

Shimojo et al. show that Synaptotagmin-11 is an essential component of a neuronal vesicular trafficking pathway that differs from the well-characterized synaptic vesicle trafficking pathway but is also essential for life.
Keywords: development, membrane trafficking, memory, mouse genetics, neuron, secretion, synaptic plasticity, synaptotagmin
Abstract
Synaptotagmin-11 (Syt11) is a Synaptotagmin isoform that lacks an apparent ability to bind calcium, phospholipids, or SNARE proteins. While human genetic studies have linked mutations in the Syt11 gene to schizophrenia and Parkinson's disease, the localization or physiological role of Syt11 remain unclear. We found that in neurons, Syt11 resides on abundant vesicles that differ from synaptic vesicles and resemble trafficking endosomes. These vesicles recycle via the plasma membrane in an activity-dependent manner, but their exocytosis is slow and desynchronized. Constitutive knockout mice lacking Syt11 died shortly after birth, suggesting Syt11-mediated membrane transport is required for survival. In contrast, selective ablation of Syt11 in excitatory forebrain neurons using a conditional knockout did not affect life span but impaired synaptic plasticity and memory. Syt11-deficient neurons displayed normal secretion of fast neurotransmitters and peptides but exhibited a reduction of long-term synaptic potentiation. Hence, Syt11 is an essential component of a neuronal vesicular trafficking pathway that differs from the well-characterized synaptic vesicle trafficking pathway but is also essential for life.
Neurons employ a variety of membrane organelles for secretion, transport of cargo between specific intracellular compartments, dynamic experience-dependent control of synaptic strength, and autophagy. These organelles may recruit unique sets of proteins that regulate their trafficking, fusion, or retrieval. Synaptotagmins (Syts) are secretory proteins composed of a short N-terminal region, a single transmembrane domain, a variable linker sequence, and two C2 domains (Sudhof 2002; Gustavsson and Han 2009). The prototypical member of the Syt family, Syt1, was identified nearly three decades ago as a calcium sensor for fast exocytosis of synaptic neurotransmitter and neuroendocrine vesicles (Perin et al. 1991a,b; Geppert et al. 1994). Extensive studies have shown that, upon binding free calcium ions via its C2 domains, Syt1 accelerates vesicle fusion by interacting with assembled SNARE complexes and negatively charged phospholipids (Davletov and Sudhof 1993; Fernandez-Chacon et al. 2001; Sudhof 2002; Jahn et al. 2003; Pang et al. 2006a; Tang et al. 2006). In vertebrates, synaptic and neuroendocrine release is also driven by Syt2, Syt7, and Syt9, whereas the remaining Syt isoforms may act in other membrane trafficking pathways (Pang et al. 2006b; Sun et al. 2007; Xu et al. 2007; Gustavsson et al. 2009; Cao et al. 2011; Dean et al. 2012; Bacaj et al. 2013). Furthermore, different members of the Syt family exhibit distinct biochemical properties, and some of these proteins do not bind calcium, phospholipids, and SNAREs in vitro (Sugita and Südhof 2000; Sugita et al. 2002). However, the biological roles of these Syts are unclear.
Syt11 is a poorly characterized Syt isoform with inactive calcium binding sites whose mRNA is abundantly expressed throughout the brain (von Poser et al. 1997). The human Syt11 gene is located on the chromosome locus 1q21-q22 and may be a risk gene for schizophrenia and Parkinson's disease (Huynh et al. 2003; Inoue et al. 2007; Sesar et al. 2016). Recent studies have proposed that in the substantia nigra pars compacta, Syt11 is a mediator of neurotoxicity that interacts with Parkin and regulates the pools of dopaminergic vesicles by inhibiting endocytosis (Wang et al. 2018). Syt11 has also been proposed to inhibit cytokine secretion and phagocytosis in glia (Du et al. 2017), but its precise role in development and function of neural circuits has remained largely unknown. Curiously, constitutive knockout mice lacking Syt4, a close Syt11 homolog, have a normal life span but exhibit enhanced synaptic potentiation and deficits in motor coordination and hippocampus-dependent memory (Ferguson et al. 2004; Dean et al. 2009). When overexpressed in cultured hippocampal neurons, recombinant Syt4 is targeted to vesicles that contain the brain-derived neurotrophic factor (BDNF) and traffic in axonal and dendritic shafts (Dean et al. 2012). Though the mammalian Syt4 also fails to bind calcium due to evolutionary inactivation (Dai et al. 2004), the fly Syt4 is believed to trigger calcium-dependent release of retrograde signals that stimulate enhanced synaptic function and growth (Yoshihara et al. 2005).
Herein, we systematically analyze the subcellular localization of neuronal Syt11, identify proteins that interact with Syt11 in the brain, and examine mutant mice that lack Syt11 in all tissues or in glutamatergic neurons. Our results show that Syt11 is a component of a vesicular transport pathway that is essential for normal development, synaptic function, and memory formation.
Results
Syt11 resides on mobile vesicular organelles that are scattered in neuronal somas and processes
To detect Syt11 in mammalian neurons, we generated a polyclonal antibody that selectively recognizes the linker sequence which connects the Syt11 transmembrane and C2A domains (Sudhof 2002). The specificity of this serum was confirmed by immunoblotting of protein homogenates from COS7 cells expressing individual recombinant Syt isoforms Syt1 to Syt13 and from brains of Syt11 knockout mice (see Fig. 3C,J; Supplemental Fig. S1A,B). Immunoblot analysis of different rodent organs, immortalized cell lines, and primary cortical cultures demonstrated that Syt11 is enriched in neurons (Supplemental Fig. S1C–E). The protein was abundant across the brain and progressively increased in levels over the first 2 wk after birth (Fig. 1A,B). Consistent with these results and publicly available analysis of Syt11 mRNA expression in the adult brain (http://mouse.brain-map.org/gene/show/86883), immunofluorescent imaging revealed strong Syt11 immunoreactivity in various brain regions and neuron types (Fig. 1C).
Figure 3.

Generation and characterization of Syt11 knockout mice. (A) Schematics of gene targeting vector for homologous recombination. The second exon of Syt11 (E2) was flanked by loxP sites (black arrowheads) to allow conditional silencing. The NEO surrounded by FRT sites (black circles) and diphtheria toxin (DT) cassettes were used for positive and negative selection, respectively. The Flag epitope (F) was inserted into a coding region of E2. To obtain a floxed knock-in line (KI), mice carrying homologously recombined alleles were crossed with a transgenic line that express FLP recombinase. To obtain constitutive Syt11 knockouts (KO), knock-in founders were crossed with mice expressing Cre in the germline. (B) Percentages of surviving offspring of indicated ages and genotypes from breedings of heterozygote knock-in and knockout patents (n > 100 per line). (C) Total brain homogenates from postnatal day 1 (P1) wild-type and homozygous Syt11 knock-in and knockout mice were analyzed by immunoblotting with indicated antibodies. Note a shift in migration of Flag-tagged Syt11 in knock-ins and complete loss of the protein in knockouts. (D–K) Offspring of heterozygous Syt11 knockout parents were analyzed at P1. (D) Photographs of pups of indicated genotypes. Note the lack of milk in stomachs of homozygote knockouts (arrows). (E) Averaged latencies to suckle, as assessed in nine to 25 pups per genotype. (F) Representative images of whole brains, olfactory bulbs, and cerebella. (G) Coronal sections of olfactory bulbs were stained with DAPI and antibodies against MAP2, Tbr2, GFAP, GAD67, and TH. (H) Cortices of embryonic day 15.5 (E15.5) embryos were in utero electroporated with the plasmid that expresses GFP to assess neuron migration. Representative images of GFP fluorescence in DAPI-stained sections from three newborn mice/genotype are shown. (I) Low magnification images of DAPI-stained brain sections. (J,K) Expression levels of indicated proteins in the brain were analyzed by quantitative immunoblotting with I125-labeled secondary antibodies. Representative blots and averaged values from three littermate pairs of wild-type and constitutive knockout (−/−) mice are shown. All quantifications are represented as Mean ± SE (error bars). (*) P < 0.05, defined by Student's t-test.
Figure 1.

Expression and subcellular localization of Syt11 in central neurons. (A) Total protein extracts (20 µg/lane) from various regions of adult mouse brain were analyzed by immunoblotting with antibodies to Syt11, Syt1, and valosine-containing protein (VCP, as loading control). (Ctx) Cerebral cortex; (Crb) cerebellum; (Hip) hippocampus; (OB) olfactory bulb; (BS) brain stem; (SC) spinal cord. (B) Brains were isolated from mice of different ages, and Syt11 expression levels were analyzed by quantitative immunoblotting with I125-labeled secondary antibody. (C) Images of brain sections from adult mice that were immunostained for Syt11. (D) Subcellular fractions of detergent-free brain homogenates were separated by centrifugation in continuous sucrose gradients. Panels show measurements of total protein concentrations (top) and immunoblots with indicated antibodies (bottom). Note that Syt11 is absent from fractions enriched with synaptic vesicle (SV) proteins Syt1, Syt12, and Syb2. (E) Quantitative immunoblot analysis of expression of indicated proteins in brains of Synapsin1/2 double-knockout mice. Data from three pairs of double-knockout and wild-type animals are plotted as percentage of wild type. Mean ± SE (error bars). (*) P < 0.05, defined by Student's t-test. (F,G) Cortical neurons were cultured in vitro for 14 d (DIV14) and then labeled with antibodies against Syt11 and either MAP2 (F) or Syt1 (G). Representative immunofluorescent confocal images are shown. (H) Electron microscopy analysis of Syt11 localization in cortical neurons. Images of ultrathin sections of adult forebrains show immuno-Gold-labeled vesicles (arrows) in cell bodies, dendritic shafts, and synapses. (PSD) Postsynaptic density.
Next, we used biochemical and imaging techniques to study the localization of Syt11 at a subcellular level. Continuous sucrose density gradient fractionations of detergent-free brain homogenates showed that Syt11 is associated with light membranes. However, Syt11 was absent from fractions enriched with proteins that are known to reside on synaptic vesicles (SVs), such as Syt1, Syt12, and Syb2 (Fig. 1D; Supplemental Fig. S1F). Likewise, quantitative immunoblotting showed that Syt11 levels were unaltered in brains of Synapsin1/2 double knockout mice which exhibit a strong reduction of SV proteins due to vesicle loss at presynaptic terminals (Fig. 1E; Maximov et al. 2007).
Immunofluorescent imaging of cortical neurons in primary cultures and immunoelectron microscopy of thin cortical sections from adult mice demonstrated that native Syt11 is localized to vesicles that are broadly distributed in neuronal somas, axons, and dendrites. Again, Syt11 was largely segregated from Syt1 (Fig. 1F–H). Similar to native Syt11, virally expressed Syt11-Venus and Syt11-myc fusion proteins were targeted to vesicles that were scattered in cell bodies and neuronal processes and did not overlap with SVs. We also found no apparent colocalization of Syt11 with common markers of the endoplasmic reticulum, Golgi apparatus, dense core vesicles, or recycling endosomes (Fig. 2A; Supplemental Fig. S2). Live time-lapse imaging of neurons carrying Syt11-Venus revealed that many Syt11 vesicles traveled in axonal and dendritic shafts in both anterograde and retrograde directions, in contrast to SVs whose cycling is restricted to presynaptic terminals. The mobility of these organelles was nearly completely arrested by treatments with nocodazole, suggesting their intracellular transport depends on microtubules (Supplemental Movies S1–S4).
Figure 2.

Imaging analyses of Syt11 vesicle trafficking. (A) Images of live (first column) and fixed neurons in DIV14 cortical cultures expressing Syt11-Venus from a lentivirus (LV). In columns 2–5, cultures were labeled with antibodies against MAP2, Synapsin, and PSD95. The third column shows sparsely infected cells to demonstrate that Syt11-Venus puncta along MAP2-positive dendrites are not axonal. (B–H) Live time-lapse imaging analysis of DIV14-16 neurons carrying pH-sensitive reporters of Syt11-positive vesicles and SVs, pHluorin-Syt11 and Synaptophysin (SyP)-pHluorin. (B) Predicted topology of pHluorin-Syt11 on trafficking organelles. During exocytosis, deacidification of vesicle lumens leads to an increase in reporter fluorescence. (C,D) Pseudocolored images of pHluorin-Syt11 fluorescence (C) and quantification of fluorescent intensity (D) before and after application of buffer with ammonium chloride, which neutralizes intravesicular pH. D shows averaged signal from 50 individual 2-µm2 regions of interest (ROIs) with stationary vesicles. (E–H) Comparative analysis of vesicle exocytosis. (E,F) Pseudocolored images of SyP-pHluorin (E) and pHluorin-Syt11 (F) fluorescence in DIV14 neurons before and immediately after trains of extracellular electrical stimulation at 20 Hz. (G) Time course of changes in fluorescent intensity in 2-µm2 ROIs. Note that fusion of pHluorin-Syt11-containing vesicles is not synchronized with the onset of stimulation. (H) Averaged densities of fusion events in the absence of stimulation and during 20-sec 20-Hz stimulus trains. Data from three independent experiments are plotted as mean ± SE (error bars). (*) P < 0.05, defined by Student's t-test. See also Supplemental Movies S1–S5.
Syt11 vesicles and SVs cycle with distinct kinetics
To explore the possibility that Syt11 vesicles undergo activity-dependent exocytosis, we took advantage of pHluorin-Syt11, a fluorescent reporter that contains a pH-sensitive form of GFP (Miesenbock et al. 1998). Since most intracellular vesicular organelles have acidic lumens, intraluminal pHluorins only display strong fluorescence after vesicle fusion with the plasma membrane due to loss of the pH gradient (Fig. 2B). We attached the pH-sensitive GFP tag to the N terminus of Syt11, expressed this construct with a lentivirus under the control of the Ubc promoter in cortical cultures, and performed live time-lapse imaging 2 wk after plating when neurons established dense synaptic networks. Similar to native Syt11 and Syt11-Venus, pHluorin-Syt11 was targeted to vesicle-like organelles scattered in cell bodies and neuronal processes. The reporter was barely detectable in these structures under the control conditions but became bright immediately after bath application of a buffer containing ammonium chloride that neutralizes intraluminal pH (Fig. 2C,D). Curiously, a fraction of Syt11 also resided in the plasma membrane with exposed extracellular N termini in both axons and dendrites, as evidenced by strong pHluorin-Syt11 signal that could be abolished by brief puffs of buffer with acidic pH. The axonal pool was particularly pronounced in growth cones (Supplemental Movie S5).
We then compared the behavior of pHluorin-Syt11 and a well-characterized reporter of exocytosis of SVs, Synaptophysin (SyP)-pHluorin (Peng et al. 2012). Neurons were imaged before, during, and after electrical stimulation with a local extracellular electrode. As expected, application of high-frequency stimulus trains triggered robust exocytosis of SVs, resulting in an increase in SyP-pHluorin signal at numerous sites. Although repetitive stimulation also induced exocytosis of vesicles tagged with pHluorin-Syt11, these events were very rare. Moreover, the fusion of Syt11 vesicles was not synchronized with the onset of stimulation, indicating that no readily releasable pools of these organelles exist (Fig. 2E–H).
Global ablation of Syt11 in mice leads to early postnatal lethality
To investigate the physiological role of Syt11, we generated mutant mice carrying a “floxed” knock-in allele that allows for conditional silencing of the Syt11 gene by Cre recombinase. In this allele, two loxP sites were inserted in intronic regions upstream of and downstream from exon 2 (E2) that encodes the transmembrane region of Syt11. We also added a Flag epitope in the middle of the Syt11 linker domain following the transmembrane region to enable detection and isolation of the protein with anti-Flag antibodies. After in vivo excision of the NEO selection marker, validated heterozygous knock-in founders were crossed with mice that express Cre in the germline to produce constitutive full-body Syt11 knockout mice (Fig. 3A).
Homozygous Syt11 knock-in mice were born at the expected Mendelian ratio, had normal life spans, and did not exhibit any apparent behavioral abnormalities in a standard laboratory environment. In contrast, all homozygous knockouts died at postnatal day 1 (P1), usually between 12 and 24 h after birth (Fig. 3B). Immunoblotting confirmed that Flag-tagged Syt11 was translated as a single polypeptide in brains of knock-in mice and was absent from homozygous knockouts (Fig. 3C). The early postnatal lethality of knockout pups was at least partially attributed to inability to suckle, as evidenced by the lack of mother's milk in their stomachs and twofold higher latency to suckle in the absence of competition from littermates (Fig. 3D,E). Considering that native Syt11 is enriched in neurons, we focused the remaining studies on the brain.
When inspected at low magnification, the brains of newborn knockout pups were almost indistinguishable from brains of their wild-type littermates, with the exception of a smaller olfactory bulb (OB) (Fig. 3F). Although the olfactory bulb was markedly reduced in Syt11 knockout mice, confocal imaging analysis of coronal sections that were labeled with the nuclear DNA marker, DAPI, and antibodies against MAP2, Tbr1, GFAP, GAD67, and tyrosine hydroxylase did not reveal defects in OB lamination (Fig. 3G). The gross anatomy of the cerebral cortex and the hippocampus was also undistorted (Fig. 3I). Moreover, we found that during development, Syt11-deficient neurons can appropriately migrate to superficial layers of the developing cortex. Specifically, upon imaging cortical sections from newborn pups that were electroporated in utero at embryonic day 15.5 with the plasmid expressing GFP under the control of the CAG promoter, we observed normal neuronal migration (Fig. 3H). Lastly, quantitative immunoblotting with a battery of antibodies did not uncover significant changes in expression of proteins other than Syt11 in brains of P1 knockout mice (Fig. 3J,K).
Syt11-deficient neurons have unaltered secretion of fast neurotransmitters
To determine whether early death of Syt11 knockout mice is attributed to abnormalities in neurotransmission, we systematically analyzed intrinsic excitability and synaptic properties of Syt11-deficient neurons in culture. We performed whole-cell current and voltage clamp recordings from primary cultures that were isolated from cortices of newborn wild-type and homozygous Syt11 knockout pups and maintained in vitro for 14–16 d (DIV14-16). In parallel, we knocked out Syt11 with lentivirally expressed Cre-recombinase in cultures from homozygous knock-in mice, using truncated catalytically inactive ΔCre as a negative control. In these experiments, Cre constructs were expressed as GFP fusions to enable monitoring of the efficiency of lentiviral infection. The Cre-dependent silencing of Syt11 was also confirmed by immunoblotting (Supplemental Fig. S3A,B).
Wild-type and Syt11-deficient neurons had comparable membrane resistances, resting membrane potentials, and thresholds for action potential firing (Supplemental Fig. S3C,D). Furthermore, neither constitutive nor conditional ablation of Syt11 prevented synaptogenesis or had detectable effects on vesicular release of glutamate and GABA. Indeed, we found no significant changes in (1) the amplitudes of correlated inhibitory and excitatory postsynaptic currents (IPSCs and EPSCs) that were triggered by single action potentials, (2) paired-pulse IPSC ratios, and (3) the kinetics of synaptic depression during high-frequency stimulus trains (Fig. 4A–H). In addition, loss of Syt11 did not abolish activity-dependent homeostatic scaling of AMPA receptors at excitatory synapses (Turrigiano et al. 1998), as assessed by recordings of spontaneous quantal EPSCs from neurons that were pretreated with Tetrodotoxin and the NMDA receptor blocker, APV (Fig. 4I–K). While glutamatergic transmission between Syt11-deficient neurons declined as the cultures aged, this effect was attributed to moderate cell loss rather than changes in synaptic function. Taken together, these results imply that Syt11 is not involved in regulation of synaptic vesicle exocytosis.
Figure 4.
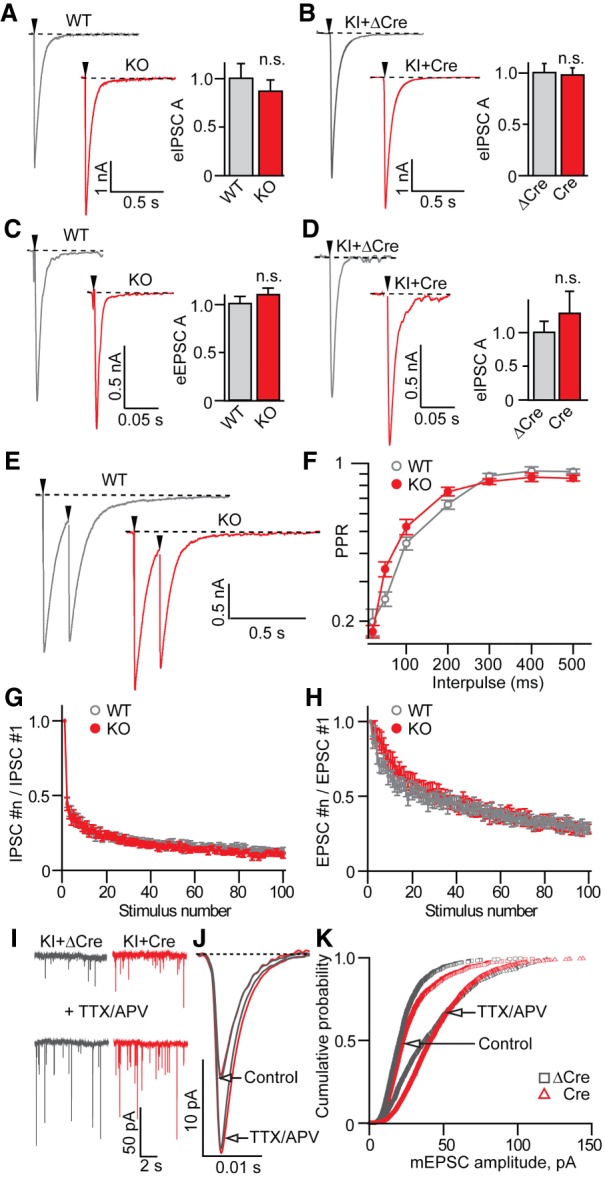
Synaptic properties of cultured Syt11-deficient neurons. Neurotransmission was monitored in whole-cell voltage clamp mode. Experiments were performed with DIV14–16 cultures isolated from cortices of P1 Syt11 knockout or homozygous knock-in mice. To silence the Syt11 gene in knock-in neurons, cultures were infected at DIV5 with an LV that expresses Cre under the control of the Ubc promoter. Truncated catalytically inactive ΔCre was used a control. Synaptic responses were triggered by injection of current through a local extracellular electrode. Each synaptic parameter was measured in at least three independent culture preparations. (A,B) Sample traces and averaged amplitudes of evoked IPSCs (eIPSCs) recorded at −70 mV in the presence of AMPA/NMDA receptor blockers CNQX (50 µM) and APV (10 µM). Wild-type: n = 11; knockout: n = 15; knock-in + ΔCre: n = 11; knock-in + Cre: n = 11. (C,D) Sample traces and averaged amplitudes of evoked excitatory postsynaptic currents (eEPSCs) recorded at −70 mV in the presence of the GABA receptor blocker picrotoxin (100 µM). Wild-type: n = 15; knockout: n = 21; knock-in + ΔCre: n = 11; knock-in + Cre: n = 11. (E,F) Paired pulse eIPSC ratios (PPR) in wild-type and Syt11 knockout neurons. Sample traces (E) and quantifications of PPR at different interpulse intervals (F) are shown. Wild-type: n = 31; knockout: n = 15. (G,H) Depression of synchronous eIPSCs (G) and eEPSCs (H) during 10-Hz stimulus trains. For EPSCs, wild-type: n = 15; knockout: n = 21. For IPSCs, wild-type: n = 11; knockout: n = 15. (I–K) Homeostatic plasticity of excitatory synapses. Spontaneous AMPA-type EPSCs were recorded in the presence of 100 µM picrotoxin and 1 µM tetrodotoxin (TTX) under the normal conditions, or after pretreating cultures with TTX and APV for 72 and 24 h, respectively. Panels display raw current traces (I), averaged quantal sEPSCs (J; five cells per >100 events per group), and cumulative probability plots of sEPSC amplitudes (K). All graphs are plotted as mean ± SE (error bars). (*) P < 0.05, defined by Student's t-test.
Identification of proteins that interact with Syt11 in the brain
As the next step, we carried out a biochemical screen to identify proteins that interact with Syt11 in the brain as a possible clue to its function and mechanism of action. To this end, we immunoprecipitated tagged Syt11 from the brains of adult knock-in mice with Flag antibody-coated sepharose beads in buffers containing a detergent and either 2 mM EDTA or 2 mM calcium. After extensive washes, attached proteins were eluted, digested, and detected by mass spectrometry (Pankow et al. 2015, 2016). To subtract “background” from proteins that nonselectively bind to beads and/or antibody, we performed similar immunoprecipitations with samples from wild-type brains, which lacked the expression of Flag-tagged Syt11 (Fig. 5A).
Figure 5.

Identification of proteins that interact with Syt11. Proteins from brains of adult wild-type and Syt11 knock-in mice were extracted with TX100 in the presence of 2 mM EDTA or 2 mM calcium. Epitope-tagged Syt11 was immunoprecipitated with monoclonal Flag antibody-coated sepharose beads, and attached proteins were detected by immunoblotting or mass spectrometry (LC/MS). (A) Input, unbound, and immunoprecipitated fractions from mice of indicated genotypes were probed by immunoblotting with a polyclonal antibody against Syt11. (B,C) Summary of LC/MS analysis of immunoprecipitations from three pairs of mice. Total numbers of identified proteins (including Syt11) and corresponding gene names are shown. (D) Input and immunoprecipitated fractions were probed by immunoblotting with antibodies against Syt11, Kif1a, Syntaxin1a, and Syb2.
Under these conditions, we identified 23 and eight proteins that specifically associated with Syt11 in the absence and presence of free calcium, respectively. Three of these proteins were present at measurable levels in both groups of samples (Fig. 5B). Several genes encoding candidate Syt11-binding partners have been shown previously to be essential for membrane trafficking, including Kif1a, Ap1b, and Stxbp5 (Gonzalez and Rodriguez-Boulan 2009; Hirokawa et al. 2009; Zhu et al. 2014). However, we did not detect vesicular or plasma–membrane SNAREs and other key regulators of exocytosis that associate with Syt isoforms residing on SVs (Fernandez-Chacon et al. 2001; Jahn et al. 2003; Tang et al. 2006). Intriguingly, the pool of Syt11 interacting partners contained several RNA-binding proteins, raising the possibility that Syt11 is involved in RNA transport (Fig. 5C). We confirmed the coimmunoprecipitation (co-IP) of Syt11 with Kif1a and lack of interaction with SNAREs Syntaxin1a and Syb2 by immunoblotting (Fig. 5D).
Generation and analysis of forebrain-specific conditional Syt11 knockout (cKO) mice
To further investigate the role of Syt11 in the CNS, we silenced the “floxed” Syt11 gene in excitatory glutamatergic neurons across the forebrain. This was accomplished by crossing the knock-in founders with well-characterized Nex:Cre, which drives recombination in differentiating neurons as early as embryonic day 11.5 (Fig. 6A,B; Goebbels et al. 2006). Unlike constitutive Syt11 knockouts, homozygous forebrain-specific cKOs were viable, fertile, and had normal life spans (Fig. 6C). In agreement with the previously described pattern of Nex:Cre recombination in mice (Goebbels et al. 2006), immunoblotting demonstrated a loss of Syt11 protein expression in the cerebral cortex and hippocampus but not in the cerebellum (Fig. 6D). However, imaging of Nissl and antibody-stained sections did not reveal obvious morphological defects in cKO forebrains (Fig. 6E,F).
Figure 6.

Characterization of Syt11 cKO mice. The Syt11 gene was silenced in excitatory forebrain neurons during embryogenesis by crossing the “floxed” knock-in allele with Nex:Cre. (A,B) Recombinase activity of Nex:Cre, as assessed with Ai9 tdTomato reporter. Images show a coronal brain section at low magnification and Topro3-stained sections of the hippocampus. (C) Homozygous knock-in and cKO mice at P60. (D) Immunoblot analysis of expression of indicated proteins in different brain regions of knock-in and cKO mice. (OB) Olfactory bulb; (Ctx) cerebral cortex; (Hip) hippocampus; (Crb) cerebellum. (E,F) Brain sections from adult mice were stained with Nissl or antibodies against Cux1 and NF. (G–O) Behavior of P60 knock-in and cKO mice. (G–I) Analysis of spatial memory in the Barnes maze. Graphs show latencies to escape during each training session (G), numbers of errors during training (H), and percentages of time spent in target and other quadrants of the maze in the Probe test (I). Averaged values are from seven pairs of males. (K) Novel object recognition test. Graph shows averaged numbers of contacts with the same object during familiarization trials (T1–T3), old object in the novel location (ONL), new object in the novel location (NNL), new object in old location (NOL), and new object in the novel location (NNL). (L) Olfaction, as assessed by numbers of approaches of the same object with distinct orders (O) in two separate trials (T). (M) Open field test of locomotor activity. Averaged numbers of beam breaks for ambulation, rearing, and center activity are shown. (N) Dark-light transfer test of anxiety-like behavior. Graph shows averaged numbers of transitions to light (TTL) and time in light per transmission (TIL; in seconds). (O) Associative fear memory was assessed using contextual and cued fear conditioning. In K–O, quantifications are from five males and six females per genotype. All graphs are plotted as mean ± SE (error bars). (*) P < 0.05, defined by Student's t-test.
To assess the capacity of Syt11-deficient animals to process sensory information and to perform cognitive and motor tasks, we interrogated them using a battery of behavioral tests. cKO mice had no detectable defects in vision, ability to recognize novel objects, olfaction, locomotor activity in open fields, and anxiety-like behavior but exhibited an abnormal performance in a Barnes maze, a setup that is designed for quantitative analysis of spatial learning and memory (Fig. 6G–N). In this test, mice were subjected to four sequential training sessions in the circular maze to memorize the location of the hole that is connected with an escape tunnel. During each session, the latencies to escape and numbers of errors were recorded. The mice were then subjected to a Probe test of spatial memory with the escape tunnel removed. When compared with their control knock-in littermates, cKOs had significantly longer escape latencies, made more errors, and did not show clear target discrimination in the Probe test, suggesting their learning and memory were impaired (Fig. 6G–I). Nonetheless, ablation of Syt11 did not affect associative fear memory (Fig. 6O), making it unlikely that the Barnes maze phenotypes reflect a global impairment of neural circuits essential for memory storage.
Syt11 cKO mice display impaired synaptic plasticity in the hippocampus
To test whether memory loss of Syt11 cKO mice is attributed to changes in experience-dependent plasticity, we monitored excitatory postsynaptic potentials (fEPSP) in area CA1 of the hippocampus in acute slices. Similar to analyses of neurotransmission in primary cultures, slice recordings showed that Syt11 does not regulate basal synaptic strengths. CA1 circuits of cKO mice and their knock-in controls had comparable dependencies of fEPSP slopes on intensity of extracellular stimulation of the Schaffer collateral path with virtually indistinguishable PPRs and depression profiles during high-frequency stimulation (Fig. 7A–C). In contrast, ablation of Syt11 in excitatory neurons impaired long-term synaptic potentiation (LTP). When LTP was induced by θ bursts, a significant difference in fEPSP slopes was observed immediately after conditioning, and this effect persisted over the course of recording (Fig. 7D,E). Yet, pyramidal cells of cKO mice had unaltered densities of dendritic spines, and we found that LTP was normal after stronger tetanic stimulation (1 sec at 100 Hz) (Fig. 7F,G; Supplemental Fig. S4).
Figure 7.

Synaptic transmission and plasticity in the hippocampus of Syt11 cKO mice. (A–E) Extracellular recordings of evoked excitatory postsynaptic potentials (fEPSP) in the CA1. Experiments were performed with acute hippocampal slices from P20 Syt11 cKO mice and their control Cre-negative knock-in littermates. (A) fEPSP slopes were measured at different intensities of extracellular stimulation of the Schaffer collateral path. (B) fEPSP PPRs were measured at different interpulse intervals. (C) fEPSPs were monitored during 10-Hz stimulus trains. Graph shows profiles of synaptic depression, represented as the fEPSPn/fEPSP1 ratio. In A–C, averaged values are from four pairs of mice. Knock-in = 18 slices, cKO = 19 slices. (D) Samples of fEPSPs recorded before and after application of θ burst stimulus (TBS) trains. (E) fEPSPs slopes were measured continuously before and after TBS-induced long-term potentiation (LTP). Averaged values are from seven pairs of mice. Knock-in = 18 slices, cKO = 20 slices. (F) Images of spines on apical dendrites of CA1 pyramidal neurons, visualized by Golgi staining. (G) Averaged spine densities. Data are from three pairs of mice, nine neurons per genotype. All graphs are plotted as mean ± SE (error bars). (*) P < 0.05, defined by Student's t-test.
Since θ-burst LTP depends on BDNF/TrkB signaling (Patterson et al. 1996; Barco et al. 2005; Edelmann et al. 2014; Park et al. 2014), and the close Syt11 homolog, Syt4, has been shown to control the secretion of BDNF (Dean et al. 2009), we asked whether Syt11 regulates neural circuits via a similar mechanism. As the first step, we overexpressed BDNF-GFP in wild-type cortical neurons from a lentivirus under the control of the Synapsin promoter and compared the distribution of the fusion protein and native Syt11 by confocal imaging. These experiments revealed that Syt11 vesicles incorporate recombinant peptides. However, we could not detect abnormalities in activity-dependent release of BDNF in neurons from Syt11 knockout mice by using a previously characterized pH-sensitive reporter, BDNF-pHluorin (Shimojo et al. 2015) as a readout (Supplemental Fig. S5A–D). To explore the possibility that observed colocalization with BDNF reflects the role of Syt11 in intracellular trafficking of ligand-bound TrkB receptors, we induced receptor endocytosis by briefly incubating live neurons at physiological temperature with anti-TrkB antibody and also examined the localization of TrkB in normal and Syt11-deficient neurons. Remarkably, Syt11 vesicles incorporated acutely internalized TrkB, but ablation of Syt11 had no apparent effect on surface receptor levels (Supplemental Fig. S5E,F).
Discussion
Our study demonstrates that Syt11 is essential for survival and important for normal development, synaptic plasticity, and memory formation in mice. Syt11 vesicles are not required for synapse formation, neurotransmitter release, and activity-dependent secretion of peptides. Rather, this pathway is likely involved in endosomal signaling that mediates long-term changes in circuit function in response to specific input.
Immunofluorescent imaging, immunoelectron microscopy, and biochemical experiments presented here consistently demonstrate that native Syt11 is absent from presynaptic neurotransmitter vesicles. Likewise, overexpressed fluorescent Syt11 fusion proteins are targeted to vesicular organelles whose properties are clearly different from SVs. These organelles are broadly distributed in neuronal somas and dendritic processes, traffic in axonal and dendritic shafts in a bidirectional manner, and, evidently, do not undergo synchronized exocytosis during synaptic excitation. Our results are at odds with a prior report suggesting that Syt11 is enriched at synapses and localized to synaptic plasma membranes and SVs (Yeo et al. 2012). This discrepancy may be attributable to the limited specificity of the commercial antibody used by Yeo et al. (2012), since our conclusions are supported by analyses of native and recombinant proteins with multiple validated readouts. Although the properties of Syt11 vesicles are reminiscent of vesicles that recruit Syt4 (Dean et al. 2012), the consequences of genetic ablation of these two Syt isoforms on neural circuit function are different, as discussed below.
In agreement with our assessment of subcellular localization of native and exogenously expressed Syt11, we found that loss of Syt11 does not lead to measurable abnormalities in synaptic transmission and short-term plasticity of neurons in primary cultures and acute brain slices. We also did not detect changes in the activity-dependent exocytosis of BDNF in Syt11-deficient neurons which, together with results of live imaging of pHluorin-Syt11, makes it unlikely that Syt11 is necessary for regulated secretion of fast neurotransmitters and peptides (note that fluorescent reporters of peptide release are not as sensitive as electrophysiology). The presence of Syt11 on the cell surface is consistent with the notion that Syt11 may be involved in endocytosis (Wang et al. 2016, 2018). Nonetheless, our measurements of synaptic responses during prolonged high-frequency stimulus trains indicate that ablation of Syt11 does not preclude endocytosis of SVs.
Despite a high degree of sequence similarity between vertebrate Syt4 and Syt11, these homologous genes may be functionally distinct or, alternatively, Syt4 may have a role that is similar to Syt11 but only activated in more restricted circumstances. Constitutive Syt4 knockout mice have normal life spans and do not exhibit major abnormalities in development or behavior, whereas Syt11 knockouts die at birth. Syt4 knockout mice have been reported to have enhanced LTP (Dean et al. 2009), a phenotype that is opposite to the phenotype of Syt11 cKO mice, which exhibit impaired LTP. Note, however, that given the often finicky nature of LTP induction in acute slices, LTP induction phenotypes are difficult to assess whether they do not constitute a complete loss of LTP. Moreover, it is essential to note that cKO mice characterized in the present study were excitatory neuron-specific, and the role of Syt11 in inhibitory circuits remains entirely unknown. It is also noteworthy that, in contrast to Syt11, transcription of Syt4 in the mammalian brain is regulated by neuronal activity (Vician et al. 1995).
It is reasonable to hypothesize that impairments of spatial learning and memory in Syt11 cKO mice are attributed to disruption of experience-dependent plasticity. Indeed, these animals performed normally in other behavioral trials, and we found no evidence that loss of Syt11 affects brain anatomy and basal neurotransmission. However, the potential role of Syt11 in neural circuit assembly cannot be ruled out at this point. Future studies will also need to define the molecular architecture of Syt11 vesicles, as well as the precise role of Syt11 in membrane trafficking. One could speculate that LTP deficits of Syt11 cKO mice reflect abnormal endosomal signaling of activated ligand-bound TrkB receptors. Our proteomic analyses also raise the possibility that Syt11 regulates the transport of transcripts for constitutive or activity-dependent local protein synthesis.
Materials and methods
Mice
The “floxed” Syt11 allele described in this paper has been deposited with Jackson Labs and is freely available. To generate this allele, the targeting vector (Fig. 3A) for the mouse Syt11 gene (chromosome 3; NC_000069.6; ID: 229521) was constructed containing the following elements: (1) A reporter tag composed of two Flag epitopes and a tetracysteine motif (DYKDDDDKCCPGCCDYKDDDDK) was inserted in-frame into second exon (E2), (2) two loxP sites were inserted in intronic regions upstream of and downstream from E2, (3) a neomycin gene cassette (for positive selection after homologous recombination) surrounded by FRT sites (to allow removal of the neomycin gene) was also inserted into the 5′ intron of E2, and (4) a diphtheria toxin gene cassette was placed next to the homologous long-arm sequence for negative selection.
The targeting vector was electroporated into embryonic R1 stem cells, positive clones were selected in medium containing G418, clones harboring homologously recombined Syt11 mutant alleles were identified by Southern blotting, and mice were generated from these clones by blastocyst injections. All experiments were performed according to the protocols approved by the Institutional Animal Care and Use Committee.
Antibodies
The following commercial antibodies were used for immunofluorescent imaging and immunoblotting: Cux1 (Santa Cruz Biotechnology, sc13024), Neurofilament (Covance, SMI-32R), βTubulin III (Sigma, T2200), Flag clone M2 (Sigma, F1804), MAP2 clone HM2 (Sigma, M4403), Tbr2 (Millipore, AB2283), Kif1a (BD Bioscience, 612094), and EGFP (AVES, GFP-1010). All other antibodies were custom-made in the Südhof laboratory.
Neuronal cultures
Cortices of P1 pups were dissociated by trypsin digestion; cells were plated on circular glass coverslips coated with poly-D-Lysine (Sigma). Cultures were maintained for 4 d in MEM (Invitrogen) supplemented with FBS, B-27 (Invitrogen), glucose, transferrin, and Ara-C (Sigma) followed by incubation in the serum-free medium.
Virus infection
Recombinant lentiviruses were produced by cotransfection of human embryonic kidney 293T cells with corresponding shuttle vectors and pVSVg and pCMVΔ8.9 plasmids that encode the elements essential for packaging of viral particles. Transfections were performed using the FuGENE reagent (Promega). Secreted viruses were harvested 48 h later and cleared by brief centrifugation. For uniform expression of fluorescent reporters and Cre recombinase, cultures were infected at DIV4-5 with 100 µL of viral supernatants per 1 mL of medium. This protocol was optimized to achieve >95% infection efficiency.
Immunocytochemistry
Neurons attached to the glass coverslips were rinsed once in PBS, fixed on ice in 4% paraformaldehyde (PFA) and 4% sucrose in PBS, and permeabilized for 5 min at room temperature in 0.2% Triton X-100 (Roche). Permeabilized neurons were incubated for 30 min in blocking solution containing 5% BSA (Sigma, fraction V) and 5% normal donkey serum followed by an overnight incubation with primary and corresponding fluorescently labeled secondary antibodies diluted in the same solution. The coverslips were mounted on glass slides with Aqua-Poly/Mount medium (Polysciences, Inc.).
Histology
Adult mice were deeply anesthetized and sacrificed by perfusion of 4% PFA. Brains were removed and further fixed in PFA for 3 h. Ninety-micron coronal sections were cut with a vibratome VT1000 (Leica). For histological analysis of neonates, pups were quickly dissected in ice-cold HBS and brains were fixed in PFA followed by cryoprotection in 30% sucrose. Twenty-micron frozen coronal sections were cut in a cryostat (Leica). Sections were blocked and stained with antibodies essentially as described above. For analysis of dendritic spines, samples were stained with Rapid Golgi stain kit (FD NeuroTechnologies). To examine cortical neuron migration, a polycistronic expression vector encoding IRES-EGFP was introduced into a side of the lateral ventricle of E15.5 embryos via in utero electroporation. EGFP signal was imaged in sections from P1 pups after enhancement with anti-EGFP antibody.
Image acquisition and data analysis
Conventional images were collected under the Olympus AX70, Zeiss, and Nikon C2 confocal microscopes using 10× (N.A. 0.45), 20× (N.A. 0.75), and 60× (N.A. 1.40) planapo objectives. Live time-lapse imaging was performed using Nikon inverted microscopes with CCD cameras controlled by the Nikon NIS-Elements or Metamorph software. Frames were sampled at 1 Hz using 10× planapo (N.A. 0.45), 20× planapo (N.A. 0.75), and 60× planapo (N.A. 1.40) objectives. Neurons were bathed in the NRS solution containing 10 mM HEPES-NaOH, 140 mM NaCl, 5 mM KCl, 0.8 mM MgCl2, 2 mM CaCl2, and 10 mM glucose (pH 7.4). Action potentials were elicited with a pair of parallel platinum electrodes connected to model 2100 Isolated Pulse Stimulator (A-M Systems, Inc.). Toxins and inhibitors were diluted in the NRS. Vesicle deacidification was induced by application of buffer containing 10 mM HEPES-NaOH, 50 mM NH4Cl, 90 mM NaCl, 5 mM KCl, 0.8 mM MgCl2, 2 mM CaCl2, and 10 mM glucose (pH 7.4). Digital images were analyzed with the Metamorph and Nikon Elements software packages. Detailed protocols for time-lapse imaging, including imaging of BDNF-pHluorin, can be found in Shimojo et al. (2015).
Electron microscopy
Brains were perfusion-fixed in 4% PFA and 0.1% glutaraldehyde in PBS (pH 7.4), followed by overnight immersion fixation in 2% PFA and 0.1% glutaraldehyde. One-hundred-twenty-micron sections were permeabilized in 0.2% Triton X-100, blocked in 4% normal goat serum, and incubated overnight with primary antibody. The sections were then incubated with the secondary antibody conjugated with 1.4-nm gold particles (1:100 dilution; Nanoprobes) for 24 h, and immunogold signal was enhanced with the an HQ silver enhancement kit (Nanoprobes). Sections were further fixed with 0.5% osmium tetroxide, dehydrated through a graded series of ethanol, and embedded in Poly/Bed 812 epoxy resin (Polysciences, Inc.). Ultrathin sections (65 nm) were stained with 5% uranyl acetate solution and examined under a transmission electron microscope (FEI Tecnai; FEI) at 120 kV accelerating voltage.
Biochemistry
For immunoprecipitations, brains were homogenized in 20 mM HEPES-NaOH, 320 mM sucrose, 5 µg/mL leupeptin, 2 µg/mL aprotinin, and 1 µg/mL pepstatin (pH 7.4). Membrane fractions were isolated by two sequential steps of centrifugation at 1500 and 100,000g. Membranes were then solubilized for 1 h in the buffer containing 10 mM Tris, 150 mM NaCl, and 1% Triton X-100 (pH 7.4). Protein extracts were cleared by centrifugation at 100,000g, split into two samples, and supplemented with either 2 mM CaCl2 or 2 mM EDTA. Anti-Flag M2 antibodies were immobilized onto protein G sepharose beads (Pierce) by cross-linking with dimethyl pimelimidate-dihydrochloride (Pierce). These beads were incubated with protein extracts overnight at 4°C. Beads were then washed three times in the extraction buffer. Attached proteins were eluted with 0.1 M glycine-HCl (pH 2.0) with 0.5% IGEPAL CA630 (Sigma) and detected by immunoblotting or LC/MS. Subcellular fractionations were performed with detergent-free brain samples homogenized in 20 mM HEPES-NaOH and 320 mM sucrose (pH 7.4). One-milliliter samples were loaded on top of continuous sucrose density gradients in 15-mL tubes. After centrifugation at 100,000g, 0.5-mL fractions were sequentially collected from bottoms of tubes and analyzed by immunoblotting.
Multidimensional chromatography and mass spectrometry
Eluted proteins were precipitated and digested with 3 μg of sequencing-grade trypsin according to Pankow et al. (2016). Resulting peptides were loaded onto a triphasic MudPIT column and separated into multiple dimensions with a modified six-step gradient containing 0%, 20%, 40%, 60%, and 100% of buffer C (500 mM ammonium acetate, 5% acetonitrile, 0.1% formic acid) and analyzed by nanoelectrospray ionization on an LTQ-Orbitrap XL by placing the triphasic MudPIT column in line with an Agilent 1200 quaternary HPLC pump. Each full scan mass spectrum (400–2000 m/z) was followed by six data-dependent MS/MS scans at 35% normalized collisional energy and an ion count threshold of 1000. A dynamic exclusion list of 500, repeat time 60 sec, and asymmetric exclusion window of −0.51 and +1.5 Da were applied. MS/MS spectra were searched with ProLuCID against the human International Protein Index database version 3.23 using a target-decoy approach, with search parameters set to no enzyme specificity, 50 ppm precursor mass tolerance, and carboxyamidomethylation (m = 57.021464 Da) as a static modification. Search results were filtered with DTASelect version 2.1, allowing for fully tryptic peptides only, mass tolerance of <5 ppm, and a peptide false discovery rate of <0.5%, corresponding to a protein false discovery rate of <1.0%.
Electrophysiological recordings from neurons in primary cultures
Evoked synaptic release was triggered by 1-msec current injections with a local extracellular stimulating electrode (FHC, Inc.). EPSCs and IPSCs were monitored in whole-cell mode from randomly selected nearby neurons using a Multiclamp 700B amplifier (Molecular Devices). The frequency, duration, and magnitude of extracellular stimuli were controlled with Model 2100 Isolated Pulse Stimulator (A-M Systems, Inc.). The whole-cell pipette solution contained 135 mM CsCl2, 10 mM HEPES-NaOH, 1 mM EGTA, 1 mM Na-ATP, 0.4 mM Na-GTP, and 1 mM QX-314 (pH 7.4). The resistance of filled pipettes varied between 3 and 5 mOhm. The bath solution contained 140 mM NaCl, 5 mM KCl, 2 mM CaCl2, 0.8 mM MgCl2, 10 mM HEPES-NaOH, and 10 mM glucose (pH 7.4). EPSCs and IPSCs were separated pharmacologically by addition of 100 µM picrotoxin or 50 µM APV and 10 µM CNQX, respectively, to the bath solution. Spontaneous EPSCs were monitored under the same conditions but in the presence of 1 µM Tetrodotoxin. The currents were sampled at 10 kHz and analyzed offline using pClamp10 (Molecular Devices) and Origin8 (Origin Lab) software.
Slice physiology
All experiments were performed “blind” to genotype. Mice were anesthetized with isoflurane, and brains were removed and incubated in ice-cold buffer comprised of 125 mM NaCl, 2.5 mM KCl, 7 MgCl2, 1.25 mM NaH2PO4, 25 mM NaHCO3, 10 mM glucose, 75 mM sucrose, 1.3 mM ascorbic acid, and 0.5 mM CaCl2, saturated with 95% O2/5% CO2 (pH 7.4; 310–320 mOsm/L). Transverse 350-µM hippocampal slices were cut in chilled sucrose solution with a Leica VT1000S vibratome (Leica Instruments), transferred to a holding chamber, and incubated at room temperature for at least 1 h in carboxygenated (95% O2/5% CO2) artificial cerebrospinal fluid (aCSF) containing 124 mM NaCl, 0.18 mM KCl, 1 mM MgCl2, 1.25 mM NaH2PO4, 26 mM NaHCO3, 10 mM glucose, and 2 mM CaCl2. Next, a slice was transferred to the recording chamber fixed to the stage of a Zeiss Axioskop 2 (Carl Zeiss, Inc.) microscope fitted with a 10× (0.25 NA) objective and a 40× (0.8 NA) water immersion objective lens. Field potentials were recorded with glass electrodes that were filled with aCSF and placed in the stratum radiatum of area CA1. Synaptic responses were triggered with a concentric bipolar electrode (FHC, catalog no. CBAEC75) placed in the Schaffer-collateral region. The recording chamber was continuously perfused (2–3 mL/min) with carboxygenated aCSF at room temperature, and test pulses were elicited at 0.33 Hz for a period of >30 min to establish a stable baseline. To induce LTP, the θ-burst stimulation (TBS) was given at the test pulse intensity and consisted of 10 bursts (five pulses at 100 Hz) delivered at an interburst interval of 150 msec, and repeated four times at 15 sec. Signals were recorded with a Multiclamp 700B amplifier and analyzed offline in pClamp10 (Molecular Devices) and Origin8 (Origin Lab).
Behavior
For analysis of suckling behavior, newborn pups were separated from nests and starved on a heating pad for 3 h. A lactating female was deeply anesthetized, and the fur was shaved to expose nipples. Pups were placed near the female's nipple to measure response time (latency) to start spontaneous suckling. The latency was counted as maximin (120 sec) if pups completely failed to suckle within this time interval. Other behavioral studies were performed at the TSRI core facility, as we described previously (Sando et al. 2012).
Statistical analyses
Means and standard deviations/errors were calculated using Prism (Graphpad, Inc.) and Origin8 (Origin Lab) software packages. Statistical significances were determined using Student's t-test.
Supplementary Material
Acknowledgments
We thank Dr. Ulrich Mueller for sharing Cre driver mice and antibodies, and Dr. Yuji Tsunekawa for help with analysis of cortical neuron migration. These studies were supported by the National Institutes of Health R01 grants MH085776, NS087026, and GM117049 (to A.M.) and the Japanese Society for Promotion of Science (to M.S.).
Author contributions: A.M. and T.C.S. conceived the study. A.M. generated Syt11 knick-in and knockout mice. M.S. performed imaging, biochemical, and behavioral studies. J.M. carried out electrophysiological recordings from brain slices. S.P. and J.Y. identified Syt11-binding partners by mass spectrometry. X.L. performed EM. A.M. and T.C.S. wrote the manuscript.
Footnotes
Supplemental material is available for this article.
Article published online ahead of print. Article and publication date are online at http://www.genesdev.org/cgi/doi/10.1101/gad.320077.118.
References
- Bacaj T, Wu D, Yang X, Morishita W, Zhou P, Xu W, Malenka RC, Südhof TC. 2013. Synaptotagmin-1 and synaptotagmin-7 trigger synchronous and asynchronous phases of neurotransmitter release. Neuron 80: 947–959. 10.1016/j.neuron.2013.10.026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barco A, Patterson SL, Alarcon JM, Gromova P, Mata-Roig M, Morozov A, Kandel ER. 2005. Gene expression profiling of facilitated L-LTP in VP16-CREB mice reveals that BDNF is critical for the maintenance of LTP and its synaptic capture. Neuron 48: 123–137. 10.1016/j.neuron.2005.09.005 [DOI] [PubMed] [Google Scholar]
- Cao P, Maximov A, Sudhof TC. 2011. Activity-dependent IGF-1 exocytosis is controlled by the Ca2+-sensor synaptotagmin-10. Cell 145: 300–311. 10.1016/j.cell.2011.03.034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai H, Shin OH, Machius M, Tomchick DR, Südhof TC, Rizo J. 2004. Structural basis for the evolutionary inactivation of Ca2+ binding to synaptotagmin 4. Nat Struct Mol Biol 11: 844–849. 10.1038/nsmb817 [DOI] [PubMed] [Google Scholar]
- Davletov BA, Sudhof TC. 1993. A single C2 domain from synaptotagmin I is sufficient for high affinity Ca2+/phospholipid binding. J Biol Chem 268: 26386–26390. [PubMed] [Google Scholar]
- Dean C, Liu H, Dunning FM, Chang PY, Jackson MB, Chapman ER. 2009. Synaptotagmin-IV modulates synaptic function and long-term potentiation by regulating BDNF release. Nat Neurosci 12: 767–776. 10.1038/nn.2315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dean C, Liu H, Staudt T, Stahlberg MA, Vingill S, Buckers J, Kamin D, Engelhardt J, Jackson MB, Hell SW, et al. 2012. Distinct subsets of Syt-IV/BDNF vesicles are sorted to axons versus dendrites and recruited to synapses by activity. J Neurosci 32: 5398–5413. 10.1523/JNEUROSCI.4515-11.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du C, Wang Y, Zhang F, Yan S, Guan Y, Gong X, Zhang T, Cui X, Wang X, Zhang CX. 2017. Synaptotagmin-11 inhibits cytokine secretion and phagocytosis in microglia. Glia 65: 1656–1667. 10.1002/glia.23186 [DOI] [PubMed] [Google Scholar]
- Edelmann E, Lessmann V, Brigadski T. 2014. Pre- and postsynaptic twists in BDNF secretion and action in synaptic plasticity. Neuropharmacology 76 Pt C: 610–627. 10.1016/j.neuropharm.2013.05.043 [DOI] [PubMed] [Google Scholar]
- Ferguson GD, Wang H, Herschman HR, Storm DR. 2004. Altered hippocampal short-term plasticity and associative memory in synaptotagmin IV (-/-) mice. Hippocampus 14: 964–974. 10.1002/hipo.20013 [DOI] [PubMed] [Google Scholar]
- Fernandez-Chacon R, Konigstorfer A, Gerber SH, Garcia J, Matos MF, Stevens CF, Brose N, Rizo J, Rosenmund C, Sudhof TC. 2001. Synaptotagmin I functions as a calcium regulator of release probability. Nature 410: 41–49. 10.1038/35065004 [DOI] [PubMed] [Google Scholar]
- Geppert M, Goda Y, Hammer RE, Li C, Rosahl TW, Stevens CF, Sudhof TC. 1994. Synaptotagmin I: a major Ca2+ sensor for transmitter release at a central synapse. Cell 79: 717–727. 10.1016/0092-8674(94)90556-8 [DOI] [PubMed] [Google Scholar]
- Goebbels S, Bormuth I, Bode U, Hermanson O, Schwab MH, Nave KA. 2006. Genetic targeting of principal neurons in neocortex and hippocampus of NEX-Cre mice. Genesis 44: 611–621. 10.1002/dvg.20256 [DOI] [PubMed] [Google Scholar]
- Gonzalez A, Rodriguez-Boulan E. 2009. Clathrin and AP1B: key roles in basolateral trafficking through trans-endosomal routes. FEBS Lett 583: 3784–3795. 10.1016/j.febslet.2009.10.050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gustavsson N, Han W. 2009. Calcium-sensing beyond neurotransmitters: functions of synaptotagmins in neuroendocrine and endocrine secretion. Biosci Rep 29: 245–259. 10.1042/BSR20090031 [DOI] [PubMed] [Google Scholar]
- Gustavsson N, Wei SH, Hoang DN, Lao Y, Zhang Q, Radda GK, Rorsman P, Sudhof TC, Han W. 2009. Synaptotagmin-7 is a principal Ca2+ sensor for Ca2+-induced glucagon exocytosis in pancreas. J Physiol 587: 1169–1178. 10.1113/jphysiol.2008.168005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirokawa N, Nitta R, Okada Y. 2009. The mechanisms of kinesin motor motility: lessons from the monomeric motor KIF1A. Nat Rev Mol Cell Biol 10: 877–884. 10.1038/nrm2807 [DOI] [PubMed] [Google Scholar]
- Huynh DP, Scoles DR, Nguyen D, Pulst SM. 2003. The autosomal recessive juvenile Parkinson disease gene product, parkin, interacts with and ubiquitinates synaptotagmin XI. Hum Mol Genet 12: 2587–2597. 10.1093/hmg/ddg269 [DOI] [PubMed] [Google Scholar]
- Inoue S, Imamura A, Okazaki Y, Yokota H, Arai M, Hayashi N, Furukawa A, Itokawa M, Oishi M. 2007. Synaptotagmin XI as a candidate gene for susceptibility to schizophrenia. Am J Med Genet B Neuropsychiatr Genet 144B: 332–340. 10.1002/ajmg.b.30465 [DOI] [PubMed] [Google Scholar]
- Jahn R, Lang T, Sudhof TC. 2003. Membrane fusion. Cell 112: 519–533. 10.1016/S0092-8674(03)00112-0 [DOI] [PubMed] [Google Scholar]
- Maximov A, Shin OH, Liu X, Sudhof TC. 2007. Synaptotagmin-12, a synaptic vesicle phosphoprotein that modulates spontaneous neurotransmitter release. J Cell Biol 176: 113–124. 10.1083/jcb.200607021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miesenbock G, De Angelis DA, Rothman JE. 1998. Visualizing secretion and synaptic transmission with pH-sensitive green fluorescent proteins. Nature 394: 192–195. 10.1038/28190 [DOI] [PubMed] [Google Scholar]
- Pang ZP, Melicoff E, Padgett D, Liu Y, Teich AF, Dickey BF, Lin W, Adachi R, Sudhof TC. 2006a. Synaptotagmin-2 is essential for survival and contributes to Ca2+ triggering of neurotransmitter release in central and neuromuscular synapses. J Neurosci 26: 13493–13504. 10.1523/JNEUROSCI.3519-06.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pang ZP, Sun J, Rizo J, Maximov A, Sudhof TC. 2006b. Genetic analysis of synaptotagmin 2 in spontaneous and Ca2+-triggered neurotransmitter release. EMBO J 25: 2039–2050. 10.1038/sj.emboj.7601103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pankow S, Bamberger C, Calzolari D, Martínez-Bartolomé S, Lavallée-Adam M, Balch WE, Yates JR. 2015. ΔF508 CFTR interactome remodeling promotes rescue of cystic fibrosis. Nature 528: 510–516. 10.1038/nature15729 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pankow S, Bamberger C, Calzolari D, Bamberger A, Yates JR. 2016. Deep interactome profiling of membrane proteins by co-interacting protein identification technology (CoPIT). Nature Protoc 11: 2515–2528. 10.1038/nprot.2016.14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park H, Popescu A, Poo MM. 2014. Essential role of presynaptic NMDA receptors in activity-dependent BDNF secretion and corticostriatal LTP. Neuron 84: 1009–1022. 10.1016/j.neuron.2014.10.045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patterson SL, Abel T, Deuel TA, Martin KC, Rose JC, Kandel ER. 1996. Recombinant BDNF rescues deficits in basal synaptic transmission and hippocampal LTP in BDNF knockout mice. Neuron 16: 1137–1145. 10.1016/S0896-6273(00)80140-3 [DOI] [PubMed] [Google Scholar]
- Peng X, Parsons TD, Balice-Gordon RJ. 2012. Determinants of synaptic strength vary across an axon arbor. J Neurophysiol 107: 2430–2441. 10.1152/jn.00615.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perin MS, Brose N, Jahn R, Sudhof TC. 1991a. Domain structure of synaptotagmin (p65). J Biol Chem 266: 623–629. [PubMed] [Google Scholar]
- Perin MS, Johnston PA, Ozcelik T, Jahn R, Francke U, Sudhof TC. 1991b. Structural and functional conservation of synaptotagmin (p65) in Drosophila and humans. J Biol Chem 266: 615–622. [PubMed] [Google Scholar]
- Sando R III, Gounko N, Pieraut S, Liao L, Yates J III, Maximov A. 2012. HDAC4 governs a transcriptional program essential for synaptic plasticity and memory. Cell 151: 821–834. 10.1016/j.cell.2012.09.037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sesar A, Cacheiro P, López-López M, Camiña-Tato M, Quintáns B, Monroy-Jaramillo N, Alonso-Vilatela ME, Cebrián E, Yescas-Gómez P, Ares B, et al. 2016. Synaptotagmin XI in Parkinson's disease: new evidence from an association study in Spain and Mexico. J Neurol Sci 362: 321–325. 10.1016/j.jns.2016.02.014 [DOI] [PubMed] [Google Scholar]
- Shimojo M, Courchet J, Pieraut S, Torabi-Rander N, Sando R III, Polleux F, Maximov A. 2015. SNAREs controlling vesicular release of BDNF and development of callosal axons. Cell Rep 11: 1054–1066. 10.1016/j.celrep.2015.04.032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sudhof TC. 2002. Synaptotagmins: why so many? J Biol Chem 277: 7629–7632. 10.1074/jbc.R100052200 [DOI] [PubMed] [Google Scholar]
- Sugita S, Südhof TC. 2000. Specificity of Ca2+-dependent protein interactions mediated by the C2A domains of synaptotagmins. Biochemistry 39: 2940–2949. 10.1021/bi9920984 [DOI] [PubMed] [Google Scholar]
- Sugita S, Shin OH, Han W, Lao Y, Südhof TC. 2002. Synaptotagmins form a hierarchy of exocytotic Ca2+ sensors with distinct Ca2+ affinities. EMBO J 21: 270–280. 10.1093/emboj/21.3.270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun J, Pang ZP, Qin D, Fahim AT, Adachi R, Südhof TC. 2007. A dual-Ca2+-sensor model for neurotransmitter release in a central synapse. Nature 450: 676–682. 10.1038/nature06308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang J, Maximov A, Shin OH, Dai H, Rizo J, Sudhof TC. 2006. A complexin/synaptotagmin 1 switch controls fast synaptic vesicle exocytosis. Cell 126: 1175–1187. 10.1016/j.cell.2006.08.030 [DOI] [PubMed] [Google Scholar]
- Turrigiano GG, Leslie KR, Desai NS, Rutherford LC, Nelson SB. 1998. Activity-dependent scaling of quantal amplitude in neocortical neurons. Nature 391: 892–896. 10.1038/36103 [DOI] [PubMed] [Google Scholar]
- Vician L, Lim IK, Ferguson G, Tocco G, Baudry M, Herschman HR. 1995. Synaptotagmin IV is an immediate early gene induced by depolarization in PC12 cells and in brain. Proc Natl Acad Sci 92: 2164–2168. 10.1073/pnas.92.6.2164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Poser C, Ichtchenko K, Shao X, Rizo J, Südhof TC. 1997. The evolutionary pressure to inactivate. A subclass of synaptotagmins with an amino acid substitution that abolishes Ca2+ binding. J Biol Chem 272: 14314–14319. 10.1074/jbc.272.22.14314 [DOI] [PubMed] [Google Scholar]
- Wang C, Wang Y, Hu M, Chai Z, Wu Q, Huang R, Han W, Zhang CX, Zhou Z. 2016. Synaptotagmin-11 inhibits clathrin-mediated and bulk endocytosis. EMBO Rep 1717: 47–63. 10.15252/embr.201540689 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang C, Kang X, Zhou L, Chai Z, Wu Q, Huang R, Xu H, Hu M, Sun X, Sun S, et al. 2018. Synaptotagmin-11 is a critical mediator of parkin-linked neurotoxicity and Parkinson's disease-like pathology. Nat Commun 9: 81 10.1038/s41467-017-02593-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu J, Mashimo T, Südhof TC. 2007. Synaptotagmin-1, -2, and -9: Ca2+ sensors for fast release that specify distinct presynaptic properties in subsets of neurons. Neuron 54: 567–581. 10.1016/j.neuron.2007.05.004 [DOI] [PubMed] [Google Scholar]
- Yeo H, Kim HW, Mo J, Lee D, Han S, Hong S, Koh MJ, Sun W, Choi S, Rhyu IJ, et al. 2012. Developmental expression and subcellular distribution of synaptotagmin 11 in rat hippocampus. Neuroscience 225: 35–43. 10.1016/j.neuroscience.2012.08.062 [DOI] [PubMed] [Google Scholar]
- Yoshihara M, Adolfsen B, Galle KT, Littleton JT. 2005. Retrograde signaling by Syt 4 induces presynaptic release and synapse-specific growth. Science 310: 858–863. 10.1126/science.1117541 [DOI] [PubMed] [Google Scholar]
- Zhu Q, Yamakuchi M, Ture S, de la Luz Garcia-Hernandez M, Ko KA, Modjeski KL, LoMonaco MB, Johnson AD, O'Donnell CJ, Takai Y, et al. 2014. Syntaxin-binding protein STXBP5 inhibits endothelial exocytosis and promotes platelet secretion. J Clin Invest 124: 4503–4516. 10.1172/JCI71245 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.