

SUMMARY
Ribosomes undergo substantial conformational changes during translation elongation to accommodate incoming aminoacyl-tRNAs and translocate along the mRNA template. We used multiple elongation inhibitors and chemical probing to define ribosome conformational states corresponding to different sized ribosome-protected mRNA fragments (RPFs) generated by ribosome profiling. We show using various genetic and environmental perturbations that short 20–22 or classical 27–29 nucleotide RPFs correspond to ribosomes with open or occupied ribosomal A sites, respectively. These distinct states of translation elongation are readily discerned by ribosome profiling in all eukaryotes tested including fungi, worms and mammals. This high-resolution ribosome profiling approach reveals mechanisms of translation elongation arrest during distinct stress conditions. Hyperosmotic stress inhibits translocation through Rck2-dependent eEF2 phosphorylation whereas oxidative stress traps ribosomes in a pre-translocation state, independent of Rck2-driven eEF2 phosphorylation. These results provide insights and approaches for defining the molecular events that impact translation elongation throughout biology.
Keywords: translation elongation, ribosome profiling, ribosome functional states, eEF2 phosphorylation
eTOC Blurb
Cellular stress triggers the regulation of protein synthesis at the level of initiation and elongation. Wu et al. developed high-resolution ribosome profiling methodology to resolve ribosome functional states during translation elongation and revealed distinct mechanisms of elongation arrest in response to various stress conditions.
Graphical Abstract
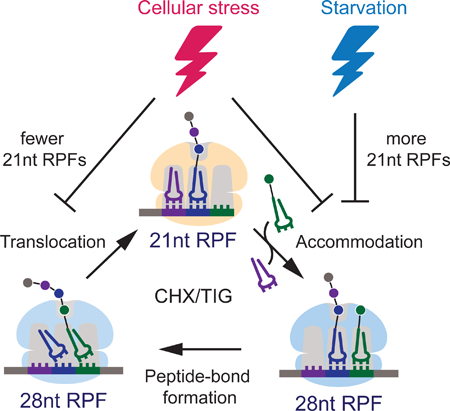
INTRODUCTION
Translation elongation is a highly coordinated process involving iterative cycles of three major reactions: aminoacyl-tRNA (aa-tRNA) decoding, peptide bond formation, and translocation; these steps are catalyzed by eEF1A, the large ribosomal subunit (60S), and eEF2, respectively (Figure 1A) (Dever et al., 2018). Ribosomes undergo substantial conformational rearrangements during elongation, alternating between two major states corresponding to post- and pre-translocation, or non-rotated and rotated (initially called the classical and hybrid tRNA states) (Agirrezabala et al., 2008; Behrmann et al., 2015; Moazed and Noller, 1989). A recent study in Saccharomyces cerevisiae revealed the potential for ribosome profiling to decipher these conformational states, showing that elongating ribosomes generate two distinct ribosome-protected footprint (RPF) sizes, 21 nts and 28 nts in length (Lareau et al., 2014). In this study, pretreatment of cells with different elongation inhibitors changed the distribution of footprint sizes: cycloheximide favors 28 nt RPFs while anisomycin favors 21 nt RPFs. It remains unclear, however, which ribosome conformations correspond to these two populations of footprints, as they were not systematically assigned in the previous study.
Figure 1. Assigning ribosome functional states to distinct footprint sizes (21 and 28 nt RPFs) from ribosome profiling samples.

(A) Schematic representation of the eukaryotic elongation cycle. PreAcc: pre-accommodation; PrePT: Pre-peptide bond formation; PreTrans: Pre-translocation. (B and C) Scatter plots showing mutation rates of 25S rRNA (a function of DMS reactivity) comparing CHX-pretreated (B) or ANS-pretreated (C) relative to mock pretreatment. Nucleotides protected by CHX or ANS are color-coded and labeled. (D) Fold change in mutation rates of nucleotides A1755 and A1756 for CHX-pretreatment with in vivo DMS modification (CHX in vivo), ANS-pretreatment with in vivo DMS modification (ANS in vivo), ANS-pretreatment with DMS modification in lysate (ANS lysate), and TIG with DMS modification in lysate (TIG lysate) (n=2, ±SD). (E) Average ribosome occupancies aligned at stop codons using 21 nt RPFs (top), 28 nt RPFs (middle) and both RPFs (bottom) for WT and eRF1-depleted (eRF1d) cells. (F) Scatter plot comparing 64 codon-specific ribosome occupancies (pause scores) for 21 nt RPFs in WT and eRF1d cells.
Cellular stresses are well known to trigger the regulation of both translational initiation and elongation through diverse pathways (Dever et al., 2018; Proud, 2018; Wek, 2018). Phosphorylation of eEF2, a highly conserved GTPase that mediates translocation during elongation, has been shown to play an important role in regulating translation elongation (Dever et al., 2018; Proud, 2018), leading to impairment of poly(U)-directed poly-phenylalanine synthesis in vitro and reduction of protein synthesis in vivo (Ryazanov et al., 1988; Teige et al., 2001). In mammals, eEF2 is phosphorylated at Thr56 by eukaryotic elongation factor 2 kinase (eEF2K); this phosphorylation is critical for cell survival under conditions of nutrient starvation (Leprivier et al., 2013; Proud, 2018). Under hyperosmotic stress, eEF2 in budding yeast is phosphorylated by Rck2 (Melcher and Thorner, 1996; Teige et al., 2001), a Ser/Thr kinase that is critical for fitness in response to this stress (Warringer et al., 2010). Rck2 is a downstream effector of the well-characterized hyperosmotic stress response pathway in budding yeast involving the highosmolarity glycerol (HOG) mitogen-activated protein kinase (MAPK) cascade (de Nadal et al., 2011). The components of this pathway are homologous to the mammalian p38 stress-activated protein kinase (SAPK) pathway activated by extracellular stimuli such as UV light, heat shock, growth factors, and inflammatory cytokines (Brewster and Gustin, 2014). While there are clear similarities in these pathways, there is much to be learned about the cellular stresses that activate them and their discrete outputs.
In this study, we systematically assign the states of translation elongation that correspond to 21 and 28 nt RPFs through a combination of DMS probing and ribosome profiling experiments. We provide evidence that 21 nt RPFs correspond to ribosomes with an open A site while 28 nt RPFs correspond to ribosomes with an occupied A site. We develop systematic approaches to effectively trap the in vivo distribution of ribosome functional states (PreAcc, PrePT and PreTrans in Figure 1A) by the use of a cocktail of antibiotics that target distinct steps in elongation. This improved protocol provides the means to isolate 21 nt RPFs that correspond to ribosomes in a pre-accommodation state and exhibit the strongest observed correlation with tRNA abundance to date. We further show that the same distinct functional states of the ribosome are revealed by ribosome profiling in fungi, worms and mammals. Finally, with our high-resolution ribosome profiling approach, we reveal specific features of translation elongation regulation under hyperosmotic (inhibition of translocation by eEF2) and oxidative (inhibition of translocation by eEF2 and decoding by proline tRNAs) stress conditions in S. cerevisiae.
RESULTS
DMS probing identifies distinct ribosome functional states stabilized by elongation inhibitors
To probe the distinct states of the ribosome trapped by cycloheximide (CHX) and anisomycin (ANS), we used dimethyl sulfate (DMS) to examine differences in the accessibility of rRNA nucleotides as a proxy for determining functional/conformational states. We pretreated S. cerevisiae cultures with CHX or ANS at 0.1 mg/mL and then added DMS to either living cells (in vivo) or to lysates prepared to mimic the sample-handling procedures of ribosome profiling (lysate). From these samples, we sequenced libraries derived from total RNA and analyzed the results using a high throughput DMS-MaPseq procedure (Zubradt et al., 2017). Increased mutation rates indicate sites of DMS modification in rRNA (i.e. accessibility) because reverse transcriptase misreads A or C bases that are modified by DMS (McClary et al., 2017; Zubradt et al., 2017). The effectiveness of this strategy is illustrated by the observation that CHX strongly protects C2764 in 25S rRNA (Figure 1B) from DMS modification, as determined by the lowered mutation rate at this position; likewise, ANS protects A2401 and A2821 (Figures 1C and S1A, in vivo and in lysate, respectively). These protections are in strong agreement with previously defined binding sites for these antibiotics on the ribosome (Garreau de Loubresse et al., 2014; Schneider-Poetsch et al., 2010).
The binding of tRNA in the A site of the small subunit protects nucleotides A1755 and A1756 in the 18S rRNA (A1492 and A1493 in E. coli) (Moazed and Noller, 1986), allowing us to identify how antibiotics change the proportion of ribosomes in the pre-accommodation (PreAcc) state (Figure 1A). A1755/56 are protected in the CHX-pretreated samples (Figure 1D), consistent with the presence of a tRNA substrate in the 40S A site. These data suggest that 28 nt RPFs isolated in ribosome profiling experiments with CHX correspond to ribosomes with an occupied 40S A site, consistent with the fact that CHX blocks translocation and traps peptidyl-tRNA in the 40S A site (Budkevich et al., 2011). In addition, pretreatment of cells with CHX or addition of CHX in lysate both yield almost exclusively 28 nt RPFs in ribosome profiling experiments (Figure S1B), consistent with the idea that the A sites are occupied in both conditions regardless of when CHX is added to block elongation. The ANS-pretreated samples show a similar protection of A1755/56 from DMS modification in vivo but these nucleotides become more accessible (i.e. show increased reactivity) when probed in lysate (Figure 1D). Our data support a model in which ribosomal A sites are mostly occupied by tRNAs upon ANS pretreatment in the cell, but that unreacted tRNAs dissociate from the A site during sample handling, since ANS binds in the peptidyl-transferase center (PTC) and blocks peptide bond formation (Barbacid and Vazquez, 1974; Garreau de Loubresse et al., 2014). These data suggest that 21 nt RPFs isolated in ribosome profiling experiments correspond to ribosomes with an unoccupied 40S A site. This hypothesis was tested further by DMS probing of ribosomes in lysates treated with tigecycline (TIG), a tetracycline-like antibiotic known to block tRNA accommodation (Jenner et al., 2013). As anticipated, TIG enhances the reactivity of A1755/56, consistent with decreased tRNA binding and a more accessible A site (Figure 1D). Taken together, these structural probing experiments with CHX, ANS, and TIG show that 21 nt RFPs correspond to ribosomes with open 40S A sites whereas 28 nt RFPs correspond to those with occupied 40S A sites (with tRNAs bound).
21 nt RPFs are enriched at open A sites in vivo
To further evaluate our findings from these structural probing studies, we performed ribosome profiling to look for enrichment of 21 nt RPFs at positions in mRNAs where we expect the 40S A site to be open. To do this, we prepared ribosome profiling libraries with CHX in the lysate (McGlincy and Ingolia, 2017) from S. cerevisiae in which one of the core termination factors, eRF1, was depleted (Figure S1C). eRF1 normally binds in the A site where it recognizes stop codons and releases the nascent peptide. If our model is correct, then we should see an increase in 21 nt RPFs specifically at stop codons in the absence of eRF1. To obtain a genome-wide view of translation termination, we aligned genes at their stop codons and calculated the average ribosome occupancy using either exclusively 28 nt RPFs, exclusively 21 nt RPFs, or both RPFs combined (Figure 1E). As predicted, we observe a dramatic accumulation of 21 nt RPFs specifically at stop codons in eRF1depleted cells (Figures 1E, top trace and S1D). In contrast, there is no enrichment at stop codons in the 28 nt RPFs when compared to WT cells (Figure 1E, middle trace), although a queue of stacked ribosomes is seen upstream of the stop codon. When the 21 and 28 nt RPFs are combined (Figure 1E, bottom trace), we see that the average ribosome occupancy at stop codons exhibits the magnitude and phasing of ribosome density that would be expected upon the depletion of a core termination factor. By comparing the ribosome occupancies (i.e. pause scores) at all 64 codons, we show that 21 nt RPFs are specifically enriched at stop codons in eRF1-depleted cells (Figure 1F and replicates in S1E). These data provided us with compelling evidence that 21 nt RPFs are indeed derived from ribosomes with an open A site in vivo.
Codon-specific occupancy of 21 nt RPFs is highly anti-correlated with tRNA abundance
The connection between two distinct RPF sizes and discrete states of the ribosome increases the resolution and utility of ribosome profiling. While ribosome profiling experiments aim to capture the exact state of ribosomes in cells at the time of lysis, it has been difficult to measure the degree to which this goal is met. One prediction is that rare codons that are decoded by low-abundance tRNAs will be translated more slowly and therefore show relatively high ribosome occupancy (i.e. an inverse correlation). Previous studies have shown at best a modest correlation of ribosome occupancy with tRNA abundance (Gardin et al., 2014; Hussmann et al., 2015; Lareau et al., 2014; Pop et al., 2014; Weinberg et al., 2016). Given that 21 nt RPFs represent ribosomes with an open A site, we wondered whether these footprints would show a stronger correlation with tRNA abundance than the 28 nt RPFs. Indeed, using the tRNA adaptation index (tAI), which accounts for tRNA copy number, codon usage, and wobble pairing constraints (dos Reis et al., 2004), we observe a slightly higher correlation between codon-specific occupancy and 1/tAI for 21 nt RPFs than for 28 nt RPFs for libraries prepared either in the absence of any antibiotics or with CHX in the lysate (Figure 2A). However, given that these correlations were modest, we wondered whether our methods for preparing ribosome footprints effectively trap ribosomes as they were in vivo or if ongoing reactions occurring in the lysate might confound this analysis.
Figure 2. 21 nt RPFs correlate with the tRNA abundance metrics.

(A) Spearman rank correlations of A-site codon-specific occupancies with the inverse of tRNA adaptation index (tAI) from ribosome profiling libraries prepared with CHX (our samples or (Matsuo et al., 2017)) or no elongation inhibitor in the lysates (n≥2, ±SD). (Lareau et al., 2014; Tunney et al., 2017) (B) Length distributions of ribosome footprints comparing libraries prepared with CHX only (grey), CHX/TIG (purple), and CHX/ANS (green) in the lysates. (C) Spearman rank correlations of A-site codon-specific occupancies with 1/tAI from samples described in (B) (n≥2, ±SD). p-values for 21 nt RPFs from Student’s t-test after Fisher’s z-transformation is indicated by asterisks. ****, p< 0.0001. (D and E) Scatter plots of codon-specific ribosome occupancies for 21 nt RPFs comparing gamma-toxin treated to untreated cells from libraries prepared with CHX only (D) or CHX/TIG (E) in the lysates. GAA codons are colored in blue and labeled. (F and G) Scatter plots of codon-specific ribosome occupancies for 21 nt RPFs comparing 3-AT treated relative to untreated cells from libraries prepared with CHX only (F) or CHX/TIG (G) in the lysates. Both histidine codons are colored in green and labeled.
Arresting ribosomes at the authentic positions on the mRNA and in the right conformation is essential to capture the in vivo translational landscape. Because the addition of elongation inhibitors to living cells perturbs the biology being studied (Gerashchenko and Gladyshev, 2014; Weinberg et al., 2016), recent profiling studies have typically only added CHX to lysates to block ongoing protein synthesis. The preponderance of 28 nt RPFs typically present in these samples (Figure 2B, light gray trace) suggests the possibility that ribosomes are accumulating in one particular state as a result of movement in the lysate. For example, ribosomes in the process of decoding (PreAcc) or making peptide bonds (pre-peptide bond formation, PrePT) might be converted in the lysate to the pre-translocation state (PreTrans) and be trapped by CHX (Figure 1A). We reasoned that an optimal procedure might use a cocktail of elongation inhibitors in the lysate to trap the multiple states of ribosomes found in an actively dividing population of cells. We prepared ribosome profiling libraries with different combinations of CHX, ANS and TIG, each of which blocks a distinct step in translation elongation. CHX/ANS and CHX/TIG libraries both yield substantially more 21 nt RPFs than CHX alone (Figure 2B), supporting the argument that there indeed is movement between states in the lysate. We observe that CHX/ANS yields relatively more 21 nt RPFs than CHX/TIG. This observation can be explained by the fact that 21 nt RPFs are derived in this latter sample from ribosomes in PreAcc and PrePT (as tRNA is lost from PrePT in the lysate), while CHX/TIG yields 21 nt RPFs that derive exclusively from PreAcc (as ribosomes in PrePT will proceed to PreTrans in lysate).
An understanding of the modes of action of CHX and TIG thus allows us to generate a population of 21 nt RFPs that derive exclusively from one specific functional state (PreAcc) where ribosomes are caught in the process of decoding. As expected, we see that 21 nt RPFs from samples prepared with CHX/TIG show the strongest observed correlation of codon-specific occupancies with 1/tAI. Samples prepared with CHX/ANS show a somewhat weaker correlation (Figure 2C) with 1/tAI in line with the expectation that these 21 nt RPFs are derived from a more diverse population comprised of both PreAcc and PrePT states. Reassuringly, in both CHX/TIG and CHX/ANS treated samples there is a substantially weaker correlation between the 28 nt RPFs and 1/tAI (Figure 2C) since 28 nt RPFs do not represent ribosomes waiting to decode; additionally, these 28 nt RPFs show only modest correlation with 21 nt RPFs (Figure S2A). We instead note the enrichment of 28 nt RPFs on codons whose amino acids are typically slow to undergo peptidyl transfer (e.g. P, G, and D residues) (Figure S2B) (Ingolia et al., 2011; Schuller et al., 2017; Wohlgemuth et al., 2008). These data likely reflect the accumulation of ribosomes at these sites in PrePT in the cell that then proceed to PreTrans in lysate (since no ANS is included). In the experiments presented throughout the remainder of this study, we use CHX/TIG in preparing ribosome profiling libraries in order to monitor the distinct conformations of the ribosome in cells, and in particular the decoding step of translation which is so effectively visualized with this combination of antibiotics.
We next performed several ribosome profiling experiments with specific perturbations that allowed us to further evaluate the protocol using CHX/TIG. In a first experiment, we created a shortage of glutamyl-tRNA by expressing gamma-toxin, a ribonuclease from Kluyveromyces lactis, that specifically cleaves tRNA-GluUUC in the anticodon loop (for the extent of depletion see northern analysis in Figure S2C) (Lu et al., 2005). Samples prepared with CHX showed only a modest enrichment in pausing on GAA codons in the 21 nt RPFs (Figures 2D), whereas samples prepared with CHX/TIG showed a much more substantial enrichment (Figures 2E, S2D to G). These data strengthen the idea that the relatively small 21 nt RPF population in samples treated with CHX alone are more complex and are therefore less useful for an analysis of decoding; 21 nt RPFs in the CHX/TIG treated sample, in contrast, report specifically on ribosomes in the PreAcc state. In a second experiment, we revisited the effects of 3-amino-1,2,4-triazole (3-AT) treatment, known to inhibit histidine biosynthesis (Klopotowski and Wiater, 1965) and to deplete histidyl-tRNA in the cell. Again, we found a strong enrichment of 21 nt RPFs exclusively at histidine codons (CAC and CAU) and the relative pause score was substantially higher in samples prepared with CHX/TIG as compared to those prepared only with CHX (Figures 2F, 2G and S2H to L). Together with the tAI results above, these data show that the combined CHX/TIG protocol dramatically improves our ability to monitor decoding rates using ribosome profiling in yeast.
21/28 nt RPFs also report on functional states in higher eukaryotes
The different functional states sampled by ribosomes during translation elongation are highly conserved from bacteria to budding yeast to higher eukaryotes. To test whether we can use ribosome profiling to observe specific steps in the elongation cycle in other eukaryotes, we prepared libraries from Caenorhabditis elegans embryos and from HeLa and human breast cancer cells (MDA-MB-231). In all cases, we observe a bimodal distribution of footprint length, similar to that observed in budding yeast (Figures 3A and B). We also note that we recently observed an enrichment in 21 nt RPFs in epidermal tissues lacking Pelota (Dom34 in yeast) that we suspect represent ribosomes stalled throughout the transcriptome in these rapidly differentiating cell types (Liakath-Ali et al., 2018). To verify that these two RPF sizes represent those same states that we characterized in S. cerevisiae, we starved HeLa cells for glutamine; as anticipated, we observed an increase in 21 nt RPFs specifically at the two glutamine codons (CAA and CAG) (Figures 3C, 3D and S3A to C). When comparing samples prepared in HeLa cells using CHX or CHX/TIG under glutamine starvation conditions, we found that the inclusion of TIG in these lysates did not further enrich the number of 21 nt RPFs at glutamine codons nor did it change the codon-specific occupancies (Figures S3D and E). These observations suggest that there is less active translation happening in the more dilute lysates of HeLa cells than those of yeast. As such, this antibiotic cocktail may be less critical in preparing samples from mammalian cells. Together, these data indicate that distinct functional states of the ribosome can be resolved in mammalian cells, as in yeast, using ribosome profiling.
Figure 3. Conserved ribosome functional states in metazoan cells visualized by distinct footprint sizes.

(A) Size distribution of ribosome footprints in C. elegans embryos. (B) Size distributions of ribosome footprints in HeLa and MDA-MB-231 cells. (C) Size distributions of ribosome footprints mapped to glutamine codons in untreated and starved cells. (D) Scatter plot of codon-specific ribosome occupancies for 21 nt RPFs comparing starved relative to untreated cells.
Ribosome profiling reveals mechanisms of translation elongation regulation under stress conditions
The ability of ribosome profiling to define the functional state of ribosomes within the cell provides a powerful tool for deciphering cellular responses to a variety of stresses. We prepared ribosome profiling libraries from S. cerevisiae cells subjected to hyperosmotic, oxidative or stationary phase stress; several of these treatments are known to down-regulate translation initiation and/or elongation (see Figure S4A for polysome profiles) (Jimenez-Diaz et al., 2013; Shenton et al., 2006; Teige et al., 2001; Uesono and Toh, 2002). Strikingly, both hyperosmotic and oxidative stress conditions resulted in substantial reductions in 21 nt RPFs relative to 28 nt RPFs when compared to unstressed cells (Figures 4A and replicates in S4B). In contrast, we saw no changes in the ratio of 21 nt RPFs relative to 28 nt RPFs in cells grown to stationary phase (Figures 4A and replicates in S4B).
Figure 4. Ribosome profiling reveals translation elongation regulation under hyperosmotic stress.

(A) Size distributions of ribosome footprints for WT cells under hyperosmotic (blue), oxidative (orange) and stationary phase (green) stress conditions relative to untreated (black). (B) Similar to (A), size distributions of ribosome footprints for rck2Δ cells under hyperosmotic (blue) or oxidative (orange) stress conditions compared to WT unstressed cells (black). (C) Scatter plot of codon-specific occupancies for 28 nt RPFs comparing oxidative stress relative to unstressed condition in WT cells. r, Pearson correlation coefficient. (D) Similar to (C), comparing rck2Δ cells under oxidative stress relative to WT unstressed cells. (E) Immunoblot of a Phos-tag gel for eEF2 phosphorylation under unstressed, hyperosmotic or oxidative stress conditions in WT and rck2Δ cells with or without λ phosphatase treatment. (F) Scatter plot of codon-specific ribosome occupancies for 21 nt RPFs comparing WT cells under oxidative stress relative to unstressed condition. All proline codons are colored in red and labeled. (G) Northern blots for Pro-tRNAAGG, Glu-tRNAUUC and Thr-tRNAAGU in unstressed, hyperosmotic and oxidative (10 and 45 min) stress conditions.
A reduction in 21 nt RPFs during hyperosmotic stress is consistent with translocation or peptide bond formation becoming a dominant rate limiting step in translation in these cells (since 28 nt RPFs are comprised of these two populations in samples prepared with CHX/TIG). Importantly, our data suggest that relative decoding rates are not perturbed in these perturbed conditions as codon-specific ribosome occupancy for the 21 nt RPFs is highly correlated between the untreated and stressed samples (Figure S4C). In contrast, there is a notable decrease in the correlation of the codon-specific occupancy in the 28 nt RPFs (Figure S4D) and a slight enrichment of 28 nt RPFs on codons that decode Thr, Ile, Val and Lys (Figure S4E); these latter data suggest that in stressed cells, PrePT and/or PreTrans ribosomes are preferentially found on these codons. While we initially considered both possibilities, a defect in translocation seemed a likely explanation because eEF2—the factor that promotes translocation—is known to be phosphorylated under hyperosmotic stress through a well-characterized signaling pathway (Melcher and Thorner, 1996; Teige et al., 2001). We asked whether the activity of Rck2, the previously implicated eEF2 kinase, is responsible for the observed reduction of 21 nt RPFs under hyperosmotic stress. In ribosome profiling experiments in an rck2Δ strain, the levels of 21 nt RPFs were indeed restored to levels observed in WT unstressed cells (Figures 4B and replicates in S4F). Moreover, the correlation of codon-specific occupancies for 28 nt RPFs between stressed rck2Δ cells and unstressed WT cells was restored (Figure S4G, compared to S4D). These results demonstrate that Rck2 is required for the observed defects in translation elongation under hyperosmotic stress.
Much less is known about the oxidative stress response pathway, but we hypothesized that eEF2 phosphorylation by Rck2 might similarly play a role in arresting translation elongation, especially given that oxidative stress in mammalian cells leads to eEF2 phosphorylation (Kang and Lee, 2001) and that 21 nt RPFs were decreased in our samples (Figures 4A and replicates in S4B). Oxidative stress also changed codon-specific occupancies for the 28 nt RPFs to a much greater extent than hyperosmotic stress (Figures 4C and replicates in S4H). However, unlike the case with hyperosmotic stress, deletion of Rck2 did not restore 21 nt RPF levels under oxidative stress (Figures 4B and replicates in S4F), nor did it restore the correlation of codon-specific occupancies for 28 nt RPFs (Figure 4D, compared to 4C).
To further corroborate our findings that Rck2 is critical in inhibiting the progression of elongation under hyperosmotic stress but not oxidative stress, we performed polysome run-on experiments (with no elongation inhibitors during sample preparation) in a gcn2Δ background where eIF2α can no longer be phosphorylated (Figure S4I) thus eliminating confounding known effects of initiation inhibition under these stress conditions (Jimenez-Diaz et al., 2013; Shenton et al., 2006). We assessed the extent to which translation elongation is inhibited by calculating a polysome-to-monosome ratio (P/M) (the higher the number the more elongation is inhibited) and found increased P/M ratios under both hyperosmotic and oxidative stress conditions (Figure S4J and K). As anticipated, the increased P/M ratio observed under hyperosmotic stress is reverted in the gcn2Δrck2Δ strain to levels seen in the unstressed condition (Figure S4J). In contrast, both gcn2Δ and gcn2Δrck2Δ strains show increased P/M ratios under oxidative stress (Figure S4K), indicative of elongation inhibition that is independent of Rck2 function. These results provide strong support for our ribosome profiling results documenting Rck2-independent elongation inhibition and point to the existence of other pathway(s) that may regulate eEF2 under oxidative stress.
To further explore potential molecular mechanisms that might rationalize the ribosome profiling results, we directly probed the phosphorylation state of eEF2 under conditions of hyperosmotic and oxidative stress. We monitored the phosphorylation status of eEF2 using Phos-tag gels (Kinoshita et al., 2009) that retard the mobility of phosphorylated proteins, creating a smear above the expected molecular weight of eEF2. We found that eEF2 phosphorylation levels are higher under both stress conditions as compared to unstressed cells (Figure 4E, lanes 1–3); the phosphorylation status of the shifted species was confirmed by its sensitivity to phosphatase treatment that collapses the more slowly migrating species (Figure 4E, lanes 7–9). We also confirmed that phosphorylation of eEF2 under hyperosmotic stress depends specifically on Rck2 (Figure 4E, lanes 2 and 5) while under oxidative stress it does not (Figure 4E, lanes 3 and 6), consistent with our ribosome profiling results. Although it has been suggested that yeast eEF2 is phosphorylated at the conserved Thr56 like its mammalian counterpart (Donovan and Bodley, 1991), phosphorylation site(s) of eEF2 under oxidative stress are yet to be determined. Here, the accumulation of 28 nt RPFs under oxidative stress that coincides with the apparent phosphorylation of eEF2 is consistent with the idea that translocation is indeed inhibited in these conditions.
In addition to the loss of correlation of 28 nt RPFs under oxidative stress, the codon-specific occupancies for 21 nt RPFs also showed a striking loss of correlation, with a drastic enrichment in ribosome occupancies at all four proline codons (CCA, CCU, CCC and CCG) (Figures 4F and replicates in S4L). These data suggest the possibility of prolyl-tRNA specific defects in decoding. Northern blotting experiments revealed the accumulation of shortened forms of prolyl-tRNAAGG under oxidative stress while glutamyl-tRNAUUC and threonyl-tRNAAGU remain mostly intact (Figure 4G). It is likely that prolyl-tRNAAGG is shortened from its 3’-CCA end under oxidative stress based on the inherent susceptibility of the CCA-end of the tRNA and previous reports (Chen and Tanaka, 2018). Together, these observations suggest that there may be multiple cellular responses to oxidative stress that effectively inhibit translation elongation in order to promote cellular survival. Future experiments will explore the signaling pathways responsible for triggering these events during oxidative stress in S. cerevisiae.
DISCUSSION
Our results provide compelling evidence that the two predominant footprint sizes revealed by ribosome profiling (21 nt RPFs and 28 nt RPFs) represent distinct, definable states of translation elongation: 21 nt RPFs derive from ribosomes that lack a tRNA substrate in their A site (during sample preparation) while 28 nt RPFs derive from ribosomes carrying substrates in their A site. What is complex about the situation is that there are three distinct ribosome states (PreAcc, PrePT and PreTrans in Figure 1A) but only two RPF sizes. Additionally, ribosomes move in the lysate to the next permitted elongation state (depending on the antibiotic that is included) and aminoacyl-tRNAs that have not reacted with the peptidyl-tRNA dissociate from the A site during sample preparation. Importantly, as we show, the distribution of elongating ribosomes in these distinct RPF sizes in ribosome profiling libraries can be modulated by the use of antibiotic cocktails that inhibit distinct steps in elongation. Here we focus primarily on the use of a cocktail comprised of CHX and TIG in order to isolate a pure population of 21 nt RPFs that corresponds to ribosomes waiting to decode the next incoming aminoacyl-tRNA. Using a different cocktail that contains CHX and ANS, we can in principle isolate a pure population of 28 nt RPFs that corresponds to ribosomes waiting for the translocation step of protein synthesis. As such, different cocktails may be used to optimize characterization of distinct ribosome populations. In addition to the extensive ribosome profiling work in yeast where we assign ribosome states to distinct RPF sizes, we provide evidence that these same ribosome profiling approaches can be used to follow the same populations of RPFs throughout eukaryotes including worms and humans with the same RPF sizes corresponding to the same ribosome states.
We argue that 21 nt RPFs are generated by RNase I cleavage of mRNA within the ribosome during sample preparation. 28 nt RPFs span the ribosome from its 5’ to 3’ boundary; although 21 nt RFPs also have 5’-ends that align to the 5’-boundary of the ribosome, they are 7 nt shorter at their 3’-ends, meaning that RNase I must cut the mRNA between the A-site codon and the 3’ edge of the ribosome to produce a 21 nt RPF. When aa-tRNA binds in the A site, there is an extensive conformational change in the small ribosomal subunit, referred to as domain closure, that is conserved from bacteria to humans (Ogle et al., 2002; Shao et al., 2016). We argue that the presence of tRNA in the A site and/or domain closure of the small subunit prevents nucleases from entering the ribosome, yielding 28 nt RPFs. Consistent with the known conservation of ribosome conformational changes during translation elongation (Ogle et al., 2003), two distinct sizes of footprints have been documented in wheat germ extract (Wolin and Walter, 1988), budding yeast, C. elegans embryos and human cell lines (Figures 2B and 3). These data indicate that the ribosome states monitored by ribosome profiling are not species-specific and thus that this line of investigation can be applied broadly.
We note that in earlier studies, Lareau and colleagues found that 3-AT treatment resulted in the accumulation of 28 nt RPFs rather than 21 nt RPFs at His codons; importantly, these profiling libraries were prepared in the absence of any elongation inhibitors (Lareau et al., 2014). By contrast, we observed stronger enrichment of 21 nt RPFs at His codons relative to 28 nt RPFs in samples prepared with CHX/TIG (Figures 2G and S2I to L) and thus assign 21 nt RPFs to ribosomes waiting to decode histidyl-tRNA in these conditions. The earlier experiments were likely confounded by the effects of ongoing translation in extracts prepared in the absence of antibiotics.
A large body of theoretical work has predicted that translation rates drive synonymous codon usage in a wide range of organisms and that tRNA abundance is a major determinant of translation rates (Plotkin and Kudla, 2011). While many efforts have been made to infer codon-specific translation rates and to observe a correlation with tRNA abundance using ribosome profiling experiments (Charneski and Hurst, 2013; Gardin et al., 2014; Hussmann et al., 2015; Weinberg et al., 2016), these successes have been limited. Importantly, however, the results from these earlier studies were detrimentally impacted by either pretreatment of cells with elongation inhibitors that perturb in vivo ribosome dynamics (Gerashchenko and Gladyshev, 2014; Weinberg et al., 2016) or simply by ongoing translation in the lysates (Hussmann et al., 2015). The approaches that we develop here to effectively trap ribosomes in distinct functional states in lysates prepared from rapidly frozen cells allow for higher resolution analysis of the translation status of ribosomes in various cell types; for example, these increases in resolution now reveal strong correlations of 21 nt RPFs with tRNA abundance (Figure 2).
In experiments aimed to show how our high resolution profiling approaches might help to define molecular events in translation regulation, we show that translation elongation in S. cerevisiae is regulated in response to hyperosmotic and oxidative stress resulting in both cases in the accumulation of ribosomes in a pre-translocation state (as 28 nt RPFs). For hyperosmotic stress, we show that this regulation is mediated primarily by the eEF2 kinase Rck2, as previously reported (Teige et al., 2001), while for oxidative stress we show that the Rck2 deletion has no impact on the observed elongation defects. Oxidative stress in S. cerevisiae has been shown to trigger multiple cellular responses that may impact translation elongation. For example, K63-linked polyubiquitination of several ribosomal proteins near the mRNA channel (Back et al., 2018; Silva et al., 2015) has been connected to oxidative stress and proposed to mediate elongation inhibition. Similarly, oxidative stress has been shown to stimulate tRNA cleavage in the anticodon region by Rny1, a member of the RNase T2 family (Thompson and Parker, 2009). Other studies have documented broad CCA-end degradation of tRNAs under oxidative stress (Chen and Tanaka, 2018) and ribosome pausing at specific codons as monitored by co-translational mRNA decay (Pelechano et al., 2015). Our data extend these studies by providing clear evidence for eEF2 phosphorylation and correlated increases in the ratio of 28 to 21 nt RPFs, a signature of ribosomes trapped in a pre-translocation state. In contrast to what we observed for hyperosmotic stress, the yeast eEF2 kinase Rck2 seems to be of little significance for the oxidative stress response and its regulation of translation elongation. Given that both oxidative and hyperosmotic stress conditions induce eIF2α phosphorylation (Jimenez-Diaz et al., 2013; Shenton et al., 2006), polysome profiles in a WT background are likely impacted by effects on both translation initiation and elongation. Indeed, our polysome run-on analysis in a yeast background where gcn2 was deleted (to eliminate the effects from initiation regulation) provided strong support for elongation inhibition resulting from oxidative stress, and this response being independent of Rck2 function (Figures S4J and K). In addition, our data indicate that oxidative stress induces substantial truncations of prolyl-tRNA and impacts the tRNA selection step of protein synthesis in a specific manner wherein all proline codons exhibit increased ribosome occupancy (Figures 4F and G). Our results and approaches provide a framework for understanding how translation elongation regulation is implemented at multiple steps in response to oxidative stress.
Importantly, regulation of the elongation phase of translation is broadly utilized in response to cellular stress and is known to be impacted by various mechanisms including the post-translational modification of elongation factors (eEF1A and eEF2 (Dever et al., 2018)) as well as ribosomes (Proud, 2018; Silva et al., 2015; Simsek and Barna, 2017). Our insights into the in vivo dynamics of elongating ribosomes will enable efforts to further explore the potentially broad role of elongation in the regulation of protein synthesis throughout eukaryotes.
STAR METHODS
CONTACT FOR REAGENT AND RESOURCE SHARING
Please direct any requests for further information or reagents to the lead contact, Rachel Green (ragreen@jhmi.edu).
METHOD DETAILS
Yeast growth and DMS modification conditions for DMS-MaPseq
For in vivo DMS-MaPseq experiments, WT cells were grown in YPD at 30° to OD ~0.7. Cells were then treated with CHX (0.1 mg/mL) for an additional 10 min or with ANS (0.1 mg/mL) for an additional 30 min before adding DMS (Sigma). 0, 300 or 600 μL of DMS was added to 15 mL of the cultures, and incubated at 30°C for 2 min. The reactions were quenched by adding 30mL quench buffer (30% BME, 70% PBS). Cells were harvested by centrifugation at 3,000g for 10 min at 4°C and total RNA was extracted. For DMS-MaPseq experiments in lysates (ANS), ~850 mL of ANS-pretreated and untreated cells were harvested by fast filtration and flash frozen in liquid nitrogen. Cell pellets were ground in 1 mL of modification buffer [20 mM HEPES (pH7.5), 140 mM KCl, 1.5 mM MgCl2, 1% Triton X-100] in a Spex 6870 freezer mill. ANS (0.1 mg/mL) was present in the modification buffer for the ANS-pretreated sample. For TIG DMS-MaPseq experiments, ~ 850 mL of untreated cells were harvested at OD ~0.6 by fast filtration and split into two, one ground in 1 mL of modification buffer and the other ground in 1 mL of modification buffer with TIG (0.1 mg/mL). Lyzed cell pellets were diluted to 15 mL in modification buffer with or without TIG, and clarified by centrifugation. For DMS modification in lysates, 40 μg of lysates were incubated at 25°C for 1 hr to mimic RNase I digestion. DMS was added to the lysates at 0, 40 or 90mM for 3 min at 25°C. RNA was extracted after quench with quench buffer (30% BME, 0.3 M NaOAc).
Preparation of DMS-MaPseq libraries
Isolated total RNA was fragmented with 10mM ZnCl2 at 94°C for 5 min and quenched by adding EDTA to 20mM. After precipitation, RNA was size-selected on a 10% TBE-urea denaturing gel, cutting between 60–80 nt. After dephosphorylation, an oligonucleotide adapter [oBZ91 (CACTCGGGCACCAAGGAC) for in vivo and ANS lysate modification; oBZ407 (NNNNNNCACTCGGGCACCAAGGAC) for TIG lysate modification] was ligated to the 3’ end of isolated fragments using T4 RNA ligase 2 truncated (NEB). Reverse transcription was carried out using TGIRT-III enzyme (InGex). After circularization using Circ Ligase I (Lucigen) and PCR amplification, libraries were sequenced on a HiSeq2500 machine.
Analysis of DMS-MaPseq data
Raw sequencing reads were first trimmed of their 3′ adaptor sequence (CACTCGGGCACCAAGGAC for libraries made with oBZ91, NNNNNNCACTCGGGCACCAAGGAC for libraries made with oBZ407) with skewer (Jiang et al., 2014). Shapemapper 2.0 (Busan and Weeks, 2018) was used to trim low-quality nucleotides, align reads to S. cerevisiae rRNA (Saccharomyces Genome Database), and count mutations and read coverage at each rRNA position. Mutation fractions, proportional to DMS reactivity, were defined as the number of mutations at an rRNA nucleotide position divided by the number of reads overlapping that position. The following rRNA positions are constitutively post-transcriptionally modified in vivo (McClary et al., 2017; Zubradt et al., 2017) and were thus excluded from further analysis: S. cerevisiae 18S rRNA 1191,1781,1782; S. cerevisiae 25S rRNA 645, 2634, 2843. Python code that integrates these processing steps and produces tables of mutation fractions is available on GitHub (https://github.com/borisz264/mod_seq).
Yeast growth conditions for ribosome profiling
For eRF1 depletion, yKW13 was grown in YPGR (2% galactose+ 2% raffinose) overnight at 30°C, collected by centrifugation, washed, and resuspended in YPD at OD 0.03. After 9 hr, cells were harvested at OD ~0.46. For gamma toxin experiments, WT cells carrying pRS416-GalL-gamma toxin (Dundee University) or pRS416-GalL (ATCC) vector were grown in CSM-Ura media supplemented with 2% raffinose and 2% galactose for 8 hr and harvested at OD ~0.5. For 3-AT experiments, 3-AT was added to yCW19 cultures were grown to OD ~0.5 and treated with 50mM 3-AT for 20min. For stress conditions, cells were grown to OD ~0.5 and subjected to salt treatment (0.4 M NaCl) for 45 min or H2O2 treatment (0.03%) for 10 or 45min before harvest. For stationary phase growth, cells were grown for 4 days before harvest. Cells were harvested by fast filtration and flash frozen in liquid nitrogen. In general, cell pellets were split into two, one for ribosome profiling and the other for western or northern blotting.
Preparation of libraries for yeast ribosome footprints
Cell pellets were ground with 1 mL yeast footprint lysis buffer [20 mM Tris-Cl (pH8.0), 140 mM KCl, 1.5 mM MgCl2, 1% Triton X-100 with specified antibiotics] in a Spex 6870 freezer mill. Antibiotics (CHX, ANS and TIG) were used at 0.1 mg/mL. For eRF1 depletion, 3-AT (CHX) and gamma-toxin (CHX) experiments, cells were lysed in yeast footprint lysis buffer containing CHX and clarified by centrifugation. Lysate containing 1 mg total RNA was treated with 375 units of RNase I (Ambion) for 1 hr at 25°C. For the rest of experiments, lysed cell pellets were diluted to 15 mL in yeast footprint lysis buffer containing specified antibiotics and clarified by centrifugation. The resultant supernatant was layered on a sucrose cushion [20 mM Tris-Cl (pH8.0), 150 mM KCl, 5 mM MgCl2, 0.5 mM DTT, 1M sucrose]. Polysomes were pelleted by centrifugation at 60,000 rpm for 106 min in a Type 70Ti rotor (Beckman Coulter). Ribosome pellets were gently resuspended in 1 mL footprint lysis buffer. 400 μg of isolated polysomes were treated with 500 units of RNaseI (Ambion) for 1 hr at 25°C. Monosomes were isolated by sucrose gradients (10–50%). The extracted RNA was size-selected from 15% denaturing PAGE gels, cutting between 15–34 nt. An oligonucleotide adapter was ligated to the 3’ end of isolated fragments. After ribosomal RNA depletion using RiboZero (Illumina), reverse transcription using SuperScript III reverse transcriptase (Thermo Fisher Scientific), circularization using CirLigase I (Lugicen) and PCR amplification (Schuller et al., 2017). Libraries were sequenced on a HiSeq2500 machine at facilities at the Johns Hopkins Institute of Genetic Medicine.
Preparation of libraries for C. elegans ribosome footprints
C. elegans was cultured according to standard methods (Brenner, 1974). Synchronized L1 worms were plated on HT115 bacteria transformed with pL4440 vector and cultured at 25°C for ~48 hours. Bacteria were grown at 37°C in LB media containing ampicillin (100 μg/mL) for 5–6 hr, induced with 5 mM IPTG for 30 min, plated on NNGM plates (nematode nutritional growth media) containing ampicillin (100 μg/mL) and IPTG (1 mM), and let grow overnight at RT. Early embryos were collected by bleaching gravid hermaphrodites. ~400 μL packed embryos were resuspended in 2ml worm footprint lysis buffer [20 mM Tris-Cl (pH8.0), 140 mM KCl, 1.5 mM MgCl2, 1% Triton X-100, 0.1 mg/mL CHX] and lysed in a Spex 6870 freezer mill. After clarification, lysate containing 300 μg of total RNA was treated with 100 units of RNaseI (Ambion) for 30 min at 25°C. Monosomes were isolated by sucrose gradients (10–50%). Procedure for library construction is identical to that for yeast ribosome footprints.
Preparation of libraries for mammalian ribosome footprints
HeLa and MD-MBA-231 cells were grown to ~70–80% confluence in DMEM and RPMI supplemented with 10% FBS in 10-cm dishes, respectively, washed with PBS, and lysed by vigorous scraping in 1 mL of mammalian footprint lysis buffer [20 mM Tris-Cl (pH8.0), 150 mM NaCl, 15 mM MgCl2, 1mM DTT, 1% Triton X-100, 2 unit Turbo DNase, 0.1 mg/mL CHX]. When indicated, CHX and TIG were added to 0.1 mg/mL. For amino acid starvation, cells were washed twice with DMEM without L-glutamine (Gibco, 11960–044) and cultured in the same media for 9 or 6 hr (replicate) before harvest. After clarification by centrifugation (at 15,000 rpm for 10min), lysate containing 20 μg total RNA was treated with 750 units of RNase I (Ambion) for 1 hr at 25°C. Ribonuclease treatment was quenched by adding 10 μL SUPERase*In (Thermo Fisher Scientific). Nuclease-treated lysate was layered on a sucrose cushion [20 mM Tris-Cl (pH8.0), 150 mM NaCl, 5 mM MgCl2, 1 mM DTT, 1 M sucrose]. Ribosomes were pelleted by centrifugation in a TLA100.3 rotor at 100,000 rpm for 1 hr at 4°C and extracted by miRNeasy mini kit (Qiagen). Procedure for library construction is identical to that for yeast ribosome footprints.
Analysis of ribosome profiling data
The R64–1-1 S288C reference genome assembly (SacCer3) from the Saccharomyces Genome Database Project was used for yeast genome alignment. Ce10 reference genome assembly from UCSC was used for C. elegans genome alignment. Hg19 reference genome assembly from UCSC was used for human genome alignment. A human transcriptome file was generated to include canonical transcripts of known genes from UCSC genome browser. Data shown in Figure 1 for WT are identical to those published previously (Schuller et al., 2017). Libraries for WT (CHX), eRF1 depletion (CHX), 3-AT (CHX) and pRS416-vector (CHX) were trimmed to remove ligated 3’ linker (CTGTAGGCACCATCAAT) with skewer (Jiang et al., 2014). For the rest of our libraries, 3’ adapter (NNNNNNCACTCGGGCACCAAGGA) was trimmed, and 4 random nucleotides included in RT primer (RNNNAGATCGGAAGAGCGTCGTGTAGGGAAAGAGTGTAGATCTCGGTGGTCGC/iSP18/TTCAGACGTGTGCTCTTCCGATCTGTCCTTGGTGCCCGAGTG) were removed from the 5’ end of reads. Trimmed reads longer than 15 nt were aligned to yeast, worm or human ribosomal and non-coding RNA sequences using STAR (Dobin et al., 2013) with ‘--outFilterMismatchNoverLmax 0.3’. Unmapped reads were then mapped to genome using the following options ‘--outFilterIntronMotifs RemoveNoncanonicalUnannotated --outFilterMultimapNmax 1 --outFilterMismatchNoverLmax 0.1’. All other analyses were performed using software custom written in Python 2.7 and R 3.3.1.
Ribosome profiling datasets for 21 nt RPFs from (Lareau et al., 2014; Matsuo et al., 2017; Tunney et al., 2017) were downloaded from GEO (GSE79622, GSE58321 and GSE106572).
For each dataset, the offset of the A site from the 5’ end of reads was calibrated using start codons of CDS (Schuller et al., 2017). In general, offsets (20:[16], 21:[17], 22:[17]) were used to infer the A site of 20–22 nt reads (21 nt RPFs) for yeast datasets and offsets (20:[16], 21:[16], 22:[16]) for human datasets. For 28 nt RPFs, offsets (27:[16], 28:[16], 29:[17], 30:[17], 31:[17], 32:[17]) were used for yeast datasets and offsets (28:[16], 29:[17], 30:[17]) for human datasets.
Relative ribosome occupancies (pause scores) for codons (motifs) were computed by taking the ratio of the ribosome density in a 3-nt window at the codon (motif) over the overall density in the coding sequence (excluding the first and the last 15 nt). For metagene plots, ribosome density at each position was normalized by the overall ribosome density of coding sequence (CDS) (21 nt and 28 nt RPFs combined). Genes with features that are smaller than the window size (100 nt upstream of stop codons and 50 nt of 3’UTRs for Figure 1E) were excluded from the analysis.
Polysome profiles
200 mL of yeast cultures from indicated stress conditions were harvested by fast filtration and ground with 500 μL yeast footprint lysis buffer [20 mM Tris-Cl (pH8.0), 140 mM KCl, 1.5 mM MgCl2, 1% Triton X-100, 0.1 mg/mL CHX] in a Spex 6870 freezer mill. CHX was omitted for run-on polysome profiles. After clarification (at 15,000 rpm for 10 min), lysate containing 250 μg total RNA was spun through sucrose gradients (10–50%) and fractionated on a Biocomp fractionator.
Statistical analysis
Statistical details can be found in figure legends.
Western blot
Cells from unstressed and stressed conditions were lysed in footprint lysis buffer containing protease inhibitor (Roche) and phosphatase inhibitor PhosSTOP (Roche). Proteins were precipitated with TCA and separated on SDS-PAGE or SDS-PAGE with Phos-tag (Wako) gels prior to transfer to PVDF membranes. The Phos-tag was used at 25 μM. For phosphatase treatment, lysates were incubated with lambda phosphatase (NEB) following the manufacturer’s instructions. Immunoblots were performed with eRF1 antibody (Eyler et al., 2013), eEF2 antibody (Kerafast, ED7002), eIF2alpha antibody (Lu et al., 1999), phospho-eIF2alpha (Abcam) and anti-PGK1 antibody (Life Technologies-Novex, 459250).
Northern blot
Total RNA was extracted from lysates using hot acid phenol (VWR). For gammatoxin expression experiments, 10 μg of total RNA was loaded on a 10% TBE-urea denaturing gel and blotted onto a Zeta-probe membrane (Bio-Rad). For other tRNA blotting, 5 μg of total RNA was loaded on a 12% TBE-urea denaturing gel and blotted onto an Amersham Hybond N+ membrane (GE Healthcare Life Sciences). The membrane was hybridized with 5’ labeled probes: Glu-tRNA (CTCCGATACGGGGAGTCGAAC) or (AGCCGTTACACTATATCGGA), 5S rRNA (GCTTTCTGGTAGATATGGCCGCAACC), Pro-tRNA (GCCGGGACTCGAACCCGGGAC) and Thr-tRNA (CCAATCGGATTTGAACCGATG) at 50°C in Rapid-Hyb buffer (GE Healthcare Life Sciences). Blots were washed with low stringency buffer [2x SSC, 0.1% SDS (w/v)] at 50°C for twice (30 min each time) and ending with high stringency buffer [1x SSC, 0.1% SDS (w/v)] at 50°C for twice (30 min each time). Membranes were stripped with stripping buffer [1% SDS] at 80 °C for 30 min before hybridization with different probes.
Accession number
Raw sequencing data were deposited in the GEO database under the accession number GSE115162. Gel images are deposited in the Mendeley database at the following link: http://dx.doi.org/10.17632/s8bk6dz7vr.1.
High-resolution ribosome profiling defines discrete ribosome elongation states and translational regulation during cellular stress (Wu et al.)
Supplementary Material
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| eEF2 antibody | Kerafast | ED7002 |
| PGK1 antibody | Life Technologies-Novex | 459250 |
| eRF1 antibody | (Eyler et al., 2013) | |
| Phospho-eIF2alpha | Abcam | ab32157 |
| eIF2alpha | (Lu et al., 1999) | |
| Chemicals and commercial reagents | ||
| Cycloheximide | Sigma-Aldrich | C1988 |
| Anisomycin | Sigma-Aldrich | A9789 |
| Tigecycline | Sigma-Aldrich | PZ0021 |
| Phos-tag | Wako | AAL107 |
| SuperaseIn | Ambion | AM2694 |
| RNase I | Ambion | AM2294 |
| Superscript III | Invitrogen | 56575 |
| T4 Polynucleotide Kinase | New England Biosciences | M0201L |
| T4 RNA Ligase 2, Truncated | New England Biosciences | M0242L |
| GlycoBlue | ThermoFisher | AM9515 |
| Ribo-Zero Gold rRNA Removal Kit (Yeast) | Illumina | MRZY1306 |
| CircLigase ssDNA Ligase | Epicentre | CL4115K |
| Oligonucleotides | ||
| AGCCGTTACACTATATCGGA | This paper | GluUUC-tRNAprobe1 |
| CTCCGATACGGGGAGTCGAAC | This paper | GluUUC-tRNAprobe2 |
| GCTTTCTGGTAGATATGGCCGCAACC | This paper | 5SrRNA-tRNAprobe |
| GCCGGGACTCGAACCCGGGAC | This paper | ProAGG-tRNAprobe |
| CCAATCGGATTTGAACCGATG | This paper | ThrAGU-tRNAprobe |
| CATTTAACGATTGGAAAAGACGAAAGTATTGTTAAGAGTACTGCTTATTTAGAGAGGATCAAACAAAATCTCTTCGGCATAGGCCACTAGTGGATCCTG | This paper | Rck2KO_F |
| CTTGTAGAAGGAGTTTAATGTATATATATCTTTTAAAAAGGAATAGGTAAAAAGATTGAAACAGAAGGGAAAGTTGAGCTGAAGCTTCGTACG | This paper | Rck2KO_R |
| rAppCACTCGGGCACCAAGGAC | This paper | oBZ91 |
| rAppNNNNNNCACTCGGGCACCAAGGAC | This paper | oBZ407 |
| rAppCTGTAGGCACCATCAAT–NH2 | NEB | Universal miRNA cloning linker |
| /5Phos/AGATCGGAAGAGCGTCGTGTAGGGAAAGAGTGTAGATCTCGGTGGTCGC/iSP18/ TTCAGACGTGTGCTCTTCCGATCTGTCCTTGGTGCCCGAGTG |
This paper | oBZ192 |
| /5Phos/AGATCGGAAGAGCGTCGTGTAGGGAAAGAGTGTAGATCTCGGTGGTCGC/Sp- C18/CACTCA/Sp-C18/TTCAGACGTGTGCTCTTCCGATCTATTGATGGTGCCTACAG |
(Schuller et al., 2017) | RT primer 1 |
| /5Phos/RNNNAGATCGGAAGAGCGTCGTGTAGGGAAAGAGTGTAGATCTCGGTGGTCGC/iSP18/ TTCAGACGTGTGCTCTTCCGATCTGTCCTTGGTGCCCGAGTG |
This paper | oBZ408 |
| Deposited Data | ||
| Raw and analyzed data | This paper | GEO: GSE115162 |
| R64–1–1 S288C sacCer3 Genome Assembly | Saccharomyces Genome Database Project | http://downloads.yeastgenome.org/sequence/S288C_reference/genome_releases/ |
| Hg19 genome assembly | UCSC genome browser | http://hgdownload.soe.ucsc.edu/goldenPath/hg19/bigZips/ |
| Ce10 genome assembly | UCSC genome browser | http://hgdownload.soe.ucsc.edu/goldenPath/ce10/bigZips/ |
| Mendeley Database | This paper | http://dx.doi.org/10.17632/s8bk6dz7vr.1 |
| Experimental Models: Cell Lines | ||
| T-Rex HeLa | Thermo Fisher Scientific | R71407 |
| MDA-MB-231 | ATCC | HTB-26 |
| Experimental Models: Organisms/Strains | ||
| BY4741 | Dharmacon | YSC1048 |
| Δrck2; MATa | Dharmacon | Clone ID: 5157 |
| Δgcn2; MATa | Dharmacon | Clone ID: 3642 |
| eRF1 depletion strain | This paper | yKW13 |
| His3+ strain | This paper | yCW19 |
| Gamma-toxin expression stress | This paper | yCW66 |
| Δgcn2 Δrck2; MATa | This paper | yCW72 |
| Caenorhabditis elegans | This paper | N2 |
| Software and Algorithms | ||
| skewer | (Jiang et al., 2014) | Version 0.2.2 |
| seqtk | https://github.com/lh3/seqtk | |
| STAR | (Dobin et al., 2013) | STAR_2.5.3a_modified |
| ImageQuant TL | GE Healthcare Life Sciences | Version 8.1 |
Highlights.
High-resolution ribosome profiling resolves distinct ribosome functional states
Codon occupancy of decoding ribosomes is highly anti-correlated with tRNA abundance
Cellular stress conditions trigger distinct paths of elongation regulation
ACKNOWLEDGMENTS
We would like to thank members of the Green lab for helpful discussions, Allen Buskirk and Jamie Wangen for critical reading of this manuscript, Sean Lee for collecting C. elegans embryos, Thomas Dever for eIF2alpha antibody, and David Mohr and the Johns Hopkins Genetic Resources Core Facility for sequencing assistance. This work is supported by the NIH (2R37GM059425-14 to R.G.) and HHMI (R.G. and C.C.W.). B.Z. is an HHMI fellow of the Damon Runyon Cancer Research Foundation [DRG-2250-16].
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- Agirrezabala X, Lei J, Brunelle JL, Ortiz-Meoz RF, Green R, and Frank J (2008). Visualization of the hybrid state of tRNA binding promoted by spontaneous ratcheting of the ribosome. Mol Cell 32, 190–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Back S, Vogel C, and Silva GM (2018). Site-specific K63 ubiquitinomics reveals post-initiation regulation of ribosomes under oxidative stress. bioRxiv. [Google Scholar]
- Barbacid M, and Vazquez D (1974). (3H)anisomycin binding to eukaryotic ribosomes. J Mol Biol 84, 603–623. [DOI] [PubMed] [Google Scholar]
- Behrmann E, Loerke J, Budkevich TV, Yamamoto K, Schmidt A, Penczek PA, Vos MR, Burger J, Mielke T, Scheerer P, et al. (2015). Structural snapshots of actively translating human ribosomes. Cell 161, 845–857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brenner S (1974). The genetics of Caenorhabditis elegans. Genetics 77, 71–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brewster JL, and Gustin MC (2014). Hog1: 20 years of discovery and impact. Sci Signal 7, re7. [DOI] [PubMed] [Google Scholar]
- Budkevich T, Giesebrecht J, Altman RB, Munro JB, Mielke T, Nierhaus KH, Blanchard SC, and Spahn CM (2011). Structure and dynamics of the mammalian ribosomal pretranslocation complex. Mol Cell 44, 214–224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Busan S, and Weeks KM (2018). Accurate detection of chemical modifications in RNA by mutational profiling (MaP) with ShapeMapper 2. RNA 24, 143–148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charneski CA, and Hurst LD (2013). Positively charged residues are the major determinants of ribosomal velocity. PLoS Biol 11, e1001508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen CW, and Tanaka M (2018). Genome-wide Translation Profiling by Ribosome-Bound tRNA Capture. Cell Rep 23, 608–621. [DOI] [PubMed] [Google Scholar]
- de Nadal E, Ammerer G, and Posas F (2011). Controlling gene expression in response to stress. Nat Rev Genet 12, 833–845. [DOI] [PubMed] [Google Scholar]
- Dever TE, Dinman JD, and Green R (2018). Translation Elongation and Recoding in Eukaryotes. Cold Spring Harb Perspect Biol. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, and Gingeras TR (2013). STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donovan MG, and Bodley JW (1991). Saccharomyces cerevisiae elongation factor 2 is phosphorylated by an endogenous kinase. FEBS Lett 291, 303–306. [DOI] [PubMed] [Google Scholar]
- dos Reis M, Savva R, and Wernisch L (2004). Solving the riddle of codon usage preferences: a test for translational selection. Nucleic Acids Res 32, 5036–5044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eyler DE, Wehner KA, and Green R (2013). Eukaryotic release factor 3 is required for multiple turnovers of peptide release catalysis by eukaryotic release factor 1. J Biol Chem 288, 29530–29538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gardin J, Yeasmin R, Yurovsky A, Cai Y, Skiena S, and Futcher B (2014). Measurement of average decoding rates of the 61 sense codons in vivo. Elife 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garreau de Loubresse N, Prokhorova I, Holtkamp W, Rodnina MV, Yusupova G, and Yusupov M (2014). Structural basis for the inhibition of the eukaryotic ribosome. Nature 513, 517–522. [DOI] [PubMed] [Google Scholar]
- Gerashchenko MV, and Gladyshev VN (2014). Translation inhibitors cause abnormalities in ribosome profiling experiments. Nucleic Acids Res 42, e134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hussmann JA, Patchett S, Johnson A, Sawyer S, and Press WH (2015). Understanding Biases in Ribosome Profiling Experiments Reveals Signatures of Translation Dynamics in Yeast. PLoS Genet 11, e1005732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ingolia NT, Lareau LF, and Weissman JS (2011). Ribosome profiling of mouse embryonic stem cells reveals the complexity and dynamics of mammalian proteomes. Cell 147, 789–802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jenner L, Starosta AL, Terry DS, Mikolajka A, Filonava L, Yusupov M, Blanchard SC, Wilson DN, and Yusupova G (2013). Structural basis for potent inhibitory activity of the antibiotic tigecycline during protein synthesis. Proc Natl Acad Sci U S A 110, 3812–3816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang H, Lei R, Ding SW, and Zhu S (2014). Skewer: a fast and accurate adapter trimmer for nextgeneration sequencing paired-end reads. BMC Bioinformatics 15, 182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jimenez-Diaz A, Remacha M, Ballesta JP, and Berlanga JJ (2013). Phosphorylation of initiation factor eIF2 in response to stress conditions is mediated by acidic ribosomal P1/P2 proteins in Saccharomyces cerevisiae. PLoS One 8, e84219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang KR, and Lee SY (2001). Effect of serum and hydrogen peroxide on the Ca2+/calmodulin-dependent phosphorylation of eukaryotic elongation factor 2(eEF-2) in Chinese hamster ovary cells. Exp Mol Med 33, 198–204. [DOI] [PubMed] [Google Scholar]
- Kinoshita E, Kinoshita-Kikuta E, and Koike T (2009). Separation and detection of large phosphoproteins using Phos-tag SDS-PAGE. Nat Protoc 4, 1513–1521. [DOI] [PubMed] [Google Scholar]
- Klopotowski T, and Wiater A (1965). Synergism of aminotriazole and phosphate on the inhibition of yeast imidazole glycerol phosphate dehydratase. Arch Biochem Biophys 112, 562–566. [DOI] [PubMed] [Google Scholar]
- Lareau LF, Hite DH, Hogan GJ, and Brown PO (2014). Distinct stages of the translation elongation cycle revealed by sequencing ribosome-protected mRNA fragments. Elife 3, e01257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leprivier G, Remke M, Rotblat B, Dubuc A, Mateo AR, Kool M, Agnihotri S, El-Naggar A, Yu B, Somasekharan SP, et al. (2013). The eEF2 kinase confers resistance to nutrient deprivation by blocking translation elongation. Cell 153, 1064–1079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liakath-Ali K, Mills EW, Sequeira I, Lichtenberger BM, Pisco AO, Sipila KH, Mishra A, Yoshikawa H, Wu CC, Ly T, et al. (2018). An evolutionarily conserved ribosome-rescue pathway maintains epidermal homeostasis. Nature 556, 376–380. [DOI] [PubMed] [Google Scholar]
- Lu J, Huang B, Esberg A, Johansson MJ, and Bystrom AS (2005). The Kluyveromyces lactis gamma-toxin targets tRNA anticodons. RNA 11, 1648–1654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu J, O’Hara EB, Trieselmann BA, Romano PR, and Dever TE (1999). The interferon-induced double-stranded RNA-activated protein kinase PKR will phosphorylate serine, threonine, or tyrosine at residue 51 in eukaryotic initiation factor 2alpha. J Biol Chem 274, 32198–32203. [DOI] [PubMed] [Google Scholar]
- Matsuo Y, Ikeuchi K, Saeki Y, Iwasaki S, Schmidt C, Udagawa T, Sato F, Tsuchiya H, Becker T, Tanaka K, et al. (2017). Ubiquitination of stalled ribosome triggers ribosome-associated quality control. Nat Commun 8, 159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McClary B, Zinshteyn B, Meyer M, Jouanneau M, Pellegrino S, Yusupova G, Schuller A, Reyes JCP, Lu J, Guo Z, et al. (2017). Inhibition of Eukaryotic Translation by the Antitumor Natural Product Agelastatin A. Cell Chem Biol 24, 605–613 e605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGlincy NJ, and Ingolia NT (2017). Transcriptome-wide measurement of translation by ribosome profiling. Methods 126, 112–129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melcher ML, and Thorner J (1996). Identification and characterization of the CLK1 gene product, a novel CaM kinase-like protein kinase from the yeast Saccharomyces cerevisiae. J Biol Chem 271, 29958–29968. [DOI] [PubMed] [Google Scholar]
- Moazed D, and Noller HF (1986). Transfer RNA shields specific nucleotides in 16S ribosomal RNA from attack by chemical probes. Cell 47, 985–994. [DOI] [PubMed] [Google Scholar]
- Moazed D, and Noller HF (1989). Intermediate states in the movement of transfer RNA in the ribosome. Nature 342, 142–148. [DOI] [PubMed] [Google Scholar]
- Ogle JM, Carter AP, and Ramakrishnan V (2003). Insights into the decoding mechanism from recent ribosome structures. Trends Biochem Sci 28, 259–266. [DOI] [PubMed] [Google Scholar]
- Ogle JM, Murphy FV, Tarry MJ, and Ramakrishnan V (2002). Selection of tRNA by the ribosome requires a transition from an open to a closed form. Cell 111, 721–732. [DOI] [PubMed] [Google Scholar]
- Pelechano V, Wei W, and Steinmetz LM (2015). Widespread Co-translational RNA Decay Reveals Ribosome Dynamics. Cell 161, 1400–1412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plotkin JB, and Kudla G (2011). Synonymous but not the same: the causes and consequences of codon bias. Nat Rev Genet 12, 32–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pop C, Rouskin S, Ingolia NT, Han L, Phizicky EM, Weissman JS, and Koller D (2014). Causal signals between codon bias, mRNA structure, and the efficiency of translation and elongation. Mol Syst Biol 10, 770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Proud CG (2018). Phosphorylation and Signal Transduction Pathways in Translational Control. Cold Spring Harb Perspect Biol. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryazanov AG, Shestakova EA, and Natapov PG (1988). Phosphorylation of elongation factor 2 by EF-2 kinase affects rate of translation. Nature 334, 170–173. [DOI] [PubMed] [Google Scholar]
- Schneider-Poetsch T, Ju J, Eyler DE, Dang Y, Bhat S, Merrick WC, Green R, Shen B, and Liu JO (2010). Inhibition of eukaryotic translation elongation by cycloheximide and lactimidomycin. Nat Chem Biol 6, 209–217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuller AP, Wu CC, Dever TE, Buskirk AR, and Green R (2017). eIF5A Functions Globally in Translation Elongation and Termination. Mol Cell 66, 194–205 e195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shao S, Murray J, Brown A, Taunton J, Ramakrishnan V, and Hegde RS (2016). Decoding Mammalian Ribosome-mRNA States by Translational GTPase Complexes. Cell 167, 1229–1240 e1215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shenton D, Smirnova JB, Selley JN, Carroll K, Hubbard SJ, Pavitt GD, Ashe MP, and Grant CM (2006). Global translational responses to oxidative stress impact upon multiple levels of protein synthesis. J Biol Chem 281, 29011–29021. [DOI] [PubMed] [Google Scholar]
- Silva GM, Finley D, and Vogel C (2015). K63 polyubiquitination is a new modulator of the oxidative stress response. Nat Struct Mol Biol 22, 116–123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simsek D, and Barna M (2017). An emerging role for the ribosome as a nexus for post-translational modifications. Curr Opin Cell Biol 45, 92–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teige M, Scheikl E, Reiser V, Ruis H, and Ammerer G (2001). Rck2, a member of the calmodulin-protein kinase family, links protein synthesis to high osmolarity MAP kinase signaling in budding yeast. Proc Natl Acad Sci U S A 98, 5625–5630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson DM, and Parker R (2009). The RNase Rny1p cleaves tRNAs and promotes cell death during oxidative stress in Saccharomyces cerevisiae. J Cell Biol 185, 43–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tunney RJ, McGlincy NJ, Graham ME, Naddaf N, Pachter L, and Lareau L (2017). Accurate design of translational output by a neural network model of ribosome distribution. bioRxiv. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uesono Y, and Toh EA (2002). Transient inhibition of translation initiation by osmotic stress. J Biol Chem 277, 13848–13855. [DOI] [PubMed] [Google Scholar]
- Warringer J, Hult M, Regot S, Posas F, and Sunnerhagen P (2010). The HOG pathway dictates the short-term translational response after hyperosmotic shock. Mol Biol Cell 21, 3080–3092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinberg DE, Shah P, Eichhorn SW, Hussmann JA, Plotkin JB, and Bartel DP (2016). Improved Ribosome-Footprint and mRNA Measurements Provide Insights into Dynamics and Regulation of Yeast Translation. Cell Rep 14, 1787–1799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wek RC (2018). Role of eIF2alpha Kinases in Translational Control and Adaptation to Cellular Stress. Cold Spring Harb Perspect Biol 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wohlgemuth I, Brenner S, Beringer M, and Rodnina MV (2008). Modulation of the rate of peptidyl transfer on the ribosome by the nature of substrates. J Biol Chem 283, 32229–32235. [DOI] [PubMed] [Google Scholar]
- Wolin SL, and Walter P (1988). Ribosome pausing and stacking during translation of a eukaryotic mRNA. EMBO J 7, 3559–3569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zubradt M, Gupta P, Persad S, Lambowitz AM, Weissman JS, and Rouskin S (2017). DMSMaPseq for genome-wide or targeted RNA structure probing in vivo. Nat Methods 14, 75–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.