

Abstract
In hepatocellular carcinoma (HCC), dysregulated expression of DDX5 (DEAD box protein 5) and impaired autophagy have been reported separately. However, the relationship between them has not been explored. Here we present evidence to show that, by interacting with autophagic receptor p62, DDX5 promotes autophagy and suppresses tumorigenesis. DDX5 inversely correlated with p62/sequestosome 1 (SQSTM1) expression in hepatitis B virus (HBV)-associated and non-HBV-associated HCCs. Patients with low DDX5 expression showed poor prognosis after tumor resection. We found that DDX5 overexpression induced, while DDX5 knockdown attenuated, autophagic flux in HepG2 and Huh7 cells. DDX5 promoted p62 degradation and markedly reduced the half-life of p62. Moreover, DDX5 overexpression dramatically reduced, while DDX5 knockdown promoted, cancer cell growth and tumorigenesis in vitro and in vivo. We found that DDX5 bound to p62 and interfered with p62/TRAF6 (tumor necrosis factor receptor–associated factor 6) interaction. Further findings revealed that the N-terminal domain of DDX5, involved in the interaction with p62, was sufficient to induce autophagy independent of its RNA binding and helicase activity. DDX5 overexpression decreased p62/TRAF6-mediated lysine 63-linked ubiquitination of mammalian target of rapamycin (mTOR) and subsequently inhibited the mTOR signaling pathway. Knockdown of TRAF6 blocked DDX5-induced autophagy. Furthermore, we showed that miR-17–5p downregulated DDX5 and im-paired autophagy. Inhibition of miR-17–5p promoted autophagic flux and suppressed tumor growth in HCC xeno-graft models.
Conclusion:
Our findings define a noncanonical pathway that links miR-17–5p, DDX5, p62/TRAF6, autophagy, and HCC. These findings open an avenue for the treatment of HCC.
Hepatocellular carcinoma (HCC) is one of the leading causes of cancer-related death.(1) Various risk factors contribute to HCC, such as chronic infection with hepatitis B or C viruses, inflammatory factors, and alcohol-related cirrhosis.(2) Autophagy, a ‘‘self-eating’’ process that clears intracellular waste, is crucial for cell development, differentiation, survival, and homeostasis.(3) In hepatocellular carcinoma, the autophagy level is decreased.(4) Studies assessing autophagy in HCC have clearly demonstrated in vitro, in mice, and in patients that autophagy suppresses tumorigenesis.(4,5) The most aggressive malignant HCC cell lines and HCC tissues with recurrent disease display much lower autophagic levels than less aggressive cell lines or tissues.(6) p62, encoded by SQSTM1, is a multi-functional protein involved in cell survival, growth, and death, degraded by autophagy.(7) p62 serves as an adaptor between selective autophagy and ubiquitin signaling.(8) p62 is an oncogenic protein aberrantly accumulated in HCCs,(9,10) and its overexpression in liver is sufficient to induce HCC without carcinogen administration or any other additional stimulus.(11) Importantly, p62 is upregulated not only in liver cancer but also in many other epithelial cancers including those of prostate,(12) lung,(13) glioblastoma,(14) and kidney.(15) Moreover, p62 interacts with tumor necrosis factor receptor–associated factor 6 (TRAF6) to promote nuclear factor κB (NF-κB) activation,(16) and NF-κB can also induce p62 expression.(17) Under nutrient-rich conditions, the p62/TRAF6 interaction enhances mammalian target of rapamycin complex 1 (mTORC1) activation, which inhibits autophagy.(18)
DDX5, the DEAD box protein 5, plays important roles in several cellular processes, including transcription, pre-mRNA processing, and microRNA (miRNA) processing.(19) Studies have also shown that DDX5 interacts with the noncoding RNA steroid receptor RNA activator (SRA), and co-activates estrogen receptor alpha.(20) DDX5 in association with SRA also influences transcriptional activation of MyoD during skeletal muscle differentiation.(21) Although DDX5 has been reported to promote several cancers, including colon cancer, lung cancer, and leukemia, through activating AKT, β-catenin, and NOTCH1 signaling,(22) DDX5 has potential antitumor effect in HCCs. DDX5 has been shown to co-activate the p53 tumor suppressor and to be required for the p53-dependent DNA damage response.(23) Employing a conditional DDX5KO mouse model, Nicol et al. demonstrated that, in the spleen and liver, DDX5 depletion results in the abrogation of DNA damage-induced p21 expression, indicating DDX5 is a selective regulator of the p53 DNA damage response and that it can modulate cell-cycle arrest and apoptosis in a tissue- and context-dependent manner.(24) Previously, we reported that DDX5 regulated Polycomb repressive complex 2 (PRC2)-mediated gene repression and modulated RNA-protein complexes formed with HOTAIR. Hepatitis B virus (HBV)-associated HCCs exhibited a strong negative correlation between DDX5 expression, pluripotency gene expression, and liver tumor differentiation.(25) Recently, DDX5 has been reported to act as a reprogramming roadblock, regulating pluripotency gene expression through its role in miRNA processing.(26) HBV-associated HCC patients exhibit reduced DDX5 expression(25); other studies have shown HCCs with reduced autophagy and accu-mulation of p62(3,27) to associate with poor prognosis after tumor resection.(1) These findings suggest a link between DDX5 downregulation and autophagy. However, the precise mechanism by which DDX5 regulates autophagy remains to be understood.
MiRNAs are small noncoding RNAs that play key roles in cell development and differentiation by mediating the posttranscriptional regulation of protein-coding genes.(28) MiRNAs have a well-recognized role in human carcinogenesis, including hepatocarcinogenesis, and accumulating experimental evidence indicates that they may act as oncogenes or tumor suppressor genes. MiR-17–92 cluster (miR-17–5p, miR-17–3p, miR-18, miR-19b, and miR-20) has been reported to be significantly overexpressed in HCCs,(29) and downregulation of miR-17–92 cluster has been shown to promote autophagy induction.(30,31) C-myc, an important proto-oncogene, is the most well-known transcriptional factor involved in miRNA-17–92 cluster transcriptional activation.(32) Inhibitors of miR-17–92 cluster could potentially serve as adjuvants in chemotherapy, as oncogenic miRNAs such as miR-17 are upregulated in rapamycin-resistant cells.(33) These studies indicate that the autophagic process could be regulated by miR-17–92 cluster, but little is known about how miR-17–92 cluster regulates autophagy to affect specific functions, especially tumorigenesis.
In this study, we hypothesized that DDX5 played a role in the regulation of autophagy. We found an inverse correlation between DDX5 and p62 expression levels in HCC tissues and studied the potential mechanisms. Our findings demonstrated that DDX5 promoted autophagy and reduced liver cancer cell proliferation and tumorigenesis in vitro and in vivo. DDX5 bound to p62 and interfered with p62/TRAF6 interaction, which decreased lysine 63-linked ubiquitination of mammalian target of rapamycin (mTOR) and inhibited the mTOR signaling path-way. MiR-17–5p downregulated DDX5 and inhibited autophagy. Inhibition of miR-17–5p induced autophagic flux and suppressed tumor growth in xenograft models. Taken together, our data demonstrate that DDX5-mediated regulation of autophagy machinery and miR-17–5p are linked with HCC tumorigenesis. These findings may be of value in the development of alternative therapeutics for patients with HCC.
Materials and Methods
Additional experimental procedures are described in the Supporting Information.
REAGENTS, CELL LINES, PLASMID, SMALL INTERFERING RNAS, GENERATION OF STABLE CELL LINES
HEK293T, HepG2, and Huh7 cells were obtained from the Cell Bank of the Chinese Academy of Sciences (Shanghai, China). All cells were cultured in the recommended media supplemented with 10% fetal bovine serum, 100 U/mL penicillin and streptomycin at 37°C in 5% CO2. DDX5-Flag and its deletion mutants DDX5-ΔC-Flag, DDX5-ΔN-Flag, DDX5-WT, and DDX5-K144N(25) were created in the pcDNA3.1 vector by standard subcloning. TRAF6-Myc was constructed in the pCMV- Tag3B. Beclin 1 (BECN1) was constructed in the pCMV6-AC-HA vector (Origene, PS100004) by standard subcloning. P62-Myc and its deletion mutants were gifts from Dr. Ke Li.(34) p62-HA (28027) was obtained from Addgene. Plasmids were transfected using Lipofectamine 3000 (Invitrogen, Carlsbad, CA). siRNAs were transfected using Lipofectamine RNAiMAX (Invitrogen). For shRNA-mediated knockdown experiments, we purchased a pGIPZ vector set carrying shRNA against DDX5 (V3LHS_636798, GE Dharmacon). DDX5 stable expression or knockdown cell lines, including HepG2-DDX5, HepG2-shDDX5, Huh7-DDX5, and Huh7-shDDX5, or their control cell lines were generated by stable transfection of pcDNA-DDX5 or shDDX5, respectively. Cell lines were routinely tested for mycoplasma. MiR-17–5p mimic, miR-17–5p inhibitor, miR-Ctrl for in vitro assay, and miR-Ctrl, miR-17–5p antagomir for in vivo assay, TRAF6, BECN1, ATG5, and control siRNAs were purchased from Ribo Biology, Inc. (Guangzhou, China).
IMMUNOBLOTS AND IMMUNOPRECIPITATION ASSAYS
Immunoblots and immunoprecipitations assays were performed using standard protocols; antibodies used are listed in Supporting Table S1.
TISSUE MICROARRAY AND IMMUNOHISTOCHEMISTRY
In vivo DDX5 or p62 expression was detected by immunohistochemistry using 32 pairs of cancer tissues and adjacent normal-tissue microarrays (Servicebio, China). Immunohistochemistry was performed according to standard protocols, the sections were scanned, and the images were then digitalized. The integrated optical density of DDX5 was calculated using Image-Pro plus 5.1. The clinicopathological characteristics of the 32 patients are summarized in Supporting Table S3.
We also selected published microarray gene expression profile data and accompanying prognostic data from the Gene Expression Omnibus (https://www.ncbi.nlm.nih.gov/geo/; GEO-NCBI) for the validation study. Gene expression profiles of 115 HCC patients (GSE76427) were downloaded and analyzed. Kaplan-Meier survival analyses were implemented to estimate the survival functions after the samples were classified into two groups with a median DDX5 mRNA expression cut-off point used for stratification. GSE30297 were downloaded and analyzed for the expression of miR-17–5p.
LIVE-CELL IMAGING FOR AUTOPHAGIC FLUX
The mRFP-GFP-LC3 adenoviral particles were purchased from HanBio (Shanghai, China). Cells were infected with adenoviral particles and cultured for 24 hours after infection. Images were obtained under ImageXpress® Micro Confocal (Molecular Devices, USA). All image acquisition settings were kept at the same state during the image collection.
STATISTICAL ANALYSIS
Statistical analysis was performed with ANOVA or Student t test by using GraphPad Prism version 5.0 (GraphPad Software, San Diego, CA). Survival curves were analyzed by Kaplan-Meier log-rank (Mentel-Cox) test. The data have been presented as the mean ± standard deviation (x ± SD) or as the mean ± standard error of the mean (x ± SEM). Generally, all experiments were carried out with n ≥ 3 biological replicates. P < 0.05 was statistically significant.
Results
LOW DDX5 EXPRESSION IS ASSOCIATED WITH LOW AUTOPHAGIC ACTIVITY AND POOR PROGNOSIS OF HUMAN HCC
To reveal the relationship between autophagic activity and DDX5 expression in human HCC, HCC tissue microarrays were immunohistochemically assessed using optimized anti-DDX5 and anti-p62 antibodies. The representative HBV- and non-HBV-associated HCC specimens showed low DDX5 expression but high p62 accumulation compared with the adjacent nontumor tissue (Fig. 1A). p62 accumulation indicated low autophagic activity. Automated image analysis was employed to score the percentage of positive cells for DDX5 and p62 protein expression (over total number of cells) across all samples. We found DDX5 protein expression was lower in tumor tissues than in adjacent nontumor tissues (Supporting Fig. S1A). A median cut-off point was chosen with respect to percent positivity of DDX5 to divide the data set into low and high DDX5 groups. As shown in Fig. 1B, tumor samples with low expression of DDX5 exhibited more p62 accumulation. Statistically significant negative correlation (r value) was quantified between protein levels of DDX5 versus p62 in HCC specimens (Fig. 1C), indicating an inverse correlation between p62 and DDX5. Additionally, we employed a human liver cancer tissue cDNA array containing 30 pairs of cancer tissues and adjacent normal tissues for DDX5 mRNA expression. We found DDX5 mRNA expression was lower in HCC tissues compared with adjacent normal tissues (Supporting Fig. S1B).
FIG. 1.

DDX5 negatively correlates with autophagy activity and prognosis of human HCC. (A) Expression of DDX5 and p62/SQSTM1 was detected with immunohistologic staining in tumor tissues and adjacent nontumor tissues of the representative HBV-and non-HBV-associated patients. (B) Expression of p62 in low and high DDX5 groups. A median DDX5 staining cut-off point was used for stratification of 32 HCC tumor and adjacent nontumor tissues. (C) Correlation of DDX5 with p62 in HCC specimens was analyzed, and linear regression coefficient and statistical significance are indicated. N, adjacent nontumor tissue; T, tumor tissue. (D, E) Kaplan-Meier survival analysis of GEO data sets (GSE76427). Disease-free survival (D) and overall survival (E) of HCC patients were determined by Kaplan-Meier analysis and log-rank test. HCC samples were divided into two groups at the median of DDX5 mRNA expression level.
To further validate our findings, we used microarray data obtained from Gene Expression Omnibus (GSE76427) and performed DDX5 mRNA expression and survival analyses. In the data set, HCC samples were divided into two groups, with a median DDX5 mRNA expression cut-off point used for stratification. Differences of the survival risk between the two groups were assessed using the Kaplan-Meier survival analysis. We discovered that liver cancer patients with low DDX5 expression tend to have had worse disease-free survival and overall survival rates among HCC patients after tumor resection (Fig. 1D,E). We also assessed the effect of DDX5 on survival using public database KMploter (https://kmplot.com). The results showed that reduced DDX5 did correlate with poor prognosis in liver cancer (Supporting Fig. S1C). These results suggest that DDX5 and p62 are negatively correlated and that DDX5 may regulate the autophagy process in HCC.
DDX5 STIMULATES AUTOPHAGY IN HEPATOCYTES
To directly test the effect of DDX5 on autophagy in hepatocytes, stable DDX5 overexpression and knockdown HepG2 and Huh7 cell lines were established. DDX5 overexpression strongly increased the expression of BECN1 and microtubule-associated protein 1 light chain 3 (LC3)-II conversion, two well-established markers of autophagy induction; and decreased the levels of p-mTOR/mTOR and p62 (Fig. 2A). By contrast, knockdown of DDX5 reduced the expression of BECN1 and LC3-II conversion but increased the levels of p-mTOR/mTOR and p62 accumulation in HepG2 and Huh7 cells (Supporting Fig. S2A).
FIG. 2.

DDX5 stimulates autophagy in HepG2 and Huh7 cells. (A) HepG2 and Huh7 cells stably expressing DDX5 affected the expression of autophagy-related proteins detected by immunoblotting. Data are representative immunoblots of three independent assays. (B) Control or DDX5 overexpressing HepG2 and Huh7 cells were infected with adenovirus with mRFP-GFP-LC3. The flux rate of autophagy was detected with live cell imaging microscopy and images showing red-colored autophagolysosomes or red/green double-colored autophagosomes. Scale bar = 10 μm. (C) Electron microscopy of HepG2 cells stably expressing vector or DDX5. Typical autophagolysosomes observed in DDX5 overexpressing HepG2 cells. The autophagolysosomes are indicated by the arrows. Scale bar = 1–2 μm. (D) Statistical analysis of the numbers of autophagolysosomes per 40 mm2 in panel (C). (E) HepG2 cells expressing either vector or DDX5 were treated with or without bafilomycin (100 nM) for 12 hours, and the expression of p62 and LC3B-II/I was measured by immunoblotting. **P < 0.01.
To verify the role of DDX5 in regulation of autophagic flux, the flux rate of autophagy was measured using an mRFP-GFP-LC3 reporter construct. Abundant red spots (autolysosomes) with occasional yellow spots (autophagosomes) were observed in DDX5-overexpressing cells (Fig. 2B). However, fewer autophagosomes and autolysosomes were found in DDX5-knockdown cells than in control-shRNA cells, indicating decreased autophagic flux (Supporting Fig. S2B). In addition, transmission electron microscopy revealed that overexpression of DDX5 increased the number of autophagosomes and autolysosomes in HepG2 cells (Fig. 2C,D). Bafilomycin, a later-phase autophagy inhibitor, also enhanced the DDX5-induced accumulation of LC3B-II in HepG2 cells (Fig. 2E). Taken together, these data indicate that DDX5 upregulates the autophagy activity in hepatocytes.
DDX5 ATTENUATES P62 ACCUMULATION AND SUPPRESSES TUMORIGENESIS
To investigate the mechanism by which DDX5 regulates autophagy, we assessed mRNA expression of p62 and BECN1. DDX5 did not change p62 and BECN1 transcription (Supporting Fig. S3A). However, DDX5 overexpression promoted the degradation of p62 in cycloheximide-treated HepG2 cells (Fig. 3A) and markedly decreased the half-life of p62 degradation from 8.5 hours to 2 hours (Fig. 3A). To determine the mechanism for the accelerated degradation of p62 in DDX5 overexpression cells, a turn-over assay was conducted in the presence or absence of bafilomycin and MG132. Bafilomycin treatment significantly attenuated DDX5-mediated p62 degradation, and proteasome inhibitor MG132 moderately prolonged the half-life of p62 (Supporting Fig. S3B). We further examined whether DDX5 affected the ubiquitination of p62. Using an anti-HA (to detect p62-HA), more ubiquitinated proteins were immunoprecipitated from DDX5-overexpressing cells than from control cells (Fig. 3B). These data indicate that DDX5 promotes the binding of p62 to ubiquitinated proteins, leading to the increase of p62-mediated autophagy and the degradation of ubiquitinated proteins and p62 itself. p62 accumulation is critical for tumorigenesis, as overexpression of p62 is sufficient for HCC induction and leads to increased tumor volume in mice.(11) To investigate the tumorigenic role of DDX5 in hepatoma cells, cell proliferation was exam-ined in DDX5 overexpressing, knockdown, and control HepG2 cells. The proliferation of HepG2 cells with DDX5 overexpression was markedly decreased compared with the control cells and significantly increased in DDX5 knockdown cells (Fig. 3C and Supporting Fig. S5). Furthermore, DDX5 overexpressing, knockdown, or control HepG2 cells were injected into the flanks of nude mice to examine whether DDX5 expression could regulate tumorigenesis in vivo. Indeed, DDX5 overexpression significantly inhibited tumor growth and reduced tumor weight compared with control groups, while DDX5 knockdown promoted tumor growth (Fig. 3D and Supporting Fig. S3C). We examined the expression of DDX5 and p62 in tumor tissues. Immunoblot results demonstrated that expression of p62 was significantly reduced while LC3-II conversion increased in the DDX5-overexpressing group (Fig. 3E and Supporting Fig. S3D). Immunohistochemistry analysis confirmed that p62 expression decreased in DDX5 overexpression tissues (Fig. 3F).
FIG. 3.

DDX5 attenuates p62 accumulation and suppresses HCC tumorigenesis. (A) Control or DDX5 overexpressing cells were incubated with cycloheximide (20 μmol/L) for the indicated time points. The cell lysates were then analyzed by immunoblotting. Data are mean ± SEM of three independent assays. (B) HEK 293T cells were transfected with ubiquitin-Flag, p62-HA, and pcDNA-DDX5 for 48 hours and then treated with MG132 (10 μmol/L) for 4 hours. The cell lysates were immunoprecipitated using anti-HA antibody. Data are representative immunoblots of three independent assays. (C) Cell proliferation of HepG2 cells stably expressing vector or DDX5. Data are mean ± SEM of three independent assays. (D) Control or DDX5 overexpressing HepG2 cells were inoculated in the flanks of nude mice (n = 6 for each group). Left: Tumor volumes were measured every 3 days. Right: Tumor weight was measured when mice were sacrificed. Data are mean ± SEM. (E) The expression of DDX5, p62, and LC3B-II/I in xenograft tumor tissues. (F) Representative immunohistochemistry images showed the DDX5 and p62 expression level of tumor tissues. *P < 0.05; **P < 0.01.
To further confirm that DDX5 inhibited tumor growth through autophagy promotion and p62 elimination, in vitro and in vivo tumor growth assays were conducted in DDX5-overexpressed HepG2 cells with autophagy increase or inhibition. We found that knockdown of BECN1 and ATG5, two essential autophagy gene, reversed the antitumor effect of DDX5 overexpression in tumor cells, as demonstrated by cell proliferation assay (Supporting Fig. S4A,B). In addition, overexpression of BECN1 could not further enhance the DDX5 overexpression-mediated HCC cell growth inhibition (Supporting Fig. S4C) but partially reversed DDX5 knockdown-mediated cell growth promotion (Supporting Fig. S4D). Pharmacologic inhibition of autophagy by 3-MA could overturn the antitumor actions of DDX5 over-expression as well (Supporting Fig. S4E,F).
To determine the role of p62 in DDX5-mediated HCC cell growth regulation, we overexpressed p62 in DDX5-overexpressing cells and knockdown of p62 in DDX5-knockdown HepG2 cells. Cell proliferation and colony formation assays results showed that overexpression of p62 reversed DDX5 mediated cell growth inhibition (Supporting Fig. S5A-C), and knockdown of p62 reversed DDX5 knockdown mediated cell growth promotion in HepG2 cells, (Supporting Fig. S5D-F). In summary, DDX5 sup-presses cell proliferation and tumorigenesis through autophagy induction and p62 elimination.
DDX5 IS A P62-INTERACTING PROTEIN
In addition to its role in selective autophagy, p62 is also a central interaction hub because of its ability to interact with key signaling proteins. We assumed that DDX5 mediated autophagy and reduction of p62 accumulation via interaction with p62. We found endogenous DDX5 co-immunoprecipitated with p62 (Fig. 4A). Reciprocal co-immunoprecipitation experiments confirmed that p62 bound to DDX5 (Fig. 4A). Ectopic expression of DDX5-Flag and p62-HA in HEK293T cells also demonstrated that DDX5 associated with p62 (Supporting Fig. S6A,B). To map the DDX5 interaction region, deletion mutants of Flag-tagged DDX5 were made and subjected to immunoprecipitation with p62-HA. The deletion of N-terminal domain of DDX5 (DDX5-ΔN-Flag) abolished the binding of DDX5 to p62 (Fig. 4B), suggesting that N-terminal domain of DDX5 was important for the interaction with p62. In addition, ectopic expression of N-terminal domain of DDX5 increased ubiquitinated p62, whereas C-terminal domain of DDX5 did not (Fig. 4C). Further, we found that N-terminal domain of DDX5 was sufficient to reduce p62 accumulation and increase the ratio of LC3B-II/LC3B-I (Fig. 4D) in HepG2 cells. Given that the ATP bind-ing domain and helicase motif that are important for DDX5 RNA binding and helicase activities are located at N-terminal domain, we wonder whether RNA binding and helicase activities are critical for its autophagy promotion function. We found that the DDX5-K144N mutant(25) that abolished ATPase activity and RNA binding activity also promoted autophagic flux the same as wild-type DDX5 (Fig. 4E). These data indicate that DDX5 stimulates autophagy via interaction with p62 independently of its RNA binding activity and helicase activity.
FIG. 4.

DDX5 is a p62 interacting protein. (A) HepG2 extracts were coimmunoprecipitated (IP) with anti-DDX5 antibody or IgG and followed by immunoblot (IB) analyses with anti-p62 antibody (left) and IP with anti-p62 antibody or IgG followed by blotting with anti-DDX5 antibody (right). Data are representative immunoblots of three independent assays. (B) Mapping of DDX5 regions involved in p62 binding. Top: deletion mutants of DDX5. Bottom: HEK293T cells were co-transfected with p62-HA and indicated constructs of DDX5-Flag. Cell extracts were IP with anti-HA antibody. Data are representative immunoblots of three independent assays. *, heavy chain. (C) HEK 293T cells were co-transfected with p62-HA and indicated constructs of DDX5-Flag expression plasmids for 48 hours and then treated with MG132 (10 μmol/L) for 4 hours. The cell lysates were extracted and immunoprecipitated using anti-HA antibody. Ub, ubiquitin. (D) The N-terminal domain of DDX5 affected the expression of autophagy-related proteins detected by immunoblotting. Data are representative immunoblots of three independent assays. (E) DDX5 promotes autophagy independent of its RNA binding and helicase activity. HepG2 cells were transfected with DDX5-WT and DDX5-K144N, indicated proteins were detected by immunoblotting (left), and the flux rate of autophagy was measured (right). Scale bar = 10 μm.
DDX5 STIMULATES AUTOPHAGY BY INTERFERING WITH THE BINDING OF P62 TO TRAF6
Autophagy adaptor p62 is a multidomain protein and can interact with LC3 for autophagic degradation of ubiquitinated and nonubiquitinated substrates.(7) Co-immunoprecipitation assays showed that DDX5 binds to p62 (Fig. 4). Thus, to characterize the inter-action between DDX5 and p62, domains of p62 that interacted with DDX5 were determined. Different truncated mutants of p62(34) were co-expressed with DDX5, and the integration of DDX5 with different truncated mutants of p62 was assessed by co-immunoprecipitation. We found that the TRAF6-binding (TBS) domain was necessary for p62 binding to DDX5 (Fig. 5A). TRAF6 is a p62 binding protein. We interpreted these results to mean that DDX5 binding to p62 influences the interaction between TRAF6 and p62. Indeed, DDX5 overexpression reduced association of p62 with TRAF6 in HepG2 cells (Fig. 5B). Moreover, co-expression of DDX5-Flag, TRAF6-Myc, and p62-HA in HEK293T cells significantly reduced TRAF6 binding to p62 (Fig. 5C). To deter-mine whether DDX5 promoted autophagy via disruption of p62/TRAF6 interaction, we employed siRNAs to specifically knockdown TRAF6. Indeed, silencing TRAF6 increased the ratio of LC3B-II/LC3B-I, and DDX5 overexpression was unable to further enhance autophagy (Fig. 5D). Results of tandem fluorescence and bafilomycin treatment consistently showed that silencing TRAF6 increased the autophagic flux, while DDX5 overexpression did not further increase autophagy flux (Supporting Fig. S7). These data indicate that DDX5 interacts with p62 and interferes with the binding of TRAF6 to p62, leading to dysfunction of p62/TRAF6-mediated signaling.
FIG. 5.

DDX5 promotes autophagy by interfering with the binding of p62 to TRAF6. (A) Mapping of p62 regions involved in DDX5 binding. Top: different truncated mutants of p62. Bottom: HEK 293T cells were co-transfected with indicated constructs of p62-Myc and DDX5-Flag. Cell extracts were IP with anti-Flag antibody. *, heavy chain. (B) HepG2 cells were transfected with vector or DDX5-Flag. The cell extracts were coimmunoprecipitated (IP) with anti-p62 antibody and blotted with anti-TRAF6 antibody. (C) HEK293T cells were co-transfected with p62-HA, TRAF6-Myc, and DDX5-Flag. Cell extracts were IP using anti-HA antibody and blotted with anti-Myc antibody. (D) HepG2 cells stably expressing vector or DDX5 were transfected with control-siRNA or TRAF6-siRNA. Cell lysates were collected, and the autophagy-related proteins were analyzed. Data are representative immunoblots of three independent assays. The ratio of LC3B-II/LC3B-I was presented as mean ± SEM (n = 3). NS: nonsignificant.
DDX5 INHIBITS K63 POLYUBIQUITINATION AND ACTIVATION OF MTOR
mTOR is part of two signaling complexes termed mTORC1 and mTORC2.(35) Rapamycin, the specific mTOR inhibitor, induces autophagy via mTOR inhibition. mTOR polyubiquitination is required for mTORC1 activation. It has been shown that p62/TRAF6 complex activates mTORC1 by K63-type polyubiquitination of mTOR, thereby inhibiting autophagy.(18) Our results showed DDX5 overexpression inhibited phosphorylation of mTOR (Fig. 2A) and disrupted p62/TRAF6 interaction (Fig. 5). Hence, we hypothesized that DDX5 promoted autophagy through inhibition of p62/TRAF6-induced lysine 63-linked ubiquitination of mTOR. DDX5 overexpression decreased lysine 63-linked ubiquitination (Fig. 6A), while it had no significant effect on the lysine 48-linked ubiquitination of mTOR (Fig. 6B). Ubiquitination through K48 of the ubiquitin chain generally targets proteins for degradation, whereas K63 ubiquitination often regulates signaling activation and trafficking.(36) We also detected mTOR downstream effectors S6K and 4EBP1. TRAF6 overexpression increased phosphorylated S6K and 4EBP1 levels, which indicated mTORC1 activation. By contrast, co-expression of DDX5 significantly blocked TRAF6-induced upregulation of S6K and 4EBP1 phosphorylation (Fig. 6C,D). Recent studies by others have also described that p62/TRAF6 complex activates mTORC1, which can upregulate c-Myc protein expression.(12,37) We found that DDX5 decreased mTORC1 activity, resulting in reduced c-Myc expression (Fig. 6C,D). Together, these results demonstrate that DDX5 inhibits the lysine 63-linked polyubiquitination of mTOR by interfering with p62 binding to TRAF6 and stimulates autophagy in HepG2 cells.
FIG. 6.

DDX5 inhibits K63 polyubiquitination and activation of mTOR. (A, B) HEK293T cells were co-transfected with TRAF6-Myc and DDX5-Flag. Cell extracts were coimmunoprecipitated (IP) using anti-mTOR antibody and blotted with anti-ubiquitin antibody (A) and anti-K63 or anti-K48 antibody (B). (C, D) HepG2 cells were transfected with the indicated plasmids. The mTOR signaling proteins were detected by immunoblotting (C). Immunoblot results are quantified in each group (D). Data are representative immunoblots of three independent assays. **P < 0.01; ***P < 0.001.
MIR-17–5P DOWNREGULATES DDX5 AND SUPPRESSES CELL AUTOPHAGY
To explore the mechanism that mediates the downregulation of DDX5, we hypothesized the involvement of DDX5 targeting miRNAs. Accumulating data have pointed to a central regulatory role for miRNAs in the initiation and development of HCC. Interestingly, the oncomir cluster miR-106b~25 and its paralog miR-17~92 target downregulation of DDX5, observed during viral replication HBV-associated HCC(25,38) (Andrisani et al, unpublished data). We integrated the results of the prediction software programs miRanda, TargetScan, and PicTar. Of the miRNAs predicted to target DDX5, miR-17–5p was identified as a candidate by all three programs (Fig. 7A). Previously published data has demonstrated that miR-17–5p was significantly upregulated in human HCC samples.(29) Thus, we assumed miR-17–5p might participate in DDX5 regulation and autophagy. We found that miR-17–5p mimic reduced the DDX5 mRNA expression, whereas miR-17–5p inhibitor increased the expression level (Fig. 7A). Because miRNAs downregulate protein synthesis by binding to the 3′UTR of mRNAs, we assessed whether miR-17–5p targets DDX5 using the 3′UTR reporter assays. Treatment of miR-17–5p mimic significantly downregulated luciferase activity of DDX5 3′UTRs in HepG2 cells, while miR-17–5p inhibitor increased luciferase activity (Fig. 7B). The regulation was blunted when the miR-17–5p binding sites were mutated (Fig. 7B). These findings indicate that the 3′UTR of DDX5 is targeted by miR-17–5p to suppress its expression. We further examined the miR-17–5p and DDX5 axis and found that miR-17–5p mimic reduced the protein level of endogenous DDX5 in HepG2 cells, whereas the miR-17–5p inhibitor increased the DDX5 protein level (Fig. 7C). Furthermore, the miR-17–5p mimic downregulated the ratio of LC3B-II/LC3B-I and increased p62 expression. At the same time, miR-17–5p inhibitor increased LC3-II conversion while reducing p62 accumulation (Fig. 7C). We found abundant red spots with fewer yellow spots in miR-17–5p inhibitor-treated cells (Fig. 7C), indicating increased autophagic flux. However, treatment of HepG2 cells with miR-17–5p mimic inhibited autophagic flux (Fig. 7C). Furthermore, bafilomycin enhanced miR-17–5p inhibitor-induced accumulation of LC3B-II in HepG2 cells, while miR-17–5p mimic did not (Supporting Fig. S8A,B). In addition, knockdown of DDX5 by siRNA did not augment miR-17–5p mimic-mediated autophagy inhibition (Supporting Fig. S8C) but reversed miR-17–5p inhibitor-mediated autophagy promotion, suggesting that miR-17–5p is an autophagy blocker through down-regulation of DDX5 (Fig. 7D).
FIG. 7.

MiR-17–5p downregulates DDX5 to promote tumorigenesis. (A) Top: Diagram showing the putative miR-17–5p-targeting seed sequence on DDX5–3′ UTR. Bottom: Relative DDX5 mRNA expression upon transfection with miR-17–5p mimic or miR-17–5p inhibitor in HepG2 cells. (B) Fold change in luciferase activity driven by the WT (wide-type) or Mut (mutant) DDX5–3′ UTR reporter under miR17–5p mimic or miR-17–5p inhibitor treatment in HepG2 cells. Data are presented as mean ± SD. (C) Immunoblots of autophagy-related proteins in HepG2 cells transfected with miR17–5p mimic and miR-17–5p inhibitor (left), and the flux rate of autophagy was detected by infecting adenovirus with mRFP-GFP-LC3 (right). Scale bars = 10 μm. (D) Knockdown of DDX5 reversed miR-17–5p inhibitor mediated autophagy promotion. Autophagy-related proteins were detected by immunoblotting. (E) HepG2 cells were inoculated in the flanks of nude mice. Upon the tumor xenografts reaching 100 mm3, miR-Ctrl or miR-17–5p antagomir were continuously intratumorally injected for 2 weeks. Left: tumor volumes were measured every 3 days. Right: tumor weight was measured when mice were sacrificed. Data are mean ± SEM (n = 6 per group). (F) The expression of DDX5, p62 and LC3B-II/I in xenograft tumor tissues were detected by immunoblotting (left) and immunohistochemistry (right). **P < 0.01; ***P < 0.001. NS: nonsignificant.
We next investigated the significance of miR-17–5p antagomir in HCC growth in vivo. The mice with tumor xenografts reaching 100 mm3 were randomly divided into two experimental groups: miR-Ctrl and miR-17–5p antagomir treatment. After continuous intratumoral injection of miR-Ctrl and miR-17–5p antagomir for 2 weeks, we found that tumor growth and tumor weight were significantly suppressed in the miR-17–5p antagomir treatment group (Fig. 7E). The expression of DDX5 was markedly increased, while p62 was significantly reduced in the miR-17–5p antagomir group (Fig. 7F and Supporting Fig. S8D). Moreover, immunohistochemistry analysis confirmed that DDX5 increased and p62 decreased in miR-17–5p antagomir-treated tumor tissues (Fig. 7F). We also used microarray data obtained from Gene Expression Omnibus (GSE30297) and performed miR-17–5p expression analysis. We found that miR-17–5p expression and tumor recurrence rate are positively correlated in human hepatocellular carcinoma (Supporting Fig. S8E). These results demonstrate that miR-17–5p antagomir can suppress HCC progression in vivo.
Discussion
A large number of studies have been conducted on the mechanisms underlying hepatocarcinogenesis. Accumulating evidence suggests that autophagy is involved in tumor suppression.(10,27) Impaired autophagy causes diverse pathologic conditions in humans, including liver dysfunction and tumorigenesis.(39) In this study, we provide evidence that low DDX5 accompanied by high p62 expression was correlated with liver tumor formation and poor survival in HCC patients (Fig. 1). We revealed a crucial role for DDX5 in regulating autophagy and tumorigenesis in vitro and in vivo. Furthermore, we provide evidence that DDX5 is a target gene of miR-17–5p. Inhibition of miR-17–5p induced autophagy and suppressed liver tumor growth. Thus, our mechanistic studies, together with clinical data, strongly suggest that DDX5 is an important player in hepatocarcinogenesis.
The discovery of the critical roles of cargo receptor p62 in cancers provides a strong rationale for p62-based cancer therapy.(5) Interference with autophagic flux attenuates p62 degradation, resulting in p62 accumulation and oligomerization, aggregation of ubiquitinated proteins, Keap1 sequestering, and NRF2 stabilization.(7,40) NRF2 could mediate transcriptional activation of the SQSTM1 gene, further increasing p62 accumulation through a feed-forward loop that promotes cancer initiation.(41) In HBV- and non-HBV-associated HCC tumor tissues, low autophagic activity and p62 accumulation were detected (Fig. 1). Our previous results showed that DDX5 decreased in HBx-expressing cells, HBx/c-myc mouse liver tumors, and HBV-associated HCC patient tissues.(25) This prompted us to investigate whether DDX5 had any effect on autophagy. DDX5 promoted autophagy and p62 degradation (Fig. 2) and significantly reduced the half-life of p62 (Fig. 3). p62 is the main component of Mallory-Denk bodies and hyaline granules, which are hallmarks of chronic liver diseases and greatly enhance HCC risk.(42) DDX5 expression in hepatic stellate cells was inversely correlated with fibrogenic genes collagen I, tissue inhibitor of metal-loproteinase-1, and transforming growth factor-β1.(43) In addition to recent studies showing the pathogenetic role of p62 inclusions during autophagy inhibition,(11) NASH, and HCC, our current study provides a potentially important role that DDX5 may play in NASH and NASH-associated HCC progression.
p62 is a signaling hub and scaffold protein with numerous functional domains and interacting partners.(44) p62, degraded by autophagy, also regulates autophagy, creating a feed-forward loop by which p62 activation of mTORC1 increases p62 levels, further promoting mTORC1 activity.(45) The core components of mTORC1 include the mTOR kinase, raptor, and mLST8/GbL.(35) Of special relevance for the p62-mTORC1 connection, p62 recruits TRAF6 to the mTORC1 complex, resulting in the K63-type polyubiquitination of mTOR, which is important for its efficient activation.(18,46) p62 also binds TRAF6 to promote nuclear factor κB (NF-κB) activation,(47) and NF-κB can also induce p62 expression.(17) p62-mTORC1 signaling induce c-Myc expression, which is important for cancer cell proliferation. A link between p62 accumulation and c-Myc expression is also observed in prostate cancer stromal fibroblasts and HCC.(11,12) These studies indicate that interfering with p62 binding to TRAF6, which is needed for mTORC1 activation, may be useful for preventing progression of chronic liver disease to HCC as well as attenuating recurrence of resectable HCC. Our data present evidence that DDX5 interacts with the TBS domain of p62, which interferes with p62 binding to TRAF6, decreasing K63-type polyubiquitination of mTOR and subsequently suppressing mTOR activity and downstream signaling (Figs. 5,6). Considering the various roles of DDX5 in the development of different cancers, the precise role may depend on the type of tumor and/or the expression of other proteins with which DDX5 interacts. Our results indicate that elevating DDX5 could be a promising strategy for HCC therapy.
The expression of certain miRNAs is deregulated in human HCC compared with matched nonneoplastic tissue.(48) One of the best-studied set of miRNAs so far is the miR-17–92 cluster members, which have been found overexpressed in HCC patient tissues.(29) MiR-17–5p, the most prominent member, plays an important role in liver cancer growth and metastasis by activating the p38 MAPK-HSP27 pathway by directly targeting the transcription factor E2F1.(49) MiR-17–5p expression is highly elevated in patient-derived HCC tissues, especially in metastasis-derived tissues. In addition, serum levels of circulating miR-17–5p were upregulated in a relapse group of patients and downregulated in the postoperative group. (50) The regulatory role of miR-17–5p in autophagy remains to be determined. Here, we reveal that oncogenic miR-17–5p targets and downregulates DDX5, with miR-17–5p inhibition inducing autophagic flux and inhibiting tumor growth of HCC xenograft in mice (Fig. 7). DDX5 plays an important role in miRNA processing. For example, DDX5 inhibits reprogramming to pluripotency through its role in producing mature miR-125b.(26) Further study is required to determine whether DDX5 regulation of autophagy involves miRNA processing. C-Myc, an important protooncogene, is involved in miR-17–5p transcriptional activation (Supporting Fig. S8F). (32) p62/TRAF6 induces c-Myc expression, while DDX5 could disrupt p62/TRAF6 interaction. Thus, our data reveal a potential feedback loop in DDX5 and miR-17–5p, involving the p62/TRAF6-mTORC1-c-Myc axis. In this context, miR-17–5p has emerged as target for liver cancer therapy.
In summary, the data provided herein establish a autophagy regulation mechanism (model, Fig. 8), involving the antagonism between DDX5 and TRAF6 in regulating mTOR signaling, by binding to p62. First, DDX5 expression was positively associated with autophagic activity and overall survival in HCC patients. Second, DDX5 mediated autophagy and HCC cell growth inhibition by binding to p62 and blocked p62/TRAF6-induced mTORC1 activation. Finally, miR-17–5p inhibition upregulated DDX5 and suppressed liver tumor growth in vitro and in vivo. Functional characterization of the miR-17–5p-DDX5-p62-mTORC1 axis in hepatocarcinogenesis may provide insight into the treatment of liver cancer. Accordingly, our studies identify DDX5 and miR-17–5p as important cellular targets for the design of therapies to combat liver cancer.
FIG. 8.
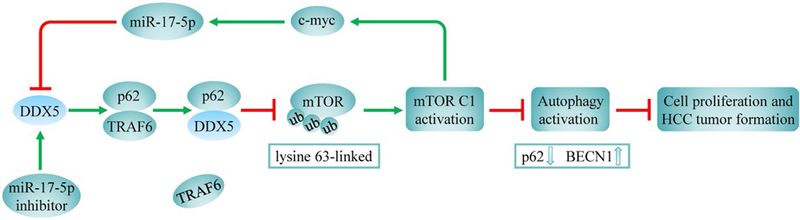
A schematic hypothetical model shows how DDX5 regulates autophagy to affect cell proliferation and tumorigenesis in HCC. DDX5 binds p62 and interferes with p62/TRAF6 interaction, suppressing mTOR signaling. MiR-17–5p inhibition upregulates DDX5 expression and promotes autophagy activity and p62 elimination, thereby suppressing cell proliferation and tumor formation.
Supplementary Material
Acknowledgments
Supported by startup funding from China Pharmaceutical University, Natural Science Foundation of Jiangsu Province (BK20181332) and National Natural Science Foundation of China (No. 81872889) to H. Zhang, NIH grant DK044533 to OA and NIH grant P30CA023168 to the Purdue Center for Cancer Research, the Priority Academic Program Development of Jiangsu Higher Education Institutions (PAPD), the 111 project (B18056), the Program for Changjiang Scholars, and the Innovative Research Team in University to LY. Kong (Program No. PCSIRT-IRT1193).
Abbreviations:
- BECN1
Beclin 1
- DDX5
DEAD box protein 5
- HBV
hepatitis B virus
- HCC
hepatocellular carcinoma
- LC3
microtubule-associated protein 1 light chain 3
- miRNA
microRNA
- mTOR
mammalian target of rapamycin
- mTORC1
mammalian target of rapamycin complex 1
- SQSTM1
sequestosome 1
- SRA
steroid receptor RNA activator
- TRAF6
tumor-necrosis factor receptor–associated factor 6.
Footnotes
Potential conflict of interest: Nothing to report.
Supporting Information
Additional Supporting Information may be found at onlinelibrary.wiley.com/doi/10.1002/hep.30300/suppinfo.
REFERENCES
- 1).Njei B, Rotman Y, Ditah I, Lim JK. Emerging trends in hepatocellular carcinoma incidence and mortality. Hepatology 2015;61:191–199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2).Bruix J, Gores GJ, Mazzaferro V. Hepatocellular carcinoma: clinical frontiers and perspectives. Gut 2014;63:844–855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3).Zhong Z, Sanchez-Lopez E, Karin M. Autophagy, inflammation, and immunity: A troika governing cancer and its treatment. Cell 2016;166:288–298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4).Rautou PE, Mansouri A, Lebrec D, Durand F, Valla D, Moreau R. Autophagy in liver diseases. J Hepatol 2010;53:1123–1134. [DOI] [PubMed] [Google Scholar]
- 5).Mathew R, Karp CM, Beaudoin B, Vuong N, Chen G, Chen HY, et al. Autophagy suppresses tumorigenesis through elimination of p62. Cell 2009;137:1062–1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6).Ding ZB, Shi YH, Zhou J, Qiu SJ, Xu Y, Dai Z, et al. Association of autophagy defect with a malignant phenotype and poor prognosis of hepatocellular carcinoma. Cancer Res 2008;68:9167–9175. [DOI] [PubMed] [Google Scholar]
- 7).Moscat J, Karin M, Diaz-Meco MT. p62 in cancer: signaling adaptor beyond autophagy. Cell 2016;167:606–609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8).Lee Y, Weihl CC. Regulation of SQSTM1/p62 via UBA domain ubiquitination and its role in disease. Autophagy 2017;13:1615–1616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9).Komatsu M Potential role of p62 in tumor development. Autophagy 2011;7:1088–1090. [DOI] [PubMed] [Google Scholar]
- 10).Wu SY, Lan SH, Wu SR, Chiu YC, Lin XZ, Su IJ, et al. Hepatocellular carcinoma-related cyclin D1 is selectively regulated by autophagy degradation system. Hepatology 2018;68:141–154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11).Umemura A, He F, Taniguchi K, Nakagawa H, Yamachika S, Font-Burgada J, et al. p62, Upregulated during preneoplasia, induces hepatocellular carcinogenesis by maintaining survival of stressed HCC-initiating cells. Cancer Cell 2016;29:935–948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12).Valencia T, Kim JY, Abu-Baker S, Moscat-Pardos J, Ahn CS, Reina-Campos M, et al. Metabolic reprogramming of stromal fibroblasts through p62-mTORC1 signaling promotes inflam-mation and tumorigenesis. Cancer Cell 2014;26:121–135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13).Duran A, Linares JF, Galvez AS, Wikenheiser K, Flores JM, Diaz-Meco MT, et al. The signaling adaptor p62 is an important NF-kappaB mediator in tumorigenesis. Cancer Cell 2008;13:343–354. [DOI] [PubMed] [Google Scholar]
- 14).Galavotti S, Bartesaghi S, Faccenda D, Shaked-Rabi M, Sanzone S, McEvoy A, et al. The autophagy-associated factors DRAM1 and p62 regulate cell migration and invasion in glioblastoma stem cells. Oncogene 2013;32:699–712. [DOI] [PubMed] [Google Scholar]
- 15).Li L, Shen C, Nakamura E, Ando K, Signoretti S, Beroukhim R, et al. SQSTM1 is a pathogenic target of 5q copy number gains in kidney cancer. Cancer Cell 2013;24:738–750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16).Moscat J, Diaz-Meco MT. p62 at the crossroads of autophagy, apoptosis, and cancer. Cell 2009;137:1001–1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17).Ling J, Kang Y, Zhao R, Xia Q, Lee DF, Chang Z, et al. KrasG12D-induced IKK2/beta/NF-kappaB activation by IL-1alpha and p62 feedforward loops is required for devel-opment of pancreatic ductal adenocarcinoma. Cancer Cell 2012;21:105–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18).Linares JF, Duran A, Yajima T, Pasparakis M, Moscat J, Diaz-Meco MT. K63 polyubiquitination and activation of mTOR by the p62-TRAF6 complex in nutrient-activated cells. Mol Cell 2013;51:283–296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19).Bourgeois CF, Mortreux F, Auboeuf D. The multiple functions of RNA helicases as drivers and regulators of gene expression. Nat Rev Mol Cell Biol 2016;17:426–438. [DOI] [PubMed] [Google Scholar]
- 20).Caretti G, Lei EP, Sartorelli V. The DEAD-box p68/p72 proteins and the noncoding RNA steroid receptor activator SRA: eclectic regulators of disparate biological functions. Cell Cycle 2007;6:1172–1176. [DOI] [PubMed] [Google Scholar]
- 21).Caretti G, Schiltz RL, Dilworth FJ, Di Padova M, Zhao P, Ogryzko V, et al. The RNA helicases p68/p72 and the noncod-ing RNA SRA are coregulators of MyoD and skeletal muscle differentiation. Dev Cell 2006;11:547–560. [DOI] [PubMed] [Google Scholar]
- 22).Cai W, Xiong Chen Z, Rane G, Satendra Singh S, Choo Z, Wang C, et al. Wanted DEAD/H or alive: helicases winding up in cancers. J Natl Cancer Inst 2017;109. [DOI] [PubMed] [Google Scholar]
- 23).Bates GJ, Nicol SM, Wilson BJ, Jacobs AM, Bourdon JC, Wardrop J, et al. The DEAD box protein p68: a novel transcriptional coactivator of the p53 tumour suppressor. EMBO J 2005;24:543–553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24).Nicol SM, Bray SE, Black HD, Lorimore SA, Wright EG, Lane DP, et al. The RNA helicase p68 (DDX5) is selectively required for the induction of p53-dependent p21 expression and cell-cycle arrest after DNA damage. Oncogene 2013;32:3461–3469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25).Zhang H, Xing Z, Mani SK, Bancel B, Durantel D, Zoulim F, et al. RNA helicase DEAD box protein 5 regulates Polycomb repressive complex 2/Hox transcript antisense intergenic RNA function in hepatitis B virus infection and hepatocarcinogenesis. Hepatology 2016;64:1033–1048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26).Li H, Lai P, Jia J, Song Y, Xia Q, Huang K, et al. RNA he-licase DDX5 inhibits reprogramming to pluripotency by miR-NA-based repression of RYBP and its PRC1-dependent and-independent functions. Cell Stem Cell 2017;20:462–477.e6. [DOI] [PubMed] [Google Scholar]
- 27).Lan SH, Wu SY, Zuchini R, Lin XZ, Su IJ, Tsai TF, et al. Autophagy suppresses tumorigenesis of hepatitis B virus-associated hepatocellular carcinoma through degradation of microRNA-224. Hepatology 2014;59:505–517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28).Bartel DP. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell 2004;116:281–297. [DOI] [PubMed] [Google Scholar]
- 29).Connolly E, Melegari M, Landgraf P, Tchaikovskaya T, Tennant BC, Slagle BL, et al. Elevated expression of the miR-17–92 polycistron and miR-21 in hepadnavirus-associated hepatocellular carcinoma contributes to the malignant phenotype. Am J Pathol 2008;173:856–864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30).Tazi MF, Dakhlallah DA, Caution K, Gerber MM, Chang SW, Khalil H, et al. Elevated Mirc1/Mir17–92 cluster expression negatively regulates autophagy and CFTR (cystic fibrosis trans-membrane conductance regulator) function in CF macrophages. Autophagy 2016;12:2026–2037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31).Xu J, Wang Y, Tan X, Jing H. MicroRNAs in autophagy and their emerging roles in crosstalk with apoptosis. Autophagy 2012;8:873–882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32).Benhamou D, Labi V, Novak R, Dai I, Shafir-Alon S, Weiss A, et al. A c-Myc/miR17–92/Pten axis controls PI3K-mediated positive and negative selection in B cell development and recon-stitutes CD19 deficiency. Cell Rep 2016;16:419–431. [DOI] [PubMed] [Google Scholar]
- 33).Totary-Jain H, Sanoudou D, Ben-Dov IZ, Dautriche CN, Guarnieri P, Marx SO, et al. Reprogramming of the microRNA transcriptome mediates resistance to rapamycin. J Biol Chem 2013;288:6034–6044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34).Hua F, Li K, Yu JJ, Lv XX, Yan J, Zhang XW, et al. TRB3 links insulin/IGF to tumour promotion by interacting with p62 and impeding autophagic/proteasomal degradations. Nat Commun 2015;6:7951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35).Guertin DA, Sabatini DM. Defining the role of mTOR in cancer. Cancer Cell 2007;12:9–22. [DOI] [PubMed] [Google Scholar]
- 36).Clague MJ, Urbe S. Ubiquitin: same molecule, different degradation pathways. Cell 2010;143:682–685. [DOI] [PubMed] [Google Scholar]
- 37).Duran A, Amanchy R, Linares JF, Joshi J, Abu-Baker S, Porollo A, et al. p62 is a key regulator of nutrient sensing in the mTORC1 pathway. Mol Cell 2011;44:134–146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38).Mani SKK, Andrisani O, Hepatitis B virus-associated hepatocellular carcinoma and hepatic cancer stem cells. Genes (Basel) 2018;9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39).Jiang P, Mizushima N. Autophagy and human diseases. Cell Res 2014;24:69–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40).Saito T, Ichimura Y, Taguchi K, Suzuki T, Mizushima T, Takagi K, et al. p62/Sqstm1 promotes malignancy of HCV-positive hepatocellular carcinoma through Nrf2-dependent metabolic reprogramming. Nat Commun 2016;7:12030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41).Jain A, Lamark T, Sjottem E, Larsen KB, Awuh JA, Overvatn A, et al. p62/SQSTM1 is a target gene for transcription factor NRF2 and creates a positive feedback loop by inducing antioxidant response element-driven gene transcription. J Biol Chem 2010;285:22576–22591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42).Zatloukal K, French SW, Stumptner C, Strnad P, Harada M, Toivola DM, et al. From Mallory to Mallory-Denk bodies: what, how and why? Exp Cell Res 2007;313:2033–2049. [DOI] [PubMed] [Google Scholar]
- 43).Guo J, Hong F, Loke J, Yea S, Lim CL, Lee U, et al. A DDX5 S480A polymorphism is associated with increased transcription of fibrogenic genes in hepatic stellate cells. J Biol Chem 2010;285:5428–5437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44).Lam HC, Baglini CV, Lope AL, Parkhitko AA, Liu HJ, Alesi N, et al. p62/SQSTM1 cooperates with hyperactive mTORC1 to regulate glutathione production, maintain mito-chondrial integrity, and promote tumorigenesis. Cancer Res 2017;77:3255–3267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45).Moscat J, Diaz-Meco MT. Feedback on fat: p62-mTORC1-autophagy connections. Cell 2011;147:724–727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46).Linares JF, Duran A, Reina-Campos M, Aza-Blanc P, Campos A, Moscat J, et al. Amino acid activation of mTORC1 by a PB1-domain-driven kinase complex cascade. Cell Rep 2015;12:1339–1352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47).Sanz L, Diaz-Meco MT, Nakano H, Moscat J. The atypical PKC-interacting protein p62 channels NF-kappaB activation by the IL-1-TRAF6 pathway. EMBO J 2000;19:1576–1586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48).Ladeiro Y, Couchy G, Balabaud C, Bioulac-Sage P, Pelletier L, Rebouissou S, et al. MicroRNA profiling in hepatocellular tumors is associated with clinical features and oncogene/tumor suppressor gene mutations. Hepatology 2008;47:1955–1963. [DOI] [PubMed] [Google Scholar]
- 49).Yang F, Yin Y, Wang F, Wang Y, Zhang L, Tang Y, et al. miR-17–5p Promotes migration of human hepatocellular carcinoma cells through the p38 mitogen-activated protein kinase-heat shock protein 27 pathway. Hepatology 2010;51:1614–1623. [DOI] [PubMed] [Google Scholar]
- 50).Chen L, Jiang M, Yuan W, Tang H. miR-17–5p as a novel prognostic marker for hepatocellular carcinoma. J Invest Surg 2012;25:156–161. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.