

Abstract
Spinocerebellar ataxia type 7 (SCA7) is an autosomal dominant neurodegenerative disorder characterized by cerebellar and retinal degeneration, and is caused by a CAG-polyglutamine repeat expansion in the ATAXIN-7 gene. SCA7 patients develop progressive cone-rod dystrophy, typically resulting in blindness. Antisense oligonucleotides (ASOs) are single-stranded chemically modified nucleic acids designed to mediate the destruction, prevent the translation or modify the processing of targeted RNAs. Here we evaluated ASOs as treatments for SCA7 retinal degeneration in representative mouse models via injection into the vitreous humor of the eye. Using Ataxin-7 aggregation, visual function, retinal histopathology, gene expression, and epigenetic dysregulation as outcome measures, we found that ASO-mediated Ataxin-7 knockdown yielded significant improvements in treated SCA7 mice. In SCA7 mice with significant retinal disease, intravitreal injection of Ataxin-7 ASO also improved visual function despite initiating treatment after symptom onset. By using color fundus photography and autofluoresence imaging, we also determined the nature of retinal degeneration in human SCA7 patients; we observed variable disease severity, and catalogued rapidly progressive degeneration. Given the accessibility of neural retina, availability of objective, quantitative read-outs for monitoring therapeutic response, and rapid disease progression, ASOs targeting ATAXIN-7 might represent a viable treatment for SCA7 retinal degeneration.
One sentence Summary:
Intravitreal injection of antisense oligonucleotides improves visual function in SCA7 mice that model retinal degeneration phenotypes in human patients.
Introduction
Spinocerebellar ataxia type 7 (SCA7) is an autosomal dominant neurodegenerative disorder characterized by cerebellar ataxia, dysarthria, ophthalmoplegia, hyperreflexia, spasticity, and retinal degeneration (1). Although the disease is rare, affecting ~1/500,000 individuals, SCA7 exhibits a wide geographic distribution, occurring in all major racial groups, and various ethnic populations (2, 3). SCA7 is caused by a CAG-polyglutamine (polyQ) repeat expansion at the 5’ end of the coding region of the ATAXIN-7 gene (4, 5). While normal individuals possess alleles ranging in size from 7 – 35 CAGs, disease-causing expanded SCA7 CAG repeats are among the most unstable of all coding repeat expansions, with patients possessing 37 to >300 repeats (5). Anticipation is striking in SCA7 pedigrees, with the longest repeat mutations producing infantile onset (6–8). Larger repeat expansions occur in male germlines, and anticipation is sometimes so pronounced that paternal transmission of SCA7 is associated with increased spontaneous miscarriage rates in affected kindreds (6, 9). There are nine recognized CAG-polyQ repeat diseases, including spinobulbar muscular atrophy (SBMA), Huntington’s disease (HD), dentatorubral-pallidoluysian atrophy (DRPLA), and six forms of spinocerebellar ataxia (SCA 1,2,3,6,7 and 17). Numerous lines of investigation have shown that the initiating event in disease pathogenesis is misfolding of the polyQ expansion tract to an altered conformation that is resistant to protein degradation (10, 11), indicating that SCA7 shares a common pathogenic basis with Alzheimer’s disease, Parkinson’s disease, amyotrophic lateral sclerosis (ALS), and tauopathies.
The ATAXIN-7 protein was identified as a novel polypeptide of unknown function with ubiquitous expression. Experiments in Saccharomyces cerevisiae revealed that mammalian ATAXIN-7 has a yeast orthologue Sgf73, which is a core component of the Spt7-Ada1-Gcn5 Acetyltransferase (SAGA) transcription co-activator complex (12); whereas, independent studies concomitantly identified ATAXIN-7 as a core component of the mammalian Spt3-Taf9-Ada-Gcn5-Acetyltransferase (STAGA) transcription co-activator complex and closely related TATA-binding protein-free TAF containing complex (TFTC) (13, 14). The STAGA complex possesses both histone acetyltransferase and histone deubiquitinase activity, and while the exact function of ATAXIN-7 is unknown, it has been shown that polyQ-ATAXIN-7 can integrate into the STAGA complex, and alter the histone acetyltransferase activity of STAGA in retinal photoreceptor cells (14, 15).
SCA7 can be distinguished clinically from the other SCAs by the presence of retinal degeneration. Although fundoscopic examination typically show degeneration of the macula, electrophysiological evaluation of SCA7 patients by electroretinogram (ERG) analysis, reveals marked cone photoreceptor cell dysfunction throughout the retina prior to any rod photoreceptor abnormality (16). As cone photoreceptor function precedes rod photoreceptor function, and this cone photoreceptor dysfunction is not restricted to one particular region of the retina, SCA7 fulfills the diagnostic criteria for cone-rod dystrophy (16). Since cone photoreceptors mediate color vision, SCA7 patients can present with asymptomatic dyschromatopsia, which prevents them from distinguishing the color yellow from the color blue (17). The cone-rich macula is located in the central region of the retina; hence, SCA7 patients first complain of problems with central vision, and often develop central scotomas (18). But, as SCA7 retinal disease progresses, rod photoreceptor cells also become affected, resulting in complete blindness.
Expression of polyQ disease proteins in cells and model organisms can disrupt a wide range of cellular processes, including proteasome-dependent protein degradation, axonal transport, mitochondrial function, macro-autophagy, neuroinflammation, glutamate transport, and synaptic transmission (19). For these reasons, the development of a meaningful therapy to treat SCA7 and related polyQ disorders has been especially challenging. However, with the realization that these diseases all stem from the production of a mutant gene product, investigators hypothesized that targeting the mutant protein for destruction or preventing its production would interrupt the pathogenic cascade at its very first step. One approach for gene silencing is use of an antisense oligonucleotide (ASO) to target the RNA. This technique employs synthetic single strands of 15–25 nucleotides of perfect (or near-perfect) sequence complementary to bind to a target RNA, and thereby produce a DNA-RNA heteroduplex that is a substrate for degradation by RNase H, or will interfere with RNA processing or translation (20).
Although ASOs were initially developed nearly three decades ago, advances in oligonucleotide synthesis and chemical modification technologies have enabled this technology to move forward as a viable therapeutic strategy in humans, with the drug Spinraza approved for use in patients with autosomal recessive spinal muscular atrophy (21). As a treatment for neurodegenerative diseases involving production of a toxic gene product, ASO therapy is emerging as an extremely promising approach, with clinical trials currently underway in adult patients with familial amyotrophic lateral sclerosis (ALS) due to dominant SOD1 mutations (NCT02623699) and a Phase 1/2a clinical trial in early-stage HD patients (NCT02519036). Furthermore, a recent study of SCA2 demonstrated that CNS delivery of Ataxin-2 ASO can effectively treat the dysfunction and degeneration of Purkinje neurons of the cerebellum in mouse models of this disorder (22).
As considerable experience with ASO knock-down in related neurodegenerative diseases augurs well for the utility and likely feasibility of this regimen in human patients, we initiated a drug discovery campaign focused on the creation of an ASO therapy for SCA7. This work pursued a two-pronged approach to reducing Ataxin-7 RNA expression, comparing an ASO targeting the Ataxin-7 mRNA versus an ASO targeting the extended CAG repeat tract. We chose to examine these different ASOs as a treatment for SCA7 retinal degeneration, as the cone-rod dystrophy phenotype of SCA7 offers the unique opportunity to evaluate this therapeutic strategy in a highly accessible neural tissue - the eye, with disease outcome measures that are highly objective and quantitative. Hence, ASO therapy development for SCA7 retinal degeneration represents an important proof-of-concept opportunity for discretely and definitively assessing the true potential of the dosage reduction paradigm as a treatment for neurodegeneration.
Results
Intravitreal injection yields safe, effective suppression of retinal Ataxin-7 RNA expression
To obtain Ataxin-7-specific ASOs with superior potency and minimal likelihood for toxicity, we synthesized ~150 Ataxin-7 ASOs directed against mouse Ataxin-7. These ASOs were synthesized as high affinity oligonucleotides, composed of constrained ethyl (cET) nucleoside analogs that lock the sugar backbone into a bridged nucleic acid (BNA) structure to promote binding affinity, and thus increase potency by a factor of 5 – 10 in comparison to the previous generation 2’-O-methoxyethyl ASO design (23). Synthesized ASOs were screened for Ataxin-7 knock-down in a mouse endothelial cell line, yielding ~25 hits that exhibited >75% knock-down at modest concentrations (fig. S1). Secondary screening of the top 12 ASOs was completed in vivo to identify the best tolerated Ataxin-7 ASO with the least toxicity, and based upon this secondary screening, ASO #144 was selected as the most promising lead for in vivo studies.
To achieve delivery of oligonucleotides to retinal neurons and to photoreceptors in particular, we performed injections of oligonucleotides into the vitreous humor, as prior work has shown that intravitreal injection (IVI) permits diffusion throughout the eye, including to retinal photoreceptors (24). To validate the IVI delivery technique, we pursued a series of proof-of-principle experiments with a Rhodopsin ASO (25). Rhodopsin is expressed in retinal photoreceptors; hence, IVI of the Rhodopsin ASO will only result in knock-down of Rhodopsin gene expression in the eye, if the oligonucleotide distributes to retinal photoreceptor neurons. We performed 1 μl IVI of Rhodopsin ASO at a concentration of 50 μg/μl in the right eye of control mice, along with 1 μl IVI of diluent (PBS) into the contralateral left eye. RT-PCR quantification of Rhodopsin expression in RNAs isolated from harvested retinas of treated mice, showed Rhodopsin knock-down ranging from 65%−95%, for an overall mean knock-down of ~70%, which was significant (P < 0.01) (fig. S2, A and B), consistent with previous work using ASOs targeting rhodopsin (25). Robust knock-down of rhodopsin upon intravitreal injection of an anti-rhodopsin ASO thus confirmed our ability to achieve delivery to retinal photoreceptors using this method.
To determine the optimal dosage for Ataxin-7 knock-down by ASO #144, we performed a series of IVI experiments in C57BL/6J mice, comparing dosages ranging from 12.5 μg to 50 μg, and observed >70% knock-down of Ataxin-7 RNA expression in the retinas of mice treated with the 50 μg dose (Fig. 1A). We selected 50 μ.g as the ideal dosage, because we observed occasional toxicity at doses of 75 μg. To evaluate the duration of oligonucleotide knock-down using the IVI delivery technique, we injected 1 μl of Ataxin-7 ASO #144 intra-vitreal at a concentration of 50 μg/μl in the right eye of 18 C57BL/6J mice and 1 μl IVI of vehicle (PBS) into the contralateral left eye of the same 18 C57BL/6J mice, and then euthanized mice at different time points after IVI. We noted >50% knock-down of Ataxin-7 RNA expression out to 4 weeks post-injection, and documented >40% knock-down at the final 6-week post-injection time point (Fig. 1B). We also examined distribution of the ASO by immunostaining retinal sections from mice subjected to IVI with an antibody directed against the oligonucleotide backbone, and detected Ataxin-7 ASO throughout the neural retina at 1 week and 6 weeks post-injection (Fig. 1, C and D). These findings suggest that ASO #144 is effective in knocking down Ataxin-7 in the retina.
Figure 1. Ataxin-7 (ATXN7) ASO achieves robust and sustained knock-down in mouse neural retina.

A)Dose response for Ataxin-7 ASO knock-down in the retinas of wild-type C57BL/6J mice. Mice received a single intravitreal injection of 12.5 μg, 25 μg, or 50 μg of Ataxin-7 ASO in the right eye, and a single intravitreal injection of an identical volume of PBS in the left eye. Ataxin-7 mRNAs were measured by qRT-PCR and expressed as the mean relative to PBS-treated control eyes (n = 7 mice / group). **P < 0.01, ***P < 0.001; t-test. B) Duration of Ataxin-7 ASO knock-down in the retinas of wild-type C57BL/6J mice. Mice received a single intravitreal injection of 50 μg of Ataxin-7 ASO in the right eye, and a single intravitreal injection of PBS in the left eye. Ataxin-7 mRNAs were measured by qRT-PCR and expressed as the mean relative to PBS-treated control eyes (n = 3 – 7 mice / time point). **P < 0.01, ***P < 0.001; t-test. C-D) Distribution of Ataxin-7 ASO in the retinas of wild-type C57BL/6J mice. Mice received a single intravitreal injection of 50 μg of Ataxin-7 ASO in the right eye, and a single intravitreal injection of PBS in the left eye. Retinal sections were immunostained with an anti-ASO antibody (red) and counterstained with Hoechst 33342 (blue) at the indicated time points after intravitreal injection. Scale bars = 20 μm. Error bars = s.e.m. n = 3 mice
Ataxin-7 ASO achieves dramatic reductions in Ataxin-7 expression and protein aggregation in the eyes of treated SCA7 knock-in mice
SCA7 is unique among the SCAs, because SCA7 patients suffer from an unusual form of retinal degeneration, known as cone-rod dystrophy. We, and others, have accurately recapitulated SCA7 cone-rod dystrophy retinal degeneration in mice (26, 27). The availability of such mouse models permits testing of potential therapies. To ascertain if ASO-mediated knock-down of Ataxin-7 might be an effective treatment for SCA7 retinal degeneration, we performed a preclinical study with Ataxin-7 ASO #144. For the study, we selected an aggressive juvenile-onset-like mouse model of SCA7, known as the SCA7 266Q “knock-in” model, because a 266 CAG repeat contained on a small DNA fragment from the human ATAXIN-7 gene was inserted into the endogenous mouse Ataxin-7 locus (27). The first signs of ERG visual impairment in SCA7 266Q knock-in mice occur at ~5 weeks of age (27). To determine if intravitreal delivery of Ataxin-7 ASO is capable of ameliorating the rapidly progressive retinal degeneration in SCA7 266Q knock- in mice, we performed one-time 1 μl IVI of Ataxin-7 ASO at a concentration of 50 μg/μl into the right eye of SCA7 266Q knock-in mice at 4 weeks of age, along with a one-time 1 |μ,l IVI injection of PBS into the left eye. Detectable visual impairment typically occurs by 6 weeks of age in SCA7 266Q knock-in mice, which succumb to disease by 10 – 11 weeks of age. For these reasons, we designed a preclinical prevention trial in which we followed cohorts of mice for 6 weeks for a variety of outcome measures (fig. S3). We began by measuring Ataxin-7 RNA expression in the retinas of treated SCA7 266Q knock-in mice at 6 weeks post-IVI, and confirmed that Ataxin-7 ASO-treated eyes displayed >60% reduction in Ataxin-7 RNA expression in comparison to vehicle-treated eyes (Fig. 2A). One important read-out for therapeutic response is the extent of aggregation of polyQ-Ataxin-7, as aggregate burden often correlates with the quantity of toxic soluble protein or oligomer species. To assess polyQ-Ataxin-7 aggregation in neural retina, we immunostained retinas from treated SCA7 266Q knock-in mice at 6 weeks post-IVI, and observed a marked reduction in aggregates in retinal neurons, especially photoreceptors (Fig. 2B). To quantify the extent of aggregate reduction in treated SCA7 266Q knock-in mice, we performed filter trap assays on protein lysates prepared from individual retinas, comparing Ataxin-7 ASO-treated retinas with vehicle-treated retinas, and noted a significant decrease (P < 0.001) in the amount of aggregated polyQ-Ataxin-7 protein in ASO-treated retinas (Fig. 2, C and D). To determine the extent to which the Ataxin-7 ASO targets the normal repeat-length Ataxin-7 allele, we performed immunoblot analysis of Ataxin-7 ASO-treated retinas of SCA7 266Q knock-in mice, and observed marked reductions of both insoluble, polyQ-expanded, aggregated Ataxin-7 and normal Q-length, soluble Ataxin-7 in comparison to diluent-treated retinas (fig. S4, A to C).
Figure 2. Ataxin-7 ASO treatment reduces expression and aggregation of polyglutamine- expanded ATAXIN-7 in retina of SCA7 knock-in mice.
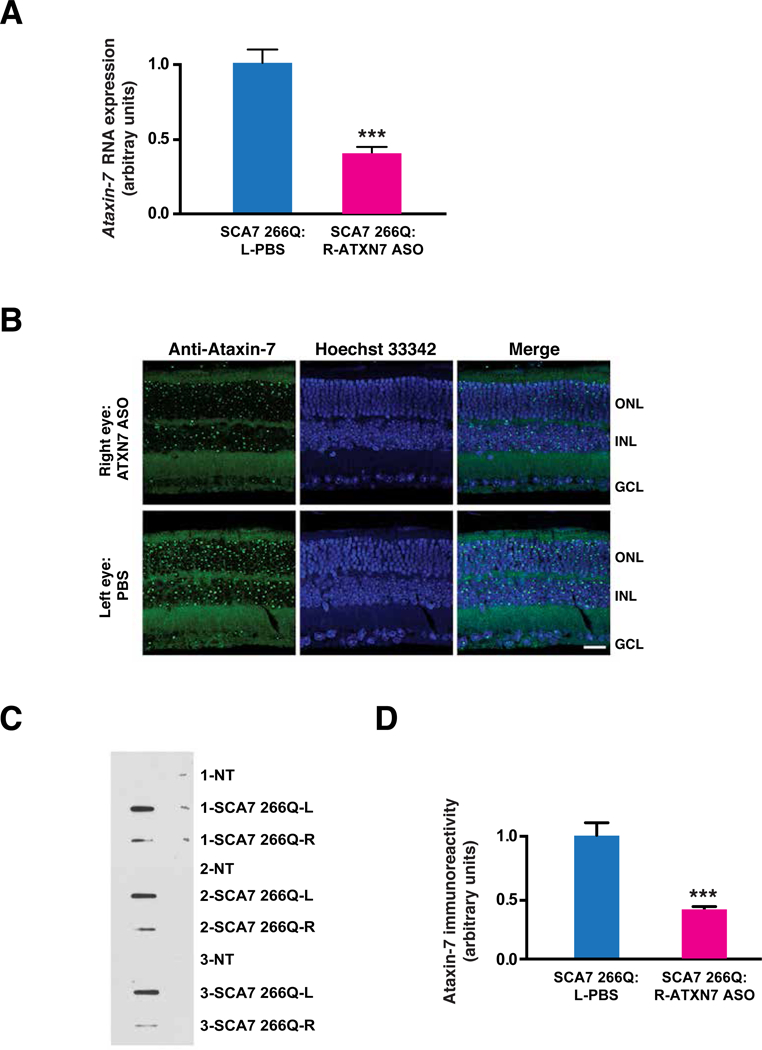
A)Ataxin-7 mRNA expression in the retinas of SCA7 266Q knock-in mice. Mice received a single intravitreal injection of 50 μg of Ataxin-7 ASO in the right eye, and a single intravitreal injection of PBS in the left eye. Ataxin-7 mRNAs were measured by qRT-PCR and expressed as the mean relative to PBS-treated control eyes (n = 7 mice / group). ***P < 0.001; t-test. B) Effect of Ataxin-7 ASO knock-down on visible aggregates in the retinas of SCA7 266Q knock- in mice. Mice received a single intravitreal injection of 50 μg of Ataxin-7 ASO in the right eye, and a single intravitreal injection of PBS in the left eye. Retinal sections were immunostained with an anti-Ataxin-7 antibody (green) and counterstained with Hoechst 33342 (blue) at 6 weeks after intravitreal injection. ONL = outer nuclear layer; INL = inner nuclear layer; GCL = ganglion cell layer. Scale bar = 20 μm C) Filter trap assay to quantify reduction of Ataxin-7 aggregates in the retinas of SCA7 266Q knock-in mice. Mice received a single intravitreal injection of 50 μg of Ataxin-7 ASO in the right eye, and a single intravitreal injection of PBS in the left eye. Here we see a representative filter trap assay, where each sample represents protein lysates from a single retina of untreated non- transgenic control mice (NT) or SCA7 266Q mice at 6 weeks after intravitreal injection. Protein lysates are filtered through a 0.2 μm pore size cellulose acetate membrane such that insoluble protein is retained on the filter because it cannot pass through the membrane. D) Densitometry quantification of Ataxin-7 immunoreactivity in filter trap assay shown in (C). ***P < 0.001; t-test. Error bars = s.e.m. (n = 3 – 4 mice / group)
Ataxin-7 ASO ameliorates vision loss in SCA7 knock-in mice
While reduction in polyQ-Ataxin-7 protein aggregation upon ASO IVI is encouraging, the status of visual function in response to ASO treatment is a crucial outcome measure for judging potential therapeutic efficacy. To assess visual function, we performed ERG analysis of ASO-treated SCA7 266Q knock-in mice at 4 weeks and 6 weeks post-IVI, comparing IVI of Ataxin-7 ASO with IVI of a scrambled control ASO or of the vehicle PBS. We began by evaluating cone-specific photopic responses, and documented enhanced cone photoreceptor function in the eyes of SCA7 266Q knock-in mice treated with Ataxin-7 ASO at both 4 weeks and 6 weeks post-IVI (Fig. 3). Based upon comparison of cone photoreceptor maximal b-wave amplitudes, Ataxin-7 ASO treated eyes of SCA7 266Q mice displayed increased visual function at 4 weeks post-IVI compared to vehicle or control-ASO-treated eyes (Fig. 3, A and B), and retained good visual function at 6 weeks post-IVI (Fig. 3, C and D). In the case of rod photoreceptor function, ERG analysis of scotopic responses revealed an even more favorable outcome, as Ataxin-7 ASO treated eyes of SCA7 266Q mice retained normal visual function at 4 weeks post-IVI (Fig. 4, A and B), and maintained excellent visual function at 6 weeks post-IVI (Fig. 4, C and D). At both time points, the maximal b-wave amplitudes of rod photoreceptors in SCA7 266Q knock-in mice were found to be markedly increased with Ataxin-7 ASO treatment in comparison to control ASO treatment or vehicle treatment (Fig. 4), although even with ASO treatment, there is a slight delay in achieving maximal b-wave amplitude in comparison to non-transgenic littermate controls, which reflects a slowed response in SCA7 retinal disease. In addition to evaluating the response to ASO IVI in the eyes of treated SCA7 266Q mice, we performed a series of IVI experiments comparing Ataxin-7 ASO treatment to vehicle treatment in normal control C57BL/6J mice to determine if knock-down of normal Ataxin-7 deleteriously affects visual function. We observed no differences in cone or rod photoreceptor responses between ASO-treated eyes and vehicle-treated eyes on ERG analysis in treated wild-type control mice (fig. S5, A and B).
Figure 3. Ataxin-7 ASO treatment improves cone visual function in SCA7 266Q knock-in mice.

A) Representative ERG recording of photopic (cone) responses in untreated wild-type (non-transgenic) and SCA7 266Q mice at 4 weeks after intravitreal injection of 50 μg of Ataxin-7 ASO in the right eye, and intravitreal injection of a control ASO or PBS in the left eye. B) Quantification of mean maximal b-wave amplitudes for ERG analysis shown in (A). *P < 0.05; ***P < 0.001; ANOVA with post-hoc Tukey test. n = 6 – 8 mice / group. NT = non-transgenic control mice. C) Representative ERG recording of photopic (cone) responses in untreated wild-type (non-transgenic) and SCA7 266Q mice at 6 weeks after intravitreal injection of 50 μg of Ataxin-7 ASO in the right eye, and intravitreal injection of a control ASO or PBS in the left eye. D) Quantification of mean maximal b-wave amplitudes for ERG analysis shown in (C). *P < 0.05; ***P < 0.001; ANOVA with post-hoc Tukey test. n = 7 – 10 mice / group. NT = non-transgenic control mice. Error bars = s.e.m.
Figure 4. Ataxin-7 ASO treatment rescues rod photoreceptor visual function in SCA7 266Q knock-in mice.

A) Representative ERG recording of scotopic (rod) responses in untreated wild-type (non-transgenic) and SCA7 266Q mice at 4 weeks after intravitreal injection of 50 μg of Ataxin-7 ASO in the right eye, and intravitreal injection of a control ASO or PBS in the left eye. B) Quantification of mean maximal b-wave amplitudes for ERG analysis shown in (A). *P < 0.05; ANOVA with post-hoc Tukey test. n = 11 – 13 mice / group. NT = non-transgenic control mice. C) Representative ERG recording of scotopic (rod) responses in untreated wild-type (non-transgenic) and SCA7 266Q mice at 6 weeks after intravitreal injection of 50 μg of Ataxin-7 ASO in the right eye, and intravitreal injection of a control ASO or PBS in the left eye. D) Quantification of mean maximal b-wave amplitudes for ERG analysis shown in (C). **P < 0.01; ***p < 0.001; ANOVA with post-hoc Tukey test. n = 11 – 12 mice / group. NT = non- transgenic control mice. Error bars = s.e.m.
Ataxin-7 ASO rescues retinal histopathology, photoreceptor gene expression defects, and altered chromatin remodeling in the eyes of treated SCA7 knock-in mice
Another important read-out of retinal degeneration is thinning of the different layers that comprise the retina. To determine if Ataxin-7 knock-down impacts this pathological process, we performed hematoxylin and eosin staining of retinal sections from treated SCA7 266Q knock-in mice. SCA7- induced thinning was reduced in the outer segments (OS) and inner segments (IS) of retinal photoreceptors, the outer nuclear layer (ONL) which comprises the nuclei of photoreceptors, and the inner plexiform layer (IPL) which comprises the axonal projections of relay neurons whose nuclei are located in the inner nuclear layer (INL) in Ataxin-7 ASO-treated eyes in comparison to vehicle treatment (Fig. 5, A and B). In addition to this histopathology, we, and others have shown that SCA7 retinal degeneration is characterized by decreased expression of certain photoreceptor- expressed genes (26, 27). To assess if Ataxin-7 knock-down ameliorates SCA7 retinal disease-associated gene expression alterations, we performed RT-PCR analysis of retinal RNAs of three genes previously shown to be downregulated in SCA-7 266Q knock-in mice, Rhodopsin, M-opsin and S-opsin. We documented an amelioration of expression reduction for all three genes in Ataxin- 7 ASO-treated eyes compared to vehicle-treated SCA-7 266Q knock-in mice (Fig. 5C), but observed no effect of Ataxin-7 ASO treatment on genes whose expression is not altered in diseased SCA7 retinas (fig. S6, A and B). As Ataxin-7 protein is a core component of a transcription coactivator complex known as STAGA (13, 14), and mutant polyQ-Ataxin-7 interferes with STAGA-dependent chromatin remodeling to impair transcription of STAGA-regulated genes, we performed chromatin immunoprecipitation (ChIP) of histone modifications subject to STAGA regulation on retinal DNAs isolated from Ataxin-7 ASO-treated and vehicle-treated eyes from SCA7 266Q knock-in mice. We measured histone modification as a function of wild-type control by qPCR analysis of ChIP’d DNAs, and documented rescue of altered histone H2B deubiquitination and histone H3 acetylation in Ataxin-7 ASO-treated eyes from SCA7 266Q knock-in mice (Fig. 5, D and E). Quantification of histone H3 acetylation at the promoter of the interphotoreceptor retinoid binding protein (IRBP) gene, whose expression is not altered in SCA7 retinal degeneration, revealed no evidence for a general effect of Ataxin-7 ASO treatment on histone H3 acetylation at retinal-expressed genes (fig. S6C). These findings indicate that Ataxin- 7 ASO-mediated knock-down ameliorates SCA7 retinal degeneration at the cellular, molecular, and biochemical level.
Figure 5. Ataxin-7 ASO treatment ameliorates retinal degeneration, photoreceptor gene expression, and epigenetic dysregulation in SCA7 266Q knock-in mice.

A) Retinal histology of untreated non-transgenic control and SCA7 266Q mice at 6 weeks after intravitreal injection of 50 μg of Ataxin-7 ASO in the right eye, and intravitreal injection of PBS in the left eye. Sections were stained with hematoxylin and eosin. OS = outer segments; IS = inner segments; ONL = outer nuclear layer; OPL = outer plexiform layer; INL = inner nuclear layer; IPL = inner plexiform layer; GCL = ganglion cell layer. Scale bar = 20 μm B) Quantification of thickness for indicated retinal layers from (A). *P < 0.05, **P < 0.01; ***P< 0.001; ANOVA with post-hoc Tukey test. n = 4 – 5 mice / group. C) Quantification of expression of indicated genes in retinal RNAs by RT-PCR analysis for SCA7 266Q mice at 6 weeks after intravitreal injection of 50 μg of Ataxin-7 ASO in the right eye, and intravitreal injection of PBS in the left eye. *P < 0.05, ***P < 0.001; ANOVA with post-hoc Tukey test. Comparison of RNA expression for PBS-treated SCA7 mice vs. Ataxin-7 ASO-treated SCA7 mice was significant by t-test for M-opsin (P < 0.001) and for S-opsin (P < 0.05). n = 4 −6 mice / group. D) Quantification of histone H2B ubiquitination at the promoters of indicated genes by real-time PCR analysis of retinal DNAs isolated by chromatin immunoprecipitation from SCA7 266Q mice at 6 weeks after intravitreal injection of 50 μ.g of Ataxin-7 ASO in the right eye, and intravitreal injection of PBS in the left eye. *P < 0.05, **P < 0.01; ***P < 0.001; ANOVA with post-hoc Tukey test. n = 5 – 7 mice / group. E) Quantification of histone H3 acetylation at the enhancers / promoters of indicated genes by real-time PCR analysis of retinal DNAs isolated by chromatin immunoprecipitation from SCA7 266Q mice at 6 weeks after intravitreal injection of 50 μg of Ataxin-7 ASO in the right eye, and intravitreal injection of PBS in the left eye. *P < 0.05, **P < 0.01; ***P < 0.001; ANOVA with post-hoc Tukey test. n = 4 – 5 mice / group. M-opsin, M-cone opsin; RER, rhodopsin enhancer region; LCR, locus control region of M-cone opsin; S-opsin, S-cone opsin; SER, S-cone opsin enhancer region. Error bars = s.e.m.
CAG repeat-targeting ASO transiently ameliorates SCA7 retinal disease phenotypes
As SCA7 is an autosomal dominant disorder in which the CAG repeat length of the normal allele is usually only 10 or 11 CAGs, and the CAG repeat length of the disease-causing expanded allele ranges from 37 to 300+ CAGs in affected patients, with many SCA7 patients carrying disease alleles with >50 CAG repeats, another possible strategy for ASO therapy would be to selectively target the expanded CAG repeat tract of the mutant Ataxin-7 gene (28). We thus synthesized a small set of CAG-repeat targeting oligonucleotides, and based upon in vivo safety testing, we narrowed down these leads to two CAG repeat-targeting ASOs, which were then tested in SCA7 patient fibroblasts. The results of these studies indicated superior knock-down of polyQ-Ataxin- 7 protein expression by CAG ASO #2 (fig. S7A-B); hence, we selected this CAG repeat-targeting ASO for therapeutic evaluation in a preclinical prevention trial, identical in design to the Ataxin-7 ASO study just described. Intra-vitreal injection of this CAG repeat-targeting ASO into the right eye of SCA7 266Q knock-in mice did not reduce endogenous normal Ataxin-7 protein (fig. S8A), as expected, since the mouse Ataxin-7 gene contains only 5 CAG repeats. IVI of the CAG repeat- targeting ASO into the right eye of SCA7 266Q knock-in mice did achieve a reduction in Ataxin- 7 protein aggregation based upon immunostaining of retinal sections and filter trap analysis (Fig.6, A and B). However, inspection of the filter trap assay results indicated that reduction of protein aggregation at 6 weeks post-IVI was often modest (fig. S8B), particularly when compared to treatment with the Ataxin-7ASO.Thus, while CAG-ASO treatment did initially yield improved visual function at 4 weeks post-IVI in SCA7 266Q knock-in mice in comparison to vehicle treatment (Fig. 6C), by 6 weeks post-IVI, visual function was not different between CAG-ASO treated eyes and vehicle-treated eyes (Fig. 6D). Results for retinal morphology analysis paralleled the findings observed for visual function, as CAG-ASO treated eyes displayed thicker outer segments and inner segments, ONL, and IPL at 4 weeks post-IVI (fig. S8C). Thus, CAG-ASO therapy could preserve the morphologies of SCA7 266Q knock-in retinas and did improve visual function in treated eyes at 4-weeks post-IVI, but less so than the Ataxin-7 ASO approach that promoted RNase H1-mediated destruction of the Ataxin-7 RNA. Moreover, by 6 weeks post-IVI, there was no benefit in the eyes of SCA7 266Q knock-in mice undergoing treatment with the CAG repeat-targeting ASO.
Figure 6. CAG-ASO treatment partially improves SCA7 retinal degeneration in mice.

A) Effect of CAG-ASO knock-down on visible aggregates in the retinas of SCA7 266Q knock-in mice. Mice received a single intravitreal injection of 50 μg of CAG-ASO in the right eye, and a single intravitreal injection of PBS in the left eye. Retinal sections were immunostained with an anti-Ataxin-7 antibody (green) and counterstained with Hoechst 33342 (blue) at 6 weeks after intravitreal injection. ONL = outer nuclear layer; INL = inner nuclear layer; GCL = ganglion cell layer. Scale bar = 20 μm B) Filter trap assay to quantify reduction of Ataxin-7 aggregates in the retinas of SCA7 266Q knock-in mice. Mice received a single intravitreal injection of 50 μg of CAG-ASO in the right eye, and a single intravitreal injection of PBS in the left eye. Here we see densitometry quantification of Ataxin-7 immunoreactivity in filter trap assays at 6 weeks post-injection. **P < 0.01; ***P < 0.001; ANOVA with post-hoc Tukey test. n = 5 mice / group. C) Quantification of mean maximal b-wave amplitudes for ERG analysis performed on untreated wild-type (non-transgenic) and SCA7 266Q mice at 4 weeks after intravitreal injection of 50 μg of CAG-ASO in the right eye, and intravitreal injection of PBS in the left eye. *P < 0.05, **P < 0.01; ***P < 0.001; ANOVA with post-hoc Tukey test. n = 8 – 11 mice / group. D) Quantification of mean maximal b-wave amplitudes for ERG analysis performed on untreated wild-type (non-transgenic) and SCA7 266Q mice at 6 weeks after intravitreal injection of 50 μg of CAG-ASO in the right eye, and intravitreal injection of PBS in the left eye. **P < 0.01; ***P <0.001; ANOVA with post-hoc Tukey test. n = 8 – 13 mice / group. Error bars = s.e.m.
Human SCA7 patients display variable severity of retinal degeneration and rapid retinal disease progression
Although ASOs achieved therapeutic efficacy in preclinical experimentation in mice where treatment is initiated prior to onset of SCA7 retinal degeneration, a crucial question is whether therapeutic benefit can be derived after SCA7 patients develop obvious disease. To better understand the clinical features of SCA7 retinal degeneration and the rapidity of its progression, we performed a series of ophthalmologic evaluations on two unrelated SCA7 patients and monitored disease progression in one patient. We began by performing color fundus photography and autofluorescence imaging on the retina of SCA7 patient #1, a 55 year-old woman with a 40 CAG repeat allele, and observed minimal foveal autofluorescence changes (Fig. 7A, top). In contrast, when we evaluated SCA7 patient #2, a 36 year-old woman with a 51 CAG repeat allele, we observed moderate foveal hypoautofluorescence at the initial evaluation (Fig. 7A, middle), which progressed to markedly increased hypoautofluorescence at her three-year follow-up examination (Fig. 7A, bottom). We also obtained cross-sectional images of the retina by optical coherence tomography (OCT), which yielded a nearly normal-appearing retina in mildly affected patient #1 (Fig 7B, top), but revealed significant retinal pathology in patient #2 at initial presentation (Fig. 7B, middle) that worsened to much more extensive retinal pathology at her three-year follow-up examination (Fig. 7B, bottom). Lastly, we performed a functional evaluation with microperimetry on these two SCA7 patients. We noted a mild deficit in retinal sensitivity in patient #1 (Fig. 7C, top), but a severe deficit in retinal sensitivity in patient #2 (Fig. 7C, bottom). These clinical studies demonstrated the variability in retinal disease in different SCA7 patients and documented the fast progression of SCA7 retinal degeneration.
Figure 7. Documentation of phenotype variability and progressive retinal degeneration in human SCA7 patients.

A) Color fundus photos (left) and blue-light autofluorescence imaging (right) representing different stages of retinal disease in patients with SCA7. The top image row is from a 55 year-old SCA7 patient (#1; 40 CAG repeat allele) with minimal changes in fundus autofluorescence. The middle and bottom rows represent testing of a 36-year-old SCA7 patient (#2; 51 CAG repeat allele) with moderate disease (middle) that progressed to advanced disease (bottom) after three years. Arrow indicates significant increase in hypo-autofluorescence within the macula, which is indicative of the retinal pigment epithelium atrophy of very severe disease. B) Corresponding ocular coherence tomography (OCT) scans of SCA7 patients #1 and #2. C) Fundus-guided microperimetry testing of SCA7 patient #1 (mild disease) and SCA7 patient #2 (severe disease). The red cross indicates the area that the patient fixates on the target, while being serially presented with points of light of varying intensities at different positions across the retina (colored, numbered dots). Green dots demarcate positions on the retina where the patient perceives a dim light stimulus, while yellow dots demarcate positions on the retina where the patient can only detect a more intense light stimulus, and red open dots indicate positions where there is no detection of the light stimulus.
Ataxin-7 ASO therapy after disease onset can ameliorate retinal degeneration in SCA7 266Q knock-in mice
Given the likelihood of significant pre-existing retinal disease at the time of presentation in SCA7 patients, we sought to perform a preclinical study initiating the treatment after the appearance of marked visual impairment. As the SCA7 266Q knock-in mice display a very severe phenotype with death as early as 10 weeks of age, the window of time for tracking therapeutic response after disease onset is too narrow in this SCA7 line for pursuit of a meaningful study. However, in the course of maintaining this line, we detected a knock-in positive individual with reduced disease severity, and upon breeding, this SCA7 knock-in subline did not display onset of ataxia until 12 – 14 weeks of age, and did not succumb to disease until 24 – 28 weeks of age. When we characterized these mice, we determined that their CAG repeat mutation had contracted to 210 CAGs (fig. S9A), and named this new subline the “SCA7 r210” knock-in model. Using a neurological screening exam, we determined that SCA7 r210 mice exhibit motor impairment by 12 weeks of age, but do not display severe neurological dysfunction until after 18 weeks of age (fig. S9, B and C). We also performed ERG analysis, and documented visual impairment at 9 weeks of age (fig. S10 and S11), in contrast to control mice (fig. S12). Hence, we chose to pursue a preclinical intervention trial with this new SCA7 knock-in model, and decided to test IVI delivery of Ataxin-7 ASO at 9 weeks of age, as we confirmed substantial vision loss at this time point in the SCA7 r210 mice (fig. S13, A and B). For the preclinical intervention trial, we employed the same outcome measures used previously, but with a different time line (fig. S14).
To determine if Ataxin-7 ASO treatment can achieve robust and sustained knock-down of polyQ- expanded Ataxin-7, we compared the extent of Ataxin-7 protein aggregation between Ataxin-7 ASO-treated eyes and vehicle-treated eyes in SCA7 r210 mice at 9 weeks post-IVI, and observed a marked reduction in Ataxin-7 aggregation in ASO-treated eyes, based upon immunostaining of retinal sections and filter trap assay (Fig. 8, A to C). We then considered visual function by tracking ERG responses at 6 and 9 weeks post-IVI. At 6 weeks post-IVI, maximal b-wave amplitudes for both cone photoreceptors and rod photoreceptors were markedly increased in the Ataxin-7 ASO treated eyes of SCA7 r210 knock-in mice (Fig. 8D, top), indicating that therapeutic benefit can be achieved even after substantive visual dysfunction is established. However, Ataxin- 7 ASO treated eyes still showed impairments compared to wild-type animals, Preservation of rod photoreceptor function in SCA7 r210 mice treated after symptom onset far exceeded that of cone photoreceptor function (Fig. 8D, bottom), which is consistent with the more rapid loss of cone function in SCA7 cone-rod dystrophy. At 9 weeks post-IVI, we observed no improvements in cone photoreceptor function with Ataxin-7 ASO treatment (Fig. 8E, top), whereas rod photoreceptor function was improved also at this later time point (Fig. 8E, bottom), compared to vehicle-treated eyes. We also measured retinal layer thickness and quantified photoreceptor gene expression at the 9 weeks post-IVI time point. At this time point the treatment did not induce improvements (fig. S15, A and B). Finally, to assess the duration of action of Ataxin-7 ASO knock-down, we repeated the Ataxin-7 ASO prevention trial, but this time in the SCA7 r210 mice, and found that visual function is improved with Ataxin-7 ASO knock-down over 11 weeks post-IVI (fig. S16, A and B).
Figure 8. Post-symptomatic treatment of SCA7 retinal degeneration with Ataxin-7 ASO achieves significant beneficial therapeutic response in mice.

A) Effect of Ataxin-7 ASO knock-down on visible aggregates in the retinas of symptomatic SCA7 r210 knock-in mice. Mice received a single intravitreal injection of 50 μg of Ataxin-7 ASO in the right eye, and a single intravitreal injection of PBS in the left eye. Retinal sections were immunostained with an anti-Ataxin-7 antibody (green) and counterstained with Hoechst 33342 (blue) at 9 weeks after intravitreal injection. ONL = outer nuclear layer; INL = inner nuclear layer; GCL = ganglion cell layer. Scale bar = 20 μm B) Filter trap assay to quantify reduction of Ataxin-7 aggregates in the retinas of symptomatic SCA7 r210 knock-in mice. Mice received a single intravitreal injection of 50 μg of Ataxin-7 ASO in the right eye, and a single intravitreal injection of PBS in the left eye. Here we see a representative filter trap assay, where each sample represents protein lysates from a single retina of untreated control mice (WT) or SCA7 r210 mice at 9 weeks after intravitreal injection. C) Densitometry quantification of Ataxin-7 immunoreactivity in filter trap assays shown in (B). ***P < 0.001; t-test. (n = 3 – 5 mice / group) D) Quantification of mean maximal b-wave amplitudes for ERG analysis performed on wild-type (non-transgenic, NT) and SCA7 r210 mice at 6 weeks after intravitreal injection of 50 μg of Ataxin-7 ASO in the right eye, and intravitreal injection of PBS in the left eye. *P < 0.05; t-test. n = 5 mice / group. E) Quantification of mean maximal b-wave amplitudes for ERG analysis performed on wild-type (non-transgenic, NT) and SCA7 r210 mice at 9 weeks after intravitreal injection of 50 μ,g of Ataxin-7 ASO in the right eye, and intravitreal injection of PBS in the left eye. *P < 0.05; t-test. n = 5 – 6 mice / group. Error bars = s.e.m.
Discussion
ASOs are chemically modified single-stranded nucleic acids that form heteroduplexes with their target RNAs, and in the case of DNA-RNA heteroduplex formation, the targeted RNA is degraded by RNAse H1 (29, 30). The production of protein from target mRNA is then typically reduced by greater than 50%. Because of impressive advances in their chemistry, ASOs exhibit dramatically increased potency and duration of action, while displaying limited toxicity, especially in the CNS. Indeed, successful development of an oligonucleotide therapy to treat autosomal recessive spinal muscular atrophy by increasing survival of motor neuron (SMN) protein expression from the paralogous gene SMN2 through alternative splicing has demonstrated that periodic CNS delivery by intrathecal injection is a viable regimen for human patients (21), heralding a powerful new approach for neurotherapeutics. While such a therapy for spinal muscular atrophy is an incredible advance, it is still unclear whether similar approaches can be applied to neurodegenerative proteinopathies, where dosage reduction of a target misfolded protein product is the treatment goal.
SCA7 is a rare neurodegenerative disorder caused by a dominant gain-of-function CAG-polyQ repeat expansion in one of the two ATAXIN-7 genes in an affected patient. In addition to significant neurological abnormalities due to dysfunction of the cerebellum, brainstem, and corticospinal tracts, SCA7 patients develop an unusual form of retinal degeneration, known as a cone-rod dystrophy (31). SCA7 is also unique among neurodegenerative repeat diseases, because of all such disorders, SCA7 displays the most dramatic anticipation, with many patients presenting before the age of 30 years, in many cases prior to the onset of disease in their affected parent, despite the fact that the affected parent is more than two or three decades older than their affected child (6). Based upon clinical experience, SCA7 patients can present with either neurological signs or vision loss, and retinal degeneration is the initial presentation in such childhood, juvenile, or young adult SCA7 patients with ≥ 59 CAG repeats (8, 9). Because of these clinical demographics, SCA7 retinal degeneration also tends to progress more rapidly in these younger SCA7 patients than in older patients who typically present with ataxia (32). Furthermore, the eye is a highly accessible neural tissue, and evaluation of retinal function can be performed in an objective, quantitative fashion. For all these reasons, SCA7 retinal disease offers an unparalleled opportunity to evaluate ASO dosage reduction therapy in a discrete and definitive manner.
Here we have taken the first steps towards developing an antisense-based therapy for SCA7 by targeting Ataxin-7 with a specific ASO. After an extensive process of oligonucleotide design, manufacture, and screening, we identified two lead ASOs, one directed against the Ataxin-7 RNA, and one directed against expanded CAG repeat-containing RNA. We validated these lead ASOs for knock-down efficacy in vitro, and tested them for potency and toxicity in vivo. To achieve delivery to retinal photoreceptors, we established intravitreal injection (IVI) as a viable and highly reproducible technique in SCA7 knock-in mice, and demonstrated that IVI could achieve marked, sustained knock-down of Ataxin-7 expression. There is good reason to expect that IVI will be an effective delivery route for ASO treatment of SCA7 retinal degeneration, as fomivirsen, the very first ASO drug ever approved by the FDA, is delivered by IVI as a treatment for CMV retinitis (33). More recently, first an aptamer and then an antibody against vascular endothelial growth factor have been developed as treatments for age-related macular degeneration and are delivered via IVI (34). To evaluate ASOs as potential therapies for SCA7 retinal degeneration, we performed a preclinical prevention trial, where we compared ASO-injected eyes with vehicle- injected eyes in a very severe SCA7 knock-in mouse model that expresses Ataxin-7 with 266 CAG repeats. Using Ataxin-7 aggregation, cone and rod photoreceptor function, retinal histopathology, and photoreceptor gene expression as read-outs, we found that Ataxin-7 ASO injection was a highly effective treatment, based upon marked long-term improvements in all of these outcome measures. CAG repeat-targeting ASO was less effective than the Ataxin-7 ASO and shorter in duration.. Although the basis for the inferior performance of the CAG repeat-targeting ASO is uncertain, there are >55 repeat tracts composed of at least 6 CAGs or more in the human and rodent genomes (35); hence, competition for the CAG repeat-targeting ASO from other CAG repeat- containing RNAs could diminish the potency of Ataxin-7 RNA knock-down. In light of these results, and because of concerns over off-target effects of CAG repeat-targeting ASOs, as some normal genes possess CAG repeat tracts numbering into the upper 30’s, we favor use of a non- CAG Ataxin-7 targeting ASO for SCA7 therapy development.
ASO dosage reduction can be “non-allele specific”, meaning that both the disease allele and the normal allele mRNAs are targeted, or “allele-specific”, meaning that the disease allele is selectively targeted, usually through a single nucleotide polymorphism that is in linkage with the disease allele. While allele-specific targeting would be ideal, studies in mouse models of Huntington’s disease have established the safety and efficacy of non-allele specific ASOs (36, 37).
We addressed the issue of whether concomitantly targeting the normal Ataxin-7 gene would produce impaired function or toxicity in the eye by performing IVI of Ataxin-7 ASO in wild-type control mice and then assessing their visual function. We did not observe any visual impairment in control mice subjected to Ataxin-7 ASO knock-down, even when Ataxin-7 mRNA expression was reduced by >70%. As ATAXIN-7 has three homologues in mammals, known as ATAXIN-7- like-1, −2, and −3, certain ATAXIN-7 functions could be compensated for by these homologues. Indeed, ATAXIN-7-like-3 is known to reside in the STAGA co-activator complex with ATAXIN-7, and has been shown to play a role in regulating the STAGA deubiquitinase module (38). Thus, while our results indicate that non-allele specific Ataxin-7 ASO therapy is likely to be well tolerated, we did not follow ASO-treated wild-type mice long enough to completely rule out potential ocular or systemic toxicity. Hence, as we observed equivalent targeting of Ataxin-7 mutant and normal alleles in treated SCA7 knock-in mice, an ideal non-allele specific ASO therapy should achieve knock-down of no more than 70% to obviate such toxicity concerns. Furthermore, our preclinical trial work relied on mouse models of SCA7 retinal degeneration, which recapitulate the cone-rod dystrophy phenotype but cannot completely represent this disease phenotype, because murine photoreceptor complexity and retinal anatomical organization differ from that of the human. These limitations of our study necessitate more detailed and extensive experimentation to rule out potential on-target or off-target toxicities of Ataxin-7 ASO knock-down, and require that we be circumspect in our extrapolation of our results to human SCA7 patients.
Although presymptomatic diagnosis by genetic testing can be pursued in at-risk children of SCA7 patients, currently most SCA7 patients do not come to diagnosis until they display ataxia or impaired vision. Hence, in many cases, initiation of therapy will not be possible until after SCA7 patients develop significant symptomatology. As part of this study, we piloted the use of three different ophthalmological testing modalities on two SCA7 patients, and observed variable retinal degeneration. We performed a follow-up examination on the more severely affected patient and documented rapid disease progression based upon increased macular autofluorescence and more extensive photoreceptor later degeneration on OCT, accompanied by very poor light detection across the retina on microperimetry. For these reasons, we pursued a preclinical intervention trial, where we tested if IVI delivery of Ataxin-7 ASO would be effective in SCA7 knock-in mice with significant retinal disease. This study illustrated that Ataxin-7 ASO therapy could reduce protein aggregation for the duration of the trial, and could ameliorate impaired visual function for both rods and cones at the initial post-treatment time point. However, at the final time point, Ataxin-7 ASO therapy only ameliorated rod photoreceptor function, but was ineffective for cone photoreceptor function, retinal histopathology, and gene expression alterations. As cone photoreceptor dysfunction begins earlier than rod photoreceptor abnormalities and proceeds much more rapidly (16), by 9 weeks of age, the time point for ASO delivery for the SCA7 r210 knock- in mice, cone photoreceptor function is reduced by ~70%. However, for rod photoreceptor function, where SCA7 r210 mice exhibit a decline of ~40% of their scotopic ERG responses by 9 weeks of age, Ataxin-7 ASO treatment did retard disease progression. Hence, given the aggressive nature of disease in the SCA7 knock-in mice, our results suggest that Ataxin-7 ASO treatment might ameliorate disease progression even when administered after symptoms develop. As we have shown that SCA7 cerebellar degeneration is reversible in mice upon termination of mutant protein expression (39), we conclude that robust knock-down of ATAXIN-7 expression might slow disease progression, even when SCA7 patients have significant disease, but we anticipate that there is likely a point at which disease becomes too severe to allow for meaningful intervention. Determining the threshold at which a beneficial response is no longer possible will be a challenge for SCA7 therapy development, as it will be for all neurodegenerative disorders.
Materials & Methods
Study design
The primary objective of this study was to determine if antisense oligonucleotide (ASO) knock-down therapy is an effective treatment for SCA7 retinal degeneration. ASOs were designed and screened for efficacy of knock-down in cell lines and in wild-type mice in experiments that used quantitative real-time PCR assays and Western blot analysis. ASOs were tested in preclinical trials in SCA7 knock-in mouse models, with aggregate accumulation, visual function, retinal histology, and gene expression as the outcome measures. The preclinical trials, which were approved by and performed in accordance with the University of California, San Diego and Duke Institutional Animal Care and Use Committees, adhered to a protocol where we arbitrarily divided littermates and balanced genders between experimental groups, with behavioral testing performed by investigators blinded to the treatment group of the mice. We based our group sizes upon power analysis to achieve 80% likelihood of detection of a 30% rescue of phenotypes, employing the G Power 3.1 software package (40). As part of this study, we performed ophthalmologic evaluations of a set of SCA7 patients, tracking disease progression over time in one case. For all experiments, replicate numbers are stated in the Figure legends.
Statistical analysis
All data were prepared for analysis with standard spreadsheet software (Microsoft Excel). Statistical analysis was done using Prism 6.0 (Graph Pad), or SigmaStat (Systat Software). For ANOVA, if statistical significance (P<0.05) was achieved, we performed post-hoc analysis to account for multiple comparisons. All t-tests were two-tailed Student’s t-test, and the level of significance (alpha) was always set to 0.05.
Supplementary Material
Acknowledgments
We are grateful to H. Kordasiewicz and T. Cole for their assistance with this project.
Funding
Supported by the National Ataxia Foundation, Harrington Discovery Initiative, Foundation Fighting Blindness, and grants from the N.I.H. (R01 EY024747 and R01 EY014061 to A.R.L., and P30EY022589 to UCSD).
Footnotes
Competing interests
The following individuals were employees of and/or shareholders in Ionis Pharmaceuticals during the conduct of these studies: C.F. Bennett, Sr. Vice President for Research, employee, shareholder with stock options; E.E. Swayze, employee, shareholder with stock options; A. Kim, employee, shareholder; T. Prakash, employee; and G. Hung, employee. All ASOs used in this study are available from Ionis Pharmaceuticals under a material transfer agreement (MTA) with Ionis Pharmaceuticals. Ionis Pharmaceuticals has filed patents covering nucleoside modifications and oligonucleotides comprising the modified nucleosides for therapeutic efficacy through the RNase H mechanism. The relevant patents for the 2’-MOE generation ASOs employed in this study are as follows: U.S. Patent No. 5,914,396: “2’-O-MODIFIED NUCLEOSIDES AND PHOSPHORAMIDITES”; U.S. Patent No. 7,015,315: “GAPPED OLIGONUCLEOTIDES”; and U.S. Patent No. 7,101,993: “OLIGONUCLEOTIDES CONTAINING 2’O-MODIFIED PURINES”.
Data and materials availability
All data are present in the paper and/or the Supplementary Materials
References
- 1.Martin JJ, Van Regemorter N, Krols L, Brucher JM, de Barsy T, Szliwowski H, Evrard P, Ceuterick C, Tassignon MJ, Smet-Dieleman H, et al. , On an autosomal dominant form of retinal-cerebellar degeneration: an autopsy study of five patients in one family. Acta Neuropathol 88, 277–286 (1994). [DOI] [PubMed] [Google Scholar]
- 2.Filla A, Mariotti C, Caruso G, Coppola G, Cocozza S, Castaldo I, Calabrese O, Salvatore E, De Michele G, Riggio MC, Pareyson D, Gellera C, Di Donato S, Relative frequencies of CAG expansions in spinocerebellar ataxia and dentatorubropallidoluysian atrophy in 116 Italian families. Eur Neurol 44, 31–36 (2000). [DOI] [PubMed] [Google Scholar]
- 3.Storey E, du Sart D, Shaw JH, Lorentzos P, Kelly L, McKinley Gardner RJ, Forrest SM, Biros I, Nicholson GA , Frequency of spinocerebellar ataxia types 1, 2, 3, 6, and 7 in Australian patients with spinocerebellar ataxia. Am J Med Genet 95, 351–357 (2000). [DOI] [PubMed] [Google Scholar]
- 4.Trottier Y, Lutz Y, Stevanin G, Imbert G, Devys D, Cancel G, Saudou F, Weber C, David G, Tora L, et al. , Polyglutamine expansion as a pathological epitope in Huntington’s disease and four dominant cerebellar ataxias. Nature 378, 403–406. (1995). [DOI] [PubMed] [Google Scholar]
- 5.David G, Abbas N, Stevanin G, Durr A, Yvert G, Cancel G, Weber C, Imbert G, Saudou F, Antoniou E, Drabkin H, Gemmill R, Giunti P, Benomar A, Wood N, Ruberg M, Agid Y, Mandel JL, Brice A, Cloning of the SCA7 gene reveals a highly unstable CAG repeat expansion. Nat Genet 17, 65–70 (1997). [DOI] [PubMed] [Google Scholar]
- 6.Benton CS, de Silva R, Rutledge SL, Bohlega S, Ashizawa T, Zoghbi HY, Molecular and clinical studies in SCA-7 define a broad clinical spectrum and the infantile phenotype. Neurology 51, 1081–1086 (1998). [DOI] [PubMed] [Google Scholar]
- 7.David G, Durr A, Stevanin G, Cancel G, Abbas N, Benomar A, Belal S, Lebre AS, Abada-Bendib M, Grid D, Holmberg M, Yahyaoui M, Hentati F, Chkili T, Agid Y, Brice A, Molecular and clinical correlations in autosomal dominant cerebellar ataxia with progressive macular dystrophy (SCA7). Hum Mol Genet 7, 165–170 (1998). [DOI] [PubMed] [Google Scholar]
- 8.Johansson J, Forsgren L, Sandgren O, Brice A, Holmgren G, Holmberg M, Expanded CAG repeats in Swedish spinocerebellar ataxia type 7 (SCA7) patients: effect of CAG repeat length on the clinical manifestation. Hum Mol Genet 7, 171–176 (1998). [DOI] [PubMed] [Google Scholar]
- 9.Gouw LG, Castaneda MA, McKenna CK, Digre KB, Pulst SM, Perlman S, Lee MS, Gomez C, Fischbeck K, Gagnon D, Storey E, Bird T, Jeri FR, Ptacek LJ, Analysis of the dynamic mutation in the SCA7 gene shows marked parental effects on CAG repeat transmission. Hum Mol Genet 7, 525–532 (1998). [DOI] [PubMed] [Google Scholar]
- 10.Paulson HL, Bonini NM, Roth KA, Polyglutamine disease and neuronal cell death. Proc Natl Acad Sci U S A 97, 12957–12958 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.A. C Ross, Intranuclear neuronal inclusions: a common pathogenic mechanism for glutamine-repeat neurodegenerative diseases? Neuron 19, 1147–1150 (1997). [DOI] [PubMed] [Google Scholar]
- 12.Timmers HT, Tora L, SAGA unveiled. Trends in biochemical sciences 30, 7–10 (2005). [DOI] [PubMed] [Google Scholar]
- 13.Helmlinger D, Hardy S, Sasorith S, Klein F, Robert F, Weber C, Miguet L, Potier N,Van-Dorsselaer A, Wurtz JM, Mandel JL, Tora L, Devys D, Ataxin-7 is a subunit of GCN5 histone acetyltransferase-containing complexes. Hum Mol Genet 13, 1257–1265 (2004). [DOI] [PubMed] [Google Scholar]
- 14.Palhan VB, Chen S, Peng GH, Tjernberg A, Gamper AM, Fan Y, Chait BT, La Spada AR, Roeder RG, Polyglutamine-expanded ataxin-7 inhibits STAGA histone acetyltransferase activity to produce retinal degeneration. Proc Natl Acad Sci U S A 102, 8472–8477 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Helmlinger D, Hardy S, Abou-Sleymane G, Eberlin A, Bowman AB, Gansmuller A, Picaud S, Zoghbi HY, Trottier Y, Tora L, Devys D, Glutamine-expanded ataxin-7 alters TFTC/STAGA recruitment and chromatin structure leading to photoreceptor dysfunction. PLoS biology 4, e67 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.To KW, Adamian M, Jakobiec FA, Berson EL, Olivopontocerebellar atrophy with retinal degeneration. An electroretinographic and histopathologic investigation. Ophthalmology 100, 15–23 (1993). [DOI] [PubMed] [Google Scholar]
- 17.Gouw LG, Digre KB, Harris CP, Haines JH, Ptacek LJ, Autosomal dominant cerebellar ataxia with retinal degeneration: clinical, neuropathologic, and genetic analysis of a large kindred. Neurology 44, 1441–1447 (1994). [DOI] [PubMed] [Google Scholar]
- 18.Enevoldson TP, Sanders MD, Harding AE, Autosomal dominant cerebellar ataxia with pigmentary macular dystrophy. A clinical and genetic study of eight families. Brain 117, 445–460 (1994). [DOI] [PubMed] [Google Scholar]
- 19.La Spada AR, Taylor JP, Repeat expansion disease: progress and puzzles in disease pathogenesis. Nat Rev Genet 11, 247–258 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Crooke ST, Progress in antisense technology. Annu Rev Med 55, 61–95 (2004). [DOI] [PubMed] [Google Scholar]
- 21.Stein CA, Castanotto D, FDA-Approved Oligonucleotide Therapies in 2017. Mol Ther 25, 1069–1075 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Scoles DR, Meera P, Schneider MD, Paul S, Dansithong W, Figueroa KP, Hung G, Rigo F, Bennett CF, Otis TS, Pulst SM, Antisense oligonucleotide therapy for spinocerebellar ataxia type 2. Nature 544, 362–366 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Seth PP, Siwkowski A, Allerson CR, Vasquez G, Lee S, Prakash TP, Wancewicz EV, Witchell D, Swayze EE, Short antisense oligonucleotides with novel 2’- 4’ conformationaly restricted nucleoside analogues show improved potency without increased toxicity in animals. J Med Chem 52, 10–13 (2009). [DOI] [PubMed] [Google Scholar]
- 24.Faktorovich EG, Steinberg RH, Yasumura D, Matthes MT, LaVail MM, Photoreceptor degeneration in inherited retinal dystrophy delayed by basic fibroblast growth factor. Nature 347, 83–86 (1990). [DOI] [PubMed] [Google Scholar]
- 25.Murray SF, Jazayeri A, Matthes MT, Yasumura D, Yang H, Peralta R, Watt A, Freier S, Hung G, Adamson PS, Guo S, Monia BP, LaVail MM, McCaleb ML, Allele- Specific Inhibition of Rhodopsin With an Antisense Oligonucleotide Slows Photoreceptor Cell Degeneration. Investigative ophthalmology & visual science 56, 6362–6375 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.La Spada AR, Fu Y, Sopher BL, Libby RT, Wang X, Li LY, Einum DD, Huang J, Possin DE, Smith AC, Martinez RA, Koszdin KL, Treuting PM, Ware CB, Hurley JB, Ptacek LJ, Chen S, Polyglutamine-expanded ataxin-7 antagonizes CRX function and induces cone-rod dystrophy in a mouse model of SCA7. Neuron 31, 913–927. (2001). [DOI] [PubMed] [Google Scholar]
- 27.Yoo SY, Pennesi ME, Weeber EJ, Xu B, Atkinson R, Chen S, Armstrong DL, Wu SM, Sweatt JD, Zoghbi HY, SCA7 knockin mice model human SCA7 and reveal gradual accumulation of mutant ataxin-7 in neurons and abnormalities in short-term plasticity. Neuron 37, 383–401 (2003). [DOI] [PubMed] [Google Scholar]
- 28.Gagnon KT, Pendergraff HM, Deleavey GF, Swayze EE, Potier P, Randolph J, Roesch E, Chattopadhyaya J, Damha MJ, Bennett CF, Montaillier C, Lemaitre M, Corey DR, Allele-selective inhibition of mutant huntingtin expression with antisense oligonucleotides targeting the expanded CAG repeat. Biochemistry 49, 10166–10178 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Martinez T, Wright N, Lopez-Fraga M, Jimenez AI, Paneda C, Silencing human genetic diseases with oligonucleotide-based therapies. Hum Genet 132, 481–493 (2013). [DOI] [PubMed] [Google Scholar]
- 30.Lu XH, Yang XW, “Huntingtin holiday”: progress toward an antisense therapy for Huntington’s disease. Neuron 74, 964–966 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Aleman TS, Cideciyan AV, Volpe NJ, Stevanin G, Brice A, Jacobson SG, Spinocerebellar ataxia type 7 (SCA7) shows a cone-rod dystrophy phenotype. Exp Eye Res 74, 737–745 (2002). [DOI] [PubMed] [Google Scholar]
- 32.Johansson J, Forsgren L, Sandgren O, Brice A, Holmgren G, Holmberg M, Expanded CAG repeats in Swedish spinocerebellar ataxia type 7 (SCA7) patients: effect of CAG repeat length on the clinical manifestation. Hum Mol Genet 7, 171–176 (1998). [DOI] [PubMed] [Google Scholar]
- 33.Marwick C, First “antisense” drug will treat CMV retinitis. JAMA 280, 871 (1998). [PubMed] [Google Scholar]
- 34.Zehetner C, Kirchmair R, Huber S, Kralinger MT, Kieselbach GF, Plasma levels of vascular endothelial growth factor before and after intravitreal injection of bevacizumab, ranibizumab and pegaptanib in patients with age-related macular degeneration, and in patients with diabetic macular oedema. Br J Ophthalmol 97, 454–459 (2013). [DOI] [PubMed] [Google Scholar]
- 35.Alba MM, Santibanez-Koref MF, Hancock JM, The comparative genomics of polyglutamine repeats: extreme differences in the codon organization of repeat- encoding regions between mammals and Drosophila. J Mol Evol 52, 249–259 (2001). [DOI] [PubMed] [Google Scholar]
- 36.Boudreau RL, Martins I, Davidson BL, Artificial microRNAs as siRNA shuttles: improved safety as compared to shRNAs in vitro and in vivo. Mol Ther 17, 169–175 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kordasiewicz HB, Stanek LM, Wancewicz EV, Mazur C, McAlonis MM, Pytel KA, Artates JW, Weiss A, Cheng SH, Shihabuddin LS, Hung G, Bennett CF, Cleveland DW, Sustained therapeutic reversal of Huntington’s disease by transient repression of huntingtin synthesis. Neuron 74, 1031–1044 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhao Y, Lang G, Ito S, Bonnet J, Metzger E, Sawatsubashi S, Suzuki E, Le Guezennec X, Stunnenberg HG, Krasnov A, Georgieva SG, Schule R, Takeyama K, Kato S, Tora L, Devys D, A TFTC/STAGA module mediates histone H2A and H2B deubiquitination, coactivates nuclear receptors, and counteracts heterochromatin silencing. Mol Cell 29, 92–101 (2008). [DOI] [PubMed] [Google Scholar]
- 39.Furrer SA, Waldherr SM, Mohanachandran MS, Baughn TD, Nguyen KT, Sopher BL, Damian VA, Garden GA, La Spada AR, Reduction of mutant ataxin-7 expression restores motor function and prevents cerebellar synaptic reorganization in a conditional mouse model of SCA7. Hum Mol Genet, (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Faul F, Erdfelder E, Lang AG, Buchner A, G*Power 3: a flexible statistical power analysis program for the social, behavioral, and biomedical sciences. Behav Res Methods 39, 175–191 (2007). [DOI] [PubMed] [Google Scholar]
- 41.Seth PP, Vasquez G, Allerson CA, Berdeja A, Gaus H, Kinberger GA, Prakash TP, Migawa MT, Bhat B, Swayze EE, Synthesis and biophysical evaluation of 2’,4’-constrained 2’O-methoxyethyl and 2’,4’-constrained 2’O-ethyl nucleic acid analogues. J Org Chem 75, 1569–1581 (2010). [DOI] [PubMed] [Google Scholar]
- 42.Guyenet SJ, Furrer SA, Damian VM, Baughan TD, La Spada AR, Garden GA, A simple composite phenotype scoring system for evaluating mouse models of cerebellar ataxia. J Vis Exp, doi: 10.3791/1787 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Marmor MF, Fulton AB, Holder GE, Miyake Y, Brigell M, Bach M, International V Society for Clinical Electrophysiology of, ISCEV Standard for full-field clinical electroretinography (2008 update). Doc Ophthalmol 118, 69–77 (2009). [DOI] [PubMed] [Google Scholar]
- 44.Schmittgen TD, Livak KJ, Analyzing real-time PCR data by the comparative C(T) method. Nat Protoc 3, 1101–1108 (2008). [DOI] [PubMed] [Google Scholar]
- 45.Chen S, Peng GH, Wang X, Smith AC, Grote SK, Sopher BL, La Spada AR, Interference of Crx-dependent transcription by ataxin-7 involves interaction between the glutamine regions and requires the ataxin-7 carboxy-terminal region for nuclear localization. Hum Mol Genet 13, 53–67 (2004). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.