

Abstract
Background:
Fetal alcohol spectrum disorders (FASD) have a strong genetic component although the genes that underlie this are only beginning to be elucidated. In the present study, one of the most common phenotypes of FASD, cell death within the early developing neural tube, was examined across a genetic reference population in a reverse genetics paradigm with the goal of identifying genetic loci that could influence ethanol-induced apoptosis in the early developing neural tube.
Methods:
BXD recombinant inbred (RI) mice as well as the parental strains were used to evaluate genetic differences in ethanol-induced cell death after exposure on embryonic day 9.5. Dams were given either 5.8 g/kg ethanol (EtOH) or isocaloric maltose-dextrin (MD) in two doses via intragastric gavage. Embryos were collected 7 hours after the initial exposure and cell death evaluated via TUNEL staining in the brainstem and forebrain. Genetic loci were evaluated using quantitative trait locus (QTL) analysis at GeneNetwork.org.
Results:
Significant strain differences were observed in the levels of ethanol-induced cell death that were due to genetic effects and not confounding variables such as differences in developmental maturity or cell-death kinetics. Comparisons between the two regions of the developing neural tube showed little genetic correlation with the QTL maps exhibiting no overlap. Significant QTLs were found on murine mid-chromosome 4 and mid-chromosome 14 only in the brainstem. Within these chromosomal loci, a number of interesting candidate genes were identified that could mediate this differential sensitivity including Nfia (nuclear factor I/A) and Otx2 (orthodenticle homeobox 2).
Conclusions:
These studies demonstrate that the levels of ethanol-induced cell death occur in strain- and region-dependent manners. Novel QTLs on mouse Chr4 and Chr14 were identified that modulate the differential sensitivity to ethanol-induced apoptosis in the embryonic brainstem. The genes underlying these QTLs could identify novel molecular pathways that are critical in this phenotype.
Keywords: Alcohol Exposure at Embryonic Day 9.5, BXD Strains, Genetics, FASD, Apoptosis
INTRODUCTION
Fetal Alcohol Spectrum Disorders (FASD) are the leading preventable neurodevelopmental disorder in the Western World, with an estimate of at least 1 to 5% of liveborn infants being affected (May et al., 2018). Approximately 5 to 10% of women who are heavy drinkers throughout their pregnancy give birth to children diagnosed with FASD (Abel, 1995). FASD is a collection of disorders with individuals at the most severe end of the spectrum characterized as having Fetal Alcohol Syndrome (FAS). There is a broad constellation of physiological and behavioral attributes that contribute to a diagnosis of FAS, including a characteristic pattern of craniofacial abnormalities, neurodevelopmental and growth delays, and intellectual disabilities (Jones et al., 1973). Additionally, there are functional deficits in a number of neurological domains, including motor, social, cognitive, and emotional, in those individuals exposed to alcohol in utero (Jacobson et al., 1998).
Multiple factors impact the severity of FASD and the variability seen between individuals. Genetic differences have been implicated as important contributors to the spectrum of possible phenotypes (Green et al., 2007, Chen et al., 2011, Eberhart and Parnell, 2016). Initial studies showing differences in the concordance of the type and severity of alcohol’s teratogenic effects between monozygotic twins compared to dizygotic twins provide evidence of genetic contributions to FASD (Christoffel and Salafsky, 1975). To this day, however, how an individual’s unique genetic makeup impacts the severity of the FASD phenotype remains largely unknown.
Mouse models have been integral to research in studying genetic candidates and pathways involved in FASD phenotypes, as mice are excellent model organisms to study the molecular effects of ethanol on the developing mammalian brain. Advantages of using mouse models include the ability to access experimentally manipulated brain tissues, an increased ability to control and manipulate environmental factors, and having access to databanks of mouse strain genomes that allow for functional characterization of candidate genes (Chesler et al., 2003, Chesler et al., 2004, Driscoll et al., 1990). There are a number of studies that have shown mice to be good models for ethanol-induced phenotypes in the developing brain with exposures at differing times and amounts that lead to morphological changes and increased rates of cell death (Dunty et al., 2001, Green et al., 2007, Chen et al., 2011, Chernoff, 1980, Boehm et al., 1997).
In order to identify genetic factors that influence mouse phenotypes, recombinant inbred (RI) strains are an excellent model system. RI strains are inbred for at least 20 generations and are homozygous at virtually all gene loci; if environmental factors are controlled for, differences between inbred strains can indicate a genetic influence (Crabbe and Belknap, 1992). Previous studies have looked at the effects of exposure to prenatal ethanol in animal models and have shown variable sensitivities in C57BL/6J (B6), DBA/2J (D2) and their RI BXD strains (Boehm et al., 1997, Downing et al., 2012). The B6 strain has shown particular sensitivity to ethanol’s teratogenic effects, and ethanol-induced apoptotic cell death was greater in the B6 strain than in the D2 strain (Dunty et al., 2001, Chen et al., 2011).
The BXD panel is one of the largest mouse RI mapping panels generated to date (Peirce et al., 2004). These strains show significant variation in ethanol preference (Cunningham, 1995), and have thus often been used to study ethanol-related phenotypes and other neuropharmacological traits (Gora-Maslak et al., 1991, Plomin et al., 1991, Chesler et al., 2003). Much of the data gathered from different BXD lines has been entered into the open-source database GeneNetwork (www.GeneNetwork.org), in which multivariate genetic analyses of complex phenotypes can be performed (Chesler et al., 2003, Chesler et al., 2004, Wang et al., 2003). Over a hundred BXD strains have been reliably genotyped, with approximately 1.8 million single nucleotide polymorphisms (SNPs) characterized to date (Peirce et al., 2004, Chesler et al., 2003, Chesler et al., 2004). Once this genotype information has been integrated with experimental data from multiple BXD strains, quantitative trait loci (QTLs) can be identified. QTLs are regions of the genome that mediate strain differences in the phenotype of interest (Wang et al., 2003).
While other mechanisms play a role in the presentation of FASD, ethanol’s ability to cause widespread cellular apoptosis throughout the developing brain is of particular interest (Olney et al., 2002b, Kotch and Sulik, 1992b, Olney et al., 2002a). The present study used TUNEL staining to detect cells undergoing apoptosis in the developing neural tube in response to early embryonic exposure to ethanol. Previous studies have reported that the brainstem and forebrain have differential susceptibility to ethanol-induced cell death at different developmental stages (Dunty et al., 2001, Kotch and Sulik, 1992a, West, 1987, Parnell et al., 2013).
A particularly sensitive time period for the teratogenic effects of ethanol is around embryonic day (E) 9 in the mouse, which is shortly after neural tube closure and is equivalent to the third to fourth week during the first trimester in humans (Dunty et al., 2001, Bannigan and Cottell, 1984, Smith, 1997). Previous studies have shown higher levels of ethanol-induced apoptotic cells compared to controls in the neuroepithelium of E9 mice within six hours of ethanol exposure (Bannigan and Cottell, 1984, Smith, 1997). The present study was conducted to evaluate genetic differences in ethanol-induced apoptosis at E9.5 in the brainstem (metencephalon, myelencephalon) and forebrain (telencephalon) using the BXD RI panel of mice, and to identify genetic loci and candidate genes using QTL analysis at GeneNetwork.org.
MATERIALS AND METHODS
I. Animals.
All animals were maintained at the University of Tennessee Health Science Center (UTHSC). Mice were maintained on a 12:12 light:dark cycle and given food and water ad libitum. Litters from twenty-six BXD RI strains and B6 and D2 parental strains were used in the current analyses. For all strains, an average of 4 litters were used per treatment group. Embryos in a few strains were difficult to obtain and therefore in those strains there were low numbers of litters particularly in the maltose dextrin control group. All experiments were conducted with the approval of the Institutional Animal Care and Use Committee at UTHSC. Females were acclimated to the handling procedure for one week prior to mating. Timed matings were used to generate embryos for this study. Males and females of the same strain were mated for only 4 hours starting between 9 and 10 a.m. daily to gain a tight window on the time of conception (determined by the presence of a vaginal plug). The day of conception was considered embryonic day 0 (E0).
II. Ethanol Exposure and Tissue Collection/Processing.
Pregnant dams were given 5.8 g/kg of EtOH, in 2 equal doses of 2.9 g/kg separated by 2 hours, by intragastric gavage at E9.0, following the protocol of Parnell et al. (2013). Two control groups were used: one given isocaloric maltose dextrin (MD) and another unhandled group to control for the stress of the gavage procedure. Initially, cell death was recorded for both the unhandled (untreated) and MD administered control groups in a subset of strains. Values were compared using a t-test and no significant difference was found between the two control groups in the forebrain or brainstem. Therefore, the MD condition was used as the only control group in the remainder of the study. The dams were sacrificed 7 hours after the initial alcohol exposure. The embryos were dissected out of the uterus into sterile saline and immersion fixed in 4% paraformaldehyde in 0.1M phosphate buffered saline (PBS). Embryos were fixed for 1–2 hours, then placed in 0.1M PBS and individually embedded in paraffin using standard protocols. Embryos were transversely sectioned at 8μm thickness and mounted on glass slides (SuperFrost Plus, Fisher Scientific).
Previous research has shown that the peak of cell death in mice at E9.5 is 6–8 hours after the initial ethanol exposure (Bannigan and Cottell, 1984, Smith, 1997). To ensure that differences in cell death were due to differential sensitivity and not due to differences in the kinetics of cell death (Dunty et al., 2001), selected strains were examined after a longer survival time. The strains examined were those that had low levels of cell death at 7 hours to ensure that peak of cell death was not at a later timepoint in these strains. Mice were treated with alcohol in the same manner as described above but the dams were sacrificed, and embryos collected at 11 hours after the initial alcohol exposure.
III. Blood Ethanol Concentrations.
Separate dams from each strain were used to determine blood ethanol concentrations (BECs; see Supplementary Table 1). Animals were exposed to ethanol as described above. Blood was collected at 1, 2, and 5 hours after the second alcohol exposure using capillary tubes obtained from Analox Instruments. BECs were measured using an Analox Alcohol Analyzer following manufacturer’s protocols.
IV. Detection and Quantification of Apoptotic Cell Death.
TUNEL staining was used to label cells undergoing apoptotic DNA fragmentation (Nikolic et al., 2000). The ApopTag In Situ Apoptosis Kit (Oncor, Gaithersburg, MD) was used to mark TUNEL positive cells following the manufacturer’s protocol. When selecting slides for analysis, slides containing sections of the developing telencephalon (neuroepithelium rostral to the optic vesicles) and developing brainstem (neuroepithelium in the region of the otic placodes) were chosen for analysis. In brief, for each staining run sections were deparaffinized and rehydrated, incubated with 20 μg/mL of proteinase K, blocked using 2% hydrogen peroxide and incubated with terminal deoxynucleotidyl transferase, followed by an anti-digoxigenin peroxidase-conjugated antibody. 3,3’-diaminobenzidine (DAB) was used as the chromogen to visualize the digoxigenin-labeled fragments, and the sections were counterstained with 0.5% methyl green. Sections were dehydrated, and coverslips were applied using Permount. Labeled cells undergoing apoptosis are brown and unlabeled cells are green.
In order to visualize the TUNEL-positive cells, a Zeiss 200 M Axiovert inverted microscope equipped with Axiovision 4.6 software was used to image and photograph the tissue. Images were taken using a 20× objective to determine the counting region and the areal measurements. TUNEL-positive cells were then manually counted within the regions of interest in the neuroepithelium under brightfield illumination (Standard Zeiss ICS Brightfield microscope) using a 40× objective. Inclusion criteria for cells undergoing apoptosis were either tightly clustered brown TUNEL staining or a more diffuse brown TUNEL staining with the critical characteristic being that the staining is found within the confines of the cell. Cells with only faint, non-confined brown stain throughout the cell were not counted as TUNEL-positive.
V. Area Measurements, Sampling, and Quantification of TUNEL-Positive Cells.
The tracing software ImageJ (National Institutes of Health, version 1.43s) was used to calculate area measurements of the E9.5 forebrain and brainstem. Telencephalon and rhombencephalon were counted using the optic vesicles and otic placodes as landmarks for those regions, respectively. Images taken from the Zeiss microscope were uploaded into the program and had preset dimensions of 1300 × 1030 pixels, which were then scaled accordingly to equivalent dimensions of mm2. The total area was calculated and the cell death count for that section was divided over this area to determine an estimate value of cell death per mm2 for each section. Average TUNEL-positive cells per mm2 values were then calculated for each embryo by averaging these sampled sections. Finally, an average value of TUNEL-positive cells per mm2 for each litter was determined to account for the individual differences across embryos. Three (± 1) sections per embryo, three (± 1) embryos per litter, and at least three (± 1) litters per treatment (ethanol (EtOH) or maltose/dextrin (MD)) were used for analysis for all strains. A subset of the sections was analyzed and chosen based on being in the region of the optic vesicle or otic placodes in the forebrain or brainstem, respectively. The litter means were used as the unit of analysis.
VI. Data Analyses.
Strain data were examined using apoptosis per unit area averages of each litter as the unit of analysis. For each region analyzed, an overall MD control treatment litter average was calculated for each strain, and this value was subtracted from each ethanol litter average and termed the ethanol-adjusted (EtOH-Adj) value. Analysis of variance (ANOVA) was performed on this data using the program StatPlus to test if there were significant differences in cell death across BXD strains. Additionally, three correlation analyses were performed: 1) to identify if there was a correlation between average cell death in the forebrain and brainstem, 2) to identify if there was a correlation between average cell death in EtOH-treated and average somite number in MD-treated litters, and 3) to determine if there was a correlation between BECs and the level of cell death. Somite pairs were counted using standard methodology (Dunty et al., 2001). Somite counts in MD controls were used as an indicator of maturation rates in each strain.
Relative to the GeneNetwork analysis discussed in the sections below, post-hoc marker regression analysis consisted of 2-sample t-tests to identify if there were significant differences in average cell death between strains carrying the B6 or D2 parental allele at the significant loci indicated by GeneNetwork after running quantitative trait loci (QTLs) halplotype analysis. An alpha level of 0.05 was used to assess significant results for ANOVA and t-test analyses.
VII. Quantitative Trait Loci Analysis.
Cell death data were uploaded into GeneNetwork.org, an open access online database which contains BXD strain genomic information. QTLs were run on both the EtOH-Adj and the unadjusted ethanol values. Genome-wide interval mapping of QTLs correlated with apoptosis was performed. Likelihood ratio statistics (LRS) were computed in order to assess the strength of any genotype-phenotype association from the genomic scans. Tests of 2000 permutations were performed to establish the significance and suggestive thresholds, where the LRS scores corresponded to genome-wide p-values of 0.05 and 0.63, respectively. Confidence intervals around the peaks identified near the significant LRS score were calculated using ±1 logarithm of the odds (LOD) score.
VIII. Identification of Candidate Genes within the Significant QTLs.
Previous studies have used bioinformatics analyses in conjunction with a specific set of criteria to narrow down the set of genes into those most likely to underlie the differential response (Baker et al., 2017, Cook et al., 2015). In the present study, genes within the significant QTLs were identified using the online tools available at GeneNetwork.org. The gene lists include expressed sequence tags and Riken clones. As it is currently unknown if these species of RNA are translated to protein, they were not evaluated further although we acknowledge that these molecules could be non-coding RNAs and therefore could have important roles. Genes were evaluated by a number of criteria including: 1) the presence of single nucleotide polymorphisms (SNPs), 2) expression at E9.5, 3) previously reported role in cell death and/or survival or in alcohol-related pathways, and 4) evidence of strain-specific expression differences. Non-synonymous SNPs in coding regions of the gene result in amino acid changes in the respective protein and were considered as good candidate genes. However, expression changes can occur via other mechanisms, e.g. methylation differences, and therefore genes without non-synonymous SNPs were still considered. To determine if the genes were expressed during the relevant developmental timepoint, the GeneAtlas GNF1M gcrma dataset containing E9.5 expression data was analyzed through BioGPS.org (Su et al., 2004). Literature searches were conducted to determine if each gene has a previously reported role in cell death or cell survival, especially during development, or involved in alcohol-related pathways or phenotypes. GeneNetwork.org contains expression analysis across BXD strains in the neocortex of mice at postnatal age 3. While expression data differs through development, this database was examined to determine whether there is evidence of strain differences in expression over developmental time.
RESULTS
Qualitative Assessment of TUNEL-Positive Cells in the EtOH- and MD-Treated Forebrain and Brainstem
The purpose of this study was to evaluate genetic differences in levels of ethanol-induced cell death using the 26 BXD RI, and B6 and D2 parental strains at E9.5. The initial examination provided a qualitative assessment to determine if differences exist in the quantity and locations of dying cells across the strains and between the ethanol (EtOH) and maltose dextrin (MD) control treatments in the brainstem and forebrain (Figure 1 and Figure 2). Generally, there were fewer cells undergoing apoptosis in the MD controls, and cells were less clustered and were more spread out throughout the neuroepithelium in both the brainstem and forebrain compared to the EtOH-treated tissue. For the majority of the strains examined, qualitative examination showed a difference in the levels of cell death between the MD and the EtOH groups. The MD-treated litters showed more limited clustering of dying cells and rather sparse cell death compared to the EtOH-treated litters in both the brainstem (Figure 1E) and forebrain (Figure 2E). After exposure to EtOH, most strains showed a higher number of TUNEL+ cells that appeared in a more clustered arrangement in both the brainstem and forebrain compared to sections in the same regions in the MD controls. In the brainstem of EtOH-treated samples, clusters of dying cells typically occurred along the narrow strips of the lateral neuroepithelium (Figure 1). In the forebrain of EtOH-treated samples across all BXD strains, the closer clustering of TUNEL+ cells appeared to preferentially occur near the junction of the neuroepithelium with the optic vesicles in the forebrain and in the neuroepithelium at the lateral edge of the neural tube (Figure 2).
Figure 1. Images of TUNEL+ sections from the brainstem.
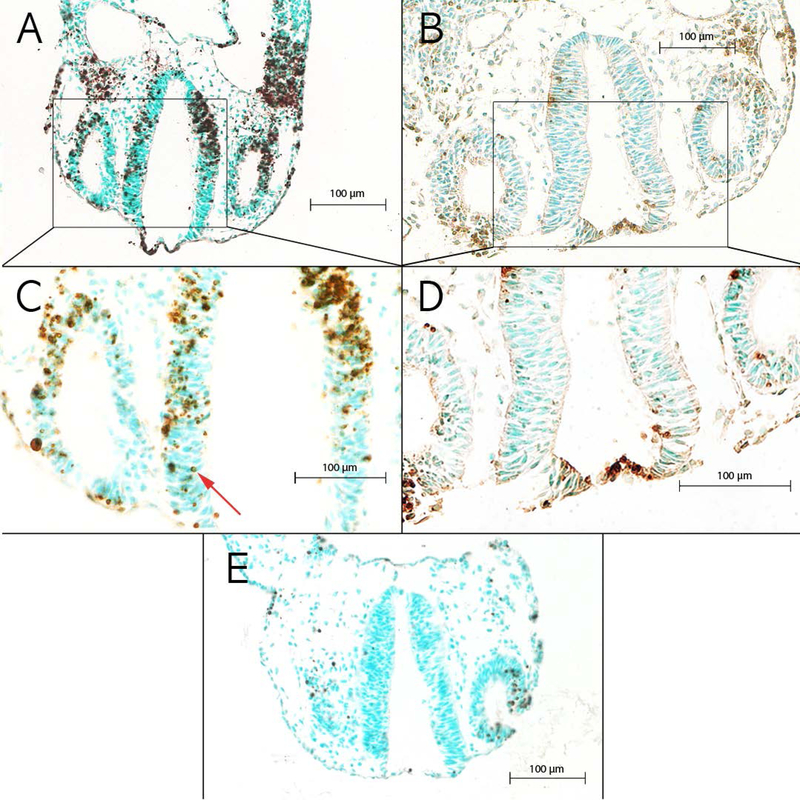
Brainstem neuroepithelium in ethanol-exposed embryos from (A) a high cell death strain, BXD51, and (B) a low cell death strain, BXD65. (C) Higher magnification of boxed region in A showing an example of TUNEL+ cells indicated by the red arrow. (D) Higher magnification of boxed region in B showing examples of the low number of TUNEL+ cells. (E) Brainstem of an MD-treated control embryo from a high cell death strain, BXD51.
Figure 2. Images of TUNEL+ sections from the forebrain.

Forebrain neuroepithelium in ethanol-exposed embryos from (A) a high cell death strain, BXD86, and (B) a low cell death strain, BXD101. (C) Higher magnification of boxed region in A showing an example of TUNEL+ cells indicated by the red arrow. (D) Higher magnification of boxed region in B showing examples of the low number of TUNEL+ cells. (E) Forebrain of an MD-treated control embryo from a high cell death strain, BXD86.
Quantitative Assessment of TUNEL-Positive Cells in EtOH-Treated Brainstem and Forebrain
The ascertainment of the number of TUNEL+ cells is critical for comparison between strains. Quantitative analyses between EtOH- and MD-treated embryos used the overall strain means that were based on the litter cell death means. The EtOH-Adj value for each strain was calculated by subtracting the MD overall strain mean from the ethanol overall mean for each litter. The EtOH-Adj values were used to control for the differences in levels of cell death in MD controls and this provided a more conservative estimate of the levels of ethanol-induced cell death. Within strains, litters whose means were a negative EtOH-Adj value were zeroed to 0.001 because biologically it is not possible to have negative cell counts. When comparing the EtOH-Adj means in Figure 3 with the MD means in Supplementary Figure 1, it is clear that both strains with high and strains with low levels of ethanol-induced cell death vary in the levels of “normal” cell death observed in the MD controls.
Figure 3. Analysis of average apoptotic cell counts in the forebrain and brainstem across BXD strains.

EtOH-Adj values were used and were computed as described in the Materials and Methods. The x-axis shows strains in ascending order and the y-axis shows the mean number of TUNEL+ apoptotic cells per mm2. Error bars represent ± SEM. (A) Mean apoptotic cell counts in the forebrain. (B) Mean apoptotic cell counts in the brainstem.
As shown in Figure 3, there was a wide variation in cell death across the strains. One-way ANOVAs on the EtOH-Adj values showed significant differences in the brainstem across strains (p = 0.0002). In the forebrain, while, there was no significant difference in the EtOH-Adj values among strains in the ANOVA, the value showed a trend towards strain differences (p = 0.0791). Moreover, comparisons between individual strains, particularly those on the extreme ends did exhibit significant differences in t-tests (p < 0.05). To assess whether the cell loss within a strain was consistent between the two regions of the developing neural tube, a correlation analysis of the EtOH-Adj strain means in the brainstem and forebrain was conducted, and the results showed a significant correlation, r2 = 0.508, p < 0.01 (Figure 4).
Figure 4. Brainstem versus forebrain TUNEL+ cells per mm2 in EtOH-Adj group.

Correlation analysis performed between strain means for TUNEL+ cells per mm2 values in the brainstem versus the forebrain in the EtOH-Adj group (r2 = 0.508, p = 0.00002).
Examination of Non-Genetic Causes of Strain Differences
In order to ensure that the strain differences are in fact due to genetic differences and not other confounding variables, two additional analyses were performed. The first analysis assessed whether differences in cell death kinetics could account for the strain differences. To determine if kinetics were a confound, three strains (BXD62, BXD65 and BXD77) with low levels of cell death at 7 hours were examined at a later time point and sacrificed at 11 hours after the initial exposure (Table 1). As levels of cell death at 11 hours were either equivalent or lower to the levels found after 7 hours, this supports the hypothesis that examination at 7 hours after initial exposure encompasses the peak level of cell death for all strains. Additionally, for most BXD strains in both treatments, somite numbers were counted in MD control litters as an indicator for developmental age to test whether differences in maturation rates across BXD strains accounted for the differential vulnerability. A correlation analysis of mean strain levels of cell death in EtOH-treated litters and mean strain somite numbers in MD-treated litters showed no significant correlations, with r2 values of 0.154 (p = 0.119) for the brainstem and 0.024 (p = 0.550) for the forebrain (Figure 5).
Table 1.
Differential susceptibility of cell death across strains BXD62, BXD65, and BXD77 is not due to differences in timing of exposure to ethanol in the brainstem or forebrain. Comparisons of number of TUNEL+ cells in embryos dissected 7 hours versus 11 hours after initial ethanol exposure.
| Brainstem | Forebrain | |||||||
|---|---|---|---|---|---|---|---|---|
| 7 hours | 11 hours | 7 hours | 11 hours | |||||
| Strain | TUNEL+/mm2 | SEM | TUNEL+/mm2 | SEM | TUNEL+/mm2 | SEM | TUNEL+/mm2 | SEM |
| BXD77 | 520 | 110 | 470 | 35 | 980 | 292 | 588 | 104 |
| BXD65 | 509 | 20 | 387 | 38 | 922 | 132 | 533 | 203 |
| BXD62 | 367 | 309 | 258 | 97 | 347 | 425 | 67 | 81 |
Figure 5. Somite counts of MD-treated animals versus Average TUNEL+ cells per mm2 in EtOH-treated animals.

Correlation analysis performed between MD-treated strain mean somite count and EtOH-treated strain mean TUNEL+ per mm2. There is no correlation between somite count at E9.5 with numbers of TUNEL+ cells in the (A) forebrain (r2 = 0.024, p = 0.550) or (B) brainstem (r2 = 0.154, p = 0.119).
Blood Ethanol Concentrations
Blood ethanol concentrations (BECs) were examined at one (m = 435 + 16.9), two (m = 374 + 15.9), and five (m = 152 + 14.6) hours after the second intubation of ethanol and the results for each strain are shown in Supplementary Table 1. Consistent with data from previous work from Grisel et al. (2002) there were strain differences in both the peak blood ethanol concentration and the rate of ethanol metabolism. A correlation analysis between BECs at each timepoint and the level of cell death showed no significant correlations, for all comparisons r2 < 0.10 (data not shown). This provides evidence that differential ethanol metabolism had minimal contributions to the strain differences in the levels of cell death.
Identification of Chromosomal Regions Associated with EtOH-Induced Cell Death
Using the quantitative differences in cell death between strains, quantitative trait loci maps (QTLs) were generated using GeneNetwork.org. QTLs for the brainstem and forebrain using EtOH-Adj values showed visually different maps, with the brainstem (Figure 6B) having several suggestive and significant QTL peaks, and the forebrain (Figure 6A) having none. For both regions, QTL analysis was conducted on the EtOH-Adj values and the unadjusted EtOH values and the QTL locations were the same using either value. Therefore, we continued to focus on the more conservative EtOH-Adj values. In the brainstem, significant QTLs were found on Chr4:95–103Mb (megabases) and Chr14:45–47.5Mb with suggestive QTLs on Chr9, Chr13, and the X chromosome. Analysis of the significant QTLs on Chr4 (Figure 6C) and Chr14 (Figure 6D), showed that the additive effect of the B6 allele closely followed the shape of the LRS score implying that if the BXD strain inherited the B6 parental allele, it carried an allelic variant that has a relationship with the observed strain differences in cell death within the brainstem. Marker regression analysis confirmed this observed trend (Figure 7), i.e.: strains carrying the B6 allele had significantly higher average cell death than those carrying the D2 allele in the brainstem, for both Chr4 (p = 0.008) and Chr14 (p = 0.037).
Figure 6. Quantitative trait loci (QTLs) map using the EtOH-Adj data in the forebrain and brainstem.

(A) Interval genome-wide mapping of the forebrain showed no suggestive or significant QTLs (Record ID: 18128). (B) Interval genome-wide mapping of the brainstem (Record ID: 18126) showed (C) a significant QTL (LRS = 14.3) on Chr4 between 95–103 Mb and (D) a significant QTL on Chr14 between 45–47.5 Mb (LRS = 16.5). For all, the x-axis denotes chromosome and megabases (Mb) with yellow dashes representing the location of SNPs and the y-axis denotes the LRS-value. The blue line represents the LRS scores of this trait. Red and green lines represent additive effects of the B6 and D2 alleles, respectively. The horizontal lines for C and D indicate genome-wide suggestive (bottom, gray line, LRS = 8.83) and significant (top, pink line, LRS = 14.21) thresholds. Green, navy, purple, red, and yellow bars in the section above the blue line represent genes.
Figure 7. Haplotype analysis of the B6 or D2 parental allele on number of TUNEL+ cells in the brainstem.

Marker regression analysis computed in GeneNetwork (Record ID: 18126) compared BXD strains carrying the B6 allele (blue) with those carrying the D2 allele (red) on Chr4 (p = 0.037) and Chr14 (p = 0.008). Shown are the mean number of TUNEL+ cells (+ SEM) for all the strains that contain the B6 allele compared to all the strains with the D2 allele.
Identification of Candidate Genes that Underlie the Significant QTLs on Chr4 and Chr14
The significant QTL on Chr4 encompassed 83 genes while the QTL on Chr14 encompassed over 200 genes. To identify potential candidate genes, a series of criteria was employed to identify the most likely candidates, as described in the Materials and Methods. Expressed sequence tags and Riken clones were eliminated from further investigations since it is currently unknown whether these species of RNA are ever translated to protein. The Chr4 QTL had thirty-one genes that contained SNPs, four of which were non-synonymous. The Chr14 QTL had fifty-four genes that contain SNPs, sixteen of which were non-synonymous (see Supplementary Table 2). While the sequence variants that alter the amino acid composition, i.e. non-synonymous SNPs, are the most likely to result in expression differences, other polymorphisms may cause expression differences in other ways such as the binding of microRNAs or transcription factors and therefore these genes were not discounted. Evaluation of expression analysis showed that fourteen genes on Chr4 and nineteen genes on Chr14 have expression at E9.5 evaluated through the GeneAtlas GNF1M gcrma database at BioGPS.org. Literature searches of publicly available databases identified genes that are shown to 1) play a role in cell death and/or cell survival or 2) be affected by alcohol exposure or contributing to ethanol-related phenotypes. On Chr4 sixteen genes were found to be related to cell death pathways in any system while eight genes were related to alcohol. On Chr14 thirty-two genes were found to be related to cell death pathways in any system while twelve genes were related to alcohol. In order to assess whether expression differences are observed across BXD strains in a developing brain region, the neocortex dataset from animals at postnatal day 3 were queried. This dataset is available at GeneNetwork.org and showed that seventeen genes on Chr4 and thirty-two genes on Chr14 have differential expression among BXD strains in the neocortex at postnatal day 3. Although expression data is likely different at this developmental stage, the database provides evidence of strain differences in expression during development. Overall, ten genes on Chr4 and ten genes on Chr14 satisfy all of the following criteria, and are therefore excellent candidates: 1) presence of a SNP, 2) expression at E9.5, 3) previously reported in relation to cell death and/or alcohol, and 4) differential expression among BXD strains. The genes within the Chr4 QTL that met all the criteria are Dnajc6, Dock7, Lepr, Nfia, Patj, Pde4b, Pgm2, Ror1, Sgip1 and Usp1 (Table 2). The genes within the Chr14 QTL that met all the criteria are Cdkn3, Cgrrf1, Gch1, Gmfb, Ktn1, Naa30, Otx2, Peli2, Styx, and Tspan14 (Table 2).
Table 2.
Candidate genes on Chr4 and Chr14 that met all of the bioinformatic criteria.
| Gene Symbol | Gene Name |
|---|---|
| Chromosome 4 | |
| Dnajc6 | DnaJ heat shock protein family (Hsp40) member C6 |
| Dock7 | Dedicator of cytokinesis 7 |
| Lepr | Leptin receptor |
| Nfia | Nuclear factor I/A |
| Patj | PATJ, crumbs cell polarity complex component |
| Pde4b | Phosphodiesterase 4B, cAMP specific |
| Pgm2 | Phosphoglucomutase 2 |
| Ror1 | Receptor tyrosine kinase-like orphan receptor 1 |
| Sgip1 | SH3-domain GRB2-like (endophilin) interacting protein 1 |
| Usp1 | Ubiquitin specific peptidase 1 |
| Chromosome 14 | |
| Cdkn3 | Cyclin-dependent kinase inhibitor 3 |
| Cgrrf1 | Cell growth regulator with ring finger domain 1 |
| Gch1 | GTP cyclohydrolase 1 |
| Gmfb | Glia maturation factor, beta |
| Ktn1 | Kinectin 1 |
| Naa30 | N(alpha)-acetyltransferase 30, NatC catalytic subunit |
| Otx2 | Orthodenticle homeobox 2 |
| Peli2 | Pellino 2 |
| Styx | Serine/threonine/tyrosine interaction protein |
| Tspan14 | Tetraspanin 14 |
DISCUSSION
In the present study, we examine BXD and parental mouse strains and the results confirm the hypothesis that there are significant strain-specific differences in cell death within the neural tube following acute ethanol exposure early in gestation that are not due to other variables such as differential cell death kinetics. Cell death is examined because it is one of the most common phenotypic outcomes following developmental alcohol exposure and because the deletion of cells early in central nervous system development can have severe consequences later in brain development and function. However, there is a regional specificity to these results. For the brainstem, the genetic differences translate into significant QTLs on murine mid-Chr4 and mid-Chr14. The Chr4 QTL is homologous to a region on human chromosome 1p32 while the Chr14 QTL is homologous to a region on human chromosome 14q22. Within the QTLs on Chr4 and Chr14, a number of viable candidate genes were identified using criteria including their functions and patterns of expression. These candidate genes could be used as markers to identify individuals at risk for ethanol-induced neuroteratogenesis or molecular pathways that could be targets for pharmacotherapeutic interventions.
Previous data have shown that the developing telencephalon and myelencephalon are regions that are sensitive to ethanol exposure at this developmental time period (Dunty et al., 2001). Generally, the results of the present study are consistent with this previous study. However, the present data adds a level of additional complexity to these findings by showing that specific strains, such as BXD73, show high levels of cell death in the forebrain but low levels of cell death in the developing brainstem. These results provide evidence that, even though the phenotypic measure is identical across regions of the neural tube, there are likely different mechanisms that underlie the ethanol-induced cell death across regions. Further, this molecular difference is consistent with the QTL analyses that differ dramatically between the regions.
It is interesting to note that there is no genetic correlation between the QTLs found in the brainstem compared to the developing forebrain even though the levels of cell death between regions are correlated. There are a number of possible reasons that could account for these differences. First, these differential results could be due to differential molecular pathways mediating cell death through different mechanisms. Further investigation of gene expression in these regions is needed to assess this possibility. Second, there are potentially other features related to differences in maturation between the two regions that could be contributing factors. For example, the brainstem is one of earlier maturing regions of the developing central nervous system and therefore a greater proportion of the neurons in this region would be developmentally advanced (Rodier, 1980, Easter et al., 1993). In contrast, the developing forebrain has a more protracted developmental trajectory and thus, more of the developing cells could be early progenitors rather than differentiating neurons. There is also more diversity in the degree of maturation between individual cells and this could mean that more strains would need to be examined to detect significant QTLs. Third, the region-specific differential sensitivity could be due to disparate mitotic rates as shown by Calegari and colleagues who demonstrated that progenitors that are set to give rise to neurons have a longer cell cycle than those that are in an early stage, i.e. giving rise to additional progenitors (Calegari et al., 2005). The different levels of development could, in turn, contribute to differential sensitivity to ethanol-induced cell death. Previous work by Downing et al. (2012) exposed BXD mice to alcohol during mid-gestation to identify QTLs associated with a range of morphologic phenotypes, such as maternal weight gain and rib abnormalities in the embryos, among others. As the same genetic reference population was studied, it is of interest to determine whether the chromosomal location identified in the present analysis replicates any of those found by Downing et al. (2012). Although the earlier publication identified a number of QTLs, none mapped to mid-Chr4 or mid-Chr14. These results demonstrate that the QTLs identified in the present study are novel chromosomal locations and further suggests that ethanol-induced cell death is mediated by different molecular mechanisms than the phenotypes examined by Downing et al. (2012).
Other work has examined specific molecular pathways, typically using knock-out or transgenic mice, to assess whether individual genes are related to differences in ethanol-induced phenotypes. Studies that evaluated the role of specific genes in ameliorating or enhancing cell death are of particular interest to determine whether any of these genes are found within the QTLs identified in the present study. A number of genes have been examined for their role in ethanol-induced cell death including Nitric Oxide Synthase 1 (Nos1 aka nNOS) (Karacay et al., 2015, Bonthius et al., 2002), Cannabinoid Receptor 1 (Cnr1) (Subbanna et al., 2013), Adenylate Cyclase 1 (Adcy1), and Adenylate Cyclase 8 (Adcy8) (Conti et al., 2009). None of the genes examined to date are located within the Chr4 or Chr14 QTLs. However, in these studies, the ethanol exposure was given at a later developmental time. Previously, we examined cell death in the same population of BXD mice following acute neonatal ethanol exposure. The QTL identified following the neonatal alcohol exposure was on Chr12 consistent with the hypothesis that a different mechanism(s) of cell death occurs in early versus late development (Goldowitz et al., 2014).
The roles of genes known to be mediators of cell death have also been examined following developmental ethanol exposure including Transformation Related Protein 53 (Trp53, aka p53), B Cell Leukemia/Lymphoma 2 (Bcl2), and BCL2-Associated X (Bax) (Camargo Moreno et al., 2017, Heaton et al., 1999, Ahlers et al., 2015). None of the genes investigated to date are present in the Chr4 or Chr14 QTLs, and thus genetic differences in expression of these candidates are unlikely to mediate the strain-specific differences in ethanol-induced cell death observed across the BXDs. However, it is possible that some of these genes interact in the same molecular pathways as genes within the Chr4 and/or Chr14 QTLs.
While we have identified two significant QTLs, the major objective is to identify the underlying genes that are responsible for the phenotypic difference across strains as well as to determine the molecular network of the relevant genes. Previously, we have used a specific set of criteria and bioinformatic analyses to determine both the genes that underlie a specific QTL and to determine the molecular network that contains the relevant gene (Cook et al., 2015, Urquhart et al., 2016, Baker et al., 2017, Zhou et al., 2017). We conducted an analogous bioinformatic analysis in the present study.
From our bioinformatic analyses a number of candidate genes look promising. These genes met all of the following criteria: 1) presence of a SNP, 2) expression at E9.5, 3) previously reported in relation to cell death and/or alcohol, and 4) differential expression among BXD strains. The most interesting and strongest candidate genes are nuclear factor I/A, Nfia, and the homeobox gene, Otx2, which have both been previously linked to FASD. Nfia has been shown to be differentially expressed in multiple brain regions after exposure to prenatal ethanol in mice and is an integral part of apoptotic pathways (Mandal et al., 2015, Tsai et al., 2014, Lee et al., 2017b, Lee et al., 2017a) Otx2 is differentially expressed in adult animals exposed to developmental alcohol and has been linked to brain malformations and mood disorders (Laufer et al., 2013, Kleiber et al., 2012).
While this bioinformatic analyses provides an excellent beginning, other genes within this region that did not meet all criteria cannot be conclusively excluded due in part to the limitations of the expression analyses. The only public database with whole-genome expression analysis at this developmental age was completed over 10 years ago and the microarrays had a more restricted gene coverage as shown by the fact that over 10% of the genes within the QTL were not present. Moreover, expression was examined from a large portion of the embryo rather than specifically any portions of the neural tube. Because of these caveats, other genes that meet a subset of the criteria warrant further evaluation including Autophagy-related 4C Cystine Peptidase (Atg4c) and Janus Kinase 1 (Jak1) from Chr4 and Neuregulin 3 (Nrg3), Prostaglandin E Receptor 2, subtype EP2 (Ptger2), and Bone Morphogenetic Protein 4 (Bmp4) from Chr14. Atg2C has been strongly linked to autophagy, another mechanism of cell death (Korkmaz et al., 2012). Jak1 is part of the apoptotic JAK/STAT1 pathway (Giunta et al., 2006). Nrg3 has previously been linked to developmental disorders such as autism and has also been reported as a genetic hub modulating differing ethanol sensitivity across BXD strains (Paterson and Law, 2014, Wolen et al., 2012). Ptger2 has been shown to play a role in both cell survival and apoptosis (Jonakait and Ni, 2009, Liou et al., 2007, Fu et al., 2015). Interestingly another gene, Bmp4, has also been implicated in FASD as a mediator of apoptosis in multiple regions of the embryo and also exerts pro-inflammatory effects that can result in cell death (Zhang et al., 2017, Kennelly et al., 2011, Trousse et al., 2001, Choy et al., 2018).
In summary, the present study has shown that exposure to ethanol just after neural tube closure in the mouse results in strain-specific differences in the level of ethanol-induced cell death within the developing brain. Furthermore, we were able to identify regions on mid-Chr4 and mid-Chr14 that mediate these strain-specific differences. There are several other factors that need to be taken into account in these results, including the effect of the maternal environment and whether epigenetic mechanisms could impact the expression of these genes and future studies will examine these factors. Interestingly, several of the genes within these QTLs are involved in epigenetic pathways and could be a putative link among these variables. Future studies are needed to both confirm the underlying quantitative trait genes (QTGs) within these newly identified QTLs as well as determine how the genes could mediate strain-specific differences in ethanol-induced cell death.
Supplementary Material
Acknowledgments
We greatly appreciate the contributions of Scott Lattimer for animal work and Aria Shokoohi, Clara van Ommen, Cheryl Tan, and Mike Wu for histological collection and staining of tissue for analysis. We are appreciative of the funding provided by the National Institute on Alcohol Abuse and Alcoholism R01AA023508 and NeuroDevNet, a Canadian Network of Centres of Excellence.
REFERENCES
- ABEL EL 1995. An update on incidence of FAS: FAS is not an equal opportunity birth defect. Neurotoxicol Teratol, 17, 437–43. [DOI] [PubMed] [Google Scholar]
- AHLERS KE, KARACAY B, FULLER L, BONTHIUS DJ & DAILEY ME 2015. Transient activation of microglia following acute alcohol exposure in developing mouse neocortex is primarily driven by BAX-dependent neurodegeneration. Glia, 63, 1694–713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BAKER JA, LI J, ZHOU D, YANG M, COOK MN, JONES BC, MULLIGAN MK, HAMRE KM & LU L 2017. Analyses of differentially expressed genes after exposure to acute stress, acute ethanol, or a combination of both in mice. Alcohol, 58, 139–151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BANNIGAN J & COTTELL D 1984. Ethanol teratogenicity in mice: an electron microscopic study. Teratology, 30, 281–90. [DOI] [PubMed] [Google Scholar]
- BOEHM SL 2ND, LUNDAHL KR, CALDWELL J & GILLIAM DM 1997. Ethanol teratogenesis in the C57BL/6J, DBA/2J, and A/J inbred mouse strains. Alcohol, 14, 389–95. [DOI] [PubMed] [Google Scholar]
- BONTHIUS DJ, TZOURAS G, KARACAY B, MAHONEY J, HUTTON A, MCKIM R & PANTAZIS NJ 2002. Deficiency of neuronal nitric oxide synthase (nNOS) worsens alcohol-induced microencephaly and neuronal loss in developing mice. Brain Res Dev Brain Res, 138, 45–59. [DOI] [PubMed] [Google Scholar]
- CALEGARI F, HAUBENSAK W, HAFFNER C & HUTTNER WB 2005. Selective lengthening of the cell cycle in the neurogenic subpopulation of neural progenitor cells during mouse brain development. J Neurosci, 25, 6533–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CAMARGO MORENO M, MOONEY SM & MIDDLETON FA 2017. Heterogeneity of p53 dependent genomic responses following ethanol exposure in a developmental mouse model of fetal alcohol spectrum disorder. PLoS One, 12, e0180873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CHEN Y, OZTURK NC, NI L, GOODLETT C & ZHOU FC 2011. Strain differences in developmental vulnerability to alcohol exposure via embryo culture in mice. Alcohol Clin Exp Res, 35, 1293–304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CHERNOFF GF 1980. The fetal alcohol syndrome in mice: maternal variables. Teratology, 22, 71–5. [DOI] [PubMed] [Google Scholar]
- CHESLER EJ, LU L, WANG J, WILLIAMS RW & MANLY KF 2004. WebQTL: rapid exploratory analysis of gene expression and genetic networks for brain and behavior. Nat Neurosci, 7, 485–6. [DOI] [PubMed] [Google Scholar]
- CHESLER EJ, WANG J, LU L, QU Y, MANLY KF & WILLIAMS RW 2003. Genetic correlates of gene expression in recombinant inbred strains: a relational model system to explore neurobehavioral phenotypes. Neuroinformatics, 1, 343–57. [DOI] [PubMed] [Google Scholar]
- CHOY KW, LAU YS, MURUGAN D, VANHOUTTE PM & MUSTAFA MR 2018. Paeonol Attenuates LPS-Induced Endothelial Dysfunction and Apoptosis by Inhibiting BMP4 and TLR4 Signaling Simultaneously but Independently. J Pharmacol Exp Ther, 364, 420–432. [DOI] [PubMed] [Google Scholar]
- CHRISTOFFEL KK & SALAFSKY I 1975. Fetal alcohol syndrome in dizygotic twins. J Pediatr, 87, 963–7. [DOI] [PubMed] [Google Scholar]
- CONTI AC, YOUNG C, OLNEY JW & MUGLIA LJ 2009. Adenylyl cyclases types 1 and 8 promote pro-survival pathways after ethanol exposure in the neonatal brain. Neurobiol Dis, 33, 111–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- COOK MN, BAKER JA, HELDT SA, WILLIAMS RW, HAMRE KM & LU L 2015. Identification of candidate genes that underlie the QTL on chromosome 1 that mediates genetic differences in stress-ethanol interactions. Physiol Genomics, 47, 308–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CRABBE JC & BELKNAP JK 1992. Genetic approaches to drug dependence. Trends Pharmacol Sci, 13, 212–9. [DOI] [PubMed] [Google Scholar]
- CUNNINGHAM CL 1995. Localization of genes influencing ethanol-induced conditioned place preference and locomotor activity in BXD recombinant inbred mice. Psychopharmacology (Berl), 120, 28–41. [DOI] [PubMed] [Google Scholar]
- DOWNING C, BALDERRAMA-DURBIN C, KIMBALL A, BIERS J, WRIGHT H, GILLIAM D & JOHNSON TE 2012. Quantitative trait locus mapping for ethanol teratogenesis in BXD recombinant inbred mice. Alcohol Clin Exp Res, 36, 1340–54. [DOI] [PubMed] [Google Scholar]
- DRISCOLL CD, STREISSGUTH AP & RILEY EP 1990. Prenatal alcohol exposure: comparability of effects in humans and animal models. Neurotoxicol Teratol, 12, 231–7. [DOI] [PubMed] [Google Scholar]
- DUNTY WC JR., CHEN SY, ZUCKER RM, DEHART DB & SULIK KK 2001. Selective vulnerability of embryonic cell populations to ethanol-induced apoptosis: implications for alcohol-related birth defects and neurodevelopmental disorder. Alcohol Clin Exp Res, 25, 1523–35. [PubMed] [Google Scholar]
- EASTER SS JR., ROSS LS & FRANKFURTER A 1993. Initial tract formation in the mouse brain. J Neurosci, 13, 285–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- EBERHART JK & PARNELL SE 2016. The Genetics of Fetal Alcohol Spectrum Disorders. Alcohol Clin Exp Res, 40, 1154–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- FU Y, YANG MS, JIANG J, GANESH T, JOE E & DINGLEDINE R 2015. EP2 Receptor Signaling Regulates Microglia Death. Mol Pharmacol, 88, 161–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GIUNTA B, OBREGON D, HOU H, ZENG J, SUN N, NIKOLIC V, EHRHART J, SHYTLE D, FERNANDEZ F & TAN J 2006. EGCG mitigates neurotoxicity mediated by HIV-1 proteins gp120 and Tat in the presence of IFN-gamma: role of JAK/STAT1 signaling and implications for HIV-associated dementia. Brain Res, 1123, 216–225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GOLDOWITZ D, LUSSIER AA, BOYLE JK, WONG K, LATTIMER SL, DUBOSE C, LU L, KOBOR MS & HAMRE KM 2014. Molecular pathways underpinning ethanol-induced neurodegeneration. Front Genet, 5, 203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GORA-MASLAK G, MCCLEARN GE, CRABBE JC, PHILLIPS TJ, BELKNAP JK & PLOMIN R 1991. Use of recombinant inbred strains to identify quantitative trait loci in psychopharmacology. Psychopharmacology (Berl), 104, 413–24. [DOI] [PubMed] [Google Scholar]
- GREEN ML, SINGH AV, ZHANG Y, NEMETH KA, SULIK KK & KNUDSEN TB 2007. Reprogramming of genetic networks during initiation of the Fetal Alcohol Syndrome. Dev Dyn, 236, 613–31. [DOI] [PubMed] [Google Scholar]
- HEATON MB, MOORE DB, PAIVA M, GIBBS T & BERNARD O 1999. Bcl-2 overexpression protects the neonatal cerebellum from ethanol neurotoxicity. Brain Res, 817, 13–8. [DOI] [PubMed] [Google Scholar]
- JACOBSON JL, JACOBSON SW, SOKOL RJ & AGER JW JR. 1998. Relation of maternal age and pattern of pregnancy drinking to functionally significant cognitive deficit in infancy. Alcohol Clin Exp Res, 22, 345–51. [DOI] [PubMed] [Google Scholar]
- JONAKAIT GM & NI L 2009. Prostaglandins compromise basal forebrain cholinergic neuron differentiation and survival: action at EP1/3 receptors results in AIF-induced death. Brain Res, 1285, 30–41. [DOI] [PubMed] [Google Scholar]
- JONES KL, SMITH DW, ULLELAND CN & STREISSGUTH P 1973. Pattern of malformation in offspring of chronic alcoholic mothers. Lancet, 1, 1267–71. [DOI] [PubMed] [Google Scholar]
- KARACAY B, MAHONEY J, PLUME J & BONTHIUS DJ 2015. Genetic absence of nNOS worsens fetal alcohol effects in mice. II: microencephaly and neuronal losses. Alcohol Clin Exp Res, 39, 221–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KENNELLY K, BRENNAN D, CHUMMUN K & GILES S 2011. Histological characterisation of the ethanol-induced microphthalmia phenotype in a chick embryo model system. Reprod Toxicol, 32, 227–34. [DOI] [PubMed] [Google Scholar]
- KLEIBER ML, LAUFER BI, WRIGHT E, DIEHL EJ & SINGH SM 2012. Long-term alterations to the brain transcriptome in a maternal voluntary consumption model of fetal alcohol spectrum disorders. Brain Res, 1458, 18–33. [DOI] [PubMed] [Google Scholar]
- KORKMAZ G, LE SAGE C, TEKIRDAG KA, AGAMI R & GOZUACIK D 2012. miR-376b controls starvation and mTOR inhibition-related autophagy by targeting ATG4C and BECN1. Autophagy, 8, 165–76. [DOI] [PubMed] [Google Scholar]
- KOTCH LE & SULIK KK 1992a. Experimental fetal alcohol syndrome: proposed pathogenic basis for a variety of associated facial and brain anomalies. Am J Med Genet, 44, 168–76. [DOI] [PubMed] [Google Scholar]
- KOTCH LE & SULIK KK 1992b. Patterns of ethanol-induced cell death in the developing nervous system of mice; neural fold states through the time of anterior neural tube closure. Int J Dev Neurosci, 10, 273–9. [DOI] [PubMed] [Google Scholar]
- LAUFER BI, MANTHA K, KLEIBER ML, DIEHL EJ, ADDISON SM & SINGH SM 2013. Long-lasting alterations to DNA methylation and ncRNAs could underlie the effects of fetal alcohol exposure in mice. Dis Model Mech, 6, 977–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LEE CC, CHEN PH, HO KH, SHIH CM, CHENG CH, LIN CW, CHENG KT, LIU AJ & CHEN KC 2017a. The microRNA-302b-inhibited insulin-like growth factor-binding protein 2 signaling pathway induces glioma cell apoptosis by targeting nuclear factor IA. PLoS One, 12, e0173890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LEE J, HOXHA E & SONG HR 2017b. A novel NFIA-NFkappaB feed-forward loop contributes to glioblastoma cell survival. Neuro Oncol, 19, 524–534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LIOU JY, ELLENT DP, LEE S, GOLDSBY J, KO BS, MATIJEVIC N, HUANG JC & WU KK 2007. Cyclooxygenase-2-derived prostaglandin e2 protects mouse embryonic stem cells from apoptosis. Stem Cells, 25, 1096–103. [DOI] [PubMed] [Google Scholar]
- MANDAL C, PARK JH, LEE HT, SEO H, CHUNG IY, CHOI IG, JUNG KH & CHAI YG 2015. Reduction of Nfia gene expression and subsequent target genes by binge alcohol in the fetal brain. Neurosci Lett, 598, 73–8. [DOI] [PubMed] [Google Scholar]
- MAY PA, CHAMBERS CD, KALBERG WO, ZELLNER J, FELDMAN H, BUCKLEY D, KOPALD D, HASKEN JM, XU R, HONERKAMP-SMITH G, TARAS H, MANNING MA, ROBINSON LK, ADAM MP, ABDUL-RAHMAN O, VAUX K, JEWETT T, ELLIOTT AJ, KABLE JA, AKSHOOMOFF N, FALK D, ARROYO JA, HERELD D, RILEY EP, CHARNESS ME, COLES CD, WARREN KR, JONES KL & HOYME HE 2018. Prevalence of Fetal Alcohol Spectrum Disorders in 4 US Communities. JAMA, 319, 474–482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- NIKOLIC P, JARLEBARK LE, BILLETT TE & THORNE PR 2000. Apoptosis in the developing rat cochlea and its related structures. Brain Res Dev Brain Res, 119, 75–83. [DOI] [PubMed] [Google Scholar]
- OLNEY JW, TENKOVA T, DIKRANIAN K, MUGLIA LJ, JERMAKOWICZ WJ, D’SA C & ROTH KA 2002a. Ethanol-induced caspase-3 activation in the in vivo developing mouse brain. Neurobiol Dis, 9, 205–19. [DOI] [PubMed] [Google Scholar]
- OLNEY JW, TENKOVA T, DIKRANIAN K, QIN YQ, LABRUYERE J & IKONOMIDOU C 2002b. Ethanol-induced apoptotic neurodegeneration in the developing C57BL/6 mouse brain. Brain Res Dev Brain Res, 133, 115–26. [DOI] [PubMed] [Google Scholar]
- PARNELL SE, HOLLOWAY HT, O’LEARY-MOORE SK, DEHART DB, PANIAQUA B, OGUZ I, BUDIN F, STYNER MA, JOHNSON GA & SULIK KK 2013. Magnetic resonance microscopy-based analyses of the neuroanatomical effects of gestational day 9 ethanol exposure in mice. Neurotoxicol Teratol, 39, 77–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- PATERSON C & LAW AJ 2014. Transient overexposure of neuregulin 3 during early postnatal development impacts selective behaviors in adulthood. PLoS One, 9, e104172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- PEIRCE JL, LU L, GU J, SILVER LM & WILLIAMS RW 2004. A new set of BXD recombinant inbred lines from advanced intercross populations in mice. BMC Genet, 5, 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- PLOMIN R, MCCLEARN GE, GORA-MASLAK G & NEIDERHISER JM 1991. Use of recombinant inbred strains to detect quantitative trait loci associated with behavior. Behav Genet, 21, 99–116. [DOI] [PubMed] [Google Scholar]
- RODIER PM 1980. Chronology of neuron development: animal studies and their clinical implications. Dev Med Child Neurol, 22, 525–45. [DOI] [PubMed] [Google Scholar]
- SMITH SM 1997. Alcohol-induced cell death in the embryo. Alcohol Health Res World, 21, 287–97. [PMC free article] [PubMed] [Google Scholar]
- SU AI, WILTSHIRE T, BATALOV S, LAPP H, CHING KA, BLOCK D, ZHANG J, SODEN R, HAYAKAWA M, KREIMAN G, COOKE MP, WALKER JR & HOGENESCH JB 2004. A gene atlas of the mouse and human protein-encoding transcriptomes. Proc Natl Acad Sci U S A, 101, 6062–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SUBBANNA S, SHIVAKUMAR M, PSYCHOYOS D, XIE S & BASAVARAJAPPA BS 2013. Anandamide-CB1 receptor signaling contributes to postnatal ethanol-induced neonatal neurodegeneration, adult synaptic, and memory deficits. J Neurosci, 33, 6350–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- TROUSSE F, ESTEVE P & BOVOLENTA P 2001. Bmp4 mediates apoptotic cell death in the developing chick eye. J Neurosci, 21, 1292–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- TSAI PC, BAKE S, BALARAMAN S, RAWLINGS J, HOLGATE RR, DUBOIS D & MIRANDA RC 2014. MiR-153 targets the nuclear factor-1 family and protects against teratogenic effects of ethanol exposure in fetal neural stem cells. Biol Open, 3, 741–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- URQUHART KR, ZHAO Y, BAKER JA, LU Y, YAN L, COOK MN, JONES BC, HAMRE KM & LU L 2016. A novel heat shock protein alpha 8 (Hspa8) molecular network mediating responses to stress- and ethanol-related behaviors. Neurogenetics, 17, 91–105. [DOI] [PubMed] [Google Scholar]
- WANG J, WILLIAMS RW & MANLY KF 2003. WebQTL: web-based complex trait analysis. Neuroinformatics, 1, 299–308. [DOI] [PubMed] [Google Scholar]
- WEST JR 1987. Fetal alcohol-induced brain damage and the problem of determining temporal vulnerability: a review. Alcohol Drug Res, 7, 423–41. [PubMed] [Google Scholar]
- WOLEN AR, PHILLIPS CA, LANGSTON MA, PUTMAN AH, VORSTER PJ, BRUCE NA, YORK TP, WILLIAMS RW & MILES MF 2012. Genetic dissection of acute ethanol responsive gene networks in prefrontal cortex: functional and mechanistic implications. PLoS One, 7, e33575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ZHANG P, WANG G, LIN Z, WU Y, ZHANG J, LIU M, LEE KKH, CHUAI M & YANG X 2017. Alcohol exposure induces chick craniofacial bone defects by negatively affecting cranial neural crest development. Toxicol Lett, 281, 53–64. [DOI] [PubMed] [Google Scholar]
- ZHOU DX, ZHAO Y, BAKER JA, GU Q, HAMRE KM, YUE J, JONES BC, COOK MN & LU L 2017. The effect of alcohol on the differential expression of cluster of differentiation 14 gene, associated pathways, and genetic network. PLoS One, 12, e0178689. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.