

Abstract
A method for direct cross coupling between unactivated C(sp3)−H bonds and chloroformates has been accomplished via nickel and photoredox catalysis. A diverse range of feedstock chemicals, such as (a)cyclic alkanes and toluenes, along with late-stage intermediates, undergo intermolecular C−C bond formation to afford esters under mild conditions using only 3 equiv of the C−H partner. Site selectivity is predictable according to bond strength and polarity trends that are consistent with the intermediacy of a chlorine radical as the hydrogen atom-abstracting species.
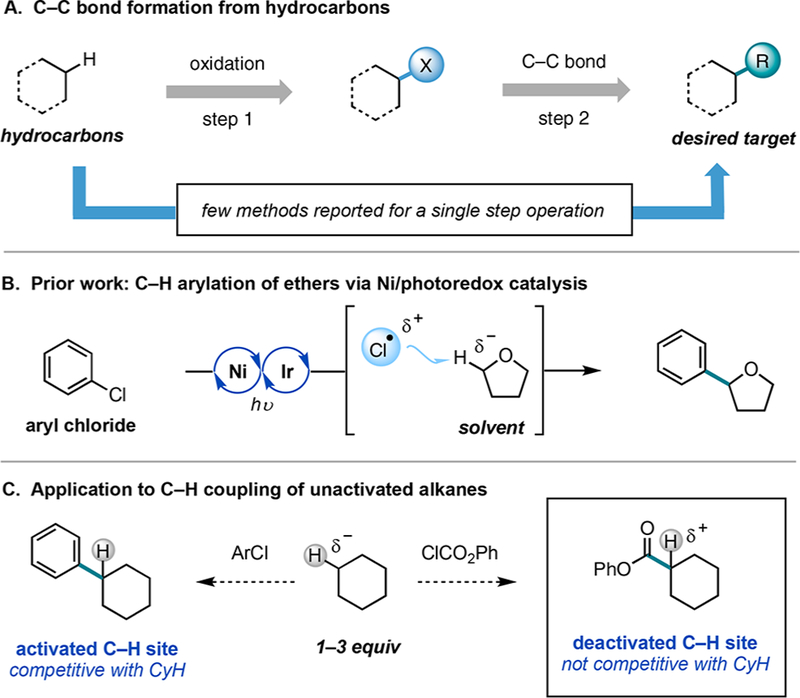
The development of catalytic methods for functionalization of unactivated aliphatic C−H bonds represents a prominent challenge in organic synthesis.1 A variety of useful strategies have been developed for selective C(sp3)−H oxidation of hydrocarbons to give C−X bonds (e.g., X = O, N, B, F, or Cl).2 While valuable in their own right, these products are also frequently used as precursors for C(sp3)−C bond-forming reactions. Direct formation of C−C bonds from unactivated hydrocarbons would thus provide an attractive strategic alternative to this multistep sequence (Figure 1A). However, most catalytic methods that effect intermolecular C−C bond formation of unactivated alkanes require the use of substrates with a coordinating directing group or the use of a substrate in large excess to impart reactivity, which limits applications at an early and late stage in synthesis.3 Alternatively, Rh-catalyzed C(sp3)−H insertion with donor−acceptor carbenoids represents a powerful methodology for nondirected C−C bond formation with alkane as the limiting reagent.4 Despite its attributes, the methodology is restricted to the installation of a specific type of C−C bond. The identification of alternative strategies that enable the modular installation of C−C bonds from unactivated alkanes could have a significant impact on organic synthesis. Here we report that Ni and photoredox catalysis represents one such strategy, enabling the generation of valuable carbonyl derivatives from unactivated C(sp3)−H bonds via catalytic C−C bond formation with chloroformates.
Figure 1.
Direct C−C bond formation from C(sp3)−H bonds.
Over the past several years, Ni and photoredox catalysis has emerged as a modular strategy for the construction of C(sp3)− C bonds from C(sp3)−H partners.5,6 Under this manifold, Ni catalysts are proposed to intercept and functionalize carbon-centered radicals generated from C(sp3)−H bonds via one of two distinct pathways: an oxidative single electron transfer (SET)/deprotonation process for electron-rich amines or a H-atom abstraction (HAT) step for electron-poor amines and ethers. Thus far, successful substrates in these coupling reactions have required C(sp3)−H bonds proximal to functional groups. For example, we recently disclosed a strategy for the arylation of ethereal C(sp3)−H bonds via the photocatalytic generation of chlorine radical from Ni (Figure 1B).5c Although chlorine radical is capable of abstracting C−H bonds of unactivated alkanes (HCl BDFE 97 kcal/mol; cyclohexane BDFE 91 kcal/mol),7 we found that the cross coupling of cyclohexane with aryl chlorides was only feasible in moderate yield using a large excess of substrate (44% yield at 10 equiv).
Guided by this result, we set out to develop a catalytic C(sp3)−H cross-coupling reaction of unactivated alkanes using substrate in stoichiometric quantities. A principal concern in the development of such a process was identifying conditions in which the unactivated alkane would undergo preferential HAT in the presence of cross-coupled product. For arylation reactions, the benzylic C(sp3)−H bond of the product is considerably weaker than the starting material C(sp3)−Hs (BDFE of 80 versus 90−95 kcal/mol),7 which could lead to overfunctionalization or unproductive consumption of the chlorine radical. Since chlorine radical is an electrophilic hydrogen atom acceptor, we anticipated that use of an electron-deficient coupling partner such as a chloroformate would discourage competitive HAT on the basis of polar effects in free-radical chlorination (Figure 1C).8 The application of chloroformates in a C(sp3)−H functionalization reaction was appealing as it would deliver direct access to carbonyl derivatives, arguably the most versatile functional groups in synthesis. Moreover, chloroformates are abundant, inexpensive reagents that undergo facile oxidative addition to Ni.9
According to our prior mechanistic studies5c,10a and literature precedent,10b–d a hypothesized catalytic cycle for the C(sp3)−H functionalization is displayed in Figure 2. We envisioned that (dtbbpy)Ni0 (1) would undergo oxidative addition to a chloroformate derivative to generate (dtbbpy)-NiII(CO2R)(Cl) (2).9b,c,11 Concurrently, visible light irradiation of the photocatalyst Ir[dF(CF3)ppy]2(dtbbpy)PF6 (3) would enable a long-lived excited species 4 that serves as a potent single electron oxidant (τ = 2.3 μs, *E1/2(IrIII/IrII) = +1.21 vs SCE in MeCN) for 2.12,13 Subsequent visible light irradiation of transient (dtbbpy)NiIII(CO2R)(Cl) (5) would affect photoelimination of chlorine radical, followed by rapid hydrogen atom abstraction of the aliphatic substrate to generate a carbon-centered radical. Addition of this radical to NiII would result in 8, followed by reductive elimination of the ester product. Single electron transfer from the reducing IrII (10) to NiI (9) would allow for regeneration of both the Ni and photoredox catalysts.13
Figure 2.
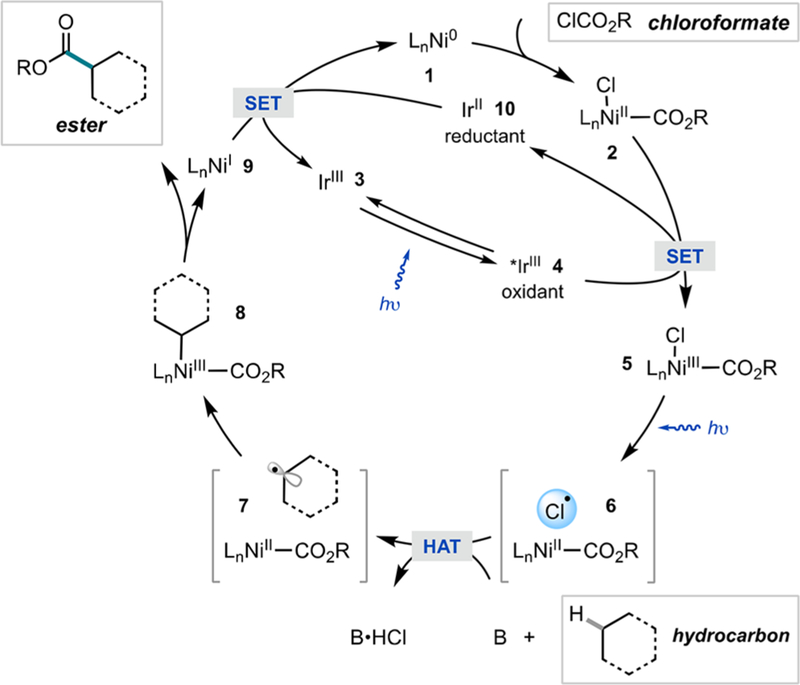
Proposed photoelimination mechanism for ester formation from hydrocarbons. 3 = Ir[dF(CF3)ppy]2(dtbbpy)PF6; dF(CF3)ppy = 2-(2,4-difluorophenyl)-5-(trifluoromethyl)pyridine; dtbbpy = 4,4′-di-tert-butyl-2,2′-bipyridine.
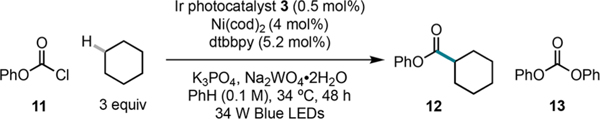
Our investigation began with the reaction of phenyl chloroformate ($0.16/g)9 with cyclohexane (Table 1). Use of this coupling partner under our previously reported conditions (10% Ni, 2% Ir, 10 equiv cyclohexane, 2 equiv K3PO4, in 0.04 M benzene) delivered 12 in only 14% yield (Figure S3). Upon extensive investigation, we identified conditions that delivered ester 12 in 66% yield after 48 h using only 0.5 mol % Ir photocatalyst 3 and 4 mol % Ni (entry 1).14 Whereas 1 equiv of alkane may be used, highest yields are obtained using a ratio of 3:1 alkane:chloroformate (entry 2). Control reactions demonstrated that the C(sp3)−H functionalization process requires the presence of both Ni and Ir catalysts, as well as light, and that consistent reaction temperatures were critical to obtaining high yields (entries 3−8). Notably, the addition of sodium tungstate, and to a lesser extent, sodium silicate, provided moderate rate accelerations (entries 1 vs 9 and 11). Preliminary data suggests that sodium tungstate is acting as a base15 rather than as a precursor to higher-order tungstates, which could function as photocatalysts and hydrogen atom acceptors.16 For example, at 72 h, a reaction without sodium tungstate proceeds to comparable yield as a reaction in its presence (entries 1 and 10). Furthermore, use of near-UV light, which enables access to the charge-transfer excited states of higher order tungstates, leads to low reaction efficiency (Table S9). In all cases, the mass balance can be attributed to the formation of diphenyl carbonate (13), a byproduct primarily generated from the reaction of chloroformate with base (Table S4).
Table 1.
Optimization of Reaction Conditionsa
 | |||
|---|---|---|---|
| Entry | Deviation from standard condition | Yield 12 (%)b | Yield 13 (%)c |
| 1 | None | 66 | 28 |
| 2 | 1 equiv of cyclohexane | 44 | 33 |
| 3 | no photocatalyst | 0 | 48 |
| 4 | no ligand | 3 | 27 |
| 5 | no nickel | 0 | 17 |
| 6 | no light | 0 | 34 |
| 7 | 26 °C | 46 | 17 |
| 8 | 40 °C | 55 | 32 |
| 9 | no Na2WO4·2H2Od | 39 | 19 |
| 10 | no Na2WO4·2H2Od, 72 h | 62 | 22 |
| 11 | Na2SiO3·5H2O instead of Na2WO4·2H2O | 57 | 25 |
| 12 | no K3PO4d | 58 | 35 |
| 13 | no K3PO4, no Na2WO4·2H2O | 17 | 11 |
K3PO4 (2 equiv); Na2WO4·2H2O (1 equiv).
Yields determined by 1H NMR using 4-fluoroanisole as an external standard.
Yield determined by GC analysis based on consumption of 2 equiv of chloroformate for production of 1 equiv of DPC.
3 equiv of base used.
With these optimized conditions established, we set out to examine the scope of the C(sp3)−H cross-coupling reaction (Figure 3). In exploring the chloride-containing coupling partner, we observed that both p-fluoro and p-methoxy derivatives of phenyl chloroformate are effective for C−H functionalization of cyclooctane (15 and 16). More notably, the use of acid chlorides in place of a chloroformate delivered aromatic and aliphatic ketones 17 and 18 in 55 and 52% yield, respectively, under identical conditions.17,18 Additionally, 4-morpholine carbonyl chloride afforded amide 19 in 15% yield, demonstrating that this methodology could be adopted for direct amidation.
Figure 3.
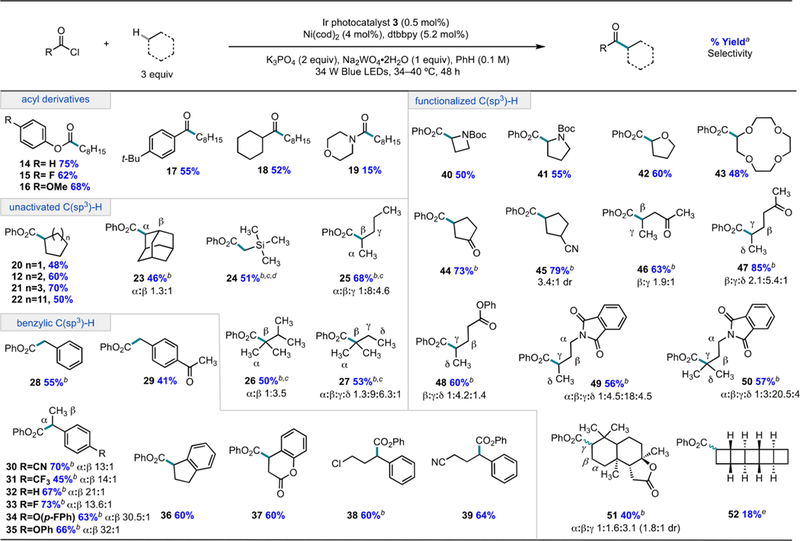
aIsolated yield of all isomers expressed as an average of two runs. Hydrocarbon substrates worked effectively at 34 °C (±5 °C), whereas hydrocarbons containing amines, ketones, or ethers, worked optimally at 40 °C (±5 °C). b 1H NMR yield of all isomers expressed as an average of two runs using 4-fluoroanisole as an external standard. c6 equiv of C−H substrate used. dYield reported postdesilylation. e 1H NMR yield expressed as a single run reported on 0.08 mmol scale with 1.8 C−H equivalents of [5]-ladderane using 12 W Blue LEDs.
In our investigation of the C(sp3)−H coupling partner, we first evaluated unfunctionalized alkanes. Cyclic alkyl hydrocarbons with ring sizes from 5 to 15 underwent C−C bond formation, transforming inert substrates to easily modified products in good yield (12, 20−22). Tetramethylsilane which possesses one of the strongest C(sp3)−H bonds (BDFE = 96 kcal/mol)7 was functionalized in 51% yield (24). Examination of the functionalization of acyclic alkanes revealed a trend among various C(sp3)−H sites consistent with the influence of BDFE on rate of C−H abstraction. Pentane was preferentially functionalized at the internal secondary positions in 68% combined yield (25 α:β:γ 1:8:4.6). Analogously, 2-methylbutane exhibited preferential functionalization at the tertiary position in 53% combined yield (27 α:β:γ:δ 1.3:9:6.3:1). An Evans−Polanyi plot (Ea = αΔG° + β) for 2-methylbutane shows a linear correlation with an α value of 0.44, comparable to tabulated α values for HAT with chlorine radical (αCl = 0.45) (Figure 4a).8c Thus, while the C(sp3)−H esterification results in mixtures of regioisomeric products for these substrates, the site selectivity is predictable.
Figure 4.
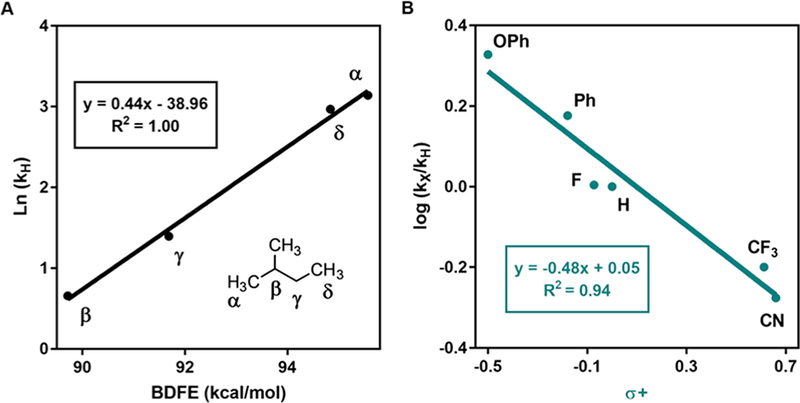
C(sp3)−H abstraction selectivity profile. (a) Evans−Polanyi relationship for 27; (b) Hammett Plot for ethylbenzene derivatives.
Because toluene and alkyl benzenes are abundant and inexpensive commodity chemicals, we also examined their use as substrates for the esterification reaction (28−39). Notably, the Stahl and Liu groups have recently reported Cu-catalyzed arylation reactions of alkyl benzenes with boronic acid derivatives that provide efficient access to diarylalkanes, albeit requiring a large excess of substrate or alkyl benzenes with extended aromatic systems.19 For the esterification reaction, we were pleased to find that toluene and its acyclic as well as cyclic derivatives underwent C−C bond formation in good yields. The influence of the polar effect is evidenced in the site selectivity of these reactions. For a series of p-substituted ethylbenzenes, the ratio of benzylic to methyl functionalization decreases with electron-withdrawing substituents (30−35). Furthermore, a Hammett-Brown analysis of the relative rate of benzylic functionalization across this series (determined by competition experiments, see SI Section III.H) reveals that electron-rich substrates undergo C−C bond formation faster than electron-poor substrates (Figure 4B). Indeed, the small ρ value is consistent with a highly exothermic abstraction step and reports on free-radical chlorination of toluenes (ρ values ranging from −0.5 to −1.0).20 These results explain the high regioselectivity in esterification for products 29, 37−39.
When functionalized cyclic and acyclic substrates were submitted to the reaction conditions, selectivity trends reflected an interplay of polarity effects and radical stability. Cyclic amides and ethers, from a 4-membered azetidine to a 12-membered crown ether were successfully converted to α-amino and α-oxy esters 40 and 43 under the dual catalytic conditions.17d,e Cyclic hydrocarbons with electron-withdrawing functionality, like cyclopentanone and cyclopentylcarbonitrile, underwent C−C bond formation with predictable and high levels of selectivity, furnishing the β-functionalized product 44 and γ-functionalized product 45 in 73 and 79% yields. For an acyclic ketone, selectivity for γ functionalization increased when that site changed from a primary to a secondary position (46 vs 47: 22% vs 54% yield). Similarly, a preference for the γ position increased between 49 and 50 when that site was changed from a secondary position to a tertiary position. Synthetically, these examples offer complementarity to arylations of similar substrates under Pd catalysis, where regioselectivity is ligand/or directing group-dependent.21 Likewise, the Martin laboratory recently described a method for Ni-catalyzed carboxylation of halogenated aliphatic hydrocarbons that affords C−C bond formation at two completely distinct positions of an aliphatic chain to that favored in this methodology.22
Application of this method to late-stage functionalization was also evaluated. Sclareolide, a model system for selective C(sp3)−H oxidations, led to 51 in 40% combined yield at the most electron-rich and sterically accessible methylene sites, which is orthogonal to reactivity exhibited by most transition metal-based catalysts.23 Additionally, a ladderane core, the derivatives of which are notoriously challenging to access, gave 52 in a modest 18% yield at 1.8 equiv of substrate. The closest reaction analog, a Mn(TMP)Cl catalyst system, delivered 40% of the chlorinated species and requires additional steps to access a C−C bond.24
In summary, this reaction provides a general strategy to access carbonyl derivatives directly from unactivated C(sp3)− H bonds that is amenable both to early and late stage synthesis. Site selectivity is predictable, arising from hydrogen atom abstraction by chlorine radical, and affords a synthetic complement to alternative approaches for achieving C−C bond formation from alkanes. Our future efforts will be directed at elucidating the mechanism of this process and developing strategies to control the site-selectivity of C(sp3)− H functionalization.
Supplementary Material
ACKNOWLEDGMENTS
The authors gratefully acknowledge Jaron Mercer and the Burns lab for their generous donation of [5]-ladderane, Dr. István Pelczer for NMR assistance, Lotus Separations, Ben J. Shields, Stephen I. Ting and Eric W. Webb for their helpful advice. Financial support was provided by NIGMS (R01 GM100985 and R35 GM126986) and an F32 Ruth L Kirschtein NRSA Fellowship under Award No. 5 F32 GM119364-02 (L.K.G.A.).
Footnotes
Notes
The authors declare no competing financial interest.
ASSOCIATED CONTENT
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/jacs.8b09191.
Experimental procedures, computational data, and characterization and spectral data for new compounds (PDF)
REFERENCES
- (1).For recent reviews: (a) Hartwig JF; Larsen MA ACS Cent. Sci 2016, 2, 281. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) He J; Wasa M; Chan KSL; Shao Q; Yu J-Q Chem. Rev 2017, 117, 8754. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Gutekunst WR; Baran PS Chem. Soc. Rev 2011, 40, 1976. [DOI] [PubMed] [Google Scholar]; (d) Yamaguchi J; Yamaguchi AD; Itami K Angew. Chem., Int. Ed 2012, 51, 8960. [DOI] [PubMed] [Google Scholar]
- (2).For selected examples: (a) Newhouse T; Baran PS Angew. Chem., Int. Ed 2011, 50, 3362. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Liu W; Groves JT Acc. Chem. Res 2015, 48, 1727. [DOI] [PubMed] [Google Scholar]; (c) Chen MS; White MC Science 2010, 327, 566. [DOI] [PubMed] [Google Scholar]; (d) Czaplyski WL; Na CG; Alexanian EJ J. Am. Chem. Soc 2016, 138, 13854. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Michaudel Q; Thevenet D; Baran PS J. Am. Chem. Soc 2012, 134, 2547. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Chiappini ND; Mack JBC; Du Bois J Angew. Chem 2018, 130, 5050. [DOI] [PubMed] [Google Scholar]; (g) Larsen MA; Cho SH; Hartwig J J. Am. Chem. Soc 2016, 138, 762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).(a) Engle KM; Mei T-S; Wasa M; Yu J-Q Acc. Chem. Res 2012, 45, 788. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Tang S; Wang P; Li H; Lei A Nat. Commun 2016, 7, 11676. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Shao B; Bagdasarian AL; Popov S; Nelson HM Science 2017, 355, 1403. [DOI] [PubMed] [Google Scholar]; (d) Choi GJ; Zhu Q; Miller DC; Gu CJ; Knowles RR Nature 2016, 539, 268. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Chu JCK; Rovis T Nature 2016, 539, 272. [DOI] [PMC free article] [PubMed] [Google Scholar]; A recent report described two examples of C−C bond formation using 1 equiv of cyclooctane: (f) Margrey KA; Czaplyski WL; Nicewicz DA; Alexanian EJ J. Am. Chem. Soc 2018, 140, 4213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).(a) Liao K; Pickel TC; Boyarskikh V; Bacsa J; Musaev DG; Davies HML Nature 2018, 551, 609. [DOI] [PubMed] [Google Scholar]; (b) Liao K; Negretti S; Musaev DG; Bacsa J; Davies HML Nature 2016, 533, 230. [DOI] [PubMed] [Google Scholar]
- (5).(a) Zuo ZW; Ahneman DT; Chu LL; Terrett JA; Doyle AG; MacMillan DWC Science 2014, 345, 437. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Shaw MH; Shurtleff VW; Terrett JA; Cuthbertson JD; MacMillan DWC Science 2016, 352, 1304. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Shields BJ; Doyle AG J. Am. Chem. Soc 2016, 138, 12719. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Heitz DR; Tellis JC; Molander GA J. Am. Chem. Soc 2016, 138, 12715. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Shen Y; Gu Y; Martin R J. Am. Chem. Soc 2018, 140, 12200. [DOI] [PubMed] [Google Scholar]
- (6).While this paper was in preparation, a method for the arylation of unactivated alkane substrates (5 equiv) using Ni/photoredox was reported by the MacMillan lab: Perry IB; Brewer TF; Sarver PJ; Schultz DM; DiRocco DA; MacMillan DWC Nature 2018, 560, 70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7). See Supporting Information for details.
- (8).(a) Free Radicals; Kochi JK, Ed.; Wiley-Interscience: New York, 1973. [Google Scholar]; (b) Tedder JM Tetrahedron 1982, 38, 313. [Google Scholar]; (c) Afanas’ev IB Russ. Chem. Rev 1971, 40, 216. [Google Scholar]
- (9).(a) (b) Zheng M; Xue W; Xue T; Gong H Org. Lett 2016, 18, 6152.(c) Otsuka S; Nakamura A; Yoshida T; Naruto M; Ataka K J. Am. Chem. Soc 1973, 95, 3180.
- (10).(a) Shields BJ; Kudisch B; Scholes GD; Doyle AG J. Am. Chem. Soc 2018, 140, 3035. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Hwang SJ; Powers DC; Maher AG; Anderson BL; Hadt RG; Zheng S-L; Chen Y-S; Nocera DG J. Am. Chem. Soc 2015, 137, 6472. [DOI] [PubMed] [Google Scholar]; (c) Hwang SJ; Anderson BL; Powers DC; Maher AG; Hadt RG; Nocera DG Organometallics 2015, 34, 4766. [Google Scholar]; (d) Mondal P; Pirovano P; Das A; Farquhar ER; McDonald AR J. Am. Chem. Soc 2018, 140, 1834. [DOI] [PubMed] [Google Scholar]
- (11). See Supporting Information for spectroscopic, computational, and stoichiometric experiments that probe this proposed catalytic cycle.
- (12). Ep = 0.4 V vs SCE in MeCN for (dtbbpy)Ni(COPh)Cl.
- (13).Lowry MS; Goldsmith JI; Slinker JD; Rohl R; Pascal RA; Malliaras GG; Bernhard S Chem. Mater 2005, 17, 5712. [Google Scholar]
- (14). When chlorobenzene was submitted to the optimized reaction conditions, no product was detected at 3 equiv. of cyclohexane. See Figure S8.
- (15).(a) Lassner E; Schubert W-D Tungsten Properties, Chemistry, Technology of the Element, Alloys, and Chemical Compounds; Kluwer Academic/Plenum Publishers: New York, 1999. [Google Scholar]; (b) Barré T; Arurault L; Sauvage FX Spectrochim. Acta, Part A 2005, 61, 551. [DOI] [PubMed] [Google Scholar]
- (16).(a) Hill CL; Prosser-McCartha CM Photocatalytic and Photoredox Properties of Polyoxometalate Systems. In Photosensitization and Photocatalysis Using Inorganic and Organometallic Compounds; Kalyanasundaram K, Graẗzel M, Eds.; Springer: Dordrecht, 1993; pp 307–330. [Google Scholar]; (b) Na2WO4 forms higher order tungstates at temperatures above 90 °C.
- (17).(a) Selected examples of ketone synthesis using acyl electrophiles: (a) Wotal AC; Weix DJ Org. Lett 2012, 14, 1476. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Wu F; Lu W; Qian Q; Ren Q; Gong H Org. Lett 2012, 14, 3044. [DOI] [PubMed] [Google Scholar]; (c) Le C; MacMillan DWC J. Am. Chem. Soc 2015, 137, 11938. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Joe CL; Doyle AG Angew. Chem., Int. Ed 2016, 55, 4040. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Amani J; Sodagar E; Molander GA Org. Lett 2016, 18, 732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Ni-photoredox catalyzed thiocarbonylation and acylation of ethers has been accomplished in solvent quantities of substrate: (a) Hong S-H; Kang B Chem. Sci 2017, 8, 6613. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Sun Z; Kumagai N; Shibasaki M Org. Lett 2017, 19, 3727. [DOI] [PubMed] [Google Scholar]
- (19).(a) Vasilopoulos A; Zultanski SL; Stahl SS J. Am. Chem. Soc 2017, 139, 7705. [DOI] [PubMed] [Google Scholar]; (b) Zhang W; Chen P; Liu G J. Am. Chem. Soc 2017, 139, 7709. [DOI] [PubMed] [Google Scholar]
- (20).Russell GA; Williamson RC Jr. J. Am. Chem. Soc 1964, 86, 2357. [Google Scholar]
- (21).(a) Zhu R-Y; Li Z-Q; Park HS; Senanayake CH; Yu JQ J. Am. Chem. Soc 2018, 140, 3564. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Li S; Zhu R-Y; Xiao KJ; Yu J-Q Angew. Chem., Int. Ed 2016, 55, 4317. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Shao Q; He J; Wu Q-F; Yu J-Q ACS Catal 2017, 7, 7777. [Google Scholar]
- (22).(a) Juliá-Hernández F; Moragas T; Cornella J; Martin R Nature 2017, 545, 84. [DOI] [PubMed] [Google Scholar]
- (23).Zou L; Paton RS; Eschenmoser A; Newhouse TR; Baran PS; Houk KN J. Org. Chem 2013, 78, 4037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Mercer JAM; Cohen CM; Shuken SR; Wagner AM; Smith MW; Moss FR III; Smith MD; Vahala R; Gonzalez-Martinez A; Boxer SG; Burns NZ J. Am. Chem. Soc 2016, 138, 15845. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.