

Abstract
Summary: Breast cancer is one of the most frequent cancers among women. Extensive studies into the molecular heterogeneity of breast cancer have produced a plethora of molecular subtype classification and prognosis prediction algorithms, as well as numerous gene expression signatures. However, reimplementation of these algorithms is a tedious but important task to enable comparison of existing signatures and classification models between each other and with new models. Here, we present the genefu R/Bioconductor package, a multi-tiered compendium of bioinformatics algorithms and gene signatures for molecular subtyping and prognostication in breast cancer.
Availability and implementation: The genefu package is available from Bioconductor. http://www.bioconductor.org/packages/devel/bioc/html/genefu.html. Source code is also available on Github https://github.com/bhklab/genefu.
Contact: bhaibeka@uhnresearch.ca
Supplementary information: Supplementary data are available at Bioinformatics online.
1 Introduction
Breast cancer is a devastating disease whose management is complicated by its high molecular heterogeneity. Breast cancer is categorized into at least four clinically relevant molecular subtypes that reflect upon expression levels of specific genes (Koboldt et al., 2012). These are ‘Her2-enriched’ (also called HER2+), ‘Triple-negative breast cancers” (ER−/HER2−/PR−, similar to basal-like) and ‘Luminal-like’ which are mainly ER+/HER2− that could be further discriminated into luminal A and B based on their low and high proliferative phenotype, respectively. These classifications have showed that breast cancer is not a single disease but is rather highly heterogeneous. Breast cancer subtypes exhibit distinct transcriptomic patterns that are associated with outcome, which emphasizes the relevance of these subtypes for basic and translational research (Sotiriou and Pusztai, 2009).
Among various molecular techniques, gene expression profiling in particular has increasingly been used to refine breast cancer stratification, and to assess prognosis and response to therapy (Prat et al., 2012). Studies to identify gene expression patterns of clinical potential have produced a considerable number of prognostic predictors and subtype-specific prognostic signatures. However, varying reports of prognostic signatures across breast cancer subtypes render comparison of these approaches difficult. Multiple meta-analyses of gene signatures indicated that the majority share similar performance, despite the limited overlap of genes (Desmedt et al., 2008; Fan et al., 2006). Importantly, the prognostic value of the vast majority of these signatures were limited to luminal-like breast tumors, calling for the development of subtype-specific prognostic models (Haibe-Kains et al., 2010). Subtype-specific prognoses however are also largely dependent on the proper identification of the major biological subtypes. The parallel development of breast cancer subtyping methods has also produced a significant number of classification algorithms, with diverse taxonomies.
The large body of literature on the molecular complexity of breast cancer has introduced a burden of computational complexity for researchers. The advent of high-throughput genomics and next-generation sequencing promises unprecedented views into the major biological breast cancer subtypes (Schnitt, 2010). However, there is a dire need for an accessible, computational platform that can easily sustain the growing variety of breast cancer classification and prognostic algorithms. This will facilitate both meta-analyses of breast cancer data as well as cross comparison of different computational methods. Here, we have developed the genefu package, a multi-leveled compendium that provides bioinformatics implementations of classification algorithms to identify molecular subtypes, as well as prognostic predictors along with their published gene signatures (Table 1, Supplementary Fig. S1). Notably, we have incorporated the most recently developed molecular subtyping algorithms in the field, including the IntClust (Curtis et al., 2012), the IHC4 prognostic scoring algorithm (Dowsett et al., 2013), the Absolute Intrinsic Molecular Subypting (AIMS) algorithm (Paquet and Hallett, 2015), as well as the classification algorithm for prediction of claudin-low breast cancer samples (Prat et al., 2010). The package also includes other functions to facilitate quick manipulation of gene expression datasets, including gene selection and probe-gene mapping across microarray platforms.
Table 1.
List of molecular classification and prognostication algorithms in genefu
| Molecular subtype classification algorithms (×9) | ||
| Single Sample Predictors (SSP2003, SSP2006 and PAM50) | ||
| Subtype Clustering Models (SCMOD1, SCMOD2 and SCMGENE) | ||
| Claudin-low | ||
| IntClust | ||
| AIMS | ||
|
| ||
| Prognostication of breast cancer (×12) | Prognostic predictor | Prognostic signature |
| EndoPredict | sig.endoPredict | |
| GENE70 | GENE70 signature | |
| GENE76 | GENE76 signature | |
| GENIUS | sig.genius signatures | |
| GGI | sig.ggi 97-gene signature | |
| Nottingham prognostic index calculation (NPI) | ||
| OncotypeDX | Oncotypedx signature | |
| PIK3CA signature | PIK3CA-GS: PIK3CA signature | |
| Risk of relapse based on subtype (RORS) | ||
| mod1 (gene modules) | Mod1: list of seven gene modules | |
| mod2 (gene modules) | Mod2: list of three gene modules | |
| IHC4 | ||
| TAMR13 predictor | TAMR13 signature | |
2 Molecular subtyping
We have implemented nine molecular subtyping algorithms within genefu, which facilitates the identification of molecular subtypes as well as assessment of stratified patient data. We compare here the molecular subtype predictions across five public datasets using the PAM50 and SCMOD2 algorithms. Both PAM50 and SCMOD2 predict patients that belong to the Basal-like, and Her2-enriched subtypes, as well as distinguish between the Luminal A (LumA) and Luminal B (LumB) subtypes. PAM50 additionally identifies ‘normal-like’ patients. Overall, there is a concordance of ∼85% between both predictors (Supplementary Data S1). Despite the lack of complete concordance between the two algorithms, patient survival across subtypes is virtually identical between the two subtyping schemes (Fig. 1A). Notably, survival is the highest for LumA patients, while patients LumB, Her2-enriched and Basal-like tumors display the poorest survival. This indicates that different molecular subtyping algorithms are consistent and yield similar prognostic value across breast cancer subtypes.
Fig. 1.
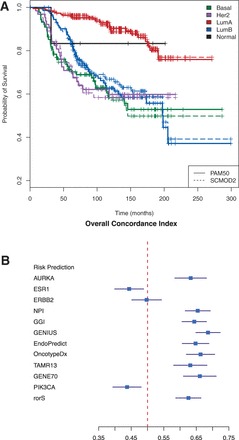
(A) Kaplan–Meier curves of breast cancer patients, stratified by molecular subtype, using predictions generated by the PAM50 and SCMOD2 predictors. (B) Overall performance estimate for each risk prediction model across five breast cancer datasets. Risk scores were first calculated for each of the prognostic predictors across 713 samples. The concordance index for the risk prediction was subsequently calculated over the risk scores using distant metastasis free survival or recurrence-free survival sample time points, dependent on the dataset. Dataset-specific concordance indices were finally combined into overall estimates for each risk prediction model. The performed analysis includes biology-driven signatures (AURKA, ESR1, ERBB2 and PIK3CA) as well as prognostic-driven signatures
3 Prognostication of breast cancer
Our implementation of numerous prognostic predictors as part of genefu facilitates meta-analysis of prognostic signatures across several breast cancer datasets. We previously demonstrated that the majority of prognostic signatures show similar performance despite limited genetic overlaps between them (Desmedt et al., 2008; Wirapati et al., 2008). We showed that many of published gene signatures were affected by the presence of proliferation-related genes, rendering the signature informative for prognosis of ER+/HER2− patients, but less informative for basal-like and HER2-enriched patients. To improve prognostication for these subtypes, we implemented a new risk prediction model, called GENIUS, which uses a fuzzy computational approach combining risk prediction models specific to each molecular subtype (Haibe-Kains et al., 2010). We have subsequently demonstrated that Gaussian-based clustering based on gene sets specifically correlated with the ER, HER2 and AURKA genes can robustly identify breast cancer molecular subtypes (Haibe-Kains et al., 2012).
In this case study, we used genefu to conduct a meta-analysis of 12 risk predictors across 713 node-negative, untreated breast cancer patients (Supplementary Data S1). For comparison, three of our predictors were based on using just one of three key breast cancer genes (represented here as AURKA, ESR1 and ERBB2). We calculated the risk score performance of each predictor, measured by the concordance index, across the five breast cancer datasets (Supplementary Fig. S2). The concordance index estimates the probability that, for a random pair of patients, the patient with earlier recurrence has a higher risk score than the patients with either later or no recurrence. Using a random-effects model, we combined the dataset-specific performance estimates into an overall performance estimate for each risk prediction model (Fig. 1B). Our findings suggest significant prognostic value for all multi-gene predictors. Notably, using a single proliferation gene (AURKA) produces a similar performance to the majority of other predictors.
4 Application across data platforms
Many of the molecular subtyping algorithms and gene expression signatures implemented within genefu have originally been derived from microarray gene expression data. This raises an important question as to whether such methods can be used on data generated on RNA sequencing platforms. We have previously compared the agreement between microarray and RNAs sequencing platforms on several prognostic signatures and classifiers from genefu (Fumagalli et al., 2014). This analysis was conducted on six molecular subtyping algorithms (SCMOD1, SCMO2, SCMGENE, SSP2003, SSP2006 and PAM50) and several prognostic-driven signatures including GENE70, GGI, RORS, ENDOPREDICT, among other gene expression signatures. We have demonstrated that the clinically relevant single genes and gene expression signatures originally defined by microarray technology can be used to reliably evaluate RNA sequencing data. The code for this analysis is additionally available on github at https://github.com/bhklab/DNA11161.
5 Conclusions
The genefu package provides a unified framework for integration of molecular subtype and survival analysis of breast cancer. We have demonstrated how the package can be utilized to perform both meta-analyses across datasets and across algorithms, to facilitate integrated analysis of breast cancer gene expression profiles.
Supplementary Material
Acknowledgements
The authors would like to thank Dr. Michael Hallet for sharing his feedback and experiences regarding implementation of some of the breast cancer molecular subtyping schemes.
Funding
CIBC-Brain Canada Brain Cancer Research Training Award to DMAG. B.H.K. is supported by the Gattuso Slaight Personalized Cancer Medicine Fund, Cancer Research Society and CIHR Grant 201412MSH-340176-229599. A.P. would like to acknowledge the Instituto de Salud Carlos III - PI13/01718, a Career Catalyst Grant from the Susan Komen Foundation (A.P.) and Banco Bilbao Vizcaya Argentaria (BBVA) Foundation.
Conflict of Interest: none declared.
References
- Curtis C., et al. (2012) The genomic and transcriptomic architecture of 2 000 breast tumours reveals novel subgroups. Nature 486, 346–352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desmedt C., et al. (2008) Biological processes associated with breast cancer clinical outcome depend on the molecular subtypes. Clin. Cancer Res. 14, 5158–5165. [DOI] [PubMed] [Google Scholar]
- Dowsett M., et al. (2013) Comparison of PAM50 risk of recurrence score with oncotype DX and IHC4 for predicting risk of distant recurrence after endocrine therapy. J. Clin. Oncol. 31, 2783–2790. [DOI] [PubMed] [Google Scholar]
- Fan C., et al. (2006) Concordance among gene-expression-based predictors for breast cancer. N Engl J Med 355, 560–569. [DOI] [PubMed] [Google Scholar]
- Fumagalli D., et al. (2014) Transfer of clinically relevant gene expression signatures in breast cancer: from Affymetrix microarray to Illumina RNA-Sequencing technology. BMC Genom. 15, 1008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haibe-Kains B., et al. (2010) A fuzzy gene expression-based computational approach improves breast cancer prognostication. Genome Biol. 11, R18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haibe-Kains B., et al. (2012) A three-gene model to robustly identify breast cancer molecular subtypes. J. Natl Cancer Inst. 104, 311–325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koboldt D.C., et al. (2012) Comprehensive molecular portraits of human breast tumours. Nature 490, 61–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paquet E.R., Hallett M.T. (2015) Absolute assignment of breast cancer intrinsic molecular subtype. J. Natl Cancer Inst. 107, 357. [DOI] [PubMed] [Google Scholar]
- Prat A., et al. (2010) Phenotypic and molecular characterization of the claudin-low intrinsic subtype of breast cancer. Breast Cancer Res. BCR 12, R68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prat A., et al. (2012) Practical implications of gene-expression-based assays for breast oncologists. Nat. Rev. Clin. Oncol. 9, 48–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schnitt S.J. (2010) Classification and prognosis of invasive breast cancer: from morphology to molecular taxonomy. Mod. Pathol. 23(Suppl 2), S60–S64. [DOI] [PubMed] [Google Scholar]
- Sotiriou C., Pusztai L. (2009) Gene-expression signatures in breast cancer. N. Engl. J. Med. 360, 790–800. [DOI] [PubMed] [Google Scholar]
- Wirapati P., et al. (2008) Meta-analysis of gene expression profiles in breast cancer: toward a unified understanding of breast cancer subtyping and prognosis signatures. Breast Cancer Res. BCR 10, R65. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.