

In this study, Zhao et al. show that in the protozoan Tetrahymena thermophila, germline-specific internally eliminated sequences (IESs) become transcriptionally activated in mutants deficient in the RNAi-dependent Polycomb repression pathway. Their findings suggest that the interplay between RNAi and Polycomb repression is a widely conserved phenomenon whose ancestral role is epigenetic silencing of TEs.
Keywords: Polycomb repression, RNAi, transposable elements
Abstract
RNAi and Polycomb repression play evolutionarily conserved and often coordinated roles in transcriptional silencing. Here, we show that, in the protozoan Tetrahymena thermophila, germline-specific internally eliminated sequences (IESs)—many related to transposable elements (TEs)—become transcriptionally activated in mutants deficient in the RNAi-dependent Polycomb repression pathway. Germline TE mobilization also dramatically increases in these mutants. The transition from noncoding RNA (ncRNA) to mRNA production accompanies transcriptional activation of TE-related sequences and vice versa for transcriptional silencing. The balance between ncRNA and mRNA production is potentially affected by cotranscriptional processing as well as RNAi and Polycomb repression. We posit that interplay between RNAi and Polycomb repression is a widely conserved phenomenon, whose ancestral role is epigenetic silencing of TEs.
Polycomb group (PcG) proteins are involved in developmentally regulated transcriptional silencing in a wide range of eukaryotic systems: Among the best characterized are Hox gene repression in Drosophila and vertebrates and X chromosome inactivation in female mammals (Di Croce and Helin 2013; Grossniklaus and Paro 2014). For example, Drosophila E(z) and its homologs are SET domain-containing histone methyltransferases specific for histone H3 Lys27 (H3K27) methylation (Cao et al. 2002; Czermin et al. 2002; Kuzmichev et al. 2002; Müller et al. 2002). This histone modification is recognized by Drosophila Pc and other chromodomain-containing proteins, leading to heterochromatin formation. Growing evidence implicates both long noncoding RNA (ncRNA) and small RNA in Polycomb repression (Brockdorff 2013; Simon and Kingston 2013; Davidovich and Cech 2015). In Drosophila, both the RNAi machinery and PcG proteins are required for silencing in somatic and germline cells (Pal-Bhadra et al. 1997, 2002; Peng et al. 2016). Many long ncRNA, including Xist RNA involved in X inactivation in mammalian cells, are associated with Polycomb-repressive complex 2 (PRC2) and implicated in PRC2-mediated transcriptional repression (Khalil et al. 2009; Tsai et al. 2010; Zhao et al. 2010). X inactivation also intersects with nuclear RNAi (Ogawa et al. 2008; Zhao et al. 2008; Kanellopoulou et al. 2009)—a conserved pathway for transcriptional silencing (Grewal and Elgin 2007; Martienssen and Moazed 2015).
In common with other ciliated protozoa, Tetrahymena thermophila contains in the same cytoplasmic compartment two types of nuclei: the germline micronucleus (MIC) and the somatic macronucleus (MAC) (Karrer 2012). MIC can differentiate into MAC during conjugation, the sexual phase of the Tetrahymena life cycle, accompanied by massive programmed genome rearrangement (Chalker et al. 2013; Yao et al. 2014). Thousands of MIC-specific internally eliminated sequences (IESs) are removed, leaving behind MAC-destined sequences (MDSs) (Fig. 1A). Studies of developmentally regulated heterochromatin formation and DNA elimination in Tetrahymena have revealed a pathway involving both the RNAi machinery and PcG proteins (Fig. 1B; Noto and Mochizuki 2017). The pathway starts with RNA polymerase II (Pol II)-catalyzed bidirectional transcription of long ncRNA in the meiotic MIC (Chalker and Yao 2001; Mochizuki and Gorovsky 2004b; Aronica et al. 2008). A special class of small RNA, referred to as scan RNA (scnRNA), accumulates in a manner dependent on the RNAi machinery, which includes DCL1, a Dicer-like protein that processes long ncRNA into scnRNA (Malone et al. 2005; Mochizuki and Gorovsky 2005), and TWI1, an Argonaute/Piwi homolog that binds scnRNA (Mochizuki et al. 2002; Mochizuki and Gorovsky 2004a; Noto et al. 2010). Conserved histone modifications, H3K27 and H3K9 methylation, are deposited in a manner dependent on both the RNAi machinery and EZL1, an E(z) homolog in Tetrahymena (Liu et al. 2004, 2007). These histone modifications are subsequently recognized by chromodomain-containing effectors like PDD1 (analogous to HP1), which help to form heterochromatic structures containing DNA sequences that are eventually eliminated (Madireddi et al. 1996; Coyne et al. 1999; Taverna et al. 2002; Liu et al. 2007; Schwope and Chalker 2014).
Figure 1.

Widespread production of IES-specific polyadenylated transcripts in mutants deficient in RNAi-dependent Polycomb repression. (A) A schematic for DNA elimination in Tetrahymena. (B) Key steps and players in the DNA elimination pathway. (C) Distribution of polyadenylated transcripts in IESs. We ranked 9878 consistently processed IESs (Supplemental File S1) from highest to lowest by their coverage of polyadenylated RNA (reads per kilobase of transcript per million mapped reads [RPKM]), in wild type and mutants deficient in RNAi-dependent Polycomb repression (ΔDCL1, ΔEZL1, and ΔPDD1, respectively). (Inset) The top 100 IESs. Note increased and widespread transcription of IES-specific sequences in the mutants. (D) Distribution of highly expressed IESs, in wild type and the mutants. Supercontig 2.1, over 3.5 Mb in length, is analyzed. Read densities (RPKM) are represented by a color scale. Note the alignment of red stripes in the mutants, indicating coregulation by the RNAi-dependent Polycomb repression pathway. (E) A representative GBrowse view illustrating RNA sequencing (RNA-seq) coverage in wild type and the mutants. (Blue bar) IES. Gene model: an IES-specific (left) and an MDS-specific (right) gene. Note dramatically increased IES transcription in the mutants, compared with wild-type cells. (F) A proportional Venn diagram representing IESs highly induced in the mutants. (G) A box plot comparing PDD1 enrichment in wild-type cells [(ChIP − input)/(ChIP + input)] in IESs induced in the mutants (ΔDCL1, ΔEZL1, and ΔPDD1 cells, respectively) with IESs not induced in any mutants. The first quartile, median, and the third quartile are marked. A Kruskal-Wallis H test was performed for all three pairwise comparisons, revealing highly significant variances. P < 2.2 × 10−16.
It has long been known that many Tetrahymena IESs contain sequences derived from transposable elements (TEs) (Wuitschick et al. 2002; Fillingham et al. 2004). Various TEs are revealed in the recently sequenced MIC genomes of ciliates, including Tetrahymena (Fass et al. 2011; Hamilton et al. 2016), Paramecium (Arnaiz et al. 2012; Guérin et al. 2017), and Oxytricha (Chen et al. 2014). Recent transposition in Tetrahymena populations is supported by TE insertion polymorphisms in certain IESs (Huvos 2004a,b), as well as purifying selection in predicted coding sequences of many potentially active TEs (Gershan and Karrer 2000; Fillingham et al. 2004; Hamilton et al. 2016). Nonetheless, a complete understanding of how TEs are propagated and controlled in the binucleated ciliates remains elusive.
Here, we show that Tetrahymena IESs—many containing TE-related sequences—are transcriptionally activated in mutants deficient in the RNAi-dependent Polycomb repression pathway. Germline mobilization of recently active TEs also increases dramatically in these mutants. Furthermore, transcriptional activation of TE-related sequences coincides with the transition from ncRNA to mRNA production, and vice versa for transcriptional silencing. The balance between ncRNA and mRNA production is tipped by cotranscriptional processing as well as RNAi and Polycomb repression. Based on conservation of key components and wide distribution of similar pathways in eukaryotes, we propose that interplay between RNAi and Polycomb repression may be a ubiquitous phenomenon utilized for TE silencing as well as transcriptional repression of developmental genes.
Results
Widespread production of IES-specific polyadenylated RNA in mutants deficient in RNAi-dependent Polycomb repression
We examined RNA transcripts from germline-specific IESs (Fig. 1A), in wild-type cells as well as three mutants deficient in different steps of the RNAi-dependent Polycomb repression pathway—ΔDCL1, ΔEZL1, and ΔPDD1 (Fig. 1B). We focused on late conjugation (10 h after mixing of complementary mating types), when IESs in the developing MAC are heterochromatinized but not yet excised. The RNA samples, after oligo-dT enrichment of polyadenylated transcripts, were analyzed by strand-specific Illumina sequencing (RNA sequencing [RNA-seq]). RNA-seq reads were mapped back to the Tetrahymena MIC reference genome (Hamilton et al. 2016). Most of the ∼10,000 IESs analyzed were covered at low levels in wild-type cells, while many of them were abundantly covered in the mutants (Fig. 1C; Supplemental File S1). Transcriptional activation was widely distributed in the genome, as illustrated by locations of IES-specific polyadenylated RNA within Supercontig_2.1, the longest assembled scaffold of the Tetrahymena MIC genome (Fig. 1D). Furthermore, transcription patterns in the mutants shared substantial similarities with each other (Fig. 1D). There were significant overlaps between the sets of IESs that were highly induced in each mutant (Fig. 1F), reflecting their coregulation by RNAi and Polycomb repression. Zooming into individual IESs, we found that IES transcripts were often clustered into distinct loci, many of which showed little or no expression in wild-type cells but were highly induced in each mutant (Fig. 1E). In contrast to IES-specific transcripts, transcripts from MDSs, corresponding to bona fide mRNA, were present at similar levels in wild-type cells and the mutants (Fig. 1E; Supplemental Fig. S1).
It should be noted that, even though all IES elimination is abolished in ΔDCL1, ΔEZL1, and ΔPDD1 cells (Feng et al. 2017), polyadenylated transcripts were not detected in all IESs, and the number of polyadenylated transcripts mapped to individual IESs could vary dramatically among the mutants (Fig. 1C). Furthermore, the IESs affected in ΔEZL1 or ΔPDD1 were essentially subsets of the IESs affected in ΔDCL1 (Fig. 1F). This containment is consistent with DCL1-mediated RNAi being upstream of EZL1 and PDD1-mediated Polycomb repression (Fig. 1B) and suggests that additional pathways may be affected in ΔDCL1. It also supports that IES processing is intrinsically robust, probably underpinned by an extensive trans-recognition network mediated by scnRNA (Noto et al. 2015; Noto and Mochizuki 2018).
We also examined distribution of PDD1 in IESs by re-analyzing published ChIP-seq (chromatin immunoprecipitation [ChIP] combined with high-throughput sequencing) data (Kataoka and Mochizuki 2015). IESs that were highly induced transcriptionally in the mutants were enriched with PDD1 in wild-type cells, while those not induced in any mutants were depleted of PDD1 (Fig. 1G; Supplemental Fig. S2), suggesting that PDD1 specifically, and this pathway in general, directly silence these transcripts. These results strongly support global transcriptional activation of IES-specific loci in the developing MAC upon disruption of the RNAi-dependent Polycomb repression pathway.
mRNA characteristics for IES-specific polyadenylated transcripts in mutants deficient in RNAi-dependent Polycomb repression
Close scrutiny of IES-specific polyadenylated transcripts (Supplemental File S2) revealed several characteristics commonly associated with mRNA. First, we observed an enrichment of poly-A-containing RNA-seq reads mapped to the 3′ termini of transcripts (Supplemental Fig. S3A,B), consistent with poly-A tailing of mRNA. Additionally, these IES-specific transcripts displayed strand-specificity, and some contained splice sites (Fig. 2A). Splicing of IES-specific transcripts was widespread (Fig. 2B). Most splice sites in IES-specific transcripts were found in the mutants exclusively, while for MDS transcripts, there was an almost complete overlap between splice sites found in wild-type cells and the mutants (Fig. 2B). The result is consistent with global transcriptional activation of IES-specific loci in the mutants, without disruption to regular mRNA production (Supplemental Fig. S1).
Figure 2.
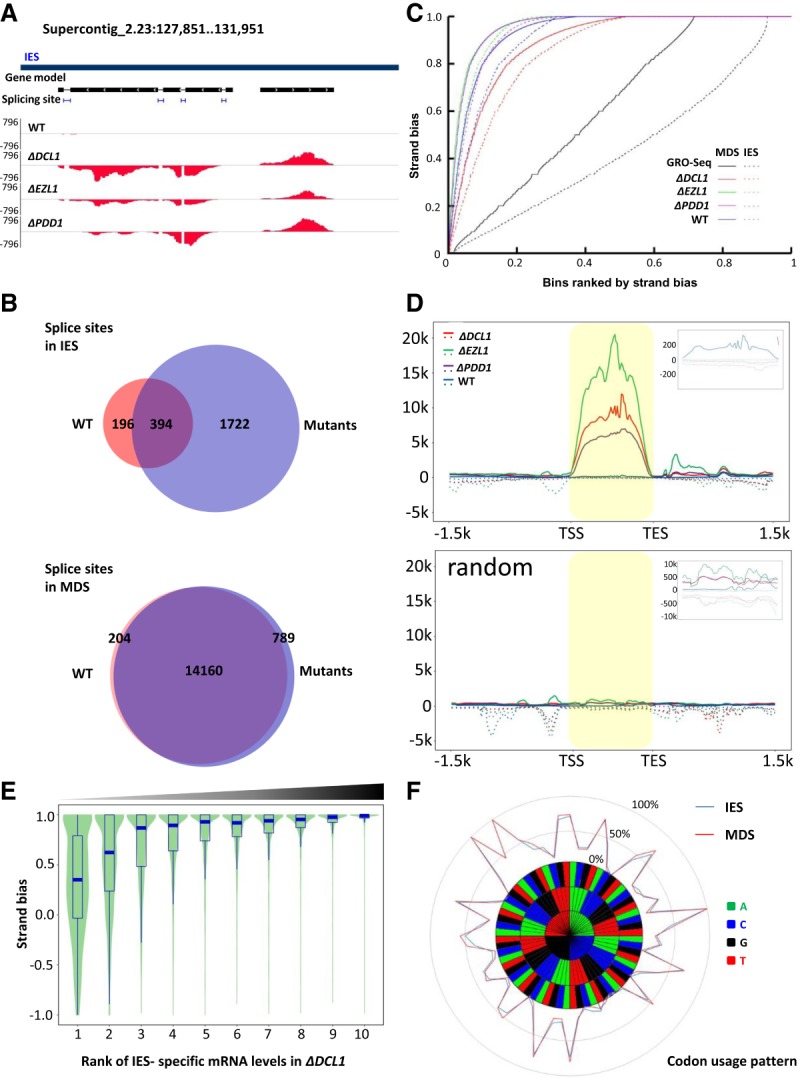
mRNA characteristics for IES-specific polyadenylated transcripts. (A) A representative GBrowse view illustrating strand bias and splice sites of IES-specific polyadenylated transcripts. Gene model: an hAT and a Tc1 family transposase. Positive values of y-axes represent normalized RNA-seq coverage (reads per million mapped reads, RPM) of the Watson strand, negative values the Crick strand. (Blue bar) IESs. (B) Venn diagrams representing splice sites associated with IES (top) and MDS (bottom) polyadenylated transcripts in wild type and mutants. (C) Strong strand bias for IES-specific polyadenylated transcripts. y-axis: Strand bias index, quantified by RNA-seq coverage of the Watson (W) and Crick strand (C) of a 200-bp bin: |W − C|/(W + C) higher value indicating stronger strand bias. x-axis: All 200-bp bins ranked by their strand bias indices, from low to high. (Solid lines) MDS bins; (dashed lines) IES bins. GRO-seq data from the meiotic MICs were used as a negative control; increases in strand bias observed for MDSs were attributable to mRNA from contaminating MACs (Schoeberl et al. 2012). (D) Composite analysis of epigenetically silenced IES-specific loci. (Top) Composite analysis of RNA-seq coverage from 2421 loci highly induced in the mutants (Supplemental File S2). Each locus is equally divided into 100 units from the 5′ to 3′ end (highlighted region) and extended by 1.5 kb in both directions (the scale is not related to the gene body length). The average RPM of each unit in all the loci were cumulated. (Inset) Zoom-in of the highlighted area. Positive values of the y-axis represent the cumulative RPM from the sense strand of the predominant transcript (solid lines), negative values the antisense strand (dashed lines). (Bottom) Control with randomized genomic locations. Note the strong strand bias of IES-specific polyadenylated transcripts and their high induction in the mutants. Both characteristics disappeared in the randomized control. (E) A positive correlation between polyadenylated transcript levels and strand bias in IES-specific loci in ΔDCL1 cells. y-axis: Strand bias index, quantified by RNA-seq coverage of the sense (S) and antisense strand (A) of an IES-specific locus: (S − A)/(S + A); values close to 1 indicate strong bias for sense strand transcripts. x-axis: Quantiles of IES-specific loci ranked by their polyadenylated transcript levels in ΔDCL1 cells; all loci expressing polyadenylated transcripts are grouped into 10 quantiles (1–10, from low to high). (F) Codon usage patterns. IES-specific loci with long ORFs (≥100 amino acids) were searched for homology by blastp (e ≤ 1 × 10−10) (Supplemental File S3). The conserved regions were translated, and the codon usage frequencies were calculated (Supplemental File S3). Codon usage patterns of IES-specific loci as well as regular genes encoded in MDS are plotted in a radar chart.
We also systematically examined strand bias. We defined the strand bias index as the normalized RNA-seq coverage difference between the Watson and Crick strands (|W − C/W + C|). The strand bias index is 0 when both strands are equally transcribed and is 1 when only one strand is transcribed; the higher the index value, the stronger the strand bias. We divided the whole MIC genome into 200-bp bins and calculated for all significantly transcribed bins their strand bias indices, which were ranked and plotted. This analysis revealed strong strand bias for most of the bins in IES regions from wild-type cells and the mutants, close to levels of strand bias observed in bona fide mRNA from MDS regions (Fig. 2C). Relative to wild-type cells, strand bias further increased in ΔPDD1 cells and even more so in ΔEZL1 cells, while it decreased in ΔDCL1 cells (Fig. 2C). Similar shifts in strand bias were observed in MDS as well as IESs (Fig. 2C). Decreased strand bias in ΔDCL1 cells coincided with widespread spurious transcription of IESs (Fig. 1C,F; Supplemental Fig. S3C), often on both strands and at low levels. In ΔEZL1, both heterochromatin marks—H3K27 and H3K9 methylations—are abolished in the developing MAC (Liu et al. 2007); in ΔPDD1 cells, H3K27 methylation is normal, while H3K9 methylation is abolished (Taverna et al. 2002; Schwope and Chalker 2014); in ΔDCL1 cells, H3K27 methylation is abolished, while H3K9 methylation is greatly increased in its levels and no longer specifically associated with IESs (Malone et al. 2005; Liu et al. 2007). Taken together, these results support a connection between heterochromatin formation and bidirectional transcription (decreased strand bias). Our RNA-seq result was in sharp contrast to the global run-on sequencing (GRO-seq) result from the wild-type MIC at early conjugation (2 h after mixing), which showed dramatically reduced levels of strand bias for both IESs and MDSs (Fig. 2C), attributable to bidirectional transcription of ncRNA from the meiotic MIC (Schoeberl et al. 2012).
We next focused on IES-specific loci silenced by the RNAi-dependent Polycomb repression pathway (i.e., epigenetically silenced loci). We performed composite analysis, in which these loci were all scaled to unit length, arranged in the 5′ to 3′ direction of the predominant transcript, and extended in both directions (Fig. 2D; Supplemental Fig. S3D). This analysis showed that transcriptional activation in the mutants was accompanied by increases in strand bias, especially in ΔEZL1 and ΔPDD1 cells (sense/antisense ratio: wild-type, 45; ΔDCL1, 83; ΔEZL1, 337; and ΔPDD1, 196). Indeed, we found that, while IES-specific loci with low levels of polyadenylated RNA exhibited a wide range of strand bias, loci with higher expression levels had a stronger sense strand bias, a trend that was particularly obvious in ΔDCL1 cells (Fig. 2E) but also detectable in other cells (Supplemental Fig. S3E).
We also analyzed potential proteins encoded by IES-specific loci. A blastp search revealed that most of the loci containing a long open reading frame (ORF; ≥100 amino acids) display significant similarities (e ≤ 1 × 10−10) to proteins typically encoded by TEs, such as transposases and reverse transcriptases (Supplemental File S3; see below). Furthermore, the codon usage pattern in these regions is very similar to that of proteins encoded by bona fide mRNA from MDS (Fig. 2F; Supplemental File S3). Taken together, these results indicate that many of the IES-specific transcripts highly induced in the mutants display mRNA hallmarks, including strand specificity, abundant and efficiently processed splice sites, poly-A tailing, and protein-coding capacity.
Broad transcriptional activation of TE-related sequences in mutants deficient in RNAi-dependent Polycomb repression
In Tetrahymena, many IESs contain TE-related sequences (Fass et al. 2011; Hamilton et al. 2016). Importantly, many of the epigenetically silenced loci have significant homology with TEs (Fig. 3A; Supplemental File S4). We found homologs to class I elements (retrotransposons; ∼10% of annotated TEs) (Supplemental Fig. S4A), represented in Tetrahymena by REP elements (Fillingham et al. 2004). We also found homologs to class II TEs (DNA transposons) even more abundant than class I TEs (∼51%), including Tc1/mariner and hAT superfamilies (Figs. 2A, 3B; Supplemental Fig. S4B–E). We also detected many sequences related to integrases (∼39%) (Supplemental Fig. S4F) and other proteins encoded by Tlr elements (Wuitschick et al. 2002), which are related to the Maverick/Polinton subclass of DNA TEs (Kapitonov and Jurka 2006; Pritham et al. 2007). As illustrated by Tc1/mariner members, many of these TE-related sequences contain long ORFs predicted to encode transposases with conserved domains and intact catalytic residues (Fig. 3B,D,E). They are often flanked by terminal inverted repeats (TIRs) and putative target site duplications (TSDs) (Fig. 3B). All of these characteristics are consistent with their presence as autonomous TEs, capable of producing transposases promoting their own mobilization and that of related nonautonomous elements.
Figure 3.

Broad transcriptional activation of TE-related sequences in mutants deficient in RNAi-dependent Polycomb repression. (A) Classification of TE-related sequences in epigenetically silenced IES-specific loci; 439 loci shared homology with interspersed repeats; 261 shared homology with various annotated TEs, as shown in the pie chart; and 178 shared homology with unclassified (Supplemental File S4). Note the predominance of DNA transposons and Tlr/viral elements. (B) A representative GBrowse view of an IES (blue bar) containing a recently duplicated Tc1/mariner DNA transposon, with intact terminal inverted repeats (TIR; magenta arrow box), target site duplications (TSD; TA in bold), and an ORF (green arrow box) encoding a putative DNA transposase featuring helix-turn-helix DNA binding (HTH1/2) and transposase (Tnp; with a DDE catalytic triad) domains. Positive values of the y-axis represent normalized RNA-seq coverage (RPM) of the Watson strand, negative values the Crick strand. Note that this gene is highly expressed only in the mutants. (C–F) Consistent transcriptional activation of putative Tc1/mariner transposases in the mutants. (C) Phylogenetic analysis of putative Tc1/mariner DNA transposases (Supplemental File S5). Tc1/mariner DNA transposases are further divided into Tc1 and Pogo families, based on protein sequence alignment of their conserved transposase domains (62 in total). Groups containing low divergence members (≥99% within-group DNA sequence identity) are marked (red star; 25 in total). The Tc1 element illustrated in Figure 3B is indicated by a blue arrow; the Tc1 element illustrated in Figure 4A is indicated by a green arrowhead. (D) Alignment of the transposase motif, containing the conserved DDD/E catalytic triad. (E) Distribution of conserved domains, including HTH DNA binding (cyan diamond [domain accession number: cl21459], green oval [smart00674] and magenta hexagon [cl09264]) and transposase (red rectangle [cl21549]) domains. (F) Transcriptional induction of putative Tc1/mariner transposases in the mutants. Transcriptional bias is quantified by RNA-seq coverage in wild type (WT) and a specified mutant (MT): (MT − WT)/(MT + WT). The values (−1 to +1) are represented by a color scale: red for induction in a mutant (>>0), green for repression (<<0), and black for little change (≈0), all relative to wild-type cells. Note that most of the 62 groups were induced in the mutants.
Elements related to the Tc1/mariner superfamily of DNA transposons are known to represent the most abundant TE-related sequences in the Tetrahymena MIC genome (∼42%) (Fass et al. 2011; Hamilton et al. 2016). To investigate how Tc1/mariner elements are controlled in Tetrahymena, we systematically identified all putative Tc1/mariner transposases in the MIC genome, as evidenced by their conserved transposase domains (Fig. 3C–E; Supplemental File S5). Phylogenetic analysis revealed 62 distinct transposases falling within either the Tc1 or Pogo subgroups, which are characterized by distinct domain architecture and catalytic triads (Fig. 3C–E; Tellier et al. 2015). Each of these subgroups was represented by a wide diversity of transposases forming multiple clades (i.e., families), and at least 25 of these clades had closely related family members in the Tetrahymena MIC genome (>99% within-group DNA sequence identity) (Fig. 3C, branches marked by red stars). Thus, a wide diversity of Tc1/mariner elements apparently underwent recent transposition and may still be capable of mobilization. Tc1/mariner members also became progressively more dominant among a diverse range of TEs induced in the mutants, as nucleotide divergence from their family consensus sequence decreases (Supplemental Fig. S5; Supplemental File S5). This reflects Tc1/mariner activities in the recent past as well as a long history of expansion and diversification in this lineage. Prolonged vertical transmission of TEs in Tetrahymena is further supported by the codon usage pattern of TE-related sequences, which is very similar to that of known Tetrahymena genes (Fig. 2F). Importantly, most of the putative Tc1/mariner transposases were transcriptionally activated in the mutants (Fig. 3F). RNAi-dependent Polycomb repression is therefore likely to play an important role in controlling TE activity in Tetrahymena and particularly that of Tc1/mariner elements.
Germline mobilization of a recently active TEs
It has long been speculated that DNA elimination in Tetrahymena and other ciliates evolved to thwart TE mobilization (Prescott 1994; Coyne et al. 1996). To test this hypothesis, we focused on a recently active Tc1 element with sequence features of an autonomous TE (Figs. 3C, 4A). Tc1/mariner elements mobilize through a “cut and paste” mechanism: after TE excision, the original genomic locus is reconnected by DNA repair, leaving behind an “empty” locus with a TE footprint (Fig. 4A; Plasterk et al. 1999; Tellier et al. 2015). We used PCR to detect the germline genomic locus after excision of the Tc1 element, which was preferentially amplified due to its much smaller size relative to the original locus (Fig. 4A). We detected a PCR product of the expected size using genomic DNA samples at the end of conjugation (24 h after mixing) as the template (Fig. 4B). Only a very small amount of the PCR product was detected from wild-type cells, reflecting the rarity of Tc1 excision. The PCR product was much more abundant in the mutants (Fig. 4B). Cloning and sequencing of the PCR product revealed the predicted TA footprint left by the excised Tc1 element (Fig. 4A). These results are consistent with the low but still significant expression levels of the Tc1 transposase in wild-type cells (RPKM: 4.6) and its dramatically increased expression in the mutants (RPKM: 136.9, 91.4, 42.7 for ΔDCL1, ΔEZL1, and ΔPDD1, respectively) (Fig. 5A). We also overexpressed the Tc1 transposase in the wild-type genetic background (Fig. 4C; Supplemental Fig. S6), controlled by the Cd2+-inducible MTT1 promoter (Shang et al. 2002). Tc1 excision was detected with Cd2+ induction but not in its absence (Fig. 4C). This confirms that the Tc1 transposase is sufficient and necessary for excision of the Tc1 element, and transcriptional silencing by the RNAi-dependent Polycomb repression pathway is critical for controlling this TE.
Figure 4.

Germline mobilization of a recently active TE. (A) A schematic for mobilization of a Tc1 element. It contains a conserved DNA transposase (ORF: green block arrow; transposase domain: indigo box; catalytic triad: DDD). The Tc1 element also features intact TIR (magenta arrow box) and TSD (TA in bold red in the top and bottom sequences), while its excision leaves a TE footprint (TA in bold blue in the bottom sequence). The red arrows represent the nested PCR primers used in the transposition assays shown in panels B–D. The boundaries of the IESs lie outside the PCR primers, so the somatically rearranged DNA was undetected by this assay. (B) Mobilization of the Tc1 element at low levels in wild-type cells but at dramatically increased levels in the mutants. To monitor excision at the Tc1 element, nested PCR was performed on total genomic DNA purified at the end of conjugation (24 h after mixing) from the specified cells. A MAC gene, JMJ1, was monitored as the loading control. (C) Tc1 mobilization by transposase overexpression. To monitor excision at the Tc1 element, nested PCR was performed on total genomic DNA purified at the end of conjugation (24 h after mixing), with or without induction of the Tc1 transposase overexpression by cadmium (±Cd2+). A MAC gene, JMJ1, was monitored as the loading control. Completion of conjugation was monitored by a PCR assay for a well-studied IES, the M element (Duharcourt and Yao 2002; Liu et al. 2004). Overexpression of the Tc1 transposase (HA-tagged) was shown by immunoblotting with an anti-HA antibody. Coomassie-stained total cellular proteins was used as the loading control. (D) Time course analysis of Tc1 mobilization. In wild-type cells, the Tc1 excision product was detected after developmentally programmed DNA elimination and persisted in conjugating cells (red brackets). Conjugation progress was monitored by a PCR assay for a well-studied IES, the M element. Its processing gives rise to the short PCR product in conjugating CU427 and CU428 cells (green brackets), distinct from the long PCR product from the mature MAC in parental cells (Duharcourt and Yao 2002; Liu et al. 2004). In ΔEZL1 cells, dramatically increased levels of the Tc1 excision product were detected, but with a temporal pattern similar to wild-type cells (red brackets). In cells overexpressing the Tc1 transposase, excision of the Tc1 element was expedited. JMJ1 was monitored as the loading control. (E) Excision frequency of the Tc1 element in ΔEZL1 cells. Genomic DNA samples were isolated from the specified conjugation time points, digested by HindIII to release the Tc1 element-containing IES fragment, and analyzed by Droplet Digital PCR. Excision frequency was quantified by dividing the number of Tc1 excision events with the number of control events. (F) Tc1 mobilization occurs in the new MIC. The developing MAC and the new MIC were purified from wild-type cells at late conjugation (24 h after mixing). The M element was monitored for the enrichment of MAC and MIC. The two bottom bands correspond to the IES-excised forms found only in MAC, while the top band corresponds to the IES-retained form found only in MIC (red arrowhead). The former were enriched in the developing MAC sample, while the latter was enriched in the new MIC sample. JMJ1 was the loading control. (G) A model for TE mobilization in Tetrahymena and other binucleated ciliates. (1) TE mRNA is generated in the developing MAC. (2) Transposases are imported into the new MIC. (3) TEs are mobilized in the new MIC: (3.1) excision from a donor locus, leaving behind a TE footprint (blue dot; see A); (3.2) insertion into a receptor locus.
Figure 5.

Alternative production of ncRNA and mRNA in IES-specific loci. (A) Active production of polyadenylated transcripts as well as scnRNA in an IES. y-axes: Normalized coverage (RPM) of polyadenylated transcripts and scnRNA in wild type and the mutants. Positive values represent coverage on the Watson strand, negative values the Crick strand. Early-scnRNA is represented by 2′-O-methylated scnRNA at early conjugation (Schoeberl et al. 2012), while late-scnRNA is represented by scnRNA associated with TWI11 expressed from the developing MAC at late conjugation (Noto et al. 2015). Note the significant levels of polyadenylated transcripts in wild-type cells and absence of early-scnRNA. (B) Active production of polyadenylated transcripts as well as scnRNA in a large cluster of IES-specific loci derived from TEs. (C) Enrichment of scnRNA in IES-specific loci. (Top) Composite analysis of the early and late scnRNA coverage of epigenetically silenced IES-specific loci. Each locus is equally divided into 100 units from the 5′ to 3′ end (highlighted region) and extended by 1.5 kb in both directions (as in Fig. 2D). The average normalized scnRNA coverage (RPM) of each unit in all the loci was cumulated. Positive values represent coverage on the sense strand of a locus (solid lines), negative values the antisense strand (dashed lines). (Bottom) Control with randomized genomic locations. (D) Relationship between polyadenylated transcript levels and the late-scnRNA bias in IES-specific loci. The late-scnRNA bias for a locus is quantified by early- (E) and late-scnRNA levels (L): (L − E)/(L + E). Values close to 1 indicate strong bias for late-scnRNAs. IES-specific loci are grouped into 10 quantiles (1–10, from low to high) according to their polyadenylated transcript levels in wild-type and ΔDCL1 cells, respectively. Note that while a positive correlation between polyadenylated transcript levels and late-scnRNA bias was found in wild-type cells, a negative correlation was found in ΔDCL1 cells.
We next performed time-course analysis of this TE excision event. In wild-type cells, Tc1 excision occurred at late conjugation (Fig. 4D); a similar pattern, with much stronger signals, was observed in ΔEZL1 cells (Fig. 4D). Tc1 excision was detected (16 h after mixing) after IES excision in wild-type cells (as shown by the excision at 10 h after mixing of a well-studied IES, the M element [Duharcourt and Yao 2002; Liu et al. 2004]) and persisted in the conjugation progeny (36 h after mixing) (Fig. 4D). Since all IESs, including the one containing the Tc1 element, are eliminated in the developing MAC of wild-type cells, these observations strongly support that transposition occurs not in the developing MAC but in the new MIC. In cells overexpressing the Tc1 transposase from the parental MAC, Tc1 excision was detected in early conjugation (6 h after mixing), probably as a direct result of the Tc1 transposase induction before initiation of conjugation. We surmise that, as it occurs before formation of the developing MAC (8 h after mixing), Tc1 excision again can only occur in the MIC, albeit during prezygotic and postzygotic divisions (Supplemental Fig. S6B). Using droplet digital PCR, we validated the conjugation time-course of Tc1 excision in ΔEZL1 (Fig 4E; Supplemental Fig. S7). We estimated that the frequency for Tc1 excision plateaued at approximately 4 × 10−4 by the end of conjugation—one to two orders of magnitude above background levels (Fig 4E; Supplemental Fig. S7). We further showed that Tc1 excision was detected in the sample enriched for the new MIC from wild-type cells at late conjugation but not in the sample enriched for the developing MAC (Fig. 4F), supporting that Tc1 excision occurs preferentially, if not exclusively, in the new MIC. We conclude that RNAi-dependent Polycomb repression is required for controlling the excision and, by inference, mobilization of the Tc1 element. Tc1 excision also proves that polyadenylated transcripts generated from Tc1 elements are bona fide mRNA encoding a functional transposase. Even though the mRNA is generated from the developing MAC, the transposase must enter the new MIC to ensure germline transposition of the element and its expansion in the Tetrahymena population (Fig. 4G). Somatic transcriptional activation coupled with germline mobilization is likely a recurring theme for TEs in binucleated ciliates, as in multicellular organisms (Wang et al. 2018).
Alternative production of ncRNA and mRNA in IESs
IESs are specifically targeted for elimination by scnRNA (Mochizuki et al. 2002). scnRNAs accumulating during early conjugation, referred to as early-scnRNAs, are derived from bidirectional ncRNA transcripts generated in the meiotic MIC (Chalker and Yao 2001; Mochizuki et al. 2002); additional scnRNAs are derived from ncRNA transcripts generated in the developing MAC during late conjugation, referred to as late-scnRNAs (Noto et al. 2015). IESs in the developing MAC generate ncRNAs (Aronica et al. 2008)—precursors to the late-scnRNA—as well as polyadenylated transcripts. Indeed, we found substantial overlap in genomic locations between late-scnRNAs and IES-specific polyadenylated transcripts (Fig. 5A,B), while early-scnRNAs were excluded from some IES-specific loci (Fig. 5A). Composite analysis of scnRNA distribution around IES-specific loci silenced by the RNAi-dependent Polycomb repression pathway (Fig. 2D) revealed that both early- and late-scnRNAs were enriched therein, compared with a control generated from randomized genomic locations (Fig. 5C; Supplemental Fig. S8A). Analysis of total scnRNA at late conjugation (10 h after mixing; a mixture of early- and late-scnRNA) also revealed similar enrichment (Supplemental Fig. S9A). However, only moderate strand bias was observed in scnRNA (Fig. 5A–C; Supplemental Figs. S8A, S9A,B), in striking contrast to the sense strand specificity of the associated polyadenylated transcripts (Fig. 2D). This result suggests that, in both the meiotic MIC and the developing MAC, there is bidirectional transcription of ncRNA around the IES-specific loci, which subsequently undergo cleavage by DCL1 (Malone et al. 2005; Mochizuki and Gorovsky 2005) and random selection of the guide strand by TWI1 (Mochizuki and Kurth 2013). Bidirectional transcription of ncRNA in the meiotic MIC was further corroborated by the GRO-seq data showing no strand specificity in IESs (Fig. 2C).
In wild-type cells, IES-specific loci with significant mRNA levels, including the aforementioned active Tc1 element (Fig. 4), were predominantly associated with late-scnRNA but not early-scnRNA (Fig. 5A,D). For IES-specific loci with decreasing mRNA levels, the late-scnRNA bias was progressively reduced (Fig. 5D, top panel). The strong positive correlation between mRNA levels and the late-scnRNA bias can be interpreted in the context of epigenetic regulation of transcriptional activation in the developing MAC. We propose that high levels of early-scnRNA target homologous sequences in the developing MAC to nucleate the formation of heterochromatin, which in turn favors the production of ncRNA over mRNA. In the absence of early-scnRNA, some IES-specific loci can produce mRNA instead of ncRNA in the developing MAC.
The correlation between mRNA levels and the late-scnRNA bias turned negative in ΔDCL1 cells, as many IES-specific loci associated with early-scnRNA were highly induced (Fig. 5D, bottom panel). Similar analyses revealed intermediate states in ΔEZL1 and ΔPDD1 cells (Supplemental Fig. S8B), as fewer IES-specific loci were induced and many of them were expressed at lower levels in these two mutants compared with in ΔDCL1 cells (Supplemental Fig. S3C,D). Global increase in IES-specific polyadenylated transcripts coincided with compromised production of late-scnRNA in all three mutants. Previous studies strongly support that DCL1 is required for both early- and late-scnRNA (Malone et al. 2005; Mochizuki and Gorovsky 2005), while EZL1 and PDD1 are implicated in late-scnRNA production (Noto et al. 2015). Our analysis confirms that scnRNA production from IES-specific loci is abolished in ΔDCL1 cells (Supplemental Fig. S9A,B). In ΔEZL1 and ΔPDD1 cells, even though global scnRNA levels were not significantly affected (Supplemental Fig. S9A,B), IES-specific loci with strong late-scnRNA bias were preferentially depleted of scnRNA at late conjugation (Supplemental Fig. S9C,D). Taken together, these results strongly support that epigenetic factors, including the RNAi machinery and PcG proteins, regulate the balance between ncRNA or mRNA production in the developing MAC.
The balance between ncRNA and mRNA production is a critical aspect of the balance between transcriptional silencing and activation
To further investigate how the alternative production of mRNA and long ncRNA is regulated, we compared polyadenylated transcript levels at different conjugation stages (3, 6, and 10 h after mixing) (Fig. 6A,B). In contrast to high levels of IES-specific polyadenylated transcripts at late conjugation, very few, if any, were detected in wild-type cells or the mutants at early conjugation (3 and 6 h after mixing) (Fig. 6A,B); as controls, abundant reads corresponding to bona fide mRNA (mapped to MDS) were found (Fig. 6A,B). During early conjugation, the transcriptionally active meiotic MIC is the sole source for IES-specific transcripts (Chalker and Yao 2001; Schoeberl et al. 2012); however, they are exclusively ncRNAs, which in Tetrahymena are distinguished from mRNA by their lack of polyadenylation, in line with the previous observation for transcripts from the M element (Chalker and Yao 2001).
Figure 6.

The balance between ncRNA and mRNA production is a critical aspect of the balance between transcriptional silencing and activation. (A) IES-specific polyadenylated transcripts are only produced during developing MAC formation. (3) 3 h after mixing: meiosis, (6) 6 h after mixing: gametogenesis, (10) 10 h after mixing: developing MAC formation, (blue bar) IES. RNA-seq was performed after oligo-dT enrichment of polyadenylated transcripts. Note that essentially no RNA-seq reads were mapped to the IES region at the two early conjugation time points. (PM) Parental MAC, (DM) developing MAC, (OM) old MAC. (B) Percentage of RNA-seq reads mapped to consistently processed IESs, relative to total mappable reads. Note that IES-specific polyadenylated transcripts were abundantly produced during developing MAC formation (10 h after mixing) but rarely detected before that (3 and 6 h after mixing). (C) Localization of the transcriptional and cotranscriptional machineries in early (3 h after mixing) and late (10 h after mixing) conjugating cells. RPB3-HA, CBP20-HA, PRP19-HA, and THO2-HA cells were stained with an anti-HA antibody (red) and counterstained with DAPI (blue). Parental MAC (PM): green arrowhead, developing MAC (DM): green arrows, MIC: white arrowheads, old MAC (OM): white arrow. (D) Interaction between TWI1 and the transcriptional/cotranscriptional machineries. The designated cells were processed for crosslink-immunoprecipitation with the anti-HA antibody at late conjugation (10 h after mixing). The anti-HA and anti-TWI1 antibodies were used for immunoblotting. Note that similar amounts of bait proteins were recovered, as shown by the anti-HA immunoblotting. (E) Production of scnRNA from intronic as well as exonic regions. Distributions of polyadenylated transcripts (dotted lines) and scnRNA (solid lines) in introns and exons of IES-specific loci. Each intron is equally divided into 10 units from the 5′ to 3′ end; the two flanking exons are also equally divided into 10 units, respectively. The average RPM of each unit in all the loci were cumulated. Double counting for exons is avoided. (F) Alternative production of ncRNA and mRNA in the meiotic MIC and the developing MAC. In this simplified schematic, transcription from IES-specific loci is emphasized, while genic transcription from MDS is omitted. See text for details.
We next investigated why the meiotic MIC can only produce ncRNA, while the developing MAC can produce both ncRNA and mRNA. In Tetrahymena, transcription in the meiotic MIC is catalyzed by RNA polymerase II (Mochizuki and Gorovsky 2004b), which is probably also responsible for IES transcription in the developing MAC, conforming with Pol II-catalyzed ncRNA production in other eukaryotes (Castel and Martienssen 2013). Pol II-driven mRNA biogenesis involves a cascade of cotranscriptional events, including the addition and recognition of the 5′ cap structure, splicing, and packaging and exporting (Aguilera 2005). We HA-tagged key components of the corresponding molecular machineries: RPB3 for Pol II, CBP20 for the cap binding complex, PRP19 for the splicing complex, and THO2 for the RNA packaging and exporting complex (Fig. 6C,D). By immunofluorescence staining, RPB3 and CBP20 were localized in the meiotic MIC during early conjugation and in the developing MAC during late conjugation, while PRP19 and THO2 were only detected in the developing MAC but were excluded from the meiotic MIC (Fig. 6C). On the other hand, all four components were detected in the parental MAC generating bona fide mRNA (Fig. 6C). We conclude that mRNA production is precluded in the meiotic MIC due to the lack of critical cotranscriptional factors.
To understand how the cotranscriptional processes are coordinated with RNAi-dependent transcriptional silencing in the developing MAC, we examined interactions of RPB3, CBP20, PRP19, or THO2 with TWI1, an Argonaute/Piwi family member that is a critical player in the pathway (Fig. 6D; Mochizuki et al. 2002; Noto et al. 2010, 2015). All these components are present in the developing MAC (Fig. 6C; Mochizuki et al. 2002), allowing physiologically relevant interactions. TWI1 was coimmunoprecipitated with RPB3 (Fig. 6D), supporting a role of Pol II in ncRNA production. Even stronger coimmunoprecipitation was detected between TWI1 and CBP20, while only weak coimmunoprecipitation was detected with PRP19, and none with THO2 (Fig. 6D). This result suggests that 5′ end processing of transcripts plays a critical role in recruiting the RNAi machinery for ncRNA production. In contrast, cotranscriptional processes that occur further downstream are progressively segregated from ncRNA production. We also found that in IES-specific loci, both early- and late-scnRNA were detected in introns at levels comparable with those of the flanking exons, in contrast to the depletion of polyadenylated transcripts in introns (Fig. 6E). This result provides further support for the lack of splicing in ncRNA produced in both the meiotic MIC and the developing MAC and precludes the possibility that scnRNA is produced by post-transcriptional processing of mRNA.
We conclude that ncRNA and mRNA can be alternatively produced from the same genomic locus under the influence of different trans-acting factors (Fig. 6F). In the meiotic MIC, lack of PRP19 and THO2, required for mRNA biogenesis but minimally involved in ncRNA biogenesis, precludes mRNA production and entails exclusive ncRNA production. In the developing MAC, many IES-specific loci can potentially generate both. Epigenetic factors regulate the transition between ncRNA and mRNA production, at least partially through their differential interactions with cotranscriptional processing factors. Transcriptional silencing of TEs is accompanied and reinforced by the transition from mRNA to ncRNA production and vice versa for transcriptional activation.
Discussion
Alternative production of ncRNA and mRNA as a critical aspect of the host–TE relationship
We have demonstrated that RNAi-dependent Polycomb repression controls TEs in Tetrahymena. Intriguingly, TE-related sequences can alternatively produce ncRNA or mRNA. As both are transcribed by Pol II, additional factors—including the RNA splicing and exporting machinery, the RNAi machinery, and the chromatin environment—dictate alternative outcomes (Fig. 6E). In the meiotic MIC, lack of key cotranscriptional processing machineries entails the exclusive production of ncRNA. Indeed, regulating the accessibility of cotranscriptional processing machineries or utilizing alternative transcriptional machineries may be a recurring theme for TE silencing during meiosis, which has also been implicated in the biogenesis of piRNA (Le Thomas et al. 2014; Mohn et al. 2014; Zhang et al. 2014; Senti et al. 2015; Andersen et al. 2017)—the metazoan equivalent of scnRNA (Gao and Liu 2012; Mochizuki 2012). In the developing MAC, the RNAi machinery targets nascent RNA transcripts and disrupts mRNA production, possibly by cleavage with the slicer activity of TWI1 (Noto et al. 2010) or interference with mRNA processing (Perales and Bentley 2009). The balance may also be affected by histone modifications associated with a locus. In particular, scnRNA production and EZL1-catalyzed histone methylation may potentially form a positive feedback loop (Noto et al. 2015). Based on all the evidence, we propose that the transition between ncRNA and mRNA production is a critical aspect underlying transcriptional silencing and activation, especially for TE-related sequences (Fig. 6E). Our results strongly supports TE-derived sequences as the template for many long ncRNA, which in other eukaryotic systems may or may not be cotranscriptionally processed or converted into small RNA (Kelley and Rinn 2012; Kapusta et al. 2013; Ha et al. 2014). We also show that the balance of transcriptional silencing and activation of TEs is closely linked to strand bias of their transcripts, implying a transition from unidirectional transcription for mRNA to bidirectional transcription for ncRNA. In several eukaryotic systems, antisense transcription has been shown to initiate from the 3′ end of a transcriptional unit or the divergent promoter of a neighboring transcriptional unit, frequently derived from a TE nested within or downstream of a gene (Conley et al. 2008) and controlled by various mechanisms including chromatin structure and RNA processing-based surveillance (Lee et al. 2013; Schulz et al. 2013; Marquardt et al. 2014).
In Tetrahymena, enrichment of scnRNA specific for IESs underlies RNAi-guided heterochromatin formation and DNA elimination (Noto and Mochizuki 2017). Although some specificity can be attributed to the scanning process depleting scnRNA homologous to MDS (Mochizuki et al. 2002; Mochizuki and Gorovsky 2004a; Schoeberl et al. 2012), a strong bias for IESs in early scnRNA and ncRNA transcripts from the meiotic MIC has not been accounted for. Based on our results, we propose that TEs’ capability for mRNA production (essential for their mobilization) can be diverted for ncRNA production (required for TE silencing and ultimately their elimination in Tetrahymena and other ciliates). Indeed, high levels of early-scnRNA, derived from ncRNA in the meiotic MIC, are a strong predictor of complete silencing of mRNA production in the developing MAC. Early-scnRNAs are primarily generated from peri-centromeric and subtelomeric regions of the MIC chromosomes (Hamilton et al. 2016), which are associated with heterochromatin in a wide range of eukaryotes (Slotkin and Martienssen 2007). When a TE is inserted into these regions, early-scnRNA production is promoted, leading to silencing of all closely related TEs in trans. This may be an important mechanism for a newly invaded TE to stabilize its copy number in the host genome and the population.
In binucleated ciliates, in a brief window after transcriptional activation but before DNA elimination in the developing MAC, TE-encoded proteins may be expressed and subsequently imported into the germline MIC, allowing TE mobilization and persistence in the germline MIC (Fass et al. 2011; Arnaiz et al. 2012; Chen et al. 2014; Hamilton et al. 2016). On the other hand, ncRNA-mediated transcriptional silencing and subsequent DNA elimination of almost all TE-related sequences from the transcriptionally active somatic MAC reduce the host's fitness cost. Alternative production of ncRNA and mRNA of TE-related sequences therefore represents a critical balancing act allowing TEs to persist in the genome of a host species—be it a protozoan or a metazoan (Levin and Moran 2011).
In mutants deficient in RNAi-dependent Polycomb repression, we find abundant mRNA from numerous TE-related sequences, as well as evidence for increased mobilization of at least one recently active Tc1/mariner element, supporting epigenetic instability as a cause for TE mobilization. Diverse Tc1/mariner elements and other TEs have been propagated recently in Tetrahymena (Hamilton et al. 2016). Under sporadic conditions of epigenetic instability, many TEs may be mobilized en masse in episodes that have the potential to dramatically alter a genome, drive a population to traverse the fitness landscape, and even lead to speciation (Zeh et al. 2009; Oliver and Greene 2012), thus providing an underlying molecular mechanism for evolution by punctuated equilibria (Eldredge and Gould 1972). Although limited, previous studies of IES positions and sequences across Tetrahymena species point to high levels of polymorphism (Huvos 1995). Future characterization of the Tetrahymena mobilome and its epigenetic regulation will provide deeper understanding of the intricate relationship between the host genome/epigenome and TEs and how they work together to shape the course of evolution.
Polycomb repression as an ancient pathway for RNAi-dependent TE silencing
RNAi-guided transcriptional silencing of TE-related sequences generally involves three ancient pathways: H3K9 methylation catalyzed by histone methyltransferases homologous to Drosophila Su(var)3-9; H3K27 methylation catalyzed by histone methyltransferases homologous to Drosophila E(z); and DNA cytosine methylation catalyzed by DNA methyltransferases (Malone and Hannon 2009; Moazed 2009). Based on their widespread distribution in all the major eukaryotic branches, there is an emerging consensus that these pathways were already present in the last eukaryotic common ancestor (Fig. 7; Supplemental File S6; Aravind et al. 2011; Iyer et al. 2011). RNAi-dependent Polycomb repression may be present in a wide range of eukaryotic organisms, even though its role is sometimes obscured by the other two pathways. In mouse embryonic stem cells, PcG proteins have been shown to act coordinately to transcriptionally repress genomic repeats, including retroviral elements (Leeb et al. 2010), a function often solely assigned to H3K9 methylation and DNA methylation. In Arabidopsis, loss of DNA methylation and accompanying H3K9 methylation in TE-related sequences can lead to redistribution of H3K27 methylation (Mathieu et al. 2005; Weinhofer et al. 2010; Deleris et al. 2012), which indicates a complex interplay between these epigenetic marks and TEs. Intriguingly, one or more of the heterochromatin formation pathways have been lost in particular branches of evolution (Fig. 7; Supplemental File S6). The DNMT DNA methylation pathway is conspicuously absent in ciliates and nematodes. There is only the Su(var)3-9 pathway in Schizosaccharomyces Pombe, and none of the three pathways is in Saccharomyces cerevisiae, even though all three are present in related fungi Neurospora and Cryptococcus (Jamieson et al. 2013; Dumesic et al. 2015). Only PcG proteins are found in Tetrahymena (Liu et al. 2007), making it an ideal system to dissect the function and molecular mechanism of RNAi-dependent Polycomb repression. To the best of our knowledge, our study is the first to demonstrate that defects in Polycomb repression lead to not only transcriptional TE activation but also germline mobilization of TEs. Combined with the phylogenetic profile, our results argue strongly that, although PcG proteins are adapted to various functions during evolution, their ancestral role may have been to control TEs.
Figure 7.
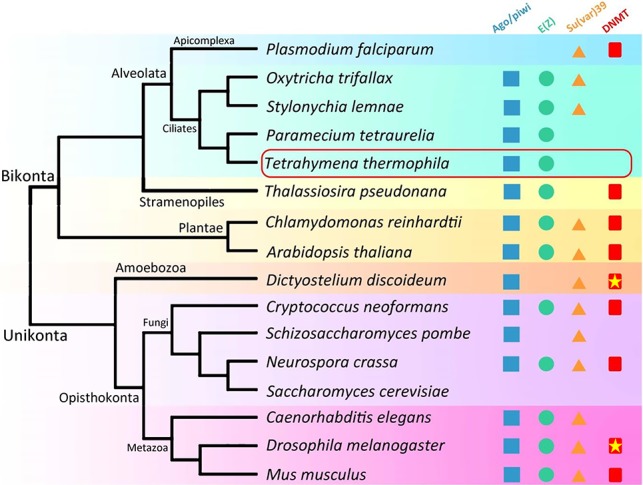
Polycomb repression as an ancient pathway for RNAi-dependent TE silencing. Distribution of RNAi-dependent heterochromatin formation pathways in eukaryotes (Supplemental File S6). Asterisks point out organisms in which DNA cytosine methylation levels are very low. See text for details.
Materials and methods
Additional details are available in Supplemental Material.
Strains and culture conditions
Tetrahymena strains were produced using fusion PCR-generated constructs (Supplemental File S7), as described previously (Gao et al. 2013). To overexpress the Tc1 transposase, its coding region was used to replace the MTT1 coding region and HA-tagged. Somatic transformants were selected for paromomycin resistance, conferred by the neo gene inserted into the nearby MTT3 locus. RPB3, CBP20, PRP19, and THO2 were HA-tagged at the endogenous locus.
Tetrahymena cells were grown at 30°C in SPP medium (Sweet and Allis 1998). To initiate conjugation, log-phase growing cells (∼2 × 105/mL) of two different mating types were washed, starved, and mixed in 10 mM Tris (pH 7.4) or Dryl's buffer (2 mM sodium citrate, 1 mM NaH2PO4, 1 mM Na2HPO4, 1.5 mM CaCl2, pH 6.8) at 30°C (Sweet and Allis 1998; Cassidy-Hanley 2012). Overexpression of the Tc1 transposase was induced with 1.5 µg/mL Cd2+ overnight in SPP medium. Cells were then washed, starved, and mated with wild-type CU427.
RNA-seq and analysis
Total RNA was extracted from Tetrahymena cells using the RNeasy Protect Cell minikit (Qiagen), as described (TetraFGD, http://tfgd.ihb.ac.cn/index/smphelp). Polyadenylated transcripts were enriched using Sera-Mag magnetic oligo-dT beads (GE). First strand-specific libraries were constructed using the Illumina TruSeq Stranded mRNA sample preparation kit (RS-122-2101). Small RNA was enriched by a mirVana™ miRNA isolation kit (Ambion). Small RNA libraries were constructed using the Illumina TruSeq Small RNA Sample Prep kit (RS-200-0012). Sequencing was performed using an Illumina HiSeq-2500 sequencer. Analysis of polyadenylated transcripts was performed as described previously (Xiong et al. 2012; Feng et al. 2017). Analysis of small RNA was performed as described (Schoeberl et al. 2012).
Selecting consistently processed IESs for analysis
IESs were identified by comparing the Tetrahymena MIC and MAC genome assemblies, as previously described (Hamilton et al. 2016). We manually removed all known nonmaintained chromosomes and exonic IESs, which either contain or are associated with abundant mRNA transcripts (Cheng et al. 2016; Hamilton et al. 2016; Lin et al. 2016; Feng et al. 2017). Some IESs are variably processed, as they are partially or even fully retained in the MAC of some conjugation progeny. We avoided this complication by focusing on consistently processed IESs (Supplemental File S1), which are covered at low levels by genomic DNA sequencing of the developing MAC and the mature MAC in wild-type cells but at high levels in the developing MAC of ΔDCL1 cells (Feng et al. 2017).
Selecting IES-specific loci for analysis
Gene models in IESs were generated using the RNA-seq data of polyadenylated transcripts in wild-type, ΔDCL1, ΔEZL1, and ΔPDD1 cells. After removing those with many reads also mapped outside of the consistently processed IESs (multimappers), we obtained a list of IES-specific loci (Supplemental File S2, Sheet 1). A subset of the IES-specific loci were silenced by the RNAi-dependent Polycomb repression pathway (RPKMMT ≥ 1 and RPKMWT/RPKMMT ≤ 0.5) (Supplemental File S2, Sheet 2).
Long ORFs in IES-specific loci (≥100 amino acids) were searched for homology by blastp. Putative conserved coding regions were identified by merging significant hits (e ≤ 1 × 10−10), and their codon usage frequencies were calculated (Supplemental File S3). IES-specific loci were further annotated for TE-related sequences (Supplemental File S4) by BLAST search against a library of interspersed repeats obtained from the Tetrahymena MIC genome (Hamilton et al. 2016). The library was built by combining known TEs from Repbase (Jurka et al. 2005) and repeats identified de novo. Repeats were classified into TE families using multiple lines of evidence, including conserved TE protein domains (Marchler-Bauer et al. 2011), homology to known elements, presence of terminal inverted repeats, and putative target site duplications. With similar methods, we also performed a systematic search for putative Tc1/mariner elements in the Tetrahymena MIC genome (Supplemental File S5).
TE mobilization assay
Two pairs of primers (Supplemental File S7) were designed for amplification by nested PCR of the truncated genomic region after excision of the Tc1 element in Supercontig_2.6:532,001..534,000. All primers anneal within the IES region; therefore, they cannot amplify the IES excision product. Tc1 excision frequency was quantified by Droplet Digital PCR, using HindIII-digested genomic DNA as the template.
Immunofluorescence staining, immunoprecipitation, and immunoblotting
For immunofluorescence staining, Tetrahymena cells were fixed in PBS with 2% paraformaldehyde for 10 min, permeabilized in 0.4% Triton X-100 for 3 min, and probed with the anti-HA antibody (Cell Signaling Technology). For immunoprecipitation, Tetrahymena cells were fixed in PBS with 0.1% paraformaldehyde at room temperature for 5 min. After washing (50 mM Tris [pH 8.0], 1 mM MgCl2, 10 mM KCl), cells were resuspended with 10 mL ice-cold immunoprecipitation buffer and sonicated (Branson Sonifier 250, 90% duty cycle, output 4, 4 × 15 sec burst). The solubilized fraction was recovered after centrifugation and filtration, and incubated with anti-HA agarose (Sigma) for 4 h at 4°C. For immunoblotting, the anti-HA antibody (Cell Signaling Technology) and the anti-TWI1 antibody (a gift from Kazufumi Mochizuki) were used.
Identification of Ago/piwi, E(z), Su(var)39, and DNMT homologs
We first searched the literature concerning RNAi, H3K27 methylation, H3K9 methylation, and 5-cytosine DNA methylation pathways in 16 organisms representing major branches of eukaryotic evolution (Supplemental File S6). In the absence of experimental evidence, we searched genome sequences for homologs, defined as reciprocal best BLAST hits of human AGO1, EZH2, SUV39H1, and DNMT genes (Supplemental File S6).
Accession numbers
RNA-seq and small RNA-seq data have been deposited at the NCBI Gene Expression Omnibus under accession GSE118200.
Supplementary Material
Acknowledgments
Wild-type Tetrahymena strains CU427 and CU428 were obtained from the Tetrahymena Stock Center. ΔDCL1 and ΔPDD1 strains were kindly provided by Douglas L. Chalker. The anti-TWI1 antibody was kindly provided by Kazufumi Mochizuki. The compiled annotations for Tetrahymena MAC and MIC genomes were obtained from the Tetrahymena Genome Database (http://www.cilate.org). Illumina sequencing was performed at the DNA Sequencing Core of the University of Michigan. J.X. was supported by the National Natural Science Foundation of China (no. 31301930). C.F. was supported by the National Institutes of Health (NIH; R01 GM077582). R.S.C. was supported by the National Science Foundation (NSF; 1158346). W.M. was supported by the Projects of International Cooperation and Exchanges Ministry of Science and Technology of China (no. 2013DFG32390). S.G. was supported by the National Natural Science Foundation of China (31522051) and the Natural Science Foundation of Shandong Province (JQ201706). Y.L. was supported by the NIH (R01 GM087343), the NSF (MCB 1411565), and the Department of Pathology at the University of Michigan.
Author contributions: S.G. and Y.L. conceived the study and experiments. S.G., L.F., and W.D. generated Illumina sequencing data. X.Z., S.G., and Y.L. performed the cell biology experiments. X.Z. performed the transposition assay. X.Z. and Y.L. performed immunoprecipitation. J.X., F.M., X.C., W.Y., Y.S., and W.M. performed/oversaw bioinformatic analysis of Illumina sequencing data. J.X., F.M., A.K., C.F., R.S.C., and W.M. performed/oversaw bioinformatic analysis of TEs. Y.L. prepared the manuscript.
Footnotes
Supplemental material is available for this article.
Article published online ahead of print. Article and publication date are online at http://www.genesdev.org/cgi/doi/10.1101/gad.320796.118.
References
- Aguilera A. 2005. Cotranscriptional mRNP assembly: from the DNA to the nuclear pore. Curr Opin Cell Biol 17: 242–250. 10.1016/j.ceb.2005.03.001 [DOI] [PubMed] [Google Scholar]
- Andersen PR, Tirian L, Vunjak M, Brennecke J. 2017. A heterochromatin-dependent transcription machinery drives piRNA expression. Nature 549: 54–59. 10.1038/nature23482 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aravind L, Abhiman S, Iyer LM. 2011. Natural history of the eukaryotic chromatin protein methylation system. Prog Mol Biol Transl Sci 101: 105–176. 10.1016/B978-0-12-387685-0.00004-4 [DOI] [PubMed] [Google Scholar]
- Arnaiz O, Mathy N, Baudry C, Malinsky S, Aury JM, Denby WC, Garnier O, Labadie K, Lauderdale BE, Le Mouël A, et al. 2012. The Paramecium germline genome provides a niche for intragenic parasitic DNA: evolutionary dynamics of internal eliminated sequences. PLoS Genet 8: e1002984 10.1371/journal.pgen.1002984 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aronica L, Bednenko J, Noto T, DeSouza LV, Siu KW, Loidl J, Pearlman RE, Gorovsky MA, Mochizuki K. 2008. Study of an RNA helicase implicates small RNA-noncoding RNA interactions in programmed DNA elimination in Tetrahymena. Genes Dev 22: 2228–2241. 10.1101/gad.481908 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brockdorff N. 2013. Noncoding RNA and Polycomb recruitment. RNA 19: 429–442. 10.1261/rna.037598.112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao R, Wang L, Wang H, Xia L, Erdjument-Bromage H, Tempst P, Jones RS, Zhang Y. 2002. Role of histone H3 lysine 27 methylation in Polycomb-group silencing. Science 298: 1039–1043. 10.1126/science.1076997 [DOI] [PubMed] [Google Scholar]
- Cassidy-Hanley DM. 2012. Tetrahymena in the laboratory: strain resources, methods for culture, maintenance, and storage. Methods Cell Biol 109: 237–276. 10.1016/B978-0-12-385967-9.00008-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castel SE, Martienssen RA. 2013. RNA interference in the nucleus: roles for small RNAs in transcription, epigenetics and beyond. Nat Rev Genet 14: 100–112. 10.1038/nrg3355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chalker DL, Yao MC. 2001. Nongenic, bidirectional transcription precedes and may promote developmental DNA deletion in Tetrahymena thermophila. Genes Dev 15: 1287–1298. 10.1101/gad.884601 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chalker DL, Meyer E, Mochizuki K. 2013. Epigenetics of ciliates. Cold Spring Harb Perspect Biol 5: a017764 10.1101/cshperspect.a017764 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X, Bracht JR, Goldman AD, Dolzhenko E, Clay DM, Swart EC, Perlman DH, Doak TG, Stuart A, Amemiya CT, et al. 2014. The architecture of a scrambled genome reveals massive levels of genomic rearrangement during development. Cell 158: 1187–1198. 10.1016/j.cell.2014.07.034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng CY, Young JM, Lin CG, Chao JL, Malik HS, Yao MC. 2016. The piggyBac transposon-derived genes TPB1 and TPB6 mediate essential transposon-like excision during the developmental rearrangement of key genes in Tetrahymena thermophila. Genes Dev 30: 2724–2736. 10.1101/gad.290460.116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conley AB, Miller WJ, Jordan IK. 2008. Human cis natural antisense transcripts initiated by transposable elements. Trends Genet 24: 53–56. 10.1016/j.tig.2007.11.008 [DOI] [PubMed] [Google Scholar]
- Coyne RS, Chalker DL, Yao MC. 1996. Genome downsizing during ciliate development: nuclear division of labor through chromosome restructuring. Annu Rev Genet 30: 557–578. 10.1146/annurev.genet.30.1.557 [DOI] [PubMed] [Google Scholar]
- Coyne RS, Nikiforov MA, Smothers JF, Allis CD, Yao MC. 1999. Parental expression of the chromodomain protein Pdd1p is required for completion of programmed DNA elimination and nuclear differentiation. Mol Cell 4: 865–872. 10.1016/S1097-2765(00)80396-2 [DOI] [PubMed] [Google Scholar]
- Czermin B, Melfi R, McCabe D, Seitz V, Imhof A, Pirrotta V. 2002. Drosophila enhancer of Zeste/ESC complexes have a histone H3 methyltransferase activity that marks chromosomal Polycomb sites. Cell 111: 185–196. 10.1016/S0092-8674(02)00975-3 [DOI] [PubMed] [Google Scholar]
- Davidovich C, Cech TR. 2015. The recruitment of chromatin modifiers by long noncoding RNAs: lessons from PRC2. RNA 21: 2007–2022. 10.1261/rna.053918.115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deleris A, Stroud H, Bernatavichute Y, Johnson E, Klein G, Schubert D, Jacobsen SE. 2012. Loss of the DNA methyltransferase MET1 Induces H3K9 hypermethylation at PcG target genes and redistribution of H3K27 trimethylation to transposons in Arabidopsis thaliana. PLoS Genet 8: e1003062 10.1371/journal.pgen.1003062 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Croce L, Helin K. 2013. Transcriptional regulation by Polycomb group proteins. Nat Struct Mol Biol 20: 1147–1155. 10.1038/nsmb.2669 [DOI] [PubMed] [Google Scholar]
- Duharcourt S, Yao MC. 2002. Role of histone deacetylation in developmentally programmed DNA rearrangements in Tetrahymena thermophila. Eukaryot Cell 1: 293–303. 10.1128/EC.1.2.293-303.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dumesic PA, Homer CM, Moresco JJ, Pack LR, Shanle EK, Coyle SM, Strahl BD, Fujimori DG, Yates JR III, Madhani HD. 2015. Product binding enforces the genomic specificity of a yeast Polycomb repressive complex. Cell 160: 204–218. 10.1016/j.cell.2014.11.039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eldredge N, Gould SJ. 1972. Punctuated equilibria: an alternative to phyletic gradualism. In Models in paleobiology (ed. Schopf JM), pp. 82–115. W.H. Freeman, San Francisco. [Google Scholar]
- Fass JN, Joshi NA, Couvillion MT, Bowen J, Gorovsky MA, Hamilton EP, Orias E, Hong K, Coyne RS, Eisen JA, et al. 2011. Genome-scale analysis of programmed DNA elimination sites in Tetrahymena thermophila. G3 1: 515–522. 10.1534/g3.111.000927 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng L, Wang G, Hamilton EP, Xiong J, Yan G, Chen K, Chen X, Dui W, Plemens A, Khadr L, et al. 2017. A germline-limited piggyBac transposase gene is required for precise excision in Tetrahymena genome rearrangement. Nucleic Acids Res 45: 9481–9502. 10.1093/nar/gkx652 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fillingham JS, Thing TA, Vythilingum N, Keuroghlian A, Bruno D, Golding GB, Pearlman RE. 2004. A non-long terminal repeat retrotransposon family is restricted to the germ line micronucleus of the ciliated protozoan Tetrahymena thermophila. Eukaryot Cell 3: 157–169. 10.1128/EC.3.1.157-169.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao S, Liu Y. 2012. Intercepting noncoding messages between germline and soma. Genes Dev 26: 1774–1779. 10.1101/gad.199992.112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao S, Xiong J, Zhang C, Berquist BR, Yang R, Zhao M, Molascon AJ, Kwiatkowski SY, Yuan D, Qin Z, et al. 2013. Impaired replication elongation in Tetrahymena mutants deficient in histone H3 Lys 27 monomethylation. Genes Dev 27: 1662–1679. 10.1101/gad.218966.113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gershan JA, Karrer KM. 2000. A family of developmentally excised DNA elements in Tetrahymena is under selective pressure to maintain an open reading frame encoding an integrase-like protein. Nucleic Acids Res 28: 4105–4112. 10.1093/nar/28.21.4105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grewal SI, Elgin SC. 2007. Transcription and RNA interference in the formation of heterochromatin. Nature 447: 399–406. 10.1038/nature05914 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grossniklaus U, Paro R. 2014. Transcriptional silencing by Polycomb-group proteins. Cold Spring Harb Perspect Biol 6: a019331 10.1101/cshperspect.a019331 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guérin F, Arnaiz O, Boggetto N, Denby Wilkes C, Meyer E, Sperling L, Duharcourt S. 2017. Flow cytometry sorting of nuclei enables the first global characterization of Paramecium germline DNA and transposable elements. BMC Genomics 18: 327 10.1186/s12864-017-3713-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ha H, Song J, Wang S, Kapusta A, Feschotte C, Chen KC, Xing J. 2014. A comprehensive analysis of piRNAs from adult human testis and their relationship with genes and mobile elements. BMC Genomics 15: 545 10.1186/1471-2164-15-545 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamilton EP, Kapusta A, Huvos PE, Bidwell SL, Zafar N, Tang H, Hadjithomas M, Krishnakumar V, Badger JH, Caler EV, et al. 2016. Structure of the germline genome of Tetrahymena thermophila and relationship to the massively rearranged somatic genome. Elife 5: e19090 10.7554/eLife.19090 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huvos P. 1995. Developmental DNA rearrangements and micronucleus-specific sequences in five species within the Tetrahymena pyriformis species complex. Genetics 141: 925–936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huvos P. 2004a. A member of a repeat family is the source of an insertion-deletion polymorphism inside a developmentally eliminated sequence of Tetrahymena thermophila. J Mol Biol 336: 1061–1073. 10.1016/j.jmb.2003.12.064 [DOI] [PubMed] [Google Scholar]
- Huvos P. 2004b. Modular structure in developmentally eliminated DNA in Tetrahymena may be a consequence of frequent insertions and deletions. J Mol Biol 336: 1075–1086. 10.1016/j.jmb.2003.12.065 [DOI] [PubMed] [Google Scholar]
- Iyer LM, Abhiman S, Aravind L. 2011. Natural history of eukaryotic DNA methylation systems. Prog Mol Biol Transl Sci 101: 25–104. 10.1016/B978-0-12-387685-0.00002-0 [DOI] [PubMed] [Google Scholar]
- Jamieson K, Rountree MR, Lewis ZA, Stajich JE, Selker EU. 2013. Regional control of histone H3 lysine 27 methylation in Neurospora. Proc Natl Acad Sci 110: 6027–6032. 10.1073/pnas.1303750110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jurka J, Kapitonov VV, Pavlicek A, Klonowski P, Kohany O, Walichiewicz J. 2005. Repbase Update, a database of eukaryotic repetitive elements. Cytogenet Genome Res 110: 462–467. 10.1159/000084979 [DOI] [PubMed] [Google Scholar]
- Kanellopoulou C, Muljo SA, Dimitrov SD, Chen X, Colin C, Plath K, Livingston DM. 2009. X chromosome inactivation in the absence of Dicer. Proc Natl Acad Sci 106: 1122–1127. 10.1073/pnas.0812210106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kapitonov VV, Jurka J. 2006. Self-synthesizing DNA transposons in eukaryotes. Proc Natl Acad Sci 103: 4540–4545. 10.1073/pnas.0600833103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kapusta A, Kronenberg Z, Lynch VJ, Zhuo X, Ramsay L, Bourque G, Yandell M, Feschotte C. 2013. Transposable elements are major contributors to the origin, diversification, and regulation of vertebrate long noncoding RNAs. PLoS Genet 9: e1003470 10.1371/journal.pgen.1003470 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karrer KM. 2012. Nuclear dualism. Methods Cell Biol 109: 29–52. 10.1016/B978-0-12-385967-9.00003-7 [DOI] [PubMed] [Google Scholar]
- Kataoka K, Mochizuki K. 2015. Phosphorylation of an HP1-like protein regulates heterochromatin body assembly for DNA elimination. Dev Cell 35: 775–788. 10.1016/j.devcel.2015.11.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelley D, Rinn J. 2012. Transposable elements reveal a stem cell-specific class of long noncoding RNAs. Genome Biol 13: R107 10.1186/gb-2012-13-11-r107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khalil AM, Guttman M, Huarte M, Garber M, Raj A, Rivea Morales D, Thomas K, Presser A, Bernstein BE, van Oudenaarden A, et al. 2009. Many human large intergenic noncoding RNAs associate with chromatin-modifying complexes and affect gene expression. Proc Natl Acad Sci 106: 11667–11672. 10.1073/pnas.0904715106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuzmichev A, Nishioka K, Erdjument-Bromage H, Tempst P, Reinberg D. 2002. Histone methyltransferase activity associated with a human multiprotein complex containing the Enhancer of Zeste protein. Genes Dev 16: 2893–2905. 10.1101/gad.1035902 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee NN, Chalamcharla VR, Reyes-Turcu F, Mehta S, Zofall M, Balachandran V, Dhakshnamoorthy J, Taneja N, Yamanaka S, Zhou M, et al. 2013. Mtr4-like protein coordinates nuclear RNA processing for heterochromatin assembly and for telomere maintenance. Cell 155: 1061–1074. 10.1016/j.cell.2013.10.027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leeb M, Pasini D, Novatchkova M, Jaritz M, Helin K, Wutz A. 2010. Polycomb complexes act redundantly to repress genomic repeats and genes. Genes Dev 24: 265–276. 10.1101/gad.544410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Thomas A, Stuwe E, Li S, Du J, Marinov G, Rozhkov N, Chen YC, Luo Y, Sachidanandam R, Toth KF, et al. 2014. Transgenerationally inherited piRNAs trigger piRNA biogenesis by changing the chromatin of piRNA clusters and inducing precursor processing. Genes Dev 28: 1667–1680. 10.1101/gad.245514.114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levin HL, Moran JV. 2011. Dynamic interactions between transposable elements and their hosts. Nat Rev Genet 12: 615–627. 10.1038/nrg3030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin CG, Lin IT, Yao MC. 2016. Programmed minichromosome elimination as a mechanism for somatic genome reduction in Tetrahymena thermophila. PLoS Genet 12: e1006403 10.1371/journal.pgen.1006403 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Mochizuki K, Gorovsky MA. 2004. Histone H3 lysine 9 methylation is required for DNA elimination in developing macronuclei in Tetrahymena. Proc Natl Acad Sci 101: 1679–1684. 10.1073/pnas.0305421101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Taverna SD, Muratore TL, Shabanowitz J, Hunt DF, Allis CD. 2007. RNAi-dependent H3K27 methylation is required for heterochromatin formation and DNA elimination in Tetrahymena. Genes Dev 21: 1530–1545. 10.1101/gad.1544207 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madireddi MT, Coyne RS, Smothers JF, Mickey KM, Yao MC, Allis CD. 1996. Pdd1p, a novel chromodomain-containing protein, links heterochromatin assembly and DNA elimination in Tetrahymena. Cell 87: 75–84. 10.1016/S0092-8674(00)81324-0 [DOI] [PubMed] [Google Scholar]
- Malone CD, Hannon GJ. 2009. Small RNAs as guardians of the genome. Cell 136: 656–668. 10.1016/j.cell.2009.01.045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malone CD, Anderson AM, Motl JA, Rexer CH, Chalker DL. 2005. Germ line transcripts are processed by a Dicer-like protein that is essential for developmentally programmed genome rearrangements of Tetrahymena thermophila. Mol Cell Biol 25: 9151–9164. 10.1128/MCB.25.20.9151-9164.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marchler-Bauer A, Lu S, Anderson JB, Chitsaz F, Derbyshire MK, DeWeese-Scott C, Fong JH, Geer LY, Geer RC, Gonzales NR, et al. 2011. CDD: a conserved domain database for the functional annotation of proteins. Nucleic Acids Res 39: D225–D229. 10.1093/nar/gkq1189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marquardt S, Escalante-Chong R, Pho N, Wang J, Churchman LS, Springer M, Buratowski S. 2014. A chromatin-based mechanism for limiting divergent noncoding transcription. Cell 157: 1712–1723. 10.1016/j.cell.2014.04.036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martienssen R, Moazed D. 2015. RNAi and heterochromatin assembly. Cold Spring Harb Perspect Biol 7: a019323 10.1101/cshperspect.a019323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathieu O, Probst AV, Paszkowski J. 2005. Distinct regulation of histone H3 methylation at lysines 27 and 9 by CpG methylation in Arabidopsis. EMBO J 24: 2783–2791. 10.1038/sj.emboj.7600743 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moazed D. 2009. Small RNAs in transcriptional gene silencing and genome defence. Nature 457: 413–420. 10.1038/nature07756 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mochizuki K. 2012. Developmentally programmed, RNA-directed genome rearrangement in Tetrahymena. Dev Growth Differ 54: 108–119. 10.1111/j.1440-169X.2011.01305.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mochizuki K, Gorovsky MA. 2004a. Conjugation-specific small RNAs in Tetrahymena have predicted properties of scan (scn) RNAs involved in genome rearrangement. Genes Dev 18: 2068–2073. 10.1101/gad.1219904 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mochizuki K, Gorovsky MA. 2004b. RNA polymerase II localizes in Tetrahymena thermophila meiotic micronuclei when micronuclear transcription associated with genome rearrangement occurs. Eukaryot Cell 3: 1233–1240. 10.1128/EC.3.5.1233-1240.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mochizuki K, Gorovsky MA. 2005. A Dicer-like protein in Tetrahymena has distinct functions in genome rearrangement, chromosome segregation, and meiotic prophase. Genes Dev 19: 77–89. 10.1101/gad.1265105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mochizuki K, Kurth HM. 2013. Loading and pre-loading processes generate a distinct siRNA population in Tetrahymena. Biochem Biophys Res Commun 436: 497–502. 10.1016/j.bbrc.2013.05.133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mochizuki K, Fine NA, Fujisawa T, Gorovsky MA. 2002. Analysis of a piwi-related gene implicates small RNAs in genome rearrangement in Tetrahymena. Cell 110: 689–699. 10.1016/S0092-8674(02)00909-1 [DOI] [PubMed] [Google Scholar]
- Mohn F, Sienski G, Handler D, Brennecke J. 2014. The rhino-deadlock-cutoff complex licenses noncanonical transcription of dual-strand piRNA clusters in Drosophila. Cell 157: 1364–1379. 10.1016/j.cell.2014.04.031 [DOI] [PubMed] [Google Scholar]
- Müller J, Hart CM, Francis NJ, Vargas ML, Sengupta A, Wild B, Miller EL, O'Connor MB, Kingston RE, Simon JA. 2002. Histone methyltransferase activity of a Drosophila Polycomb group repressor complex. Cell 111: 197–208. 10.1016/S0092-8674(02)00976-5 [DOI] [PubMed] [Google Scholar]
- Noto T, Mochizuki K. 2017. Whats, hows and whys of programmed DNA elimination in Tetrahymena. Open Biol 7: 170172 10.1098/rsob.170172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noto T, Mochizuki K. 2018. Small RNA-mediated trans-nuclear and trans-element communications in Tetrahymena DNA elimination. Curr Biol 28: 1938–1949 e1935. 10.1016/j.cub.2018.04.071 [DOI] [PubMed] [Google Scholar]
- Noto T, Kurth HM, Kataoka K, Aronica L, DeSouza LV, Siu KW, Pearlman RE, Gorovsky MA, Mochizuki K. 2010. The Tetrahymena argonaute-binding protein Giw1p directs a mature argonaute-siRNA complex to the nucleus. Cell 140: 692–703. 10.1016/j.cell.2010.02.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noto T, Kataoka K, Suhren JH, Hayashi A, Woolcock KJ, Gorovsky MA, Mochizuki K. 2015. Small-RNA-mediated genome-wide trans-recognition network in Tetrahymena DNA elimination. Mol Cell 59: 229–242. 10.1016/j.molcel.2015.05.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogawa Y, Sun BK, Lee JT. 2008. Intersection of the RNA interference and X-inactivation pathways. Science 320: 1336–1341. 10.1126/science.1157676 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oliver KR, Greene WK. 2012. Transposable elements and viruses as factors in adaptation and evolution: an expansion and strengthening of the TE-Thrust hypothesis. Ecol Evol 2: 2912–2933. 10.1002/ece3.400 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pal-Bhadra M, Bhadra U, Birchler JA. 1997. Cosuppression in Drosophila: gene silencing of Alcohol dehydrogenase by white-Adh transgenes is Polycomb dependent. Cell 90: 479–490. 10.1016/S0092-8674(00)80508-5 [DOI] [PubMed] [Google Scholar]
- Pal-Bhadra M, Bhadra U, Birchler JA. 2002. RNAi related mechanisms affect both transcriptional and posttranscriptional transgene silencing in Drosophila. Mol Cell 9: 315–327. 10.1016/S1097-2765(02)00440-9 [DOI] [PubMed] [Google Scholar]
- Peng JC, Valouev A, Liu N, Lin H. 2016. Piwi maintains germline stem cells and oogenesis in Drosophila through negative regulation of Polycomb group proteins. Nat Genet 48: 283–291. 10.1038/ng.3486 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perales R, Bentley D. 2009. “Cotranscriptionality”: the transcription elongation complex as a nexus for nuclear transactions. Mol Cell 36: 178–191. 10.1016/j.molcel.2009.09.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plasterk RH, Izsvák Z, Ivics Z. 1999. Resident aliens: the Tc1/mariner superfamily of transposable elements. Trends Genet 15: 326–332. 10.1016/S0168-9525(99)01777-1 [DOI] [PubMed] [Google Scholar]
- Prescott DM. 1994. The DNA of ciliated protozoa. Microbiol Rev 58: 233–267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pritham EJ, Putliwala T, Feschotte C. 2007. Mavericks, a novel class of giant transposable elements widespread in eukaryotes and related to DNA viruses. Gene 390: 3–17. 10.1016/j.gene.2006.08.008 [DOI] [PubMed] [Google Scholar]
- Schoeberl UE, Kurth HM, Noto T, Mochizuki K. 2012. Biased transcription and selective degradation of small RNAs shape the pattern of DNA elimination in Tetrahymena. Genes Dev 26: 1729–1742. 10.1101/gad.196493.112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schulz D, Schwalb B, Kiesel A, Baejen C, Torkler P, Gagneur J, Soeding J, Cramer P. 2013. Transcriptome surveillance by selective termination of noncoding RNA synthesis. Cell 155: 1075–1087. 10.1016/j.cell.2013.10.024 [DOI] [PubMed] [Google Scholar]
- Schwope RM, Chalker DL. 2014. Mutations in Pdd1 reveal distinct requirements for its chromodomain and chromoshadow domain in directing histone methylation and heterochromatin elimination. Eukaryot Cell 13: 190–201. 10.1128/EC.00219-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Senti KA, Jurczak D, Sachidanandam R, Brennecke J. 2015. piRNA-guided slicing of transposon transcripts enforces their transcriptional silencing via specifying the nuclear piRNA repertoire. Genes Dev 29: 1747–1762. 10.1101/gad.267252.115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shang Y, Song X, Bowen J, Corstanje R, Gao Y, Gaertig J, Gorovsky MA. 2002. A robust inducible-repressible promoter greatly facilitates gene knockouts, conditional expression, and overexpression of homologous and heterologous genes in Tetrahymena thermophila. Proc Natl Acad Sci 99: 3734–3739. 10.1073/pnas.052016199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simon JA, Kingston RE. 2013. Occupying chromatin: Polycomb mechanisms for getting to genomic targets, stopping transcriptional traffic, and staying put. Mol Cell 49: 808–824. 10.1016/j.molcel.2013.02.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slotkin RK, Martienssen R. 2007. Transposable elements and the epigenetic regulation of the genome. Nat Rev Genet 8: 272–285. 10.1038/nrg2072 [DOI] [PubMed] [Google Scholar]
- Sweet MT, Allis CD. 1998. Culture and biochemical analysis of cells. In: Cells: a laboratory manual (ed. Spector DL, et al.). Cold Spring Harbor Laboratory Press, Woodbury, NY. [Google Scholar]
- Taverna SD, Coyne RS, Allis CD. 2002. Methylation of histone h3 at lysine 9 targets programmed DNA elimination in Tetrahymena. Cell 110: 701–711. 10.1016/S0092-8674(02)00941-8 [DOI] [PubMed] [Google Scholar]
- Tellier M, Bouuaert CC, Chalmers R. 2015. Mariner and the ITm superfamily of transposons. Microbiol Spectr 3: MDNA3-0033-2014 10.1128/microbiolspec.MDNA3-0033-2014 [DOI] [PubMed] [Google Scholar]
- Tsai MC, Manor O, Wan Y, Mosammaparast N, Wang JK, Lan F, Shi Y, Segal E, Chang HY. 2010. Long noncoding RNA as modular scaffold of histone modification complexes. Science 329: 689–693. 10.1126/science.1192002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L, Dou K, Moon S, Tan FJ, Zhang ZZ. 2018. Hijacking oogenesis enables massive propagation of LINE and retroviral transposons. Cell 174: 1082–1094. 10.1016/j.cell.2018.06.040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinhofer I, Hehenberger E, Roszak P, Hennig L, Köhler C. 2010. H3K27me3 profiling of the endosperm implies exclusion of Polycomb group protein targeting by DNA methylation. PLoS Genet 6: e1001152 10.1371/journal.pgen.1001152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wuitschick JD, Gershan JA, Lochowicz AJ, Li S, Karrer KM. 2002. A novel family of mobile genetic elements is limited to the germline genome in Tetrahymena thermophila. Nucleic Acids Res 30: 2524–2537. 10.1093/nar/30.11.2524 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiong J, Lu X, Zhou Z, Chang Y, Yuan D, Tian M, Zhou Z, Wang L, Fu C, Orias E, et al. 2012. Transcriptome analysis of the model protozoan, Tetrahymena thermophila, using deep RNA sequencing. PLoS One 7: e30630 10.1371/journal.pone.0030630 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao MC, Chao JL, Cheng CY. 2014. Programmed genome rearrangements in Tetrahymena. Microbiol Spectr 2 10.1128/microbiolspec.MDNA3-0012-2014 [DOI] [PubMed] [Google Scholar]
- Zeh DW, Zeh JA, Ishida Y. 2009. Transposable elements and an epigenetic basis for punctuated equilibria. Bioessays 31: 715–726. 10.1002/bies.200900026 [DOI] [PubMed] [Google Scholar]
- Zhang Z, Wang J, Schultz N, Zhang F, Parhad SS, Tu S, Vreven T, Zamore PD, Weng Z, Theurkauf WE. 2014. The HP1 homolog rhino anchors a nuclear complex that suppresses piRNA precursor splicing. Cell 157: 1353–1363. 10.1016/j.cell.2014.04.030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao J, Sun BK, Erwin JA, Song JJ, Lee JT. 2008. Polycomb proteins targeted by a short repeat RNA to the mouse X chromosome. Science 322: 750–756. 10.1126/science.1163045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao J, Ohsumi TK, Kung JT, Ogawa Y, Grau DJ, Sarma K, Song JJ, Kingston RE, Borowsky M, Lee JT. 2010. Genome-wide identification of Polycomb-associated RNAs by RIP-seq. Mol Cell 40: 939–953. 10.1016/j.molcel.2010.12.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.