

Abstract
The mammalian secondary palate forms from shelves of epithelia-covered mesenchyme that meet at midline and fuse. The midline epithelial seam (MES) is thought to degrade by apoptosis, epithelial-to-mesenchymal transition (EMT), or both. Failure to degrade the MES blocks fusion and causes cleft palate. It was previously thought that transforming growth factor β3 (Tgfβ3) is required to initiate fusion. Members of the Eph tyrosine kinase receptor family and their membrane-bound ephrin ligands are expressed on the MES. We demonstrated that treatment of mouse palates with recombinant EphB2/Fc to activate ephrin reverse signaling (where the ephrin acts as a receptor and transduces signals from its cytodomain) was sufficient to cause mouse palatal fusion when Tgfβ3 signaling was blocked by an antibody against Tgfβ3 or by an inhibitor of the TgfβrI serine/threonine receptor kinase. Cultured palatal epithelial cells traded their expression of epithelial cell markers for that of mesenchymal cells and became motile after treatment with EphB2/Fc. They concurrently increased their expression of the EMT-associated transcription factors Snail, Sip1, and Twist1. EphB2/Fc did not cause apoptosis in these cells. These data reveal that ephrin reverse signaling directs palatal fusion in mammals through a mechanism that involves EMT but not apoptosis and activates a gene expression program not previously associated with ephrin reverse signaling.
The secondary palate in humans and mice forms from shelves of mesenchyme covered by epithelium. These shelves grow out bilaterally from the internal surfaces of the maxillary processes, elongate on each side of the tongue and become horizontal above the tongue as it descends. As soon as the opposing shelves reach each other, the lateral surfaces of the medial edge epithelia (MEE) cells form the midline epithelial seam (MES) (Murray and Schutte, 2004). Complete disintegration of the MES is essential to form a confluent structure, and failure of palatal fusion causes cleft palate, one of the most common birth defects(Croen et al., 1998). Thus, understanding the mechanism of fusion is an important goal of craniofacial biology.
Palatal fusion has been thought to require Transforming Growth Factor β-3 (Tgfβ3) because Tgfβ3 knockout mice, as well as naturally TGFβ3-null avian systems, display cleft palate, and treatment of either with exogenous Tgfβ3 rescues palatal fusion (Martínez-Alvarez et al., 1996; Sun et al., 1998; Taya et al., 1999). Genetic and phamacological studies have shown that the Tgfβ3 signal, acting through serine/threonine kinase Tgfβ receptors (Tgfβr) on MEE cells, activates Smad, p38 mitogen-activated protein kinase (MAPK), and phosphotidyl inositol 3 kinase (PI3K) pathways in palate epithelium (Kang and Svoboda, 2002; Xu et al., 2008). Fusion requires PI3K and either (but not necessarily both) the Smad or p38 pathways (Xu et al., 2008). However, the mechanism of MES degradation is still in question. Numerous studies suggest that the epithelial cells undergo epithelial-to-mesenchymal transition (EMT), apoptosis, or both (thoroughly reviewed in (Nawshad, 2008)). Recent work on cultured primary MEE cells indicates that Tgfβ3 causes these cells to shift gene expression patterns away from epithelial markers to fibroblastic ones, while assuming a migratory phenotype. They then initiate caspase-dependent apoptosis. This entire process occurs in culture over the same 72 hour time frame as does fusion in the mouse embryo, consistent with a mechanism that is reflective of the actual process in vivo (Ahmed et al., 2007).
We recently reported a role for ephrin signaling in palatal fusion. The Ephs are the largest family of receptor tyrosine kinases. They are classified as A or B based on sequence homology and on their binding preference for the transmembrane B ephrin or the glysosyl phosphotidyl inositol linked A ephrin ligands (Orioli and Klein, 1997). Eph-ephrin systems control a number of contact-dependent processes in development, including cell migration, boundary formation, and proliferation (Davy et al., 2004; Davy and Soriano, 2005; Davy and Soriano, 2007). Ephs function as traditional receptor tyrosine kinases when bound by their ephrin ligands, but they can also act as ligands that activate signaling downstream of the ephrin, which assumes the role of receptor in what is called “reverse signaling” (Murai and Pasquale, 2004). We reported EphB and ephrin-B expression in the MEE during fusion, and we found that ephrin-B reverse signaling is required for palatal fusion in mice and is sufficient to cause fusion in chicken palates without the addition of Tgfβ3 (San Miguel et al., 2011). This finding was supported by a report of cleft palate in ephrin-B2 reverse signaling-deficient mutant mice (Dravis and Henkemeyer, 2011). Interestingly, we discovered that the ephrin reverse signal passes through PI3K, a signaling pathway not previously associated with reverse signaling (San Miguel et al., 2011).
Here we report our most recent study of the cellular mechanism of ephrin reverse signaling in palatal fusion. We found that activation of reverse signaling in mouse palates is sufficient to cause fusion independently of Tgfβr, and that the ephrin signal activates an EMT-like program in palatal epithelial cells, but does not cause apoptosis in these cells. Our data describe a novel role for ephrins in craniofacial development, and clarify their role in palatal fusion.
Materials And Methods
Chemicals and reagents
Anti-Tgfβ3 (Cat#AF-243-NA) and anti-EphB2 (Cat#AF467) were obtained from R&D Systems (Minneapolis, MN). The TgfβrI Kinase Inhibitor VI (SB431542) was from Calbiochem (EMD Millipore Cat#616465) (Billerica, MA). EphB2 ectodomain Fc fusion protein was from R&D Systems (Cat #467-B2) (Minneapolis, MN). IgG Fc protein was from Calbiochem (EMD Millipore Cat #401104) (Billerica, MA). Recombinant Tgfβ3 was purchased from R&D systems, CA. For Immunofluorescence, primary antibodies used (and their source) included the following:, E-Cadherin, Desmoplakin, and Plakoglobin (kindly provided by Dr. James Wahl, University of Nebraska Medical Center), Vimentin (Sigma-Aldrich, MO), Fibronectin (Abcam, MA), ZO-1 (Invitrogen, CA). All antibodies and inhibitors were used at the concentration and time point recommended by the respective manufacturer/provider.
Embryonic palate culture
All animal care and experiments were performed under protocols approved by the Institutional Animal Care and Use Committees of the Baylor College of Dentistry and the University of Nebraska Medical Center. Mouse palate culture was performed as previously described (Kang and Svoboda, 2002; Yu et al., 2008; San Miguel et al., 2011). In brief: Palatal shelves were dissected from e13.5 CD1 mouse embryos and placed nasal side down on polycarbonate membranes (Nucleopore Corp.) with their medial edges in contact. The tissues were cultured with BGJb medium (Gibco) for 72 h. Medium was replaced every 24 h with fresh treatments. Anti-Tgfβ3 was used at concentration of 10 μM. TgfβrI Kinase Inhibitor VI (SB431542) was used at a concentration of 25 μM. Based on our initial dose-response experiments (not shown), this was the concentration of kinase inhibitor that abolished MES degradation in cultured palates while showing no signs of altered cell morphology. EphB2/Fc and control IgG Fc proteins were used at 5 μg/mL, as in our previously published studies. Fc proteins were clustered by mixing with anti-human Fc in a 4 to 1 w/w ratio and incubated at 22°C for 1 h or overnight at 4°C. This treatment allows the soluble Fc proteins to mimic the clustering that occurs on cell membranes and is required to initiate biologically relevant signaling.
Histological analysis
Cultured palates were fixed in 4% formaldehyde/phosphate buffered saline, stabilized in low melting point agarose, and processed for paraffin embedding. Serial 6-μm sections were collected in the coronal orientation from anterior to posterior. Sections were stained with hematoxylin and eosin (H&E) and scored for fusion by at least two independent blinded observers using the previously described scale as follows (Kang and Svoboda, 2002): A score of 5 denotes complete fusion with no epithelia persisting in the midline. A score of 4 means epithelial triangle or islands remain, but they are less than 1/3 the total width of the palatal shelf interface. A score of 3 signifies mesenchymal confluence was achieved in places, but over 1/3 or less of the palatal shelf interface, with large epithelial islands or triangles remaining. A score of 2 means that a continuous epithelial seam persisted in the midline. Palatal shelves that were not touching each other in the midline received a score of 1. Palatal shelves with a score of 1 were cultured in contact with each other but came apart during processing and embedding due to lack of adhesion. Any palates that were not in contact for the entire culture period were discarded and not scored.
Statistical analysis
Palate culture experiments
All palate fusion experiments were performed at least three times for a total n =12–18 for each treatment group. Fusion scores reported are the mean ± standard error of the mean (SEM) of the pooled scores across all experiments. Statistical analyses were made using SPSS software. Mean Fusion Scores were analyzed using Kruskal–Wallis test with the Mann-Whitney U test used to analyze specific sample pairs for significant differences. Differences in fusion score between groups with P <0.01 were considered to be statistically significant. The statistical power of the samples in experiments was evaluated by G*POWER software (Version 3.1). The power with respect to the seriousness of types I and type II errors rate was calculated with the settings type I error, α =0.01 and type II error, β =0.05. We expected that the power analysis under these settings and with a sample size large enough would yield a statistically significant effect.
Cell culture experiments
Data from at least three replicates for each parameter were evaluated and analyzed for significance by SPSS 14.0. The treatment groups included TGFβ3, EphB2/Fc and the control groups (IgG Fc). The observation times were collapsed due to the convenience of the study, and one-way ANOVA was conducted. The significance level was set as 0.05. AP-value of ≤0.05 was considered significant. The one-way ANOVA indicated that the values differ significantly across the treatment groups. Bonferroni post-hoc comparisons of the treatment groups indicated that the negative control control group significantly differ from each other (P ≤0.005). The comparison of each treatment group (time and dose) showed EphB2/Fc treatments groups also differed significantly from the negative control groups, (P ≤0.005).
Culture of isolated primary MEE cells
Embryonic MEE cell culture was performed as previously described (Ahmed et al., 2007; Nawshad et al., 2007; Iordanskaia and Nawshad, 2011; Jalali et al., 2012). The single cell thick periderm covering on each shelf was removed by incubating the shelves with Proteinase K for 1hr at 37°C. The shelves were then cultured at 37°C for 12 h to allow brief adherence to the corresponding opposite shelf (adhered). Adhered shelves in organ culture were then cut close to the seam to ensure limited or no mesenchymal tissues attached to isolated seam. The shelves were then separated and treated with Dispase II for 30 min to allow the primary MES cells to separate from the underlying basement membrane so that epithelial cells could be collected without any mesenchymal contamination. Cells were then cultured in flasks and harvested at the exponential growth stage (~80% confluence) before any exogenous treatment began.
Apoptosis assay
MEE cells were treated in culture with clustered IgG/Fc (negative control), EphB2/Fc, or Cisplatin (positive control) for 24 or 48 h. Cells were then fixed and underwent in situ terminal deoxynucleotidyl transferase (TdT) to transfer biotin-dUTP to the free 3′-OH of cleaved DNA. The biotin-labeled cleavage sites were then visualized by reaction with fluorescein conjugated avidin (avidin-FITC)(TUNEL Apoptosis Detection Kit, Millipore, MA # 17-141f). The same samples underwent a second step of immuno-labeling for tubulin (Cell Signal, MA# 2148) with Alex Flour 488 conjugated secondary Antibody (Invitrogen, CA), followed by mounting with with DAPI (Vectashiel, CA, H1200).
FACS analysis
MEE cells were grown in 10% FBS containing DMEM in T-25 flasks. Approximately 60% confluent cells were treated with 6.0 μM Aphidicholin for 16 h, washed with HBSS and released into complete medium for 30 min. Cells were then treated with complete medium containing clustered IgG/Fc, EphB2/Fc, or cisplatin. Cells were collected every 24 h for live and dead cell stain analysis with a BD FACSArray Bioanalyzer. Vibrant cell metabolic assay kit and Sytox red dead cell stain were purchased from Invitrogen. Cells were stained according to the manufacturer’s protocol. In brief, floating cells were collected and resuspended in PBS with 2 μM C12-resazurin, followed by incubation for 15 min at 37°C. Cells were then detached by trypsin, pelleted, resuspended in 5 nM Sytox Red stain/mL, and incubated for a minimum 15 min at room temperature in the dark. The stained cells were analyzed on a BD FACSArray Bioanalyzer using a green laser at 532 nm to detect C12-resazurin and a red laser at 635 nm to detect Sytox Red stain.
Scratch-wound assay
The scratch-wound assay was conducted as previously described (Nawshad et al., 2007). MES cells were grown to 80% confluency in 6-well culture plates, and a uniform straight line scratch was made with a sterile pipette tip. Scratches in EphB2/Fc (2, 5 and 10 μg/mL) treated and IgG Fc (control) wells were examined for 48h. The migration of cells (or gap filling) was monitored every 12h with phase contrast microscopy where cells were morphologically assessed for the migratory phenotype.
Cell Motility Assay
The Cell Motility Assay was conducted as reported (Nawshad et al., 2007). 8 μm pore size Transwell migration chambers of a 6-well plate (BD BioCoat, MA) were used for migration analyses. 5 × 105 MES cells were seeded in the presence of 5 mg/mL EphB2/Fc in 8 μm pore size Transwell migration upper chambers of a 6-well plate. Treated and control (Ig Fc) MES cells were allowed to migrate through the filter toward media containing serum (10%) for 24–48 h at 37°C. Cells that did not migrate through the filter were removed with a cotton swab from inside the upper chamber. Each filter was fixed in 4% Paraformaldehyde for 10 min, washed three times, each time for 5 min with 1× PBS, placed in Hematoxylin stain (Dako, Mayer’s hematoxylin) for 20 min, rinsed with water, and placed in bluing reagent (alkaline solution such as a weak ammonia solution, 0.08% in water) until the stain turned blue. Subsequently, the filters were washed again using deionized water. Migrating MES cells on the lower side of the filter were randomly counted at 10 areas per field by phase-contrast microscopy. The mean of the 10 areas was determined and is represented in the bar graph in Fig. 6B.
Fig. 6.

Ephrin reverse signaling induces migration of mouse palatal MEE cells. (A) Embryonic MEE cells were grown to confluence and then scratched with a needle to create a cleared area with uniform borders. The cells were treated with IgG Fc or EphB2/Fc for 48 h. (B) The number of cells that migrated across an 8 μm membrane in a transwell chamber was counted at 24 and 48 h. The change in the number of migrating cells was determined by comparison to control (IgG Fc) and plotted as numbers of migrating cells (mean ± SD.; n =3; *P <0.005 compared with controls AP-value of ≤0.05 was considered significant. The one-way ANOVA indicated that the values differ significantly across the treatment groups. All EphB2 treatment (time dependent) differed significantly (*P ≤0.005) from the control groups (IgG Fc).
Immunohistochemistry, immunofluorescence, and immunobloting
The MES cells and embryonic palates from 14.0 to 16.5 dpc underwent Immunohistochemistry, Immunofluorescence and Immunoblotting techniques as described by us previously (Ahmed et al., 2007; Nawshad et al., 2007; Iordanskaia and Nawshad, 2011; Jalali et al., 2012), For protein expression of MES cells by western blot, the cells were grown to confluence in 10% FBS and serum starved in 1% FBS for 24 h, followed by treatment with TGFβ3 (2 and 5 ng/mL) and EphB2 (2 and 5 μg/mL) in 1.0% FBS DMEM for 0–24 h for total protein extraction. For total proteins, we used the nuclear extraction kit from Chemicon total protein Extraction Kit (Millipore) as done by us previously (Ahmed et al., 2007; Iordanskaia & Nawshad 2011). The concentration of total protein was obtained with the Genesys 10 UV scanner (Thermoscientific) at 595 nm. 25 μg of protein extract was electrophoresed on a 10% denaturing gel and transferred onto a nitrocellulose membrane. The membranes were blocked with gelatin, washed with PBS-Tween, incubated with the EphB2 and TGFβ3 antibodies and reacted with anti-goat (1:1000) and anti-rabbit (1:2000) secondary antibodies (Cell Signaling). The bands were then visualized by using an odyssey scanner (Li-Cor). Intensity of the band was measured using the Carestream Molecular Imaging Software version 5.3.1 (Rochester). To perform a t-test analysis of mean intensity measurements, an ROI analysis was done from the data to Microsoft Excel software from the exported “.txt” files. Data points for all samples are paired by spatial arrangement on gel and compared pairwise to minimize the impact of subtle background artifacts on image analysis. MES cells or 8μm sections of 14.5 dpc palates from WT and TGFβ3 knockout mice underwent Immunofluorescence or immnohistochemistry, respectively, as described by us previously (Nawshad and Hay, 2003; Nawshad et al., 2007; Ahmed et al., 2007). Immunofluorescence secondary antibodies were obtained from Invitrogen (Rhodamine, 1:100) and Jackson Immunoresearch (FITC, 1:200).
Gene expression
As described previously, (LaGamba et al., 2005; Zhu et al., 2012) RNA from MEE cells treated with clustered EphB2/Fc (1, 2, or 5 μg/mL) for 48 h, was harvested using the RNeasy Mini Kit (Qiagen, CA) according to the manufacturer’s instructions. RNA integrity was assessed using formaldehyde gels in1XTAE buffer, and RNA purity and concentration were determined by the 260/280 ratio on a Nanodrop 2000C (Thermoscientific, MA). The Ct values were exported into a Microsoft Excel Spreadsheet and analysed according to the ΔCt system. The −ΔΔCt (Snail, Sip1, Twist and E-Cadherin/vs IgG Fc control) values were plotted to show the genes that are up or downregulated in fold/s increase.
The sequences of primers were obtained from the Invitrogen online PCR primer design site, and were synthesized at the Molecular Biology Core Facility, UNMC.
| Mouse Snail | 5′-TGAGGTACAACAGACTATGCAATAGTTC-3′ 5′-CCTGCTGAGGCATGGTTACA-3′ |
| Mouse Twist | 5′-TCCGCGTCCCACTAGCA -3′ 5′-TTCTCTGGAAACAATGACATCTAGGT -3′ |
| Mouse Sip1 | 5′-TTGTGCCCATCACGAAAAAG -3′ 5′-GTGCACAGTTTGACAATTTAATTGAA -3′ |
| Mouse E-cadherin, | 5′-AAGTGACCGATGATGATGCC-3′ 5′-CTTCTCTGTCCATCTCAGCG-3′. |
Gene expression was determined by normalization with the control gene, GAPDH. Each RT-PCR experiment was performed in triplicate.
Results
Ephrin reverse signaling mediates mammalian palatal fusion independently of Tgfβ3 and Tgfβr kinase
We previously reported that exogenous ephrin activation causes fusion in chicken palates without the need for Tgfβ3 (San Miguel et al., 2011). The chicken palate has long been used as a convenient and naïve system to examine Tgfβ3 signaling because it does not produce endogenous Tgfβ3. However, the chicken palate does not fuse naturally in development. We therefore asked whether activation of the ephrin signal would also cause fusion of mammalian palates in the absence of their endogenous Tgfβ signal. We answered this question in two ways using our mouse palate culture system. We performed these experiments by placing embryonic mouse palatal shelves in contact on a support, and observing MES degradation and fusion over 72 h. After histological processing, each palate was scored for fusion on a one to five scale to generate a mean fusion score (MFS) for anterior, middle, and posterior regions. First, we cultured a set of embryonic mouse palates in the presence of a blocking antibody against Tgfβ3 with or without clustered EphB2/Fc protein to activate ephrin-B reverse signaling (preclustering with anti-Fc is necessary to induce signaling, whereas unclustered Eph/Fc acts as a competitive inhibitor of signaling). The use of neutralizing antibodies has long been an accepted way to effectively block Tgfβ action in tissue culture (Martínez-Alvarez et al., 1996; Neptune et al., 2003), and we chose this method as more practical than generating, culturing, and treating large numbers of Tgfβ3 knockout embryos. Second, we cultured another set of palates with a chemical inhibitor of the Tgfβr kinase (SB 431542), again with or without EphB2/Fc. We evaluated the palates for fusion in the anterior, middle, and posterior region of each using a 1–5 scale in which a score of 1 or 2 indicates failure to degrade the MES, while a score of 3 or above indicates significant epithelial degradation and mesenchymal confluence (see Materials and Methods and Fig. 1A). Control palates in the anti-Tgfβ3 experiment fused normally over the three-day time window of these experiments. Fusion was incomplete in the posterior region of these palates (the last part to fuse developmentally), averaging a 3.0 that nevertheless indicates substantial fusion. The anterior and middle regions had average score above 4, signifying near complete fusion (Table 1 and Fig. 1B and C). Antibody treatment abolished MES degradation and palatal fusion such that the epithelial layers in the MES remained almost entirely intact, and no area averaged above a MFS of 2 (Table 1 and Fig. 1B and C). This result validated our use of neutralizing antibody to block Tgfβ3 activity. Kinase inhibitor treatments to block Tgfβr kinase-based signaling also abrogated fusion (Table 1 and Fig. 1D and E). Addition of recombinant EphB2/Fc restored wide-spread seam degradation and largely rescued fusion in antibody-treated palates. Anterior and posterior regions had average scores of 2.6, meaning that, although several of the palates showed significant mesenchymal confluence in these areas, the epithelial layers remained intact on average. However, the middle portion averaged an MFS of 3.9, indicating near complete fusion (Table 1 and Fig. 1B and C). EphB2/Fc addition to inhibitor-treated palates largely rescued fusion in the middle and posterior regions with MFS of 3.8 and 3.0, respectively, but the anterior remained essentially unfused with MFS of 1.9 (Table 1 and Fig. 1D and E). The fact that EphB2/Fc treatment restored fusion in the presence of SB431542 demonstrates that the kinase inhibitor did not impair fusion through non-specific or toxic effects on the tissue. Therefore, activation of ephrin reverse signaling rescued overall palatal fusion in the absence of Tgfβ signaling, but the anterior palate was particularly resistant to this rescue.
Fig. 1.

Ephrin reverse signaling induces palatal fusion without Tgfβ3. Mouse e13.5 palatal shelves were dissected and grown with their medial edges in contact for 72h in the presence of treatments as indicated. All samples received either EphB2/Fc or IgG Fc protein at 5 μg/mL. Tissues were then fixed, paraffin-embedded and sectioned in the coronal orientation from anterior to posterior for histological analysis. Anterior, medial, and posterior regions were scored for fusion based on our one to five scale (see Materials and Methods). Values shown are mean ±SEM with n =12 to 18 palates for each group pooled from three independent experiments. (A) Diagram of palate scoring. (B) Control palates were treated with IgG Fc control protein and fused normally, with a slight decrease in posterior score indicative of the incomplete fusion commonly observed in some embryos during the 72 h experimental period (MFS =4.5 anterior, 4.6 middle, 3.0 posterior). Palates treated with 10 μM anti-Tgfβ3 failed to fuse (MFS =1.4 anterior, 2 middle, 1.3 posterior) and displayed intact MES. Palates treated with anti-Tgfβ3 antibody +EphB2/Fc fused substantially better, especially in the middle region, displaying significant MES degradation (MFS =2.6 anterior, 3.9 middle, 2.6 posterior). (C) Example H&E stained sections from each experimental group in A. (D) Experimental conditions were the same as in A, except that the SB431542 inhibitor of the Tgfβr kinase was used at 25 μM instead of anti-Tgfβ3. IgG Fc control palates fused normally (MFS =3.5 anterior, 4.7 middle, 3.4 posterior), and SB431542 abolished fusion (MFS =1.3 anterior, 1.7 middle, 1.1 posterior). EphB2/Fc largely rescued fusion in the presence of kinase inhibitor (MFS =2.0 anterior, 3.8 middle, 3.1 posterior). (E) Example H&E stained sections from each experimental group in C. Differences between antibody or inhibitor treated groups and their corresponding EphB2/Fc treated groups were statistically significant as determined by Mann Whitney U Test (**P <0.001). Arrows denote midline epithelial cells. Scale bars =100 μm.
TABLE 1.
Mean fusion scores of palates used in this study. Scores are shown ± SEM
| Treatment group | Anterior | Middle | Posterior |
|---|---|---|---|
| IgG Fc | 4.5 ±0.08 | 4.6 ±0.09 | 3.0 ±0.24 |
| IgG +anti-Tgfβ3 | 1.4 ±0.08 | 2.0 ±0.10 | 1.3 ±0.23 |
| EphB2/Fc +anti-TGFβ3 | 2.6 ±0.17 | 3.9 ±0.11 | 2.6 ±0.08 |
| IgG Fc | 3.5 ±0.17 | 4.7 ±0.22 | 3.4 ±0.10 |
| IgG Fc +SB431542 | 1.2 ±0.13 | 1.7 ±0.20 | 1.1 ±0.11 |
| EphB2/Fc +SB431542 | 1.9 ±0.08 | 3.8 ±0.11 | 3.0 ±0.20 |
One concern with our use of clustered EphB2/Fc to activate reverse signaling is that this reagent could also block forward signaling by binding to B ephrins and blocking them from binding to endogenous Eph receptors. Two pieces of data rule this possibility out as an explanation for our results. First, we previously cultured mouse palates with an unclustered EphA4/Fc. This reagent inhibits reverse signaling because it binds B ephrins without activating signaling while acting as a competitive inhibitor to prevent binding of endogenous Ephs. We showed that this treatment blocked fusion, the opposite of the result we report here with clustered EphB2/Fc. Second, we showed that treatment with clustered ephrin-B2/Fc to activate forward signaling was unable to cause fusion in chicken palates, even though clustered EphB2/Fc did cause fusion (San Miguel et al., 2011). Together, these data demonstrate that ephrin reverse signaling is required for mouse palatal fusion and that exogenous activation of this ephrin signal is capable of causing MES degradation and fusion in the absence of a Tgfβ signal.
Our results indicate that ephrins are downstream of Tgfβ3 in palatal fusion, and so we investigated the possibility that Tgfβ3 may simply activate Eph expression in the MEE to cause the fusion signal. Because we know that EphB2, at least, is capable of acting as a ligand to induce fusion, we examined its expression in the palatal MEE in the absence of Tgfβ3. We found that EphB2 protein expression levels in the palates of Tgfβ3 knock out mice were comparable to those in wild type mice, as assessed by immunohistochemical stain (Fig. 2A). Further, when we cultured primary palatal MEE cells in the presence of either Tgfβ3 or clustered EphB2/Fc, we found that Tgfβ3 did not appreciably increase EphB2 levels on Western blot (Fig. 2B). Interestingly, EphB2/Fc did cause a noticeable increase in Tgfβ3 protein, suggesting that the ephrin signal may feedback to stimulate Tgfβ-related pathways during fusion. From these data, we conclude that it is likely not the role of Tgfβ3 to simply induce EphB expression and thereby initiate fusion.
Fig. 2.
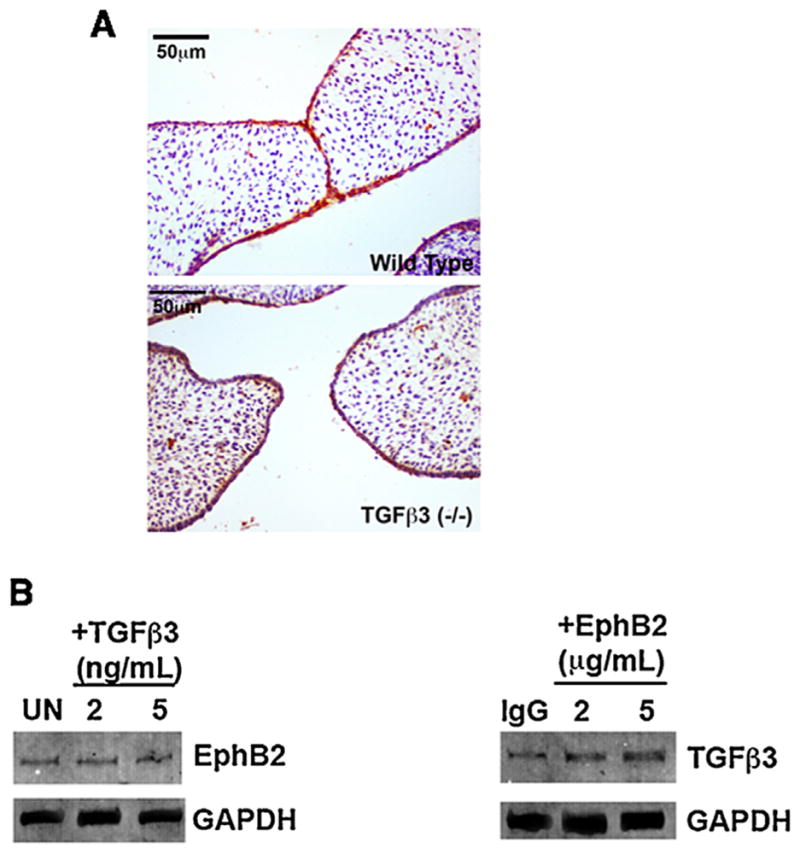
Tgfβ3 is not required for EphB2 expression. (A) Sections of palates from wild type and Tgfβ3 knockout mice were stained with antibody against EphB2. Staining (reddish-brown, DAB) is apparent in the MEE with both genotypes. (B) Mouse palatal MEE cells were grown in the presence of the indicated doses of either 10 μM Tgfβ3 or 5 μg/mL EphB2/Fc for 48 h before being harvested for Western analysis with anti-Tgfβ3 or anti-EphB2. UN =untreated; IgG =IgG Fc treated control. Tgfβ3 treatment did not increase EphB2 levels while EphB2 treatment increased Tgfβ3 levels modestly. Thus, the ability of Tgfβ3 to cause palatal fusion cannot be explained by simple stimulation of EphB expression.
Ephrin reverse signaling does not cause apoptosis in palatal MEE cells
Many studies support the theory that elimination of the palatal MES occurs by programmed cell death, and that the apoptotic signal comes, at least in part, from Tgfβ3 (Glücksmann, 1965; Martínez-Alvarez et al., 2000; Cuervo and Covarrubias, 2004; Ahmed et al., 2007). Because ephrins are known to signal apoptosis in other mammalian systems during development (Depaepe et al., 2005a), we hypothesized that the role of ephrin reverse signaling in the palate is to initiate apoptosis in MEE cells. To test this hypothesis, we cultured primary mouse MEE cells in the presence of EphB2/Fc over 48 h and then looked for apoptosis using the terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) method. Even though Tgfβ3 typically causes widespread cell death in these cultures by 48 h (Ahmed et al., 2007), we were surprised to find no evidence of apoptosis in EphB2/Fc-treated cells compared to IgG Fc negative controls at either 24 or 48 h (Fig. 3). To confirm this result, we quantified apoptosis in cultured MEE cells using a fluorescent live/dead assay with fluorescence activated cell sorting (FACS). Treatment with cisplatin over 48 h (as a positive control) activated apoptosis in 30% of cells (Fig. 4C). By contrast, treatment with clustered EphB2/Fc over the same period generated less than 0.5% apoptotic cells (Fig. 4B), and no more than in the IgG Fc negative control (Fig. 4A).
Fig. 3.

MEE cells were isolated from the mouse palatal MES and grown to 80% confluence, then treated with 5 μg/mL IgG Fc (−ve control) or clustered EphB2/Fc. At 24 or 48 h, cells were labeled with anti-tubulin (Alexa Fluor 488, red), avidin-FITC for TUNEL (green), and DAPI for nuclei (blue). IgG Fc-treated MEE showed strong tubulin expression without any TUNEL signal throughout the experiment. EphB2/Fc-treated cells showed no substantial change in Tubulin expression, and very few cells undergoing apoptosis at either 24 or 48 h. Cisplatin treated MEE cells (+ve control) underwent apoptosis within 24 h with reduced tubulin on average, and showing a characteristic rounded and clumped morphology.
Fig. 4.

Activation of ephrin reverse signaling in does not increase cell death in FACS-sorted MEE cells. MEE cells grown in culture to 80% confluence were treated for 48 h with IgG Fc (−ve control), EphB2/Fc, or cisplatin (+ve control), then incubated with the nucleic acid dyes C12-resazurin (excitation at 488nm) and Sytox Red (at 633 nm) to label live and dead cells, respectively, and analyzed by flow cytometry. Live cells are shown in green (Q1). Dead cells are shown in red (Q4). Cell cycle-arrested cells are shown in pink (Q2) and cellular debris in blue (Q3). (A) 87.7% of IgG Fc-treated cell were alive and viable with only 0.6% intact cells dead. (B) EphB2/Fc treatment showed no difference in the number of dead cells compared to the negative control (86.5% viable and 0.4% dead cells). (C) Cisplatin treatment caused cycle arrest in 19.1% and death in 30.2% of total cells.
Ephrin reverse signaling induces mesenchymal traits in MEE cells
We previously observed some ephrin-B2-expressing MEE cells in the lateral mesenchyme of fusing palates, suggesting that they may have migrated away from the midline (San Miguel et al., 2011). This is reminiscent of the report by Jin and Ding that found genetically labeled MEE cells in similar positions and interpreted them as evidence of EMT and migration (Jin and Ding, 2006). Other studies have also reported evidence that these cells undergo EMT as part of the mechanism of MES degradation, possibly migrating into the adjacent mesenchyme prior to undergoing apoptosis (Fitchett and Hay, 1989; Shuler et al., 1992; Nawshad et al., 2004; Jalali et al., 2012). In support of this theory, Tgfβ3 added to MEE cells in culture causes EMT-like phenotypic changes, cell migration, and gene expression before initiating apoptosis (Ahmed et al., 2007). We therefore used the MEE culture system to test the hypothesis that ephrin reverse signaling causes EMT-like changes in these cells. Cultured MEE cells grown to confluence exhibit the hallmarks of epithelial cells: tightly packed, cuboidal cells joined in a sheet by desmosomes and tight junctions. E-cadherin, desmoplakin, plakoglobin, and zona occludens-1 (ZO-1) are among the proteins that are conspicuously and highly expressed in these epithelia-specific junctions. We observed that the expression of these proteins, while maintained in IgG Fc-treated control cells, was markedly diminished in MEE cells after 24 h of exposure to EphB2/Fc, and largely disappeared by 48 h (Fig. 5A and B). At the same time, expression of the fibroblast markers fibronectin and vimentin increased in these cells (Fig. 5B). These data indicate the disassembly of desmosomes and tight junctions in favor of the assembly of the focal adhesions more suited for mobility. Consistent with this shift, EphB2/Fc-treated cells also lost their tight packing over this time period and assumed a looser, mesenchymal shape (Fig. 5A and B).
Fig. 5.

Ephrin reverse signaling causes EMT-like marker changes in mouse palatal MEE cells. Embryonic mouse MEE cells were cultured for 48 h in either IgG Fc or EphB2/Fc protein at 5 ng/mL, then fixed and processed for immunofluorescent detection of epithelial or mesenchymal markers. (A) Expression of the epithelia-specific cell junction markers E-cadherin, demosplakin, and plakoglobin (green) virtually disappeared after 48 h of EphB2/Fc treatment. (B) Expression of the mesenchymal markers fibronectin (green) and vimentin (red) increased dramatically after 48 h of EphB2/Fc exposure while expression of epithelia-associated proteins E-cadherin (red) and Z01 (green) essentially disappeared.
We tested whether Eph/B2/Fc treatment caused MEE cells to become more motile, as their marker expression suggested, using a scratch-wound assay. Monolayers of MEE cells were scratched with a pipet to create a cell-free zone, and then treated with EphB2/Fc or control IgG Fc for 48 h. Substantial numbers of EphB2/Fc treated cells moved into the cleared scratch area over this period, whereas control cells moved very little (Fig. 6A). We quantified the effect of EphB2 on motility with a transwell assay. MEE cells were placed in the transwell chambers and allowed to cross a filter in the presence of either IgG Fc or EphB2/Fc. After 48 h, the number of cells migrating through the filter was six-fold higher in EphB2/Fc cultures over that observed in controls (Fig. 6B). From our immunofluorescence and scratch-would data, we concluded that activation of ephrin reverse signaling in MEE cells causes them to assume a phenotype indicative of EMT.
Ephrin reverse signaling induces EMT-associated gene expression in MEE cells
EMT requires a shift in gene expression, and so we examined the levels of some key transcription factors associated with gene expression profile changes in EMT. Both the basic helix-loop-helix (bHLH) transcription factor Snail and the zinc-finger Smad-interacting protein 1 (Sip1) are upregulated during developmental EMT and have been shown to repress E-cadherin expression (Jalali et al., 2012). The EMT-associated bHLH factor Twist1 is also upregulated during palatal fusion and plays a role in MES degradation (Yu et al., 2008; Yu et al., 2009; Micalizzi et al., 2010). We quantified the mRNA levels of these three genes in MEE cells after 48 hours of EphB2/Fc treatment using real-time PCR. The messages for these genes increased in a dose-dependent manner. Snail mRNA doubled at the 5 μg/mL dose of EphB2/Fc that was used for all of our palate and MEE culture experiments, and Sip1 increased more than five-fold versus control at the same dose. Although Twist1 mRNA increased only 30%, the change was significant and reproducible. At the same time, E-cadherin mRNA was reduced 60% compared to control (Fig. 7). This result is consistent with a role for ephrin reverse signaling in activation of the EMT gene expression program in MEE cells, although the final determination of the extent of that program will await a more complete gene expression profile.
Fig. 7.
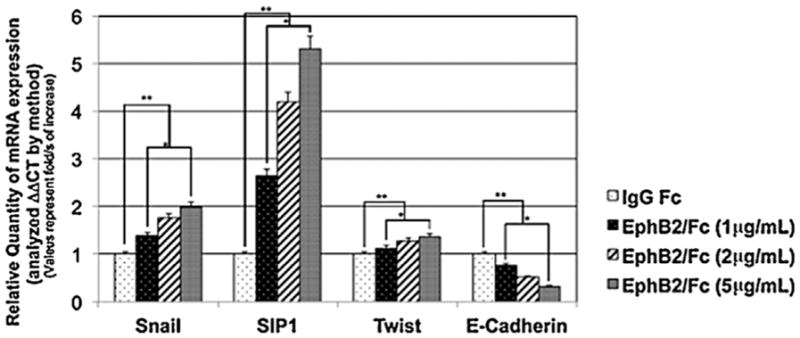
Ephrin reverse signaling induces EMT-associated gene expression in palatal MEE cells. RNA was harvested from mouse palatal MEE cells cultured for 48 h with 1, 2, or 5 μg/mL EphB2/Fc as indicated. Quantitative real time PCR analysis showed that messages for Snail, Sip1, and Twist1 were all significantly increased by EphB2/Fc treatment, demonstrating that ephrin reverse signaling activated expression of EMT-associated transcription factors. The change in mRNA levels was determined by comparison to control (IgG Fc) and plotted as fold change/s (mean ± SD.; n =3; *P <0.005 compared with controls; AP-value of ≤0.05 was considered significant. The one-way ANOVA indicated that the values differ significantly across the treatment groups. All EphB2 treatment (dose dependent) differed significantly from the control groups (IgG Fc) **P <0.0005.
Discussion
The results in this study, along with our published data (San Miguel et al., 2011), show that ephrin reverse signaling is necessary and sufficient to cause mouse palatal fusion, even in the absence of Tgfβ3, a growth factor that was previously considered indispensible for fusion. Further, we show that the ephrin signal causes an EMT-like change in palatal epithelial cells, but does not cause them to undergo apoptosis. Our findings are significant for three reasons. First, the fact that ephrins cause EMT in palatal epithelial cells adds weight to the argument that palatal fusion proceeds through an EMT mechanism. Second, the discovery that ephrin signaling during fusion is separate from, and can supersede, Tgfβ3 shifts the focus of intracellular signaling away from purely those pathway intermediates affiliated with the Tgfβr serine/threonine kinase receptor. Third, the association of ephrin reverse signaling with EMT reveals a previously unknown role for ephrins in activation of a gene expression program.
There are two prevailing theories of the mechanism of MES degradation in palatal fusion. One argues that the MEE cells proceed through EMT to achieve mesenchymal confluence in the palate (Shuler et al., 1991; Shuler et al., 1992; Sun et al., 1998; Kang and Svoboda, 2005; Nawshad et al., 2005; Yu et al., 2009). The other says that these cells are removed by apoptosis to allow the mesenchyme to join (Glücksmann, 1965; Martínez-Alvarez et al., 2000; Cuervo and Covarrubias, 2004). Both of these views have been supported with strong evidence. Recent data suggest that these theories are not mutually exclusive. Ahmed et al. reported that MEE cells in culture exposed to Tgfβ3 undergo EMT, with appropriate changes in morphology and gene expression, followed by apoptosis (Ahmed et al., 2007). Their studies are consistent with genetic evidence from mouse studies of palatogenesis. Jin and Ding showed that Apaf1 knockout mice, while deficient in apoptosis, developed fused palates, indicating that fusion does not rely on apoptosis alone. However, histological examination revealed that the triangles of epithelial cells normally found at the oral and nasal edges of fusing palates persisted in Apaf1 knockouts, whereas they eventually disappear in wild type animals (Jin and Ding, 2006). Ahmed et al observed these same triangles in cultured palates treated with a caspase inhibitor. Of course, caspase-independent apoptosis may still be involved in the process (Dormann and Bauer, 1998; Arnoult et al., 2002). On balance however, the data suggest that both EMT and apoptosis combine to remove the MEE cells and complete palatal fusion. Ephrin-B signaling has been shown to induce apoptosis in other systems (Depaepe et al., 2005b; Park et al., 2013), but we were unable to demonstrate such a role in palatal epithelial cells. Our finding that EphB2 treatment both induces fusion and initiates EMT in MEE cells independently of Tgfβ3 supports a hypothesis in which ephrin induction of EMT is a part of the fusion mechanism, but perhaps leaves the job of programmed cell death to Tgfβ3. Our EphB2 treatments did not completely rescue fusion in Tgfβ-blocked palates, and this observation could be explained by an insufficiency of ephrin reverse signaling to activate a specific part of the fusion program, such as a Tgfβr-dependent apoptotic activity that removes remaining MEE cells. Alternatively, it could be that there is a Tgfβr-specific signal (e.g: one that is Smad-associated) that, while not formally required for fusion, combines with the ephrin signal to complete fusion in the observed time window. In either model, ephrin and Tgfβ signals would collaborate to complete the fusion process, with some signaling branches in common and some unique to each.
The B ephrin cytodomain contains docking sites for a number of signaling proteins. Conserved tyrosines can be phosphorylated and function as SH2 domain binding sites (Holland et al., 1996; Brückner et al., 1997). The SH2/SH3 adaptor protein Grb4/Nckβ was shown to bind to activated ephrin-B1 and signal the disassembly of actin cytoskeletal elements (Cowan and Henkemeyer, 2001). The C-terminal end also carries a PDZ domain binding motif (Lu et al., 2001). Any of these signaling motifs may participate in signaling fusion in the palate. However, the Henkemeyer group demonstrated that mutation in mice of all known conserved ephrin-B2 tyrosines and the PDZ binding domain does not produce cleft palate, even though homozygous deletion of the entire cytodomain in ephrin-B2/LacZ mice does (Dravis and Henkemeyer, 2011). This means that ephrin-B2 contains an as yet unidentified signaling domain that is crucial for palatal seam degradation. We previously published that PI3K signaling is required for ephrin-mediated fusion (San Miguel et al., 2011). This pathway has not previously been associated with reverse signaling and represents uncharted territory in the ephrin field. We are focusing our efforts on identification of the ephrin-B domain responsible for the PI3K signal and its binding proteins.
PI3K phosphorylates Akt, which in turn activates mTor complexes to induce cell migration (Gulhati et al., 2011). Activation of mTor is associated with carcinoma EMT and metastasis, and so the connection of ephrin-Bs to PI3K provides an explanation for why Eph/ephrin signal activation is so often associated with tumor metastases. The PI3K/Akt/mTor axis also connects to the EMT transcriptional program. The mTor kinase phosphorylates the transcriptional activator Stat3 (Yokogami et al., 2000; Zhou et al., 2007), which in turn activates expression of Twist1 and Snail as part of the EMT transcriptional program (Yamashita et al., 2004; Qin et al., 2012), and both Twist1 and Snail are important for palatal fusion (Yu et al., 2008; Yu et al., 2013). Although ephrin-B reverse signaling was previously shown to associate with both Stat3 and the Groucho repressor of Stat3 (Bong et al., 2007; Kamata et al., 2011), very little is known about the potential for reverse signaling to access a gene expression program. The connection of ephrin-B signals to the PI3K pathway in our previous work showed that a connection to transcriptional activation in EMT is plausible. Our data presented here indicate that such a connection exists and is functional during the developmental process of palatal fusion. It also implies that the same connection functions in cases of metastatic EMT, and suggests that ephrin-mediated pathways may be valid targets for cancer therapies.
Acknowledgments
Contract grant sponsor: NIH/NIDCR;
Contract grant numbers: R01DE022804, R03-DE020119, R03-DE020119, R01-DE017986.
The authors would like to thank Isra Abraham and Jeffrey Dyke for help with histology and palate scoring. This work was supported by the National Institutes of Health (R01DE022804 and R03-DE020119 to M. D. B., and R01-DE017986 to A. N.) and by the Baylor Oral Health Foundation (to K. K. H. S.).
Footnotes
Conflict of interest: The authors state that no competing financial interests exist.
Literature Cited
- Ahmed S, Liu C-C, Nawshad A. Mechanisms of palatal epithelial seam disintegration by transforming growth factor (TGF) beta3. Dev Biol. 2007a;309:193–207. doi: 10.1016/j.ydbio.2007.06.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnoult D, Parone P, Martinou JC, Antonsson B, Estaquier J, Ameisen JC. Mitochondrial release of apoptosis-inducing factor occurs downstream of cytochrome c release in response to several proapoptotic stimuli. J Cell Biol. 2002;159:923–929. doi: 10.1083/jcb.200207071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bong Y-S, Lee H-S, Carim-Todd L, Mood K, Nishanian TG, Tessarollo L, Daar IO. EphrinB1 signals from the cell surface to the nucleus by recruitment of ST AT3. Proc Natl Acad Sci USA. 2007;104:17305–17310. doi: 10.1073/pnas.0702337104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brückner K, Pasquale EB, Klein R. Tyrosine phosphorylation of transmembrane ligands for Eph receptors. Science (New York, NY) 1997;275:1640–1643. doi: 10.1126/science.275.5306.1640. [DOI] [PubMed] [Google Scholar]
- Cowan CA, Henkemeyer M. The SH2/SH3 adaptor Grb4 transduces B-ephrin reverse signals. Nature. 2001;413:174–179. doi: 10.1038/35093123. [DOI] [PubMed] [Google Scholar]
- Croen LA, Shaw GM, Wasserman CR, Tolarová MM. Racial and ethnic variations in the prevalence of orofacial clefts in California, 1983–1992. Am J Med Genet. 1998;79:42–47. doi: 10.1002/(sici)1096-8628(19980827)79:1<42::aid-ajmg11>3.0.co;2-m. [DOI] [PubMed] [Google Scholar]
- Cuervo R, Covarrubias L. Death is the major fate of medial edge epithelial cells and the cause of basal lamina degradation during palatogenesis. Development (Cambridge. 2004;131:15–24. doi: 10.1242/dev.00907. [DOI] [PubMed] [Google Scholar]
- Davy A, Aubin J, Soriano P. Ephrin-B1 forward and reverse signaling are required during mouse development. Genes Dev. 2004;18:572–583. doi: 10.1101/gad.1171704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davy A, Soriano P. Ephrin signaling in vivo: Look both ways. Dev Dyn : An official publication of the American Association of Anatomists. 2005;232:1–10. doi: 10.1002/dvdy.20200. [DOI] [PubMed] [Google Scholar]
- Davy A, Soriano P. Ephrin-B2 forward signaling regulates somite patterning and neural crest cell development. Dev Biol. 2007;304:182–193. doi: 10.1016/j.ydbio.2006.12.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Depaepe V, Suarez-Gonzalez N, Dufour A, Passante L, Gorski JA, Jones KR, Ledent C, Vanderhaeghen P. Ephrin signalling controls brain size by regulating apoptosis of neural progenitors. Nature. 2005a;435:1244–1250. doi: 10.1038/nature03651. [DOI] [PubMed] [Google Scholar]
- Depaepe V, Suarez-Gonzalez N, Dufour A, Passante L, Gorski JA, Jones KR, Ledent C, Vanderhaeghen P. Ephrin signalling controls brain size by regulating apoptosis of neural progenitors. Nature. 2005b;435:1244–1250. doi: 10.1038/nature03651. [DOI] [PubMed] [Google Scholar]
- Dormann S, Bauer G. TGF-beta and FGF-trigger intercellular induction of apoptosis: Analogous activity on non-transformed but differential activity on transformed cells. Int J Oncol. 1998;13:1247–1252. doi: 10.3892/ijo.13.6.1247. [DOI] [PubMed] [Google Scholar]
- Dravis C, Henkemeyer M. Ephrin-B reverse signaling controls septation events at the embryonic midline through separate tyrosine phosphorylation-independent signaling avenues. Dev Biol. 2011;355:138–151. doi: 10.1016/j.ydbio.2011.04.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fitchett JE, Hay ED. Medial edge epithelium transforms to mesenchyme after embryonic palatal shelves fuse. Dev Biol. 1989;131:455–474. doi: 10.1016/s0012-1606(89)80017-x. [DOI] [PubMed] [Google Scholar]
- Glücksmann A. Cell death in normal development. Arch biol. 1965;76:419–437. [PubMed] [Google Scholar]
- Gulhati P, Bowen KA, Liu J, Stevens PD, Rychahou PG, Chen M, Lee EY, Weiss HL, O’Connor KL, Gao T, Evers BM. MTORC1 and mTORC2 Regulate EMT, Motility, and Metastasis of Colorectal Cancer via RhoA and Rac1 Signaling Pathways. Cancer Res. 2011;71:3246–3256. doi: 10.1158/0008-5472.CAN-10-4058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holland SJ, Gale NW, Mbamalu G, Yancopoulos GD, Henkemeyer M, Pawson T. Bidirectional signalling through the EPH-family receptor Nuk and its transmembrane ligands. Nature. 1996;383:722–725. doi: 10.1038/383722a0. [DOI] [PubMed] [Google Scholar]
- Iordanskaia T, Nawshad A. Mechanisms of transforming growth factor β induced cell cycle arrest in palate development. J Cell Physiol. 2011;226:1415–1424. doi: 10.1002/jcp.22477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jalali A, Zhu X, Liu C, Nawshad A. Induction of palate epithelial mesenchymal transition by transforming growth factor β3 signaling. Dev Growth Differ. 2012;54:633–648. doi: 10.1111/j.1440-169X.2012.01364.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin J, Ding J. Analysis of cell migration, transdifferentiation and apoptosis during mouse secondary palate fusion. Development (Cambridge, England) 2006;133:3341. doi: 10.1242/dev.02520. [DOI] [PubMed] [Google Scholar]
- Kamata T, Bong Y-S, Mood K, Park MJ, Nishanian TG, Lee H-S. EphrinB1 interacts with the transcriptional co-repressor Groucho/xTLE4. BMB Reports. 2011;44:199–204. doi: 10.5483/BMBRep.2011.44.3.199. [DOI] [PubMed] [Google Scholar]
- Kang P, Svoboda KKH. PI-3 kinase activity is required for epithelial-mesenchymal transformation during palate fusion. Dev Dyn : An official publication of the American Association of Anatomists. 2002;225:316–321. doi: 10.1002/dvdy.10161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang P, Svoboda KKH. Epithelial-mesenchymal transformation during craniofacial development. J Dental Res. 2005;84:678–690. doi: 10.1177/154405910508400801. [DOI] [PubMed] [Google Scholar]
- LaGamba D, Nawshad A, Hay ED. Microarray analysis of gene expression during epithelial-mesenchymal transformation. Dev Dyn : An official publication of the American Association of Anatomists. 2005;234:132–142. doi: 10.1002/dvdy.20489. [DOI] [PubMed] [Google Scholar]
- Lu Q, Sun EE, Klein RS, Flanagan JG. Ephrin-B reverse signaling is mediated by a novel PDZ-RGS protein and selectively inhibits G protein-coupled chemoattraction. Cell. 2001;105:69–79. doi: 10.1016/s0092-8674(01)00297-5. [DOI] [PubMed] [Google Scholar]
- Martínez-Alvarez C, O’Kane S, Taya Y, Ferguson MW. Palate development in the TGF-beta 3 knockout mouse. Low vacuum scanning electron microscopy reveals changes in the medial edge epithelium. Int J Dev Biol. 1996:115S–116S. [PubMed] [Google Scholar]
- Martínez-Alvarez C, Tudela C, Pérez-Miguelsanz J, O’Kane S, Puerta J, Ferguson MW. Medial edge epithelial cell fate during palatal fusion. Dev Biol. 2000;220:343–357. doi: 10.1006/dbio.2000.9644. [DOI] [PubMed] [Google Scholar]
- Micalizzi DS, Farabaugh SM, Ford HL. Epithelial-mesenchymal transition in cancer: Parallels between normal development and tumor progression. J mammary Gland Biol Neoplasia. 2010;15:117–134. doi: 10.1007/s10911-010-9178-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murai KK, Pasquale EB. Eph receptors, ephrins, and synaptic function. Neuroscientist : A review journal bringing neurobiology, neurology and psychiatry. 2004;10:304–314. doi: 10.1177/1073858403262221. [DOI] [PubMed] [Google Scholar]
- Murray JC, Schutte BC. Cleft palate: Players, pathways, and pursuits. J Clin Invest. 2004;113:1676–1678. doi: 10.1172/JCI22154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nawshad A. Palatal seam disintegration: To die or not to die? that is no longer the question. Dev Dyn : An official publication of the American Association of Anatomists. 2008;237:2643–2656. doi: 10.1002/dvdy.21599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nawshad A, Hay ED. TGFbeta3 signaling activates transcription of the LEF1 gene to induce epithelial mesenchymal transformation during mouse palate development. J Cell Biol. 2003;163:1291–1301. doi: 10.1083/jcb.200306024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nawshad A, LaGamba D, Hay ED. Transforming growth factor beta (TGFbeta) signalling in palatal growth, apoptosis and epithelial mesenchymal transformation (EMT) Arch Oral Biol. 2004;49:675–689. doi: 10.1016/j.archoralbio.2004.05.007. [DOI] [PubMed] [Google Scholar]
- Nawshad A, LaGamba D, Polad A, Hay ED. Transforming growth factor-beta signaling during epithelial-mesenchymal transformation: Implications for embryogenesis and tumor metastasis. Cells Tissues Organs. 2005;179:11–23. doi: 10.1159/000084505. [DOI] [PubMed] [Google Scholar]
- Nawshad A, Medici D, Liu C-C, Hay ED. TGFbeta3 inhibits E-cadherin gene expression in palate medial-edge epithelial cells through a Smad2-Smad4-LEF1 transcription complex. J Cell Sci. 2007;120:1646–1653. doi: 10.1242/jcs.003129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neptune ER, Frischmeyer PA, Arking DE, Myers L, Bunton TE, Gayraud B, Ramirez F, Sakai LY, Dietz HC. Dysregulation of TGF-beta activation contributes to pathogenesis in Marfan syndrome. Nat Genet. 2003;33:407–411. doi: 10.1038/ng1116. [DOI] [PubMed] [Google Scholar]
- Orioli D, Klein R. The Eph receptor family: Axonal guidance by contact repulsion. Trends Genet : TIG. 1997;13:354–359. doi: 10.1016/s0168-9525(97)01220-1. [DOI] [PubMed] [Google Scholar]
- Park E, Kim Y, Noh H, Lee H, Yoo S, Park S. EphA/ephrin-A signaling is critically involved in region-specific apoptosis during early brain development. Cell Death Differ. 2013;20:169–180. doi: 10.1038/cdd.2012.121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin Q, Xu Y, He T, Qin C, Xu J. Normal and disease-related biological functions of Twist1 and underlying molecular mechanisms. Cell Res. 2012;22:90–106. doi: 10.1038/cr.2011.144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- San Miguel S, Serrano MJ, Sachar A, Henkemeyer M, Svoboda KKH, Benson MD. Ephrin reverse signaling controls palate fusion via a PI3 kinase-dependent mechanism. Dev dyn : An official publication of the American Association of Anatomists. 2011;240:357–364. doi: 10.1002/dvdy.22546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shuler CF, Guo Y, Majumder A, Luo RY. Molecular and morphologic changes during the epithelial-mesenchymal transformation of palatal shelf medial edge epithelium in vitro. Int J Dev Biol. 1991;35:463–472. [PubMed] [Google Scholar]
- Shuler CF, Halpern DE, Guo Y, Sank AC. Medial edge epithelium fate traced by cell lineage analysis during epithelial-mesenchymal transformation in vivo. Dev Biol. 1992;154:318–330. doi: 10.1016/0012-1606(92)90071-n. [DOI] [PubMed] [Google Scholar]
- Sun D, Vanderburg CR, Odierna GS, Hay ED. TGFbeta3 promotes transformation of chicken palate medial edge epithelium to mesenchyme in vitro. Development (Cambridge, England) 1998;125:95–105. doi: 10.1242/dev.125.1.95. [DOI] [PubMed] [Google Scholar]
- Taya Y, O’Kane S, Ferguson MW. Pathogenesis of cleft palate in TGF-beta3 knockout mice. Development (Cambridge, England) 1999;126:3869–3879. doi: 10.1242/dev.126.17.3869. [DOI] [PubMed] [Google Scholar]
- Xu X, Han J, Ito Y, Bringas P, Deng C, Chai Y. Ectodermal Smad4 and p38 MAPK are functionally redundant in mediating TGF-beta/BMP signaling during tooth and palate development. Dev Cell. 2008;15:322–329. doi: 10.1016/j.devcel.2008.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamashita S, Miyagi C, Fukada T, Kagara N, Che Y-S, Hirano T. Zinc transporter LIVI controls epithelial-mesenchymal transition in zebrafish gastrula organizer. Nature. 2004;429:298–302. doi: 10.1038/nature02545. [DOI] [PubMed] [Google Scholar]
- Yokogami K, Wakisaka S, Avruch J, Reeves SA. Serine phosphorylation and maximal activation of STAT3 during CNTF signaling is mediated by the rapamycin target mTOR. Curr Biol : CB. 2000;10:47–50. doi: 10.1016/s0960-9822(99)00268-7. [DOI] [PubMed] [Google Scholar]
- Yu W, Kamara H, Svoboda KKH. The role of twist during palate development. Dev Dyn : An official publication of the American Association of Anatomists. 2008;237:2716–2725. doi: 10.1002/dvdy.21627. [DOI] [PubMed] [Google Scholar]
- Yu W, Ruest L-B, Svoboda KKH. Regulation of epithelial-mesenchymal transition in palatal fusion. Exp Biol Med (Maywood, NJ) 2009;234:483–491. doi: 10.3181/0812-MR-365. [DOI] [PubMed] [Google Scholar]
- Yu W, Zhang Y, Ruest LB, Svoboda KKH. Analysis of Snail1 function and regulation by Twist1 in palatal fusion. Front Physiol. 2013;4:12. doi: 10.3389/fphys.2013.00012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou J, Wulfkuhle J, Zhang H, Gu P, Yang Y, Deng J, Margolick JB, Liotta LA, Petricoin E, Zhang Y. Activation of the PTEN/mTOR/STAT3 pathway in breast cancer stem-like cells is required for viability and maintenance. Proc Nat Acad Sci. 2007;104:16158–16163. doi: 10.1073/pnas.0702596104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu X, Ozturk F, Liu C, Oakley GG, Nawshad A. Transforming growth factor-β activates c-Myc to promote palatal growth. J Cell Biochem. 2012;113:3069–3085. doi: 10.1002/jcb.24184. [DOI] [PMC free article] [PubMed] [Google Scholar]