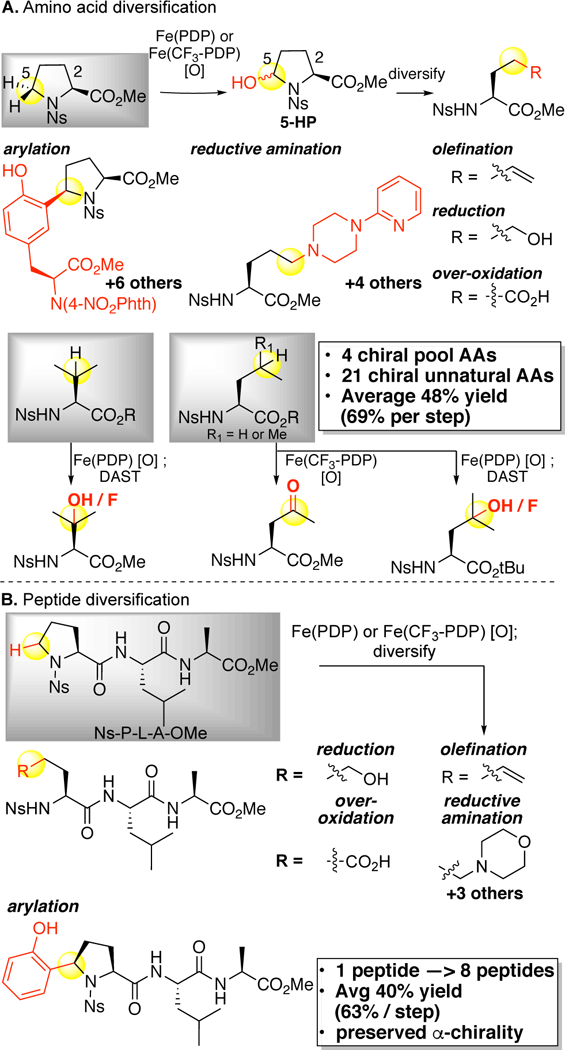
Fe(PDP)- and Fe(CF3-PDP)-catalyzed oxidative diversification of amino acids and peptides. (A) Four chiral pool amino acids are transformed into 21 chiral unnatural amino acids. (B) Late-stage oxidative derivatization of one peptide transforms it into eight peptides with preserved α-chirality.
Figure 28.
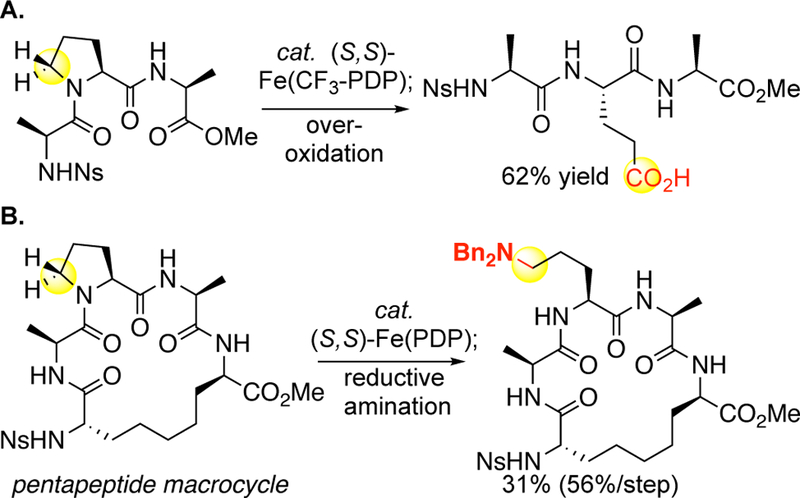
Late-stage oxidation of internal proline residues. (A) A tripeptide with an internal proline residue undergoes Fe(CF3-PDP) oxidation to a glutamic acid residue. (B) Internal prolines may be incorporated into linear peptides to facilitate cyclization and then transformed at a late-stage via Fe(PDP) oxidation to linear residues (e.g., an ornithine derivative).
The scope for such late-stage functionalization reactions where no requirement exists for a specific substrate topology or functionality has the potential to be unlimited.
Streamlining synthesis.
Prior to 2007,1, 2, 10 sporadic examples of intramolecular C—H alkylations and intramolecular C—H aminations using pre-existing functionality to direct reactivity were used in complex molecule synthesis. Novel disconnections were showcased however, a clear strategic advantage to more traditional routes was not evident. For example, in the 2003 synthesis of tetrodotoxin that uses both intramolecular C—H alkylation and amination reactions to forge key C—C and C—N bonds, the overall step count is slightly longer than the first synthesis reported in 1972 (32 steps versus 29 steps, respectively).105, 106 After early demonstrations with allylic oxi-dations,9, 20a it is now being demonstrated that applying aliphatic C— H oxidation in total synthesis can greatly streamline synthetic sequences. By enabling key oxygen installation at a late-stage, C—H oxidations circumvent wasteful protection/deprotections, oxidation state changes and other manipulations that are often necessary when carrying such reactive functionality through synthetic sequences.17, 22–23
Most recently, this strategy has been demonstrated in the total syntheses of densely functionalized polyketides and terpenes.21–23 In the total synthesis of gracilioether F, a polyketide housing 5 stereo-centers at its core, Fe(PDP) C—H hydroxylation was used as a method to install the tertiary hydroxyl in the last step of the synthesis (Figure 29A).21 Total synthesis of the scaparvins, a family of natural products featuring three contiguous stereogenic centers in a densely functionalized decalin core, featured installation of the tertiary alcohol via Fe(PDP) C—H oxidation at step 18 of a 22 or 23 step sequence for scaparvin C and scaparvin B/D, respectively.23 This late-stage oxygen installation significantly streamlines the synthesis by reducing the burden inherent to manipulating multiple oxygens in close proximity throughout the synthesis.
Figure 29.
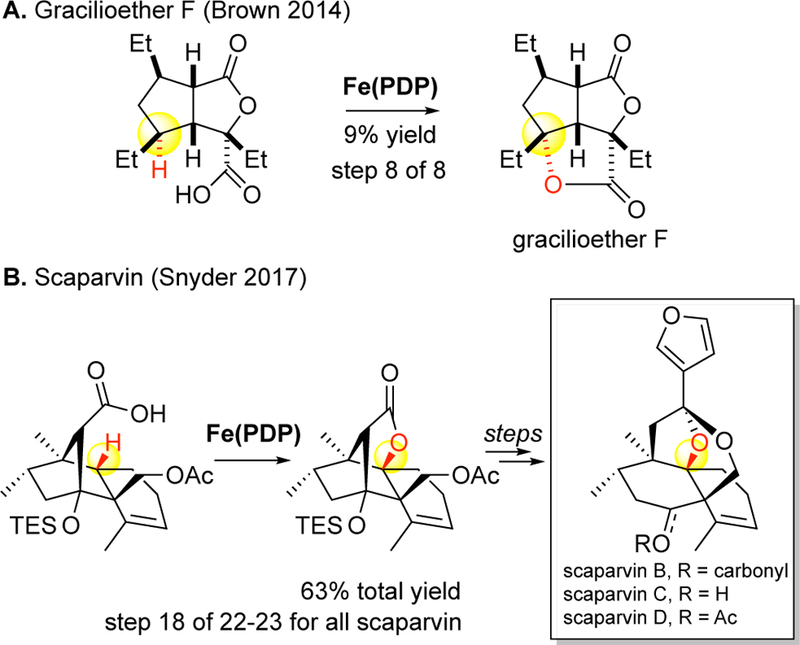
Late-stage C–H oxidations with Fe(PDP) to streamline total syntheses of densely functionalized natural products: (A) gracilioether F (Brown 2014, ref 21) and (B) scaparvin (Snyder 2017, ref 23).
Future Opportunities and Challenges
Continuing to Alter Site-Selectivity.
It has been simplistically professed that catalyst control requires overriding the inherent reactivity of C—H bonds based on steric and electron properties.97 We and others have shown that this “inherent reactivity” depends very much on the catalyst/oxidant being used. Ultimately, what will most impact chemical synthesis is to have predictable and general control over site-selectivity in C—H oxidations. Importantly, for such reactions to be maximally impacting, the catalysts must exploit selectivity modes that are general and potentially applicable to all classes of small molecules having differentiated C—H bonds. Rather than “overriding inherent reactivity” we should seek to discover catalysts that read the physical properties of C—H bonds in different ways.
We recognized in 2007 that, perhaps outside of petrochemicals, all molecules (i.e. pharmaceuticals and natural products) have C— H bonds in different electronic, steric, and stereoelectronic environments. With this realization we were able to first demonstrate and delineate how these differences could be exploited to effect preparatively useful site-selectivities.11, 12 An important feature of Fe(PDP) and Fe(CF3-PDP) catalysis is the demonstrated ability to structurally tune the catalyst to modify how it responds to these different environments within the molecule.13 In principle, it is now possible to continue to alter site-selectivities by designing catalysts that discriminate differentially between C—H bonds on the basis of varying combinations of these physical organic properties.
Increasing Chemoselectivity.
The major challenge with metal(oxo) catalysts is that they traditionally do not tolerate π-functionality such as olefins, aromatics or basic nitrogen. The exceptions to this are aromatics with very electron-withdrawing functionality (e.g. OTf, CF3, NO2, Figure 30B, C) and nitrogen bound to electronically and or sterically deactivating groups (e.g. imides, Figure 30A).14 The presence of lowlying non-bonding and p-orbitals renders such functionality easier to oxidize than aliphatic C(sp3)—H bonds. Nitrogen functionality may promote a-oxidation via hyper-conjugative activation (as seen in amino acids, Figure 27)15 or additionally bind to the catalyst and deactivate it towards oxidation. Given the prevalence of such functionality in medicinally relevant compounds and natural products (vide supra), this limitation must be overcome for widespread use in late-stage functionalization.
Figure 30.
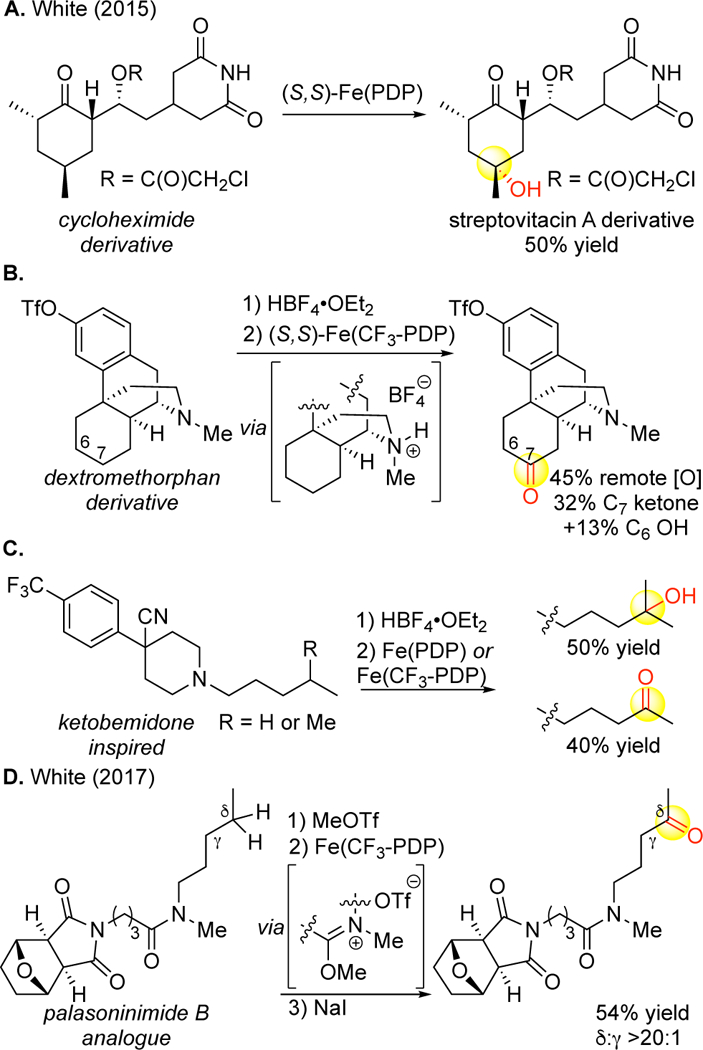
Strategies for expanding basic nitrogen tolerance in aliphatic C–H oxidations. (A) Imides are tolerated with no protection. (B,C) Tertiary amines can be masked via protonation as HBF4 salts or via BF3 complexation (not shown) to promote remote C–H oxidation. (D) Amides and lactams (not shown) can be masked as imidate salts to promote remote C–H oxidations.
We have reported a complexation strategy with Lewis acids (BF3·Et2O) and/or Brønsted acids (HBF4)14 for basic nitrogen and an imidate salt16 strategy for simple amides and lactams that enables Fe(PDP) and Fe(CF3-PDP) to perform remote tertiary and methylene aliphatic C—H hydroxylations in the presence of such functionality (Figure 30, 15B, 15C, 26D). This enables late-stage aliphatic C—H hydroxylations to be performed for the first time on alkaloid14 and β-lactam16 natural products and a wider range of pharmaceuticals (e.g. pyridines, piperidines, amides, Figure 30B-D).
Unfortunately, the vast majority of p-systems cannot be practically protected from oxidation. The development of catalyst systems that chemoselectively oxidize strong aliphatic C—H bonds in the presence a broader range of medicinally relevant aromatics (e.g. aryl halides), non-basic heterocycles, and olefins is an important future goal. Ultimately such chemoselectivity will render this methodology as powerful for the construction of C(sp3)—O bonds as cross-coupling reactions have been for C(sp2)—C(sp2) bond formation.
Metabolism.
One of the main mechanisms for clearing drugs from the body is metabolism, where the drugs are converted to new, often more hydrophilic chemicals that may be rapidly excreted from the body. Of the enzymes participating in metabolism, CYP450’s are considered the most important because of the wide range of organic functionality they are capable of metabolizing via oxidation. In addi-tion to aliphatic C—H hydroxylations these enzymes perform aromatic hydroxylations, heteroatom dealkylations, N- and S-oxygenations, dehydrogenations, and epoxidations.107, 108 Oxidative metabolism generally diminishes the activity of the parent compound, however in some cases metabolites have deleterious (e.g. off-target toxicity) or beneficial pharmacological potency. Metabolites with retained or even enhanced desired activity can result in their advancement as the clinical candidate (e.g. piragliatin for type 2 diabetes, Figure 31).109 Rapid identification of the metabolite (MetID) and its production on a preparative scale is of critical importance to the pharmaceutical industry for further biological evaluation. Currently such studies are done with biotransformations using CYPs or Fenton systems (Udenfriend system)110 that are not preparative, generally necessitating the use of multistep syntheses to provide metabolites on scale (Figure 31B). The development of chemoselective catalysts capable of enabling preparative aliphatic C—H oxidations in the presence of medicinally relevant aromatic functionality stands to streamline metabolite identification and production. Current catalyst systems that tolerate such functionality only do so in the oxidation of weaker C—H bonds (e.g. benzylic).
Figure 31.
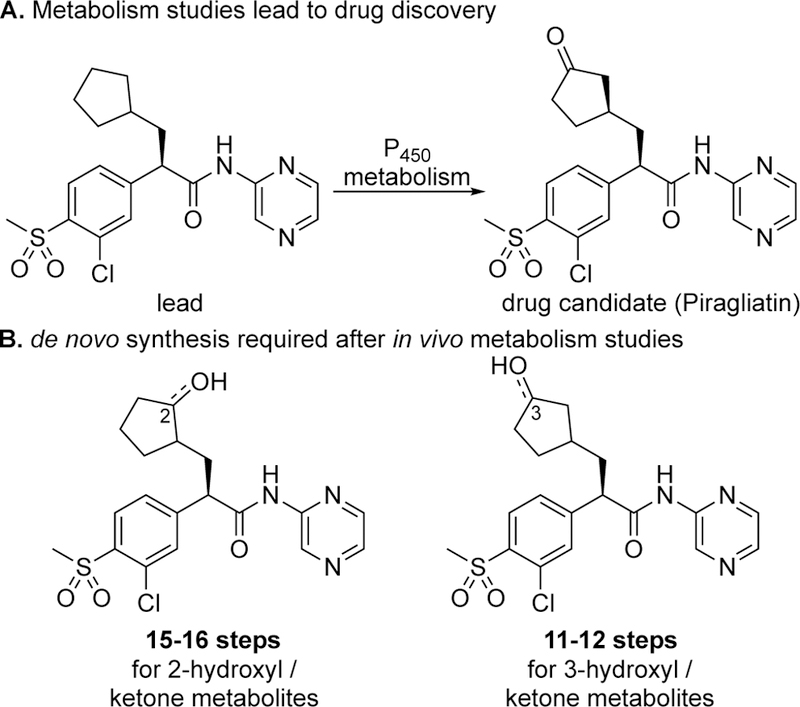
Application of late-stage C–H oxidation to rapidly identify drug metabolites and produce them on a preparative scale is a future opportunity. (A) Piragliatin, a drug candidate for diabetes, was discovered via a MetID study of the lead compound. (B) Independent total syntheses were required to identify the structures of the possible cyclopentane ring metabolites and fully evaluate their biological activities.
Natural Products as Modifiable Drug Leads.
Natural products have been the most successful single source of drugs, with a large number of currently marketed drugs being of natural product origins.111 Interestingly, the pharmaceutical industry has shifted its focus to synthetic compound libraries even though the results have not met expectations with declining numbers of new drugs reaching the market. Pharmaceutical companies have increasingly distanced themselves from natural product drug discovery research in large part because of the difficulty in modifying natural product drug leads. As with all drug leads, structural modifications may be needed to increase potency, improve solubility, decrease side-effects, improve metabolic stability. Modifications of natural products in drug discovery have primarily been done by altering existing functionality on the molecule.4a, 112 If this fails to produce the desired outcome, adding new functionality is rarely feasible in a time and cost-effective way. In order to add new functionality to the core of a complex molecule, lengthy de novo syntheses or developing enzymatic methods is necessary,113 presenting significant technical challenges to drug discovery with natural products. The advent of catalysts that pro-mote site-selective, non-directed installation of oxygen, nitrogen, and alkyl moieties on broad classes of natural products should be viewed as an opportunity to reinvigorate this rich approach to drug discovery.
Nitrogen and Carbon.
A long-term challenge for the field is to develop non-directed, aliphatic C—H amination and alkylation reactions that proceed with the same level of tunable site-selectivity, functional group tolerance, stereospecificity and reactivity as the aliphatic C—H hydroxylations. It is well established that such atomistic changes can significantly alter the chemical and medicinal properties of small molecules (Figure 32).114, 115
Figure 32.
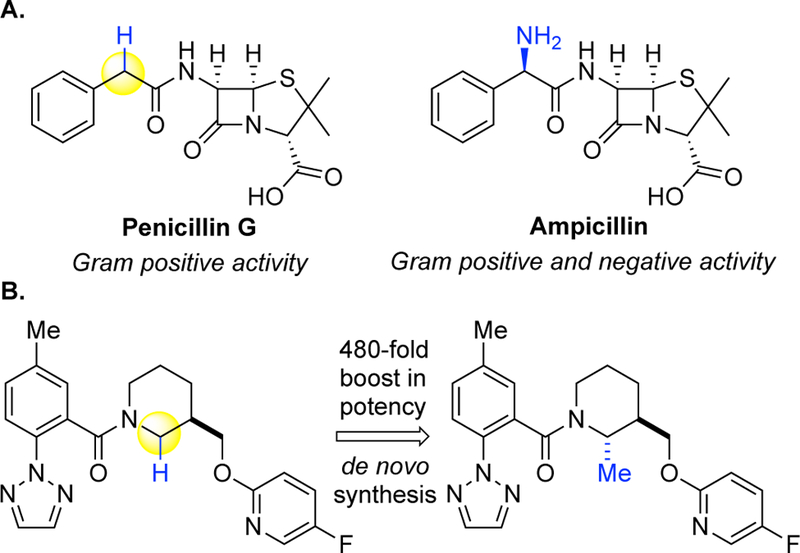
Atomistic change of C(sp3)–H to C(sp3)–N and C(sp3)–C impact a molecule’s properties. (A) The first broad-spectrum penicillin derivative (ampicillin) differs from penicillin G by the introduction of a benzylic amine. (B) The introduction of a methyl group, often alpha to heteroatoms, can significantly boost the potency of drug candidates and is referred to as the “magic methyl effect”.
Catalysts that proceed via high-valent metal nitrene and carbene species via late, product-like transition states for C—H abstraction with rapid rebound steps are excellent prospects for continuing to discover aliphatic C—H oxidation reactions that advance with catalyst tunable site-selectivity, functional group tolerance and stereo-specificity. In principle, both the C—H cleavage step and the functionalization can be tightly regulated and tuned via ligand/metal modifications. Intramolecular C—H aminations proceeding via manganese nitrenes have recently been reported showing a remarkable combination of high reactivity (aminate primary aliphatic C— H bonds) and chemoselectivity (tolerate olefin functionality).116 Thus far, preparative intermolecular C—H aminations and alkylations are limited to activated bond types (Figure 33) primarily because of low reactivity (Figure 25).91, 95, 117–118
Figure 33.
A Mn(ClPc)-catalyzed intermolecular benzylic C(sp3)–H amination that shows site-selectivity and chemoselectivity trends analogous to those established with Fe(PDP) hydroxylation catalysis.
Continued advances in chemoselectivity and divergent site-selectivity in Fe(PDP) hydroxylations as well as the discovery of analogous reactions for introduction of nitrogen and carbon are needed. Analogous to cross-coupling reactions for C(sp2) scaffolds, such reactions will ultimately empower scientists to fully explore C(sp3) complex molecule scaffolds for function.
ACKNOWLEDGMENT
We thank the National Institutes of Health NIGMS (previously R01 GM 112492; now R35 GM122525), the University of Illinois at Urbana-Champaign in the form of start-up funds, and Pfizer for their generous support of this work. We are indebted to our current and former co-workers for insightful discussions featured throughout the perspective. M.C.W. gives special thanks to Prof. Mark S. Chen, Dr. Paul E. Gormisky, and Mr. Jinpeng Zhao for seminal studies on these and future systems and Dr. William (“Rick”) Ewing and Dr. Antonia Stepan for helpful discussions and references on the role of hydroxyl functionality in drug discovery and metabolism.
Footnotes
Notes
The authors declare no competing financial interest.
REFERENCES
- (1).Bergman RG Nature 2007, 446, 391. [DOI] [PubMed] [Google Scholar]
- (2).Godula K; Sames D Science 2006, 312, 67. [DOI] [PubMed] [Google Scholar]
- (3).(a) The hydroxyl group of Lyral® imparts the Lilly of the Vally odor to this aroma chemical and removal of the hydroxyl leaves the compound odorless: Rossiter KJ Chem. Rev 1996, 96, 3201. [DOI] [PubMed] [Google Scholar]; (b) The natural product (+)-valencene that gives oranges their characteristic flavor can be enzymatically oxidized at just one site to furnish b-nootkatone, a compound that now tastes like grapefruit: Rouseff R; Perez-Cacho PR Citrus Flavour. In Flavours and Fragrances: Chemistry, Bioprocessing and Sustainability; Berger RG, Eds.; Springer-Verlag: Berlin, Heidelberg, 2007; pp 117. [Google Scholar]
- (4).(a) Paclitaxel (Taxol®) a highly oxygenated diterpenoid clinically used for the treatment of a range of aggressive cancers (ovarian, breast, lung, pancreatic) looses its ability to polymerize tubulin and to effect cytoxicity upon removal of the C2 benzoate: Chen S-H; Farina V Paclitaxel Structure-Activity Relationships and Core Skeletal Rearrangements. In Taxane Anticancer Agents: Basic Science and Current Status; Georg GI; Chen TT; Ojima I; Vyas DM, Eds.; ACS Symposium Series 583; American Chemical Society: Washington, DC, 1994; pp 247. [Google Scholar]; (b) Erythromycin A, a clinically used antibiotic, is rendered 2–4 fold less active upon removal of the C6 hydroxyl moiety: Weber JW; Leung JO; Swanson SJ; Idler KB; McAlpine JB Science 1991, 252, 114. [DOI] [PubMed] [Google Scholar]; (c) Zambias RA; Hammond ML; Heck JV; Bartizal K; Trainor C; Abruzzo G; Schmatz DM; Nollstadt KM J. Med. Chem 1992, 35, 2843. [DOI] [PubMed] [Google Scholar]; (d) The hydroxyl functionality found in HIV (human immunodeficiency virus) protease inhibitors are critical for binding to the active site via H-bonding to catalytic aspartate residues. Removal of the hydroxyl group in the HIV medication ritonavir has led to the drug cobicistat, having greatly reduced anti-HIV activity but retaining potent inhibitory activity of liver enzymes (CYP3A): Xu L; Liu H; Hong A; Vivian R; Murray BP; Callebaut C; Choi YC; Lee MS; Chau J; Tsai LK; Stray KM; Strickley RG; Wang J; Tong L; Swaminathan S; Rhodes GR; Desai MC Bioorg. Med. Chem. Lett 2014, 24, 995. [DOI] [PubMed] [Google Scholar]
- (5).Davies NM Clin. Pharmacokinet 1998, 34, 101. [DOI] [PubMed] [Google Scholar]
- (6).Jiang W; Cacho RA; Chiou G; Garg NK; Tang Y; Walsh CT J. Am. Chem. Soc 2013, 135, 4457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Rawlings BJ Nat. Prod. Rep 2001, 18, 190. [DOI] [PubMed] [Google Scholar]
- (8).Luo Y-R; Cheng J-P Bond Dissociation Energies. In CRC Handbook of Chemistry and Physics, 99th Edition (Internet Version 2018); Rumble John R., Eds.; CRC Press/Taylor & Francis: Boca Raton, FL, 2018; pp 9–72. [Google Scholar]
- (9).(a) Fraunhoffer KJ; Bachovchin DA; White MC Org. Lett 2005, 7, 223. [DOI] [PubMed] [Google Scholar]; (b) Covell DJ; Vermeulen NA; Labenz NA; White MC Angew. Chem. Int. Ed 2006, 45, 8217. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Wender PA; Hilinski MK; Mayweg AV Org. Lett 2005, 7, 79. [DOI] [PubMed] [Google Scholar]
- (10).(a) Corey EJ; Cheng X-M The Logic of Chemical Synthesis; John Wiley, 1995. [Google Scholar]; (b) Nicolaou KC; Sorensen EJ Classics in Total Synthesis: Targets, Strategies, Methods; Wiley-VCH, 1996. [Google Scholar]; (c) Nicolaou KC; Snyder SA. Classics in Total Synthesis II: More Targets, Strategies, Methods; Wiley-VCH, 2003. [Google Scholar]
- (11).Chen MS; White MC Science 2007, 318, 783. [DOI] [PubMed] [Google Scholar]
- (12).(a) Chen MS Discovery of Predictable and Selective C—H Oxidations for Complex Molecule Synthesis Ph.D. Dissertation, Harvard University, Cambridge, MA, September 2009 [Google Scholar]; (b) Chen MS; White MC Science 2010, 327, 566. [DOI] [PubMed] [Google Scholar]; (c) For a slow addition protocol: Vermeulen NA; Chen MS; White MC Tetrahedron 2009, 65, 3078. [Google Scholar]
- (13).Gormisky PE; White MC J. Am. Chem. Soc 2013, 135, 14052. [DOI] [PubMed] [Google Scholar]
- (14).Howell JM; Feng K; Clark JR; Trzepkowski LJ; White MC J. Am. Chem. Soc 2015, 137, 14590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Osberger TJ; Rogness DC; Kohrt JT; Stepan AF; White MC Nature 2016, 537, 214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Nanjo T; de Lucca EC Jr.; White MC J. Am. Chem. Soc 2017, 139, 14586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).“Our studies were influenced by those of Chen and White, who were the first to explicitly demonstrate that steric and electronic factors can be used to predict the selectivity of intermolecular tertiary C—H oxidation in complex settings using iron catalysis.”: Chen K; Baran PS Nature 2009, 459, 824. [DOI] [PubMed] [Google Scholar]
- (18).(a) White MC Science 2012, 335, 807. [DOI] [PubMed] [Google Scholar]; (b) White MC Synlett 2012, 23, 2746. [Google Scholar]
- (19).(a) Cernak T; Dykstra KD; Tyagarajan S; Vachal P; Krska SW Chem. Soc. Rev 2016, 45, 546. [DOI] [PubMed] [Google Scholar]; (b) Yamaguchi J; Yamaguchi AD; Itami K Angew. Chem. Int. Ed 2012, 51, 8960. [DOI] [PubMed] [Google Scholar]; (c) McMurray L; O’Hara F; Gaunt MJ Chem. Soc. Rev 2011, 40, 1885. [DOI] [PubMed] [Google Scholar]; (d) Malik HA; Taylor BL; Kerrigan JR; Grob JE; Houk KN; Du Bois J; Hamann LG; Patterson AW Chem. Sci 2014, 5, 2352. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Blakemore DC; Castro L; Churcher I; Rees DC; Thomas AW; Wilson DM; Wood A Nat. Chem 2018, 10, 383. [DOI] [PubMed] [Google Scholar]
- (20).(a) Stang EM; White MC Nat. Chem 2009, 1, 547. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) McCallum ME; Rasik CM; Wood JL; Brown MK J. Am. Chem. Soc 2016, 138, 2437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Rasik CM; Brown MK Angew. Chem. Int. Ed 2014, 53, 14522. [DOI] [PubMed] [Google Scholar]
- (22).Hung K; Condakes ML; Morikawa T; Maimone TJ J. Am. Chem. Soc 2016, 138, 16616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Ye Q; Qu P; Snyder SA J. Am. Chem. Soc 2017, 139, 18428. [DOI] [PubMed] [Google Scholar]
- (24).Karimov RR; Hartwig JF Angew. Chem. Int. Ed 2018, 57, 4234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).(a) Fenton HJH J. Chem. Soc., Trans 1894, 65, 899 [Google Scholar]; (b) Walling C Acc. Chem. Res 1975, 8, 125. [Google Scholar]
- (26).Haber F; Weiss J Proceedings of the Royal Society A: Mathematical, Physical and Engineering Sciences 1934, 147, 332. [Google Scholar]
- (27).(a) Bressan M; Liberatore L; d’Alessandro N; Tonucci L; Belli C; Ranalli G J. Agric. Food. Chem 2004, 52, 1228. [DOI] [PubMed] [Google Scholar]; (b) Huling SG; Pivetz BE Engineering Issue: In-situ Chemical Oxidation EPA 600-R-06-072. U. S. Environmental Protection Agency: Ada, OK, 2007. [Google Scholar]
- (28).Rüchardt C Angew. Chem. Int. Ed 1970, 9, 830. [Google Scholar]
- (29).Gozzo F J. Mol. Catal. A: Chem 2001, 171, 1. [Google Scholar]
- (30).(a) Barton DHR; Halley F; Ozbalik N; Schmitt M; Young E; Balavoine G J. Am. Chem. Soc 1989, 111, 7144 [Google Scholar]; (b) Perkins MJ Chem. Soc. Rev 1996, 25, 229. [Google Scholar]
- (31).Mello R; Fiorentino M; Fusco C; Curci R J. Am. Chem. Soc 1989, 111, 6749. [Google Scholar]
- (32).Adam W; Curci R; Gonzalez Nunez ME; Mello R J. Am. Chem. Soc 1991, 113, 7654. [Google Scholar]
- (33).D’Accolti L; Dinoi A; Fusco C; Russo A; Curci R J. Org. Chem 2003, 68, 7806. [DOI] [PubMed] [Google Scholar]
- (34).Bovicelli P; Gambacorta A; Lupattelli P; Mincione E Tetrahedron Letters 1992, 33, 7411. [Google Scholar]
- (35).Bovicelli P; Lupattelli P; Mincione E; Prencipe T; Curci R J. Org. Chem 1992, 57, 5052. [Google Scholar]
- (36).Gol’dshleger NF; Es’Kova VV; Shilov AE; Shteinman AA Zh. Fiz. Khim. (Engl. Transl.) 1972, 46, 785. [Google Scholar]
- (37).Periana RA; Taube DJ; Gamble S; Taube H; Satoh T; Fujii H Science 1998, 280, 560. [DOI] [PubMed] [Google Scholar]
- (38).Stahl SS; Labinger JA; Bercaw JE Angew. Chem. Int. Ed 1998, 37, 2180. [DOI] [PubMed] [Google Scholar]
- (39).(a) Janowicz AH; Bergman RG J. Am. Chem. Soc 1982, 104, 352 [Google Scholar]; (b) Hoyano JK; Graham WAG J. Am. Chem. Soc 1982, 104, 3723 [Google Scholar]; (c) Jones WD; Feher FJ Organometallics 1983, 2, 562 [Google Scholar]; (d) Periana RA; Bergman RG Organometallics 1984, 3, 508 [Google Scholar]; (e) Jones WD; Feher FJ Acc. Chem. Res 1989, 22, 91. [Google Scholar]
- (40).For the first report of using iodosyl acetate as oxidant in Pd(II) catalyzed acetoxylations see: (a) Yoneyama T; Crabtree RH J. Mol. Catal. A: Chem 1996, 108, 35 [Google Scholar]; (b) Desai LV; Hull KL; Sanford MS J. Am. Chem. Soc 2004, 126, 42. [DOI] [PubMed] [Google Scholar]
- (41).For early report using copper oxidant for Shilov reaction see: Lin MR; Shen CY; Garcia-Zayas EA; Sen A J. Am. Chem. Soc 2001, 123, 1000. [DOI] [PubMed] [Google Scholar]
- (42).(a) Burk MJ; Crabtree RH J. Am. Chem. Soc 1987, 109, 8025; [Google Scholar]; (b) Liu F; Pak EB; Singh B; Jensen CM; Goldman AS J. Am. Chem. Soc 1999, 121, 4086. [Google Scholar]
- (43).(a) Sakakura T; Sodeyama T; Sasaki K; Wada K; Tanaka M J. Am. Chem. Soc 1990, 112, 7221 [Google Scholar]; (b) Rosini GP; Zhu K; Goldman AS J. Organomet. Chem 1995, 504, 115. [Google Scholar]
- (44).(a) Iverson CN; Smith MR J. Am. Chem. Soc 1999, 121, 7696 [Google Scholar]; (b) Chen H; Schlecht S; Semple TC; Hartwig JF Science 2000, 287, 1995. [DOI] [PubMed] [Google Scholar]
- (45).For the Shilov hydroxylation of aliphatic amino acid derivatives using 1 equiv. of starting material see: (a) Dangel BD; Johnson JA; Sames D J. Am. Chem. Soc 2001, 123, 8149. [DOI] [PubMed] [Google Scholar]; For aliphatic amines using 5 equiv. of starting material see: (b) Lee M; Sanford MS J. Am. Chem. Soc 2015, 137, 12796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).For C—H amination using native functionality: Willcox D; Chappell BG; Hogg KF; Calleja J; Smalley AP; Gaunt MJ Science 2016, 354, 851. [DOI] [PubMed] [Google Scholar]
- (47).For a formal primary C—H carboxylation see: Julia-Hernandez F; Moragas T; Cornella J; Martin R Nature 2017, 545, 84. [DOI] [PubMed] [Google Scholar]
- (48).Ortiz de Montellano PR Cytochrome P450: Structure, Mechanism, and Biochemistry, 4th edition; Springer, Switzerland, 2015. [Google Scholar]
- (49).(a) Groves JT; Nemo TE; Myers RS J. Am. Chem. Soc 1979, 101, 1032 [Google Scholar]; (b) Groves JT; Nemo TE J. Am. Chem. Soc 1983, 105, 6243. [Google Scholar]
- (50).(a) Groves JT; Kruper WJ; Haushalter RC J. Am. Chem. Soc 1980, 102, 6375 [Google Scholar]; (b) Collman JP; Tanaka H; Hembre RT; Brauman JI J. Am. Chem. Soc 1990, 112, 3689. [Google Scholar]
- (51).Groves JT; Haushalter RC; Nakamura M; Nemo TE; Evans BJ J. Am. Chem. Soc 1981, 103, 2884. [Google Scholar]
- (52).Groves JT; Stern MK J. Am. Chem. Soc 1988, 110, 8628. [Google Scholar]
- (53).(a) Evans DA; Nelson JV; Taber TR Stereoselective Aldol Condensations. In Topics in Stereochemistry; Allinger NL; Eliel EL; Wilen SH, Eds.; Wiley, 2007;Vol 13, pp 1 [Google Scholar]; (b) Rubottom GM; Vazquez MA; Pelegrina DR Tetrahedron Letters 1974, 15, 4319 [Google Scholar]; (c) Hua DH; Huang X; Chen Y; Battina SK; Tamura M; Noh SK; Koo SI; Namatame I; Tomoda H; Perchellet EM; Perchellet JP J. Org. Chem 2004, 69, 6065. [DOI] [PubMed] [Google Scholar]; (d) Kim J; Ashenhurst JA; Movassaghi M Science 2009, 324, 238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (54).(a) Majetich G; Wheless K Tetrahedron 1995, 51, 7095 [Google Scholar]; (b) Barton DHR; Budhiraja RP; McGhie JF Journal of the Chemical Society C: Organic 1969, 0, 336. [Google Scholar]; (c) Harnik M; Lederman Y; Szpigielman R; Herling J Tetrahedron 1976, 32, 1001 [Google Scholar]; (d) Concepción JI; Francisco CG; Hernández R; Salazar JA; Suárez E Tetrahedron Letters 1984, 25, 1953. [Google Scholar]
- (55).(a) Murahashi SJ Am. Chem. Soc 1955, 77, 6403 [Google Scholar]; (b) Murahashi S; Horiie SJ Am. Chem. Soc 1956, 78, 4816. [Google Scholar]
- (56).(a) Bennett MA; Milner DL J. Am. Chem. Soc 1969, 91, 6983 [Google Scholar]; (b) Komiya S; Ito T; Cowie M; Yamamoto A; Ibers JA J. Am. Chem. Soc 1976, 98, 3874 [Google Scholar]; (c) Ibers JA; DiCosimo R; Whitesides GM Organometallics 1982, 1, 13. [Google Scholar]
- (57).Murai S; Kakiuchi F; Sekine S; Tanaka Y; Kamatani A; Sonoda M; Chatani N Nature 1993, 366, 529. [Google Scholar]
- (58).(a) Giri R; Liang J; Lei JG; Li JJ; Wang DH; Chen X; Naggar IC; Guo C; Foxman BM; Yu J-Q Angew. Chem. Int. Ed 2005, 44, 7420. [DOI] [PubMed] [Google Scholar]; (b) Wang DH; Hao XS; Wu DF; Yu J-Q Org. Lett 2006, 8, 3387. [DOI] [PubMed] [Google Scholar]
- (59).Cook BR; Reinert TJ; Suslick KS J. Am. Chem. Soc 1986, 108, 7281. [Google Scholar]
- (60).Groves JT; Neumann R J. Org. Chem 1988, 53, 3891. [Google Scholar]
- (61).(a) Breslow R; Zhang X; Huang Y J. Am. Chem. Soc 1997, 119, 4535 [Google Scholar]; (b) Breslow R; Huang Y; Zhang X; Yang J Proc. Natl. Acad. Sci 1997, 94, 11156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (62).Belvedere S; Breslow R Bioorg. Chem 2001, 29, 321. [DOI] [PubMed] [Google Scholar]
- (63).Das S; Incarvito CD; Crabtree RH; Brudvig GW Science 2006, 312, 1941. [DOI] [PubMed] [Google Scholar]
- (64).Leow D; Li G; Mei TS; Yu J-Q Nature 2012, 486, 518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (65).Shugrue CR; Miller SJ Chem. Rev 2017, 117, 11894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (66).Anslyn EV; Dougherty DA Chapter 7: Energy Surfaces and Kinetic Analyses. In Modern Physical Organic Chemistry; Unversity Science Books: Sausalito, CA, 2006; pp 355; Chapter 11: Organic Reaction Mechanisms, Part 2: Substitutions at Aliphatic Centers and Thermoal Isomerizations/Rearrangements. pp 627. [Google Scholar]
- (67).de Visser SP; Kumar D; Cohen S; Shacham R; Shaik S J. Am. Chem. Soc 2004, 126, 8362. [DOI] [PubMed] [Google Scholar]
- (68).(a) Yosca TH; Rittle J; Krest CM; Onderko EL; Silakov A; Calixto JC; Behan RK; Green MT Science 2013, 342, 825. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Krest CM; Silakov A; Rittle J; Yosca TH; Onderko EL; Calixto JC; Green MT Nat. Chem 2015, 7, 696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (69).(a) Wang W; Liang AD; Lippard SJ Acc. Chem. Res 2015, 48, 2632; [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Lee D; Lippard SJ J. Am. Chem. Soc 2001, 123, 4611. [DOI] [PubMed] [Google Scholar]
- (70).(a) Leising RA; Norman RE; Que L Jr. Inorg. Chem 1990, 29, 2553. [Google Scholar]; When t-BuOOH is used as an oxidant, there is evidence for radical autoxidation chemistry: (b) Leising RA; Kim J; Perez MA; Que L Jr. J. Am. Chem. Soc 1993, 115, 9524. [Google Scholar]
- (71).Okuno T; Ito S; Ohba S; Nishida Y J. Chem. Soc., Dalton Trans 1997, 3547.
- (72).de Vries ME; La Crois RM; Roelfes G; Kooijman H; Spek AL; Hage R; Feringa BL Chem. Commun 1997, 1549.
- (73).(a) Kim C; Chen K; Kim J; Que L Jr. J. Am. Chem. Soc 1997, 119, 5964 [Google Scholar]; (b) Chen K; Que L Jr. Chem. Commun 1999, 1375.
- (74).White MC; Doyle AG; Jacobsen EN J. Am. Chem. Soc 2001, 123, 7194. [DOI] [PubMed] [Google Scholar]
- (75).England J; Britovsek GJ; Rabadia N; White AJ Inorg. Chem 2007, 46, 3752. [DOI] [PubMed] [Google Scholar]
- (76).Denmark SE; Fu J; Lawler MJ Org. Synth 2006, 83, 121. [Google Scholar]
- (77).(a) Kharasch MS; Sosnovsky G J. Am. Chem. Soc 1958, 80, 756 [Google Scholar]; (b) Andrus MB; Lashley JC Tetrahedron 2002, 58, 845. [Google Scholar]
- (78).Bigi MA; Reed SA; White MC Nat. Chem 2011, 3, 216. [DOI] [PubMed] [Google Scholar]
- (79).Mas-Balleste R; Que L Jr. J. Am. Chem. Soc 2007, 129, 15964. [DOI] [PubMed] [Google Scholar]
- (80).Bigi MA; Reed SA; White MC J. Am. Chem. Soc 2012, 134, 9721. [DOI] [PubMed] [Google Scholar]
- (81).(a) Wang Y; Janardanan D; Usharani D; Han K; Que L Jr.; Shaik S ACS Catalysis 2013, 3, 1334 [Google Scholar]; (b) Fan R; Serrano-Plana J; Oloo WN; Draksharapu A; Delgado-Pinar E; Company A; Martin-Diaconescu V; Borrell M; Lloret-Fillol J; Garcia-Espana E; Guo Y; Bominaar EL; Que L Jr.; Costas M; Munck E J. Am. Chem. Soc 2018, 140, 3916. [DOI] [PubMed] [Google Scholar]
- (82).Chen S-H; Huang S; Gao Q; Golik J; Farina V J. Org. Chem 1994, 59, 1475. [Google Scholar]
- (83).Moteki SA; Usui A; Zhang T; Solorio Alvarado CR; Maruoka K Angew. Chem. Int. Ed 2013, 52, 8657. [DOI] [PubMed] [Google Scholar]
- (84).Kawamata Y; Yan M; Liu Z; Bao DH; Chen J; Starr JT; Baran PS J. Am. Chem. Soc 2017, 139, 7448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (85).(a) Liu W; Groves JT J. Am. Chem. Soc 2010, 132, 12847. [DOI] [PubMed] [Google Scholar]; (b) Quinn RK; Konst ZA; Michalak SE; Schmidt Y; Szklarski AR; Flores AR; Nam S; Horne DA; Vanderwal CD; Alexanian EJ J. Am. Chem. Soc 2016, 138, 696. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) N-centered radicals have been sterically tuned to afford primary C—H bond selectivities in chlorinations: Carestia AM; Ravelli D; Alexanian EJ Chem. Sci 2018, 9, 5360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (86).Schmidt VA; Quinn RK; Brusoe AT; Alexanian EJ J. Am. Chem. Soc 2014, 136, 14389. [DOI] [PubMed] [Google Scholar]
- (87).Liu W; Huang X; Cheng MJ; Nielsen RJ; Goddard WA 3rd; Groves JT Science 2012, 337, 1322. [DOI] [PubMed] [Google Scholar]
- (88).Huang X; Bergsten TM; Groves JT J. Am. Chem. Soc 2015, 137, 5300. [DOI] [PubMed] [Google Scholar]
- (89).(a) Sharma A; Hartwig JF Nature 2015, 517, 600. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Karimov RR; Sharma A; Hartwig JF ACS Cent. Sci 2016, 2, 715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (90).Czaplyski WL; Na CG; Alexanian EJ J. Am. Chem. Soc 2016, 138, 13854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (91).Liao K; Negretti S; Musaev DG; Bacsa J; Davies HM Nature 2016, 533, 230. [DOI] [PubMed] [Google Scholar]
- (92).Chen K; Richter JM; Baran PS J. Am. Chem. Soc 2008, 130, 7247. [DOI] [PubMed] [Google Scholar]
- (93).Bovicelli P; Lupattelli P; Fiorini V; Mincione E Tetrahedron Letters 1993, 34, 6103. [Google Scholar]
- (94).Zou L; Paton RS; Eschenmoser A; Newhouse TR; Baran PS; Houk KN J. Org. Chem 2013, 78, 4037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (95).Chiappini ND; Mack JBC; Du Bois J Angew. Chem. Int. Ed 2018, 57, 4956. [DOI] [PubMed] [Google Scholar]
- (96).Newhouse T; Baran PS Angew. Chem. Int. Ed 2011, 50, 3362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (97).(a) Hartwig JF J. Am. Chem. Soc 2016, 138, 2. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Hartwig JF; Larsen MA ACS Cent. Sci 2016, 2, 281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (98).Mahy J-P; Bedi G; Battioni P; Mansuy D New. J. Chem 1989, 13, 651. [Google Scholar]
- (99).Demonceau A; Noels AF; Hubert AJ; Teyssié P Bull. Soc. Chim. Belg 1984, 93, 945. [Google Scholar]
- (100).Minisci F; Galli R; Bernardi R Chem. Commun. (London) 1967, 903. [Google Scholar]
- (101).Zhang K; Shafer BM; Demars MD 2nd; Stern HA; Fasan R J. Am. Chem. Soc 2012, 134, 18695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (102).Kim KE; Adams AM; Chiappini ND; Du Bois J; Stoltz BM J. Org. Chem 2018, 83, 3023. [DOI] [PubMed] [Google Scholar]
- (103).Rasmussen K; Lönnberg H; Wheaton RJ; Tuhtar D; Sjöblom J; Strand TG; Sukhoverkhov VF Acta Chem. Scand 1980, 34a, 155. [Google Scholar]
- (104).(a) Voigt B; Porzel A; Golsch D; Adam W; Adam G Tetrahedron 1996, 52, 10653 [Google Scholar]; (b) D’Accolti L; Fusco C; Lampignano G; Capitelli F; Curci R Tetrahedron Letters 2008, 49, 5614. [Google Scholar]
- (105).Hinman A; Du Bois J J. Am. Chem. Soc 2003, 125, 11510. [DOI] [PubMed] [Google Scholar]
- (106).(a) Kishi Y; Aratani M; Fukuyama T; Nakatsubo F; Goto T; Inoue S; Tanino H; Sugiura S; Kakoi H J. Am. Chem. Soc 1972, 94, 9217. [DOI] [PubMed] [Google Scholar]; (b) Kishi Y; Fukuyama T; Aratani M; Nakatsubo F; Goto T; Inoue S; Tanino H; Sugiura S; Kakoi H J. Am. Chem. Soc 1972, 94, 9219. [DOI] [PubMed] [Google Scholar]
- (107).Walsh JS; Miwa GT Annu. Rev. Pharmacol. Toxicol 2011, 51, 145. [DOI] [PubMed] [Google Scholar]
- (108).Obach RS Pharmacol. Rev 2013, 65, 578. [DOI] [PubMed] [Google Scholar]
- (109).Sarabu R; Bizzarro FT; Corbett WL; Dvorozniak MT; Geng W; Grippo JF; Haynes NE; Hutchings S; Garofalo L; Guertin KR; Hilliard DW; Kabat M; Kester RF; Ka W; Liang Z; Mahaney PE; Marcus L; Matschinsky FM; Moore D; Racha J; Radinov R; Ren Y; Qi L; Pignatello M; Spence CL; Steele T; Tengi J; Grimsby J J. Med. Chem 2012, 55, 7021. [DOI] [PubMed] [Google Scholar]
- (110).Slavik R; Peters J-U; Giger R; Bürkler M; Bald E Tetrahedron Letters 2011, 52, 749. [Google Scholar]
- (111).(a) McGrath NA; Brichacek M; Njardarson JT J. Chem. Educ 2010, 87, 1348 [Google Scholar]; (b) Lam KS Trends Microbiol 2007, 15, 279. [DOI] [PubMed] [Google Scholar]; (c) Demain AL; Zhang L, Natural Products and Drug Discovery. In Natural Products: Drug Discovery and Therapeutic Medicine; Zhang L; Demain AL Eds; Humana Press, 2005; pp 3; [Google Scholar]; (d) Breinbauer R; Vetter IR; Waldmann H Angew. Chem. Int. Ed 2002, 41, 2878. [DOI] [PubMed] [Google Scholar]; (e) Newman DJ; Cragg GM J. Nat. Prod 2016, 79, 629. [DOI] [PubMed] [Google Scholar]
- (112).Ojima I; Das M J. Nat. Prod 2009, 72, 554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (113).Wiechert R, The History of Drospirenone. In Analogue-based Drug Discovery; Fischer J; Ganellin CR Eds.; WILEY-VCH Verlag GmbH & Co; KGaA, Weinheim, 2006; pp 395. [Google Scholar]
- (114).(a) Richter MF; Drown BS; Riley AP; Garcia A; Shirai T; Svec RL; Hergenrother PJ Nature 2017, 545, 299. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Acred P; Brown DM; Turner DH; Wilson MJ Br. J. Pharmacol. Chemother 1962, 18, 356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (115).Schonherr H; Cernak T Angew. Chem. Int. Ed 2013, 52, 12256. [DOI] [PubMed] [Google Scholar]
- (116).Paradine SM; Griffin JR; Zhao J; Petronico AL; Miller SM; White MC Nat. Chem 2015, 7, 987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (117).For seminal studies on Rhodium-catalyzed C—H alkylations via metallocarbenes see: Doyle MP; Duffy R; Ratnikonv M; Zhou L Chem. Rev 2010, 110, 704 and references therein. [DOI] [PubMed] [Google Scholar]
- (118).Clark JR; Feng K; Sookezian A; White MC Nat. Chem 2018, 10, 583. [DOI] [PMC free article] [PubMed] [Google Scholar]