

Summary
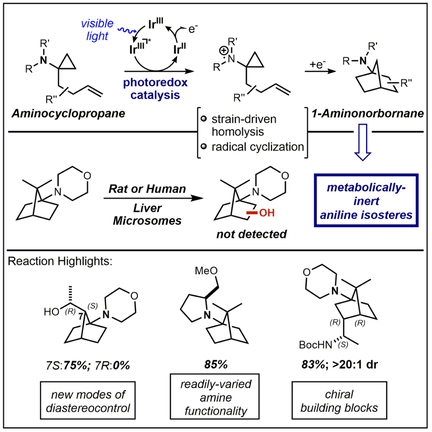
This report describes the photochemical conversion of aminocyclopropanes into 1-aminonorbornanes via formal [3+2] cycloadditions initiated by homolytic fragmentation of amine radical cation intermediates. Aligning with the modern movement toward sp3-rich motifs in drug discovery, this strategy provides access to a diverse array of substitution patterns on this saturated carbocyclic framework while offering the robust functional group tolerance (e.g. −OH, −NHBoc) necessary for further derivatization. Evaluating the metabolic stability of selected morpholine-based 1-aminonorbornanes demonstrated a low propensity for oxidative processing and no proclivity toward reactive metabolite formation, suggesting a potential bioisosteric role for 1-aminonorbornanes. Continuous flow processing allowed for efficient operation on gram-scale, providing promise for translation to industrially-relevant scales. This methodology only requires low loadings of a commercially-available, visible light-active photocatalyst and a simple salt, thus it stays true to sustainability goals while readily delivering saturated building blocks that can reduce metabolic susceptibility within drug development programs.
Keywords: 1-aminonorbornane, Aminocyclopropane, photoredox catalysis, amine radical cation, homolytic fragmentation, radical cyclization, bioisostere, metabolic stability
Graphical Abstract
3) eTOC blurb
The chemistry provided herein details an efficient and flexible route toward architecturally-distinctive 1-aminonorbornanes through the use of visible light photoredox catalysis. The incorporation of readily-diversifiable functional handles (e.g. -OH, -CO2Me, -NHBoc, -NHCbz) illustrates the potential utility of these 1-aminonorbornanes within drug discovery programs. Additionally, these motifs offer improved metabolic stability relative to their aniline congeners (as demonstrated through microsomal stability assays and metabolite identification efforts), indicating applicability of 1-aminonorbornanes as aniline bioisosteres.
Introduction
The recent discovery that approved drugs are enriched in saturated content (higher Fsp3) relative to the average high-throughput screening compound1–3 has sparked an increasing demand for new, saturated building blocks for use in drug discovery efforts.4 Further incentive arises from the fact that saturated substructures are generally less susceptible to adverse metabolic processing events than their aromatic congeners,5 while also offering more potential sites of diversification. Thus the introduction of sp3-rich motifs can not only generate new intellectual property and access unique chemical space, but also promote improved drug safety by reducing the likelihood of the metabolism-derived toxicities6–8 (e.g. drug-induced liver injury9). However, these saturated systems tend to require more involved syntheses, a central reason for the bias toward aromatic systems10 in modern commercial screening libraries. Diversifying the medicinal chemistry toolbox from its current arene-rich state therefore requires the development of efficient methods for the preparation of complex, saturated substructures. This work provides one solution to this challenge, generating the intriguing yet rarely-implemented 1-aminonorbornane (1-aminoNB; 2) motif via an operationally-simple, visible light-mediated transformation.
Interestingly, despite receiving little attention in recent times, 1-aminoNBs were central to delineating the basic principles of SN1 and SN2 reactivity,11 as illustrated by the landmark report from Bartlett and Knox12 in which bridgehead-substituted 1-apocamphanes (7,7-dimethylnorbornanes) were used as negative controls for SN2 reactivity supporting the Walden inversion mechanism.13–15 These classical accounts16–20 as well as more recent approaches21–24 largely generate the bridgehead amine via Hofmann or Curtius rearrangements of C1-CO2H motifs within terpene derivatives or from downstream products of cyclopentadiene-based Diels-Alder reactions. One radical-based closure exists from Della and Knill,25 cyclizing a C7-radical into an oxime after traditional tin-based initiation conditions. The lack of structural diversity available through these strategies has likely contributed to the minimal utilization of 1-aminoNB-based scaffolds within medicinal chemistry,21 yet these substructures aptly address the needs of Fsp3-centric drug discovery. Notably, these motifs can also be envisioned to serve as aniline bioisosteres, complementing recent work with amine-substituted [1.1.1]bicyclopentanes26 and cubanes.27 Anilines are prevalent in modern library generation owing to their ease of synthesis, despite their reputation as the most “noto rious” of all structural alerts 7 (functional groups known to predispose a lead compound toward metabolism-derived toxicities or risk of adverse drug-drug interactions). This proclivity for deleterious metabolic susceptibility thus heightens the importance of delivering saturated systems that can supplant anilines yet be accessed with equivalent efficiency.28
Our approach to the synthesis of 1-aminoNBs sought to generate the norbornane core through serialized radical cyclizations, initiating from a photochemically-generated amine radical cation intermediate (see Figure 1). Specifically, through the oxidation of 1-homoallyl-1-aminocyclopropanes (aminoCPs; 129), amine radical cations of form 3 (isoelectronic with a cyclopropylcarbinyl radical30) would undergo β-scission to generate a β-iminium radical (4), thereby enabling a 6-exo-trig radical cyclization toward distonic radical cation 5 and subsequent 5-exo-trig cyclization to forge the norbornane core. Reduction to product (2) closes the catalytic cycle (or propagates the chain) to provide a net redox-neutral transformation. Single-electron oxidation of amines can be readily-achieved through the reductive quenching of excited state photocatalysts such as [Ru(bpy)3]Cl2, reactivity that was first observed by Whitten and co-workers in 1977 for both anilines and trialkylamines.31 Kellogg32 and Mariano33 provided some of the earliest reports that capitalized on the synthetic utility of the resultant amine radical cations. The recent rise of photoredox catalysis both in academia34,35 and industry36 has led to a multitude of new methods employing amine radical cations,37–39 including our group’s 2010 report of visible light-mediated aza-Henry reactions.40 Interestingly, the large majority of these methods still operate through the classically-studied heterolytic mode of decomposition,41,42 fragmenting α-C-R bonds to access α-amino radicals (or iminium species upon further oxidation). Homolytic decomposition modes are far less common,43–45 though only small amounts of ring strain are necessary to drive C-C bond scission as seen in our efforts converting catharanthine (4.2 kcal/mol ring strain) into Aspidosperma and Iboga alkaloids.46 Zheng and co-workers have demonstrated the viability of cyclopropane fragmentations within photoredox catalysis, coupling oxidized anilinocyclopropanes with a variety of styrenes,47 arylacetylenes,48 diynes, and enynes.49 The goal of this work was to harness the unique capabilities of photoredox catalysis to generate 1-aminoNBs as a means of addressing the need for new, saturated building blocks in drug discovery. The following describes the optimization, scope, scalability, and performance in continuous flow of this formal [3+2] cycloaddition toward 1-aminoNBs, as well as preliminary investigations into the metabolic fate of these 1-aminoNBs.
Figure 1. Generating 1-aminoNBs via homolytic fragmentation.
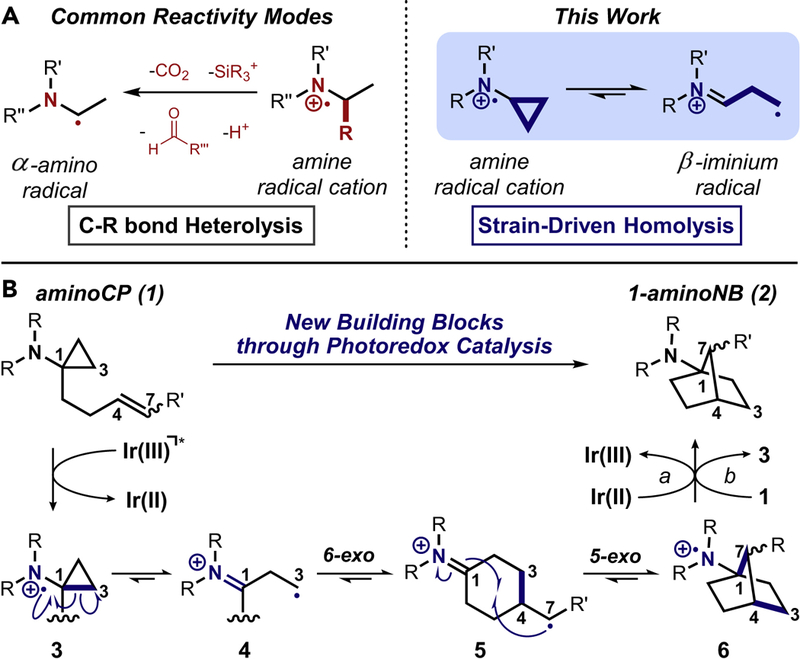
A) Amine radical cation decomposition pathways: heterolytic vs. strain-driven homolytic. B) Proposed mechanistic pathways.
Results
Initial optimization was performed with 7,7-dimethyl aminoCP 1a, prepared via Kulinkovich cyclopropanation50 of the corresponding amide (see Supplemental Information). Heteroleptic Ir(III) photocatalyst [Ir(dF[CF3]ppy)2(dtbbpy)](PF6)51 (7) in degassed acetonitrile was identified as the best starting point, affording 63% of 1-aminoNB 2a after 12 hrs of irradiation (see Figure 2, entry 1). It was found that simple Lewis acidic salts enhanced conversion to product even at sub-stoichiometric loadings (20 mol%). LiBF4 and ZnCl2 proved to be the most effective, yielding 79% and 84% of 1-aminoNB 2a, respectively (entries 2–3). The addition of ZnCl2 even enabled partial productivity when employing the less oxidizing photocatalyst Ru(bpy)3Cl2·6H2O (entries 10–11; for 8: Ru(II)*/Ru(I) E1/2 = +0.77 V vs. SCE34; for 7: Ir(III)*/Ir(II) E1/2 = +0.89 V vs. SCE51). Other Lewis acids provided little enhancement (NaBF4; entry 4) or facilitated degradation pathways (Me2AlCl, BF3·OEt2, MgBr2·OEt2; entries 5–7), while redox non-innocent metal salts inhibited reactivity (CuBr2, NiCl2; entries 8 and 9); of note, ZnCl2 is not necessarily redox-innocent under these conditions given the efforts of Bernhard and co-workers,52 though only the most inefficient substrates afforded a grey precipitate that could indicate reduction to Zn0 (see Supplemental Information). Importantly, this formal [3+2] cycloaddition methodology was readily translated to continuous flow processing (compare entry 2 and entry 15; 79% and 78%, respectively53), including efforts on gram-scale (entry 16). Of note, the reaction did not proceed to any extent in the absence of photocatalyst, without light, or under purely thermal conditions (entries 12–14).
Figure 2. Optimization of visible light-mediated formal [3+2] cycloaddition toward 1-aminoNBs and DPV measurements.
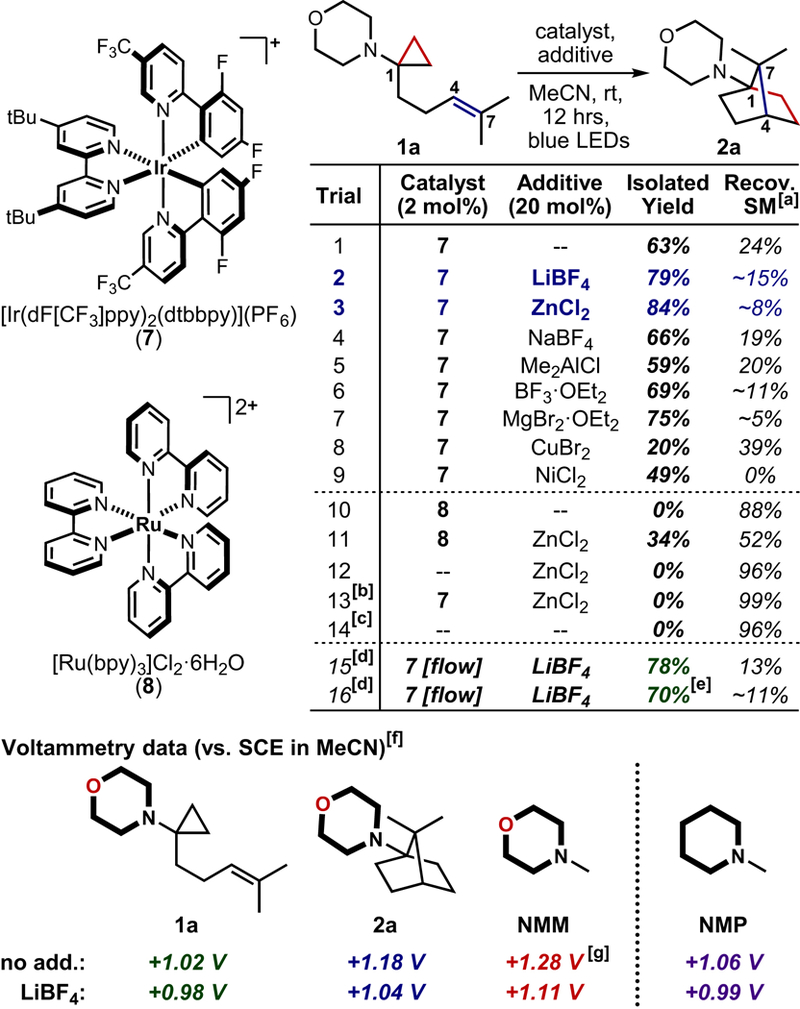
[a]Recov. SM = recovered starting material, approximate values indicate that recovered material was not entirely pure and are provided only for general comparison, [b]Reaction run in absence of light, [c]Reaction run in absence of light at 80 °C, [d]Detailed descriptions of flow and batch setups are provided in the Supplemental Information, [e]Reaction run on 1 g-scale, [f]Values obtained through DPV vs. Ag/AgCl, peak potentials were then converted to vs. SCE (VSCE = VAg/AgCl + (0.241 V −0.197 V)), see Supplemental Information for further details and additional DPV measurements, [g]Measured NMM oxidation potential closely compares to previously reported value.63
Differential pulse voltammetry (DPV) data suggests that the Lewis acidic salts are eliciting an effect on reaction outcome through modulation of amine oxidation potential. Morpholines are unusually difficult to oxidize relative to other N,N-dialkylamines (for N-methylmorpholine (NMM): Ep = +1.28 V vs. SCE, for N-methylpiperidine (NMP): Ep = +1.06 V vs. SCE; see Figure 2), thus prior morpholine oxidation efforts in the field have necessitated the use of an Ir(ppy)3/O2-based oxidative quenching cycle54 (operating via the Ir(IV) species) or the development of new Ir(III) photocatalysts with excited state oxidizing power that vastly exceeds that of photocatalyst 7.55 Alternatively, this data appears to show that morpholine oxidation is more facile in the presence of readily-available Lewis acidic salts (for NMM: ΔEp = −170 mV56). Intriguingly, the impact of the Lewis acidic salts was not limited to morpholines, facilitating conversion to product for a variety of nitrogen heterocycles, i.e. piperidine, piperazine, azetidine, pyrrolidine (1-aminoNBs 2b-2e; see Figure 3B; see Supplemental Information for corresponding DPV data). The discrepancy in oxidation potential between aminoCP 1a and NMM (ΔEp = −260 mV) can be rationalized by the facile post-oxidation fragmentation pathway, as single-electron transfer processes with amines are highly influenced by post-oxidation reactivity.41 This effect also suggests a propagative mechanistic pathway, as there is a clear enthalpic driving force for the reduction of 1-aminoNB radical cation 6 with an additional equivalent of starting aminoCP 1 (path b in Figure 1) rather than the Ir(II) species (path a);57 the latter is also enthalpically-feasible but is kinetically unfavorable, requiring the interaction of two low-concentration species. Interestingly, the quantum yield for the transformation of aminoCP 1a to 1-aminoNB 2a was determined to be just 0.34.58 The high quenching efficiency (see Supplemental Information) and the presence of facile post-oxidation fragmentation (putatively minimizing back-electron transfer) limit the impact of two common explanations for artificially low quantum yields.59 An alternative hypothesis is that the 5-exo-trig cyclization is sufficiently slow relative to the electron transfer processes that it renders the measured quantum yield <1 due to photons lost during the lifetime of distonic radical cation 5. Defining the role of these kinetic idiosyncrasies could have ramifications for a variety of photochemical transformations beyond this methodology.
Figure 3. Reaction scope.
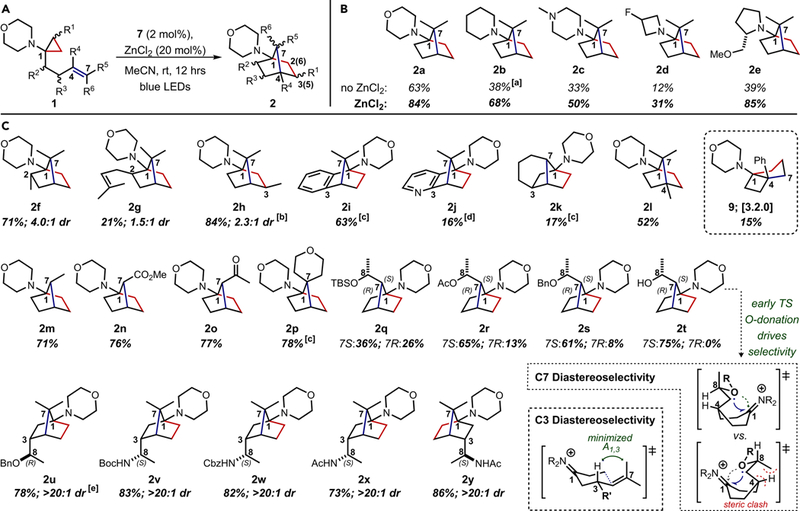
A) Generic depiction of aminoCPs converted to 1-aminoNBs, C2/C3 from cyclopropane can also be C6/C5 depending on substitution pattern. B) [a]No additive trial toward 1-aminoNB 2b was inhibited by the low solubility of the corresponding aminoCP in MeCN. C) [b]84% yield obtained with the anti-C3-Me aminoCP, the syn-C3-Me aminoCP yielded 81% 1-aminoNB 2h, also in 2.3:1 dr, [c]24 hr-time course, [d]Conditions: 7 (5 mol%), MeCN, rt, 72 hrs, blue LEDs, [e]78% corresponds to a 3.3:1 mix of the 3R,4R and 3S,4S isomers (both axial) from a 4:1 mix of the anti:syn aminoCPs.
In addition to enabling the use of various nitrogen heterocycles, the ZnCl2-assisted procedure proved effective for generating C7-, C2-, C3-, and C4-substituted systems (see Figure 3). Presence of C2-substitution led to preferential formation of the C2-axial isomer as seen with 1-aminoNB 2f. A C2-prenyl substituent was less well-tolerated (toward 2g) due to competing cyclization and C-H abstraction pathways. Selectivity for the equatorial C3-Me 1-aminoNBs 2h was observed when the methyl substitution arose from a substituted cyclopropane ring, while allylic functionality proceeded with high selectivity for axial C3-substiution (vide infra). Further annulated 1-aminoNBs were also accessible via this strategy, as evidenced by C2-C3 (hetero)aryl-fused 1-aminoNBs 2i and 2j as well as C3-C7-propyl-bridged 1-aminoNB 2k. (Hetero)aryl-fused systems necessitated a 24 hr-time course likely due to the competing photosensitization of the styrenyl-type functionality within the aminoCP, while the bridged system was low-yielding regardless of irradiation time, presumably due to unfavorable torsional strain during the radical cyclizations. C4-methyl 1-aminoNB 2l was prepared in a modest 52% yield, but the analogous C4-phenyl system afforded [3.2.0]-bicyclic core 9, preferring an initial 7-endo-trig cyclization over the anticipated 6-exo-trig. Both acrylate-and enone-based systems were efficiently converted to C7-substituted 1-aminoNBs 2n and 2o (>75% yield). C7-spirofused 1-aminoNB 2p was also produced in good yield after 24 hrs (78%).
Importantly, diastereoselective variants of this formal [3+2] methodology enables the preparation of optically-pure 1-aminoNBs. One particularly interesting discovery was the protecting group-dependent control of C7 configuration with C7-(2-hydroxyethyl) derivatives 2q-2t. The increasing availability of the oxygen lone pairs (both in steric and electronic terms; -OH > -OBn > -OAc > -OTBS) was positively correlated with selectivity for the 7S,8R diastereomer, with the C8-OH system (2t) proceeding in >20:1 dr. Our hypothesis is that the oxygen lone pairs are donating into the antibonding orbital of the iminium species in the earliest phases of the transition state for the formation of the C1-C7 bond, a donation that is readily accomplished toward the favored 7S-epimer but impeded by steric encumbrance en route to the disfavored 7R-epimer. Additionally, C3-(2-hydroxyethyl) and C3-(2-aminoethyl) aminoCPs cleanly afforded a single isomer of the resultant 1-aminoNBs (2u-2y), necessarily providing control over the absolute configuration of both C1 and C4. The divergent axial vs. equatorial selectivities of cyclopropane-substituted and allylic-substituted aminoCPs offers an inherent form of diastereocontrol thus avoiding the necessity of developing exogenous stereocontrol mechanisms. Significantly, these two series of aminoCPs also serve to demonstrate the robust functional group tolerance of this strategy, proceeding in good to high yields while incorporating alcohol, ether, acyloxy, amide, and carbamate functionality.
The synthetic flexibility shown above inherently positions the 1-aminoNB motif as a new option for discovery chemists; demonstrating the feasibility of our aniline bioisosterism hypothesis would only enhance the potential applicability. A preliminary in silico analysis indicates that the norbornane core will lead to minimal perturbation of physicochemical parameters relative to the aniline congener (as well as alternative aniline bioisosteres: [1.1.1]-bicyclopentanes and cubanes). As seen in Figure 4A, the transition from the arene system to the saturated core(s) largely maintains the overall lipophilicity. Of note, the pKa of protonated N-alkyl morpholines is comparable to physiological pH, thus supplanting a given morpholine-based aniline with a 1-morpholinonorbornane will likely lead to population of both protonated and deprotonated forms; this imbues enhanced hydrophilicity despite an overall increase in carbons, potentially providing further pharmacokinetic benefits in specific cases. Other N,N-dialkylamine-based 1-aminoNBs will be fully protonated under physiological conditions (unlike their direct aniline counterpart), and this would need to be taken into consideration in any drug design or re-engineering effort. Additionally, the norbornane core does lead to slight departure from linearity for the C4-substituted form, but given the diversity of substitution patterns that can be accessed via the above method, these 1-aminoNBs are well-suited to explore chemical space beyond simple 1,4-substituted arene modalities.
Figure 4. Metabolic susceptibility of 1-aminoNBs relative to anilines.
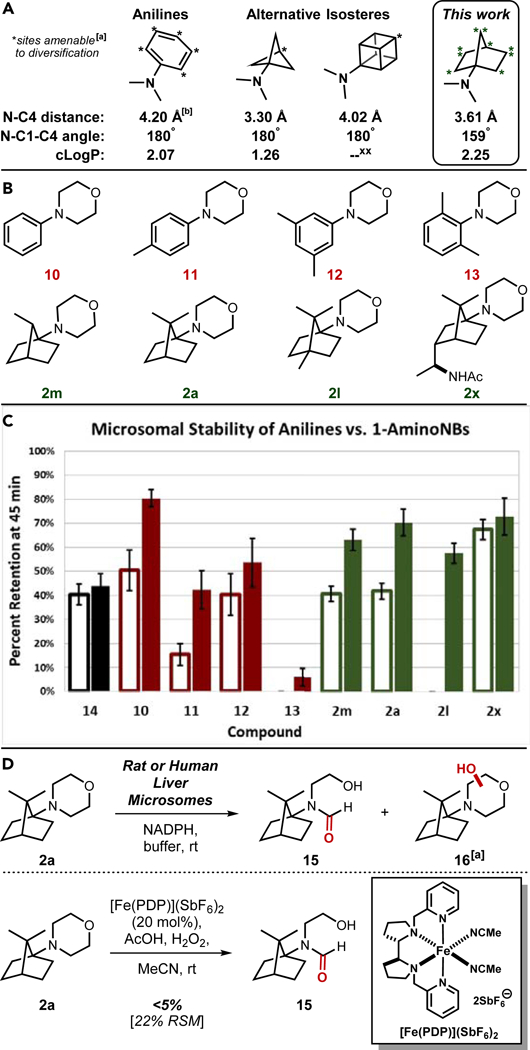
A) Fundamental physicochemical parameters of a dimethyl 1-aminoNB relative to congeners of aniline and related bioisosteres; [a]diversification sites for [1.1.1]-bicyclopentanes and cubanes chosen based on references 26 and 27; [b]data calculated using OpenEye software. B) Panel of anilines and 1-aminoNBs evaluated in the microsomal stability assay. C) Graphical depiction of compound retention after 45 min of incubation in the presence of either rat or human liver microsomes; empty bars = RLMs, filled bars = HLMs; black bars = verapamil·HCl (14; positive control), red bars = aniline compounds, green bars = 1-aminoNB compounds; all data is normalized to a negative control that was not exposed to the incubation period (see Supplemental Information for more details) and reported as ± SEM. D) Metabolite profile of 1-aminoNB 2a and synthetic recapitulation of CYP450 reactivity; [a]location of hydroxylation on the morpholine ring is unknown at this juncture.
Preliminary assessment of metabolic susceptibility was achieved through microsomal stability assays and identification of prominent metabolites. Selected compounds (see Figure 4B; 10 μM assay concentration) were incubated for 45 min with either rat or human liver microsomes (RLMs or HLMs, respectively), and the percent compound retention was determined relative to an untreated control (see Figure 4C; see Supplemental Information of additional details). In general, the RLMs were more efficient in processing both classes of compounds relative to the HLMs. Interestingly, aniline stability was highly dependent on the pattern of methyl substitution (10: H > 12: 3,5-diMe > 11: 4-Me > 13: 2,6-diMe), with the 2,6-dimethyl system (13) being all but entirely decomposed in both trials. N-Phenylmorpholine (10) appeared to be the most stable aniline system (at least, in HLMs), likely due to the lack of potential benzylic oxidation sites and reduced hydrophobicity. The decomposition of 1-aminoNBs was more consistent overall across the selected substitution patterns, especially in HLMs (all four compounds between 55–75% retention). In both RLMs and HLMs, the 1-aminoNB panel outperformed or was on par with the most robust aniline compounds. One notable exception was C4-Me 1-aminoNB 2l in the RLM assay, in which it was entirely decomposed in the 45 min. This is presumably an isoform-specific and/or species-dependent result given the 58% retention measured in HLMs. CYP450 active sites tend to be hydrophobic in nature,60 thus the trimethylated norbornane core of 2l could be serving as an optimal lipophilic anchor for a specific RLM-derived CYP450 isoform. Any given 1-aminoNB-based drug lead is likely to have additional polar functionality that would disrupt this anchoring effect, and unsurprisingly, the C3-substituted acetamide-based 1-aminoNB 2x offered the best overall retention profile between the two species (67% and 73%, respectively). Importantly, the aniline bioisostere concept does not hinge on imbuing complete inertness to metabolic processing, rather the avoidance of reactive metabolite generation is the true goal. The norbornane core provides no obvious path toward reactive metabolites, and this analysis was confirmed with metabolite profiling. Using LC-MS/MS, formamide 15 and a hydroxylated system (generically represented by 16) were identified as the primary metabolites arising from 1-aminoNB 2a (see Figure 4D). The identity of the former was confirmed through oxidation of 1-aminoNB 2a with a non-heme iron catalyst developed by White and co-workers for C-H hydroxylation,61,62 generating formamide 15 as the major isolable product. No products of core oxidation were directly observed in either system, likely owing to the increased s-character of the norbornane C-H bonds. Addition of glutathione to the microsomal incubations led to no discernable change in metabolite profile (see Supplemental Information), thus the 1-aminoNB system shows no inherent propensity to the glutathione adduction issues that can plague aniline-based drug scaffolds.7
Discussion
This report details the most efficient and flexible access to 1-aminoNBs to date in an effort to provide the medicinal chemistry community with valuable new saturated building blocks. Via this visible light-mediated formal [3+2] cycloaddition, a wide variety of substitution patterns have been readily prepared, many incorporating functional handles for further diversification. These include optically-pure 1-aminoNBs via diastereoselective variants, a central requirement for drug discovery purposes, as well as bridged, fused, and spiro-fused structures for exploration of unique chemical space. This methodology operates well in continuous flow, including gram-scale preparations, suggesting that this chemistry can translate beyond discovery scale. Microsomal stability data and preliminary metabolite profiling indicates a strong potential for these 1-aminoNB building blocks to serve as aniline bioisosteres. Future investigations will seek to demonstrate the value of these substructures for generating bioactive leads.
Experimental Procedures
General procedure of the photochemical conversion of 1-amino-1-homoallylcyclopropanes (1) to 1-aminoNBs (2):
In a dry vial under inert atmosphere, aminoCP 1 was dissolved in dry MeCN (0.1 M aminoCP) before adding [Ir(dF[CF3]ppy)2(dtbbpy)](PF6) (0.02 eq.) then ZnCl2 (1.0 M in ether; 0.2 eq.) in one portion each. Reaction mixture was degassed with three freeze-pump-thaw cycles. The reaction was stirred at room temp (as controlled with jacketed water bath, temp maintained between 18–23 °C) while irradiating with two strips of 4.4 W blue LEDs for 12 hrs. The mixture was partitioned between 1:1 saturated NaHCO3 (aq.):1 M NaOH and 1:1 ether:hexanes. Phases were separated, and the aqueous phase was further extracted with 1:1 ether:hexanes three times. Combined organics were washed with brine, dried over anhydrous magnesium sulfate, filtered to remove solids, and concentrated in vacuo. The crude residue was purified via flash chromatography over silica to afford the desired 1-aminoNB 2.
Supplementary Material
1) Bigger Picture.
Recent years have witnessed an increasing focus on saturated substructures within drug development, owing to the pharmacokinetic and toxicological benefits correlated with higher saturation content. However, the synthetic challenges presented by densely-functionalized saturated architectures generally prohibits their evaluation. The abundance of anilines within high-throughput screening libraries is demonstrative of these competing needs. Anilines are prone to adverse metabolic processing, commonly necessitating re-engineering of a given drug lead to ameliorate CYP450 inhibition and/or glutathione adduction issues, but the ease with which these systems are prepared outweighs the toxicity risks. This report contributes to the need for aniline bioisosteres through the development of a robust, photochemical methodology that supplies 1-aminonorbornanes, saturated bicyclic ring systems that offer similar spatial occupancy to anilines while improving metabolic stability.
2) Highlights.
- Strain-driven homolysis initiates radical cyclization sequence toward norbornane core
- Robust functional group tolerance provides readily-modifiable building blocks
- Unique modes of diastereocontrol afford enantiopure 1-aminonorbornanes
- 1-Aminonorbornanes are shown to offer improved metabolic stability over anilines
Acknowledgements
The authors acknowledge the financial support for this research from the NIH NIGMS (R01-GM127774) [CRJS], NIH NCI (K01CA190711) [KDJ], the Camille Dreyfus Teacher-Scholar Award Program [CRJS], and the University of Michigan [CRJS]. TMS was supported by an MCubed Grant. DS was supported by a Postdoctoral Fellowship, PF-16–236-01 -CDD, from the American Cancer Society.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
For detailed experimental procedures and characterization data for all aminoCPs and 1-aminoNBs presented above, see the Supplemental Information and Figures S1-S170.
Declaration of Interests
The authors declare no competing financial interests.
References
- 1 ).Lovering F, Bikker J, and Humblet C (2009). Escape from flatland: increasing saturation as an approach to improving clinical success. J. Med. Chem 52, 6752–6756. [DOI] [PubMed] [Google Scholar]
- 2 ).Lovering F (2013). Escape from flatland 2: complexity and promiscuity. Med. Chem. Commun 4, 515–519. [Google Scholar]
- 3 ).For a pertinent review on correlations between physical parameters and drug development, see: Meanwell N (2016). Improving drug design: an update on recent applications of efficiency metrics, strategies for replacing problematic elements, and compounds in nontraditional drug space. Chem. Res. Toxicol 29, 564–616. [DOI] [PubMed] [Google Scholar]
- 4 ).For examples of saturated ring systems that have impacted the clinic, see: Stockdale T, and Williams C (2015). Pharmaceuticals that contain polycyclic hydrocarbon scaffolds. Chem. Soc. Rev 44, 7737–7763. [DOI] [PubMed] [Google Scholar]
- 5 ).Ritchie T, Macdonald S, Young R, and Pickett S (2011). The impact of aromatic ring count on compound developability: further insights by examining carbo-and hetero-aromatic and -aliphatic ring types. Drug Disc. Today 16, 164–171. [DOI] [PubMed] [Google Scholar]
- 6 ).Orr S, Ripp S, Ballard E, Henderson J, Scott D, Obach S, Sun H, and Kalgutkar A (2012). Mechanism-based inactivation (MBI) of cytochrome P450 enzymes: structure-activity relationships and discovery strategies to mitigate drug-drug interaction risks. J. Med. Chem 55, 4896–4933. [DOI] [PubMed] [Google Scholar]
- 7 ).Kalgutkar A, and Dalvie D (2015). Predicting toxicities of reactive metabolite-positive drug candidates. Annu. Rev. Pharmacol. Toxicol 55, 35–54. [DOI] [PubMed] [Google Scholar]
- 8 ).Thompson R, Isin E, Ogese M, Mettetal J, and Williams D (2016). Reactive metabolites: current and emerging risk and hazard assessments. Chem. Res. Toxicol 29, 505–533. [DOI] [PubMed] [Google Scholar]
- 9 ).Iorga A, Dara L, and Kaplowitz N (2017). Drug-induced liver injury: cascade of events leading to cell death, apoptosis or necrosis. Int. J. Mol. Sci 18, 1018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10 ).Specific commentary can be found in Box 1 of: Scannell J, Blanckley A, Boldon H, and Warrington B (2012). Diagnosing the decline in pharmaceutical R&D efficiency. Nat. Rev. Drug. Disc 11, 191–200. [DOI] [PubMed] [Google Scholar]
- 11 ).Applequist D, and Roberts J (1954). Displacement reactions at bridgeheads of bridged polycarbocyclic systems. Chem. Rev 54, 1065–1089. [Google Scholar]
- 12 ).Bartlett P, and Knox L (1939). Bicyclic structures prohibiting the Walden inversion. Replacement reactions in 1-substituted 1-apocamphanes. J. Am. Chem. Soc 61, 3184–3192. [Google Scholar]
- 13 ).Walden P (1896). Ueber die gegenseitige Umwandlung optischer Antipoden. Ber. Dtsch. Chem. Ges 29, 133–138. [Google Scholar]
- 14 ).Kenyon J, and Phillips H (1930). Some recent developments in the study of the Walden inversion. Trans. Faraday Soc 26, 451–458. [Google Scholar]
- 15 ).Olson A, and Long F (1934). The mechanism of substitution reactions. J. Am. Chem. Soc 56, 1294–1299. [Google Scholar]
- 16 ).von Houben J, and Pfankuch E (1931). Uber den ersatz von 2,6-umlagerungen in der campherreihe, die racemization optisch aktiver camphenderivate und den ubergang von D-in L-campherderivate durch camphenumlagerungen zweiter art. (Uber campher und terpene. VII). Justus Liebigs Ann. Chem 489, 193–223. [Google Scholar]
- 17 ).Wilt J, Parsons C, Schneider C, Schultenover D, and Wagner W (1968). The preparation and study of some 1-norbornenyl and norbornenyl-1-carbinyl derivatives. J. Org. Chem 33, 694–708. [Google Scholar]
- 18 ).Nickon A, Nishida T, and Lin Y (1969). Bridgehead ketols. J. Am. Chem. Soc 91, 6860–6861. [Google Scholar]
- 19 ).Beak P, and Harris B (1974). Reactions of apotricyclyl, apocamphor, and apobornenyl chloroformates with silver(I). Generation and aromatic substitution of a bridgehead radical and a bridgehead cation. J. Am. Chem. Soc 96, 6363–6372. [Google Scholar]
- 20 ).Kropp P, Poindexter G, Pienta N, and Hamilton D (1976). Photochemistry of alkyl halides. 4. 1-Norbornyl, 1-norbornylmethyl, 1-and 1-adamantyl, and 1-octyl bromides and iodides. J. Am. Chem. Soc 98, 8135–8144. [Google Scholar]
- 21 ).Martinez A, Vilar E, Fraile A, Cerero S, Herrero M, Ruiz P, Subramanian L, and Gancedo A (1995). Synthesis of substituted 1-norbornylamines with antiviral activity. J. Med. Chem 38, 4474–4477. [DOI] [PubMed] [Google Scholar]
- 22 ).Buser S, and Vasella A (2005). 7-Oxanorbornane and norbornane mimics of a distorted β-D-mannopyranoside: synthesis and evaluation as β-mannosidase inhibitors. Helv. Chim. Acta 88, 3151–3173. [Google Scholar]
- 23 ).Dejmek M, Hrebabecky H, Sala M, Dracinsky M, Prochazkova E, Leyssen P, Neyts J, Balzarini J, and Nencka R (2014). From norbornane-based nucleotide analogs locked in south conformation to novel inhibitors of feline herpes virus. Bioorg. Med. Chem 22, 2974–2983. [DOI] [PubMed] [Google Scholar]
- 24 ).For a single example of a Rh(I)-catalyzed method from a (diallyl)vinylsulfonamide, see: Aïssa C, Ho K, Tetlow D, and Pin-Nó M (2014). Diastereoselective carbocyclizat ion of 1,6-heptadienes triggered by rhodium-catalyzed activation of an olefinic C-H bond. Angew. Chem. Int. Ed 53, 4209–4212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25 ).Della E, and Knill A (1994). Synthesis of bridgehead-substituted bicyclo[2.2.1]heptanes. Radical cyclization of an oxime ether and an α,β-unsaturated ester. Aust. J. Chem 47, 1833–1841. [Google Scholar]
- 26 ).Stepan A, Subramanyam C, Efremov I, Dutra J, O’Sullivan T, DiRico K, McDonald S, Won A, Dorff P, Nolan C, et al. (2012). Application of the bicyclo[1.1.1]pentane motif as a nonclassical phenyl ring bioisostere in the design of a potent and orally active γ-secretase inhibitor. J. Med. Chem 55, 3414–3424. [DOI] [PubMed] [Google Scholar]
- 27 ).Chalmers B, Xing H, Houston S, Clark C, Ghassabian S, Kuo A, Cao B, Reitsma A, Murray C, Stok J, et al. (2016). Validating Eaton’s hypothesis: cubane as a benzene bioisostere. Angew. Chem. Int. Ed 55, 3580–3585. [DOI] [PubMed] [Google Scholar]
- 28 ).Brink A, Pahler A, Funk C, Schuler F, and Schadt S (2017). Minimizing the risk of chemically reactive metabolite formation of new drug candidates: implications for preclinical design. Drug Disc. Today 22, 751–756. [DOI] [PubMed] [Google Scholar]
- 29 ).For a recent comprehensive review of aminocyclopropane reactivity, see: Rassadin V, and Six Y (2016). Ring-opening, cycloaddition and rearrangement reactions of nitrogen-substituted cyclopropane derivatives. Tetrahedron 72, 4701–4757. [Google Scholar]
- 30 ).Hancock A, and Tanko J (2012). Radical cation/anion and neutral radicals: a comparison. In Encyclopedia of Radicals in Chemistry, Biology and Materials, Chatgilialoglu C and Studer A, eds. (John Wiley & Sons, Inc.), doi: 10.1002/9781119953678.rad014. [DOI] [Google Scholar]
- 31 ).DeLaive P, Lee J, Sprintschnik H, Abruna H, Meyer T, and Whitten D (1977). Photoinduced redox reactions of hydrophobic ruthenium(II) complexes. J. Am. Chem. Soc 99, 7094–7097. [Google Scholar]
- 32 ).Hedstrand D, Kruizinga W, and Kellogg R (1978). Light induced and dye accelerated reductions of phenacyl onium salts by 1,4-dihydropyridines. Tetrahedron Lett 14, 1255–1258. [Google Scholar]
- 33 ).Xu W, Jeon Y, Hasegawa E, Yoon U, and Mariano P (1989). Novel electron-transfer photocyclization reactions of α-silyl amine α,β-unsaturated ketone and ester systems. J. Am. Chem. Soc 111, 406–408. [Google Scholar]
- 34 ).Douglas J, Nguyen J, Cole K, and Stephenson CRJ (2014). Enabling novel photoredox reactivity via photocatalyst selection. Aldrichim. Acta 47, 15–25. [Google Scholar]
- 35 ).Shaw M, Twilton J, and MacMillan D (2016). Photoredox catalysis in organic chemistry. J. Org. Chem 81, 6898–6926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36 ).Douglas J, Sevrin M, and Stephenson CRJ (2016). Visible light photocatalysis: applications and new disconnections in the synthesis of pharmaceutical agents. Org. Process Res. Dev 20, 1134–1147. [Google Scholar]
- 37 ).Beatty J, and Stephenson CRJ (2015). Amine functionalization via oxidative photoredox catalysis: methodology development and complex molecule synthesis. Acc. Chem. Res 48, 1474–1484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38 ).Staveness D, Bosque I, and Stephenson CRJ (2016). Free radical chemistry enabled by visible light-induced electron transfer. Acc. Chem. Res 49, 2295–2306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39 ).Morris S, Wang J, and Zheng N (2016). The prowess of photogenerated amine radical cations in cascade reactions: from carbocycles to heterocycles. Acc. Chem. Res 49, 1957–1968. [DOI] [PubMed] [Google Scholar]
- 40 ).Condie A, Gonzalez-Gomez J, and Stephenson CRJ (2010). Visible-light photoredox catalysis: aza-Henry reactions via C-H functionalization. J. Am. Chem. Soc 132, 1464–1465. [DOI] [PubMed] [Google Scholar]
- 41 ).Yoon U, Su Z, and Mariano P (2004). The Dynamics and Photochemical Consequences of Aminium Radical Reactions In CRC Handbook of Organic Photochemistry and Photobiology, 2nd Ed., Horspool W and Lenci F, eds. (CRC Press; ), ch., 101. [Google Scholar]
- 42 ).For the seminal proposal of homolytic amine radical cation decomposition pathways, see: DeLaive P, Foreman T, Giannotti C, and Whitten D (1980). Photoinduced electron transfer reactions of transition-metal complexes with amines. Mechanistic studies of alternate pathways to back electron transfer. J. Am. Chem. Soc 102, 5627–5631. [Google Scholar]
- 43 ).For the seminal use of an oxidized aminoCP in synthesis, see: Itoh T, Kaneda K, and Teranishi S (1975). Novel oxygenolysis of cyclopropylamines to epoxy ketones. Tetrahedron Lett 32, 2801–2804. [Google Scholar]
- 44 ).For the seminal formal [3+2] cycloaddition utilizing an oxidized aminoCP, see: Takemoto Y, Yamagata S, Furuse S, Hayase H, Echigo T, and Iwata C (1998). CAN-mediated tandem 5-exo-cyclisation of tertiary aminocyclopropanes: novel accelerative effect of an N-benzyl group for oxidative ring-opening. Chem. Commun 651–652. [Google Scholar]
- 45 ).For the seminal photochemical generation of an oxidized aminoCP (UV light), see: Lee J, Sun J, Blackstock S, and Cha J (1997). Facile ring opening of tertiary aminocyclopropanes. J. Am. Chem. Soc 119, 10241–10242. [Google Scholar]
- 46 ).Beatty J, and Stephenson CRJ (2014). Synthesis of (−)-pseudotabersonine, (−)-pseudovincadifformine, and (+)-coronaridine enabled by photoredox catalysis in flow. J. Am. Chem. Soc 136, 10270–10273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47 ).Maity S, Zhu M, Shinabery R, and Zheng N (2012). Intermolecular [3+2] cycloaddition of cyclopropylamines with olefins by visible-light photocatalysis. Angew. Chem. Int. Ed 51, 222–224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48 ).Nguyen T, Maity S, and Zheng N (2014). Visible light mediated intermolecular [3+2] annulation of cyclopropylamines with alkynes. Beilstein J. Org. Chem 10, 975–980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49 ).Nguyen T, Morris S, and Zheng N (2014). Intermolecular [3+2] annulation of cyclopropylanilines with alkynes, enynes, and diynes via visible light photocatalysis. Adv. Synth. Catal 356, 2831–2837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50 ).Cha J, and Kulinkovich O (2012). The Kulinkovich cyclopropanation of carboxylic acid derivatives. In Organic Reactions, Vol. 77, Denmark S, ed. (John Wiley & Sons, Inc.), pp. 1–160. [Google Scholar]
- 51 ).Lowry M, Goldsmith J, Slinker J, Rohl R, Pascal R, Malliaras G, and Bernhard S (2005). Single-layer electroluminescent devices and photoinduced hydrogen production from an ionic iridium(III) complex. Chem. Mater 17, 5712–5719. [Google Scholar]
- 52 ).Brooks A, Basore K, and Bernhard S (2013). Photon-driven reduction of Zn2+ to Zn metal. Inorg. Chem 52, 5794–5800. [DOI] [PubMed] [Google Scholar]
- 53 ).ZnCl2 results were similar (81% in flow; see Supplemental Information), but the modest heterogeneity of these reaction mixtures brought concerns of clogging, thus LiBF4 was chosen for gram-scale procedures.
- 54 ).Douglas J, Cole K, and Stephenson CRJ (2014). Photoredox catalysis in a complex pharmaceutical setting: toward the preparation of JAK2 inhibitor LY2784544. J. Org. Chem 79, 11631–11643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55 ).Musacchio A, Lainhart B, Zhang X, Naguib S, Sherwood T, and Knowles R (2017). Catalytic intermolecular hydroaminations of unactivated olefins with secondary alkyl amines. Science 355, 727–730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56 ).ZnCl2 generally led to moderate heterogeneity which was exacerbated upon addition of electrolyte, thus LiBF4 was employed for all mechanistic studies.
- 57 ).Zheng and co-workers have provided preliminary data that supports a radical chain process for their system, though the enthalpic contributions are not assessed: Cai Y, Wang J, Zhang Y, Li Z, Hu D, Zheng N, and Chen H (2017). Detection of fleeting amine radical cations and elucidation of chain processes in visible light-mediated [3+2] annulation by online mass spectrometric techniques. J. Am. Chem. Soc 139, 12259–12266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58 ).It is important to note that while a measured Φ > 1 ensures the involvement of a radical chain process, observing Φ < 1 does not guarantee a closed catalytic cycle; the efficiency with which photons are converted into chain initiation events contributes to the quantum yield measurements, thus any radiative or non-radiative relaxation pathways will reduce the measured Φ.
- 59 ).Cismesia M, and Yoon T (2015). Characterizing chain processes in visible light photoredox catalysis. Chem. Sci 6, 5426–5434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60 ).Munro A, Girvan H, Mason A, Dunford A, and McLean K (2013). What makes a P450 tick? Trends Biochem. Sci 38, 140–150. [DOI] [PubMed] [Google Scholar]
- 61 ).Chen M, and White MC (2007). A predictably selective aliphatic C-H oxidation reaction for complex molecule synthesis. Science 318, 783–788. [DOI] [PubMed] [Google Scholar]
- 62 ).Of note, addition of strong Bronsted acid was able to protect the morpholine from decomposition (in line with the reference that follows), leading to only recovered starting material: Howell J, Feng K, Clark J, Trzepkowski L, and White MC (2015). Remote oxidation of aliphatic C-H bonds in nitrogen-containing molecules. J. Am. Chem. Soc 137, 14590–14593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63 ).Tajima T, and Fuchigami T (2005). An electrolytic system that uses solid-supported bases for in situ generation of a supporting electrolyte from acetic acid solvent. Angew. Chem. Int. Ed 44, 4760–4763. [DOI] [PubMed] [Google Scholar]
- 64 ).OpenEye software package was unable to generate a reasonable cLogP for cubane-based systems. It is suspected that the fragment library employed by the software to calculate these values does not contain a cubane, leading to an erroneous value. Reference 27 provides various ChromLogD values for cubane vs. benzene comparisons, leading to an average increase of 0.28 units upon introduction of the cubane motif.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.