

SUMMARY
Virulent pathogens often cause the release of host-derived damage-associated molecular patterns (DAMPs) from infected cells. During encounters with immune-evasive viruses that block inflammatory gene expression, preformed DAMPs provide backup inflammatory signals that ensure protective immunity. Whether DAMPs exhibit additional backup defense activities is unknown. Herein, we report that viral infection of barrier epithelia (keratinocytes) elicits the release of preformed interleukin-1 (IL-1) family cytokines, including the DAMP IL-1α. Mechanistic studies revealed that IL-1 acts on skin fibroblasts to induce an interferon (IFN)-like state that restricts viral replication. We identified a branch in the IL-1 signaling pathway that induces IFN-stimulated gene expression in infected cells and found that IL-1 signaling is necessary to restrict viral replication in human skin explants. These activities are most important to control immune-evasive virus replication in fibroblasts and other barrier cell types. These findings highlight IL-1 as an important backup antiviral system to ensure barrier defense.
Graphical Abstract
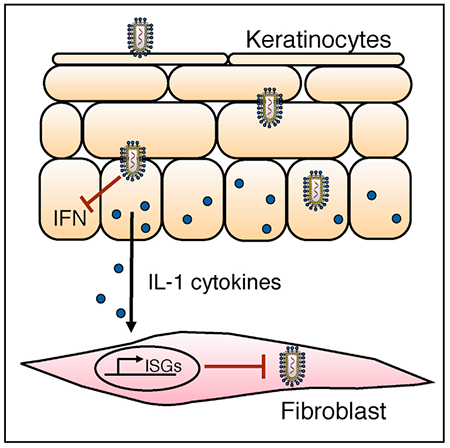
In Brief
Keratinocytes are a barrier cell type that contribute to antiviral host defense. Orzalli et al. report that IL-1 cytokines released from infected keratinocytes induce antiviral responses in stromal cells. Mechanistic studies identified a genetic requirement for an IRF1-dependent signaling axis that regulates antiviral gene expression downstream of the IL-1R.
INTRODUCTION
Barrier surfaces, including the skin and mucosa, are primary portals of entry for diverse viruses and the first site of host defense during infection. Barriers are comprised mainly of non-hematopoietic epithelial and stromal cells, which prevent viruses from gaining access to cell types and tissues that are not exposed to the external environment. Mechanical disruption of the physical barrier, or replication of viruses within cells that comprise these barriers, can facilitate spread of the infection and detection by hematopoietic cells. Despite the critical role of barrier-associated epithelial and stromal cells at the earliest stages of infection, much of our knowledge of the signaling pathways of our innate immune system has derived from studies of the later-acting immune cells (Chow et al., 2015; Iwasaki et al., 2017). We know much less about how defenses in epithelial and stromal cells are regulated.
The hallmarks of antiviral innate immunity include the ability of a cell to sense infection and subsequently initiate an antiviral program in infected and neighboring cells. Central to antiviral responses are type I and type III interferon (IFN) family members (Odendall and Kagan, 2015; Schoggins and Rice, 2011). Following viral infection, these families are expressed and secreted to induce antiviral responses in neighboring cells. The mechanisms by which IFNs prevent viral replication are numerous and explained by the collective actions of IFN-stimulated genes (ISGs), which antagonize the viral life cycle at various stages (Fensterl et al., 2015).
The best-characterized regulators of IFN expression are the pattern recognition receptors (PRRs) of the innate immune system. These receptors sense microbial products directly and promote the expression of IFNs, ISGs, and inflammatory chemokines and cytokines (Arpaia and Barton, 2011; Iwasaki, 2012). PRRs are therefore important for host defense and are commonly blocked by proteins encoded by viral pathogens (Chan and Gack, 2016; García-Sastre, 2017; Ma and Damania, 2016). The common virulence strategy to block PRR-mediated gene expression raises the question of whether backup systems are in place to promote host defense during encounters with immune-evasive pathogens. In order to operate as a backup defense strategy to PRR-mediated antiviral gene expression, these putative systems should be regulated by different means than transcriptional upregulation. We therefore considered host factors that were present in resting cells prior to infection.
When considering constitutively expressed factors that may be released from infected cells and exhibit antiviral activity, candidates were members of the damage-associated molecular pattern (DAMP) family (Kono and Rock, 2008). DAMPs are endogenous molecules that are released from dying cells under conditions where plasma membrane integrity is compromised. DAMPs are recognized to promote inflammation (Kono and Rock, 2008), and their release is often tied closely to encounters with virulent pathogens. The spectrum of backup defense benefits that DAMPs may provide to the host, however, remains unclear. In particular, it is unknown whether DAMPs can serve as a backup system to prevent replication of immune-evasive viruses. Several examples of DAMPs exist, and we were particularly interested in interleukin-1 (IL-1) family cytokines for two reasons. First, these factors can be present at high levels in resting barrier epithelial cells of human skin and mucosal surfaces. The best-defined cytokines of this family are IL-1α and IL-1β, both of which signal via the IL-1 receptor (IL-1R) to promote inflammatory chemokine and cytokine expression (Garlanda et al., 2013; Sims and Smith, 2010). The presence of IL-1 in resting cells and their ability to be released upon cell death rather than by regulated protein secretion could allow these cytokines to induce inflammation, even during encounters with pathogens that prevent PRR-induced gene expression. The second reason we were interested in the IL-1 family derives from studies that suggested an antiviral activity of recombinant IL-1β in various cell lines (Iwata et al., 1999; Randolph-Habecker et al., 2002; Van Damme et al., 1985). We know very little about the mechanisms and biological context for these potential IL-1 activities in viral infection. These collective observations prompted us to examine the function of the IL-1 family in antiviral immunity in human skin, a common site of initial viral infection.
Herein, we report that human keratinocytes contain high amounts of IL-1 family cytokines in a resting state and that these cells release IL-1 following viral infection. Notably, IL-1 release is insensitive to the immune evasion strategies used by a virus that prevents IFN gene expression. We identify human stromal fibroblasts and endothelial cells as highly sensitive sensors of IL-1 and find that IL-1 induces a robust antiviral program in these cells. We demonstrate the importance of IL-1 signaling in the control of viral infection in human foreskin explants and identify two regulators of IL-1-induced antiviral immunity that highlight a branchpoint in the IL-1 signaling pathway. This branchpoint distinguishes the expression of classic inflammatory chemokines from the expression of antiviral genes, with the latter being regulated by the transcription factors IFN regulatory factor 1 (IRF1) and IRF2. Perhaps most notably, the antiviral actions of IL-1 are particularly important for controlling infections with viruses that block IFN expression. This study therefore reveals IL-1 family cytokines as an important backup antiviral system in the skin and broadens the function of the IL-1 family in host defense.
RESULTS
Keratinocytes Release IL-1 in Response to PRR-Evasive Virus Infection
To define how barrier epithelial cells respond to PRR-evasive viruses, we utilized vesicular stomatitis virus (VSV). VSV can be transmitted to the skin via the bite of an infected sandfly. This virus is highly sensitive to the antiviral actions of IFNs and thus prevents PRR-induced IFN expression through activities of the viral matrix (M) protein (Ahmed et al., 2003). VSV is therefore a useful model to examine antiviral immune responses that operate during PRR-evasive virus infection.
We performed infections of normal oral keratinocytes (NOKs) with wild-type (WT) VSV or a recombinant virus with a loss-of-function point mutation in M (rVSV-M51R). Consistent with the immune-evasive activities of VSV M, infection with rVSV-M51R resulted in transcriptional induction of type I and type III IFNs (IFNB or IL-29, respectively) and an ISG (RSAD2; Figure 1A). By contrast, infections with WT VSV induced 10–100 times less expression of these genes (Figure 1A). Consistent with these results, IL-29 protein was released from cells infected with rVSV-M51R, but not WT VSV (Figure 1B). IL-29 release correlated with the ability to detect STAT1 phosphorylation and the ISG products RSAD2 and IFIT1 in infected cells (Figure 1C). Thus, WT VSV blocks IFN expression and responses in NOKs, as has been reported for other cell types (Ferran and Lucas-Lenard, 1997).
Figure 1. VSV Infection Elicits the Release of IL-1 Cytokines from Human Keratinocytes.
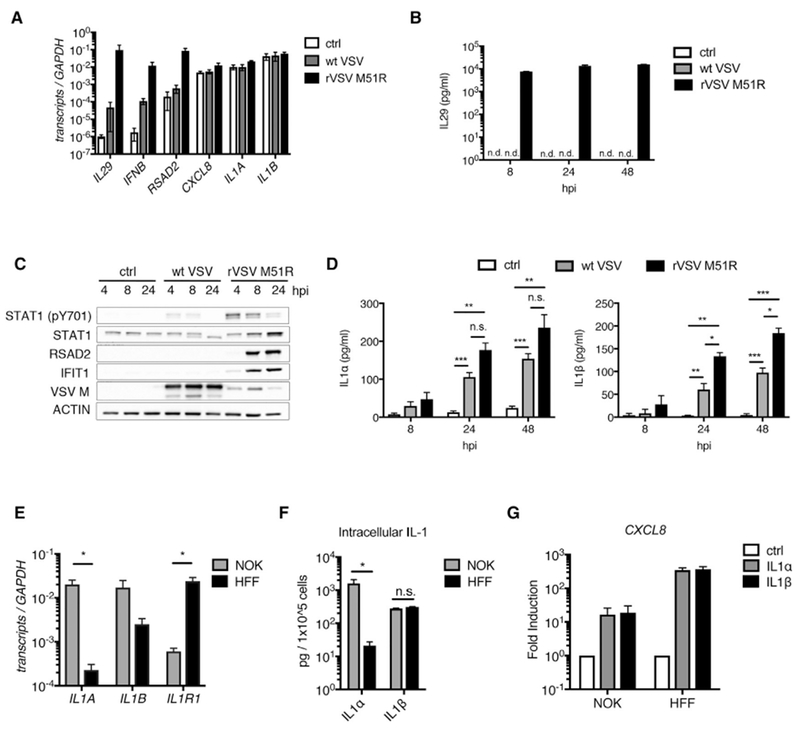
(A–D) NOKs were mock-infected (ctrl) or infected with WT VSV or rVSV-M51R (MOI 10).
(A) Total RNA was isolated at 8 hpi and analyzed by qRT-PCR.
(B and D) Cell-free supernatants were harvested at indicated time points and analyzed by ELISA for IL-29 (B) and IL-1α/IL-1β (D).
(C) Whole-cell lysates were harvested at indicated time points and separated by SDS-PAGE, and endogenous proteins were detected by immunoblot.
(E) Total RNA was isolated from NOKs and HFFs. IL1A, IL1B, and IL1R1 transcript counts were quantified by nCounter and normalized to GAPDH.
(F) 5 × 105 NOKs or HFFs were lysed in 1% NP40 buffer and clarified by centrifugation, and ELISAs were used to analyze the soluble fractions for IL-1α and IL-1β.
(G) NOKs and HFFs were stimulated with IL-1α or IL-1β (10 ng/mL). Total RNA was harvested at 6 hr and quantified by qRT-PCR. Samples are normalized to GAPDH and presented as fold induction over the control (ctrl) samples.
(A–G) Data are the average of at least three independent experiments ± SEM. Student’s t test: *p < 0.05; **p < 0.01; ***p < 0.001.
Whereas WT VSV could block IFN and ISG expression, both viral strains elicited the release of the IL-1 family members IL-1α and IL-1β from infected cells (Figure 1D). IL-1 release did not correspond with augmented IL-1 gene expression, as neither virus induced IL1A or IL1B transcription in infected NOKs (Figure 1A). However, the abundance of these transcripts was higher than IL-29, IFNB, and RSAD2 transcripts in uninfected NOKs (Figure 1A). High basal IL1A transcripts were specific to keratinocytes, as compared to amounts detected in primary human fibroblasts (HFF) (Figure 1E), a cell type that associates with keratinocytes at barrier surfaces. Consistent with our transcriptional results, resting NOKs contained 80 times more intracellular IL-1α protein than HFFs, and both cell types had similar levels of intracellular IL-1β (Figure 1F). HFFs express substantially higher IL1R1 transcripts than NOKs (Figure 1E), and IL-1 treatment in HFFs elicited higher CXCL8 expression than that observed in NOKs (Figure 1G). These results demonstrate that resting NOKs contain IL-1α and IL-1β and can respond to virus infection through release of these preformed cytokines. HFFs, in contrast, do not contain abundant IL-1 yet are highly responsive to this cytokine.
IL-1 Subfamily Cytokines Produce Antiviral Responses in Human Fibroblasts and Endothelial Cells
Our observations that NOKs produce IL-1 cytokines, and HFFs potently respond to IL-1, raised the questions of whether fibroblasts detect keratinocyte-derived IL-1 cytokines following virus infection and whether IL-1 signaling in fibroblasts contributes to antiviral immunity. To address these questions, we examined the ability of IL-1 to promote transcriptional responses in HFFs. We used nCounter analysis to examine the expression of 23 innate immune genes in HFFs stimulated with IL-1β. Consistent with IL-1R signaling activating nuclear factor κB (NF-κB), several chemokines and cytokines were upregulated in response to IL-1β (Figure 2A). IL-1β also stimulated the expression of IFNs and ISGs (Figure 2A). Similar results were observed in IL-1α-stimulated fibroblasts (Figure 2B). The kinetics and magnitude of IFNB and RSAD2 induction in IL-1-stimulated HFFs were similar to that observed in cells treated with a synthetic TLR3/MDA5 agonist, poly(I:C) (Figure 2B), an established inducer of IFN expression (Alexopoulou et al., 2001; Gitlin et al., 2006). Treatment of primary human lung fibroblasts (MRC-5) with IL-1β resulted in the induction of IFNB, RSAD2, CXCL10, and CXCL8 transcripts (Figure 2C). Notably, in MRC-5 cells, the induction of RSAD2 by IL-1β approached the levels induced by treatment with IFNβ, and IL-1β induced a greater increase in IFNB expression than poly(I:C) in these cells (Figure 2C). IL-1 treatment also resulted in detection of STAT1 phosphorylation and the ISG product interferon regulatory factor 1 (IRF1) in cell lysates (Figure 2D).
Figure 2. IL-1 Cytokines Induce an Antiviral Response in Human Fibroblasts and Endothelial Cells.
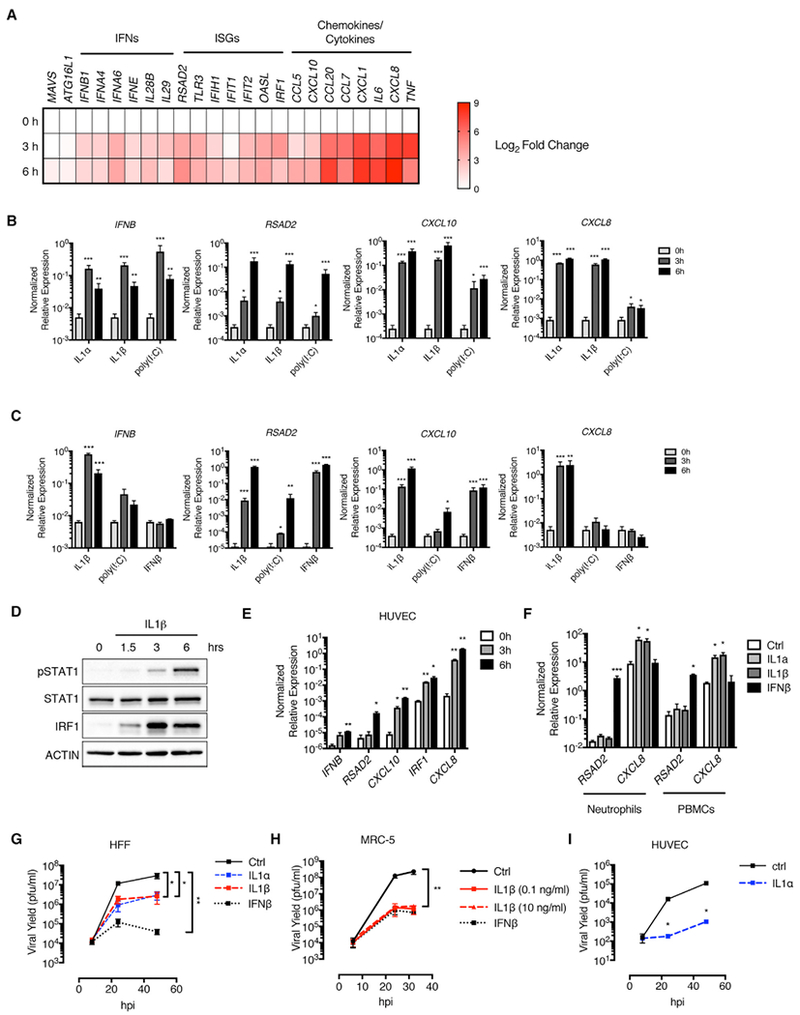
(A) HFFs were stimulated with recombinant IL-1β, and total RNA was isolated at 0, 3, and 6 hr post-stimulation. Absolute transcript counts were measured by nCounter and normalized to GAPDH. Data are presented as a fold induction over the 0 hr time point.
(B and C) HFFs (B) and MRC-5 lung fibroblasts (C) were stimulated with IL-1α, IL-1β, poly I:C, or IFN β. Total RNA was isolated at indicated time points and analyzed by qRT-PCR.
(D) Whole-cell lysates from IL-1β-stimulated HFFs were separated by SDS-PAGE and analyzed by immunoblot.
(E) HUVECs were stimulated with IL-1α. Total RNA was isolated 0, 3, and 6 hr post-stimulation and analyzed by qRT-PCR.
(F) Human neutrophils or peripheral blood mononuclear cells (PBMCs) were stimulated with IL-1α, IL-1β, or IFN β for 6 hr. Total RNA was isolated and analyzed by qRT-PCR.
(G–I) HFFs (G), MRC-5 lung fibroblasts (H), and HUVECs (I) were infected with WT VSV (MOI 0.1) in the presence of IL-1α, IL-1β, or IFNβ. Cell-free supernatants were collected at 8, 24, 32, and/or 48 hpi, and viral yields were determined by plaque assay.
(A–I) IL-1 cytokine stimulations were performed at 10 ng/mL unless otherwise stated. Poly I:C or IFN β stimulations were performed at 1 μg/mL or 10 U/mL, respectively. Data are the average of at least three independent experiments ± SEM. (E and I) Data are the average of two independent experiments ± SEM. Student’s t test: *p < 0.05; **p < 0.01; and ***p < 0.001.
To determine whether the ability of IL-1 to induce antiviral genes was restricted to fibroblasts, we performed similar studies in cells associated with other barriers in the human body. We examined IL-1-mediated responses in endothelial cells, an important cell type that lines blood and lymph vessels. Using primary human umbilical vein endothelial cells (HUVECs) (Figure 2E), we found that IL-1α induced a similar antiviral transcriptional response to that observed in fibroblasts (Figure 2B). By contrast, IL-1 treatment of human neutrophils or peripheral blood mononuclear cells elicited modest CXCL8 but no RSAD2 expression (Figure 2F). IFNβ treatment, however, induced RSAD2 expression in these cell types (Figure 2F). These data suggest that the ability of IL-1 to induce an antiviral transcriptional signature may be limited to non-hematopoietic cells.
We examined whether additional IL-1 family members could induce antiviral transcriptional responses in human cells. Neither IL-33, IL-36α, nor IL-18 elicited RSAD2 expression in HFFs or NOKs (Figures S1A–S1C). However, these cytokines also did not induce CXCL8 expression in either cell type (Figures S1A–S1C), suggesting that these cells may not express the receptors for these cytokines.
To determine whether IL-1 subfamily cytokines are sufficient to prevent viral infection, we examined the replication of VSV in HFFs, MRC-5 cells, and HUVECs over 48 hr. IL-1 treatments reduced VSV replication by 1 to 2 logs in HFFs (Figure 2G) and 2 to 3 logs in MRC-5 cells (Figure 2H) and HUVECs (Figure 2I). IFNβ treatment was highly antiviral in HFFs and MRC-5 cells, as expected (Figures 2G and 2H). Interestingly, IL-1β was as capable of restricting VSV replication as IFNβ in MRC-5 cells, and as little as 100 pg of IL-1β displayed antiviral activity in these cells (Figure 2H). These data indicate that IL-1 family cytokines exhibit functional antiviral responses that restrict the replication of VSV.
The Antiviral Activity of IL-1 Is Evident in Human Skin, but Not Murine Fibroblasts or Mice
To determine whether IL-1 can induce antiviral responses in stromal cells from other species, we examined IL-1 responses in murine fibroblasts that were isolated from anatomical locations corresponding to the human cells we have studied. Specifically, we examined primary mouse lung fibroblasts (MLFs), dermal fibroblasts (MDFs), as well as immortal embryonic fibroblasts (MEFs). IL-1 treatment elicited Cxcl1 expression from all cell types (Figures 3A, 3C, and 3E). However, Rsad2 was only induced in IL-1-stimulated MLFs and MDFs. The ISG Ifit2 was weakly induced in MDFs (Figure 3C). Notably, the fold induction of Rsad2 in IL-1-treated MLFs was 1,000-fold less than that observed in IL-1-treated MRC-5 cells (Figures 3A and 2C). Functional analysis indicated that IL-1 treatment could not significantly restrict VSV replication in these cells (Figures 3B, 3D, and 3F). By contrast, IFNβ treatment inhibited VSV replication in MLFs, MDFs, and MEFs (Figures 3B, 3D, and 3F). Consistent with the inability of IL-1 to induce a functional antiviral response in murine fibroblasts, we observed equal viral titers in the skin of VSV-infected WT and Il1r1 -deficient mice (Figure 3G). Intranasal infection of these mice resulted in a similar spread of VSV to the olfactory bulb and cerebrum at 48 hr post-infection (hpi) (Figure 3H). By contrast, Ifnar1-deficient mice were more susceptible for VSV infection than WT mice under all conditions examined (Figure 3H), confirming the importance of the IFN response in restricting VSV replication in mice (Nair et al., 2014).
Figure 3. IL-1 Does Not Restrict VSV Replication in Murine Fibroblasts.
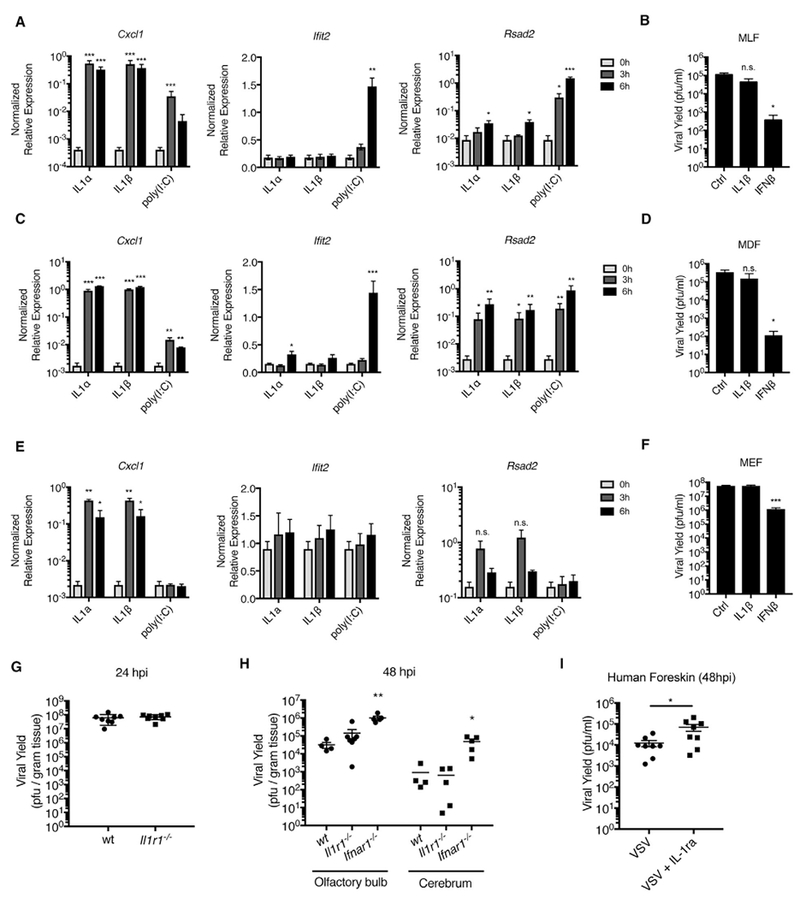
(A, C, and E) Mouse lung fibroblasts (MLFs) (A), mouse dermal fibroblasts (MDFs) (C), and mouse embryonic fibroblasts (MEFs) (E) were stimulated with IL-1α (10 ng/mL), IL-1β (10 ng/mL), or poly I:C (1 μg/mL). Total RNA was isolated at 0, 3, and 6 hr post-stimulation and analyzed by qRT-PCR.
(B, D, and F) MLFs (B), MDFs(D), and MEFs (F) were infected with WT VSV (MOI 0.1) in the presence orabsenceofIL-1β (10ng/mL) or IFNβ (10U/mL). Viral yields were determined from infected-cell supernatants at 24 hpi by plaque assay.
(G) Wild-type (wt) or Il1r1−/− mice were infected with 5 × 106 plaque-forming units (PFUs) of VSV via epidermal scarification. Viral titer within the skin was determined at 24 hpi.
(H) wt, Il1r1−/−, or Ifnar1−/− mice were infected with 5 × 106 PFUs of VSV via intranasal injection. Viral titer was determined from indicated tissues at 48 hpi.
(I) Punch biopsies (5 mm) from human newborn foreskin were infected with 2.5 × 103 PFUs of VSV in the presence or absence of IL-1ra (500 ng) by intradermal injection. Results are the combined data points from two independent experiments ± SEM.
(A–F) Results are an average of three independent experiments ± SEM. Student’s t test; *p < 0.05; **p < 0.01; and ***p < 0.01.
The IL-1R antagonist (IL-1ra) blocks activation of the IL-1R signaling pathway by IL-1α and IL-1β (Dinarello, 2009). In contrast to our observations in mice, inhibition of IL-1R signaling by treatment with recombinant IL-1ra resulted in an increase in VSV replication in human foreskin tissue explants (Figure 3I). These collective observations suggest that IL-1 is necessary and sufficient to control viral infection in human cells and tissues but that IL-1R signaling is either not required to control VSV replication in mice or is important in the context of cell types or infection models not tested here.
The Antiviral Activity of IL-1 Is Most Important during Encounters with Immune-Evasive Viruses
We speculated that IL-1 might be most important during encounters with immune-evasive viruses that prevent PRRs from inducing IFN expression. Under these conditions, the IL-1 released from infected cells may compensate for the lack of PRR-induced activities. The immuno-stimulatory rVSV-M51R virus provided an opportunity to test this hypothesis. We assessed the ability of IL-1β to prevent replication of WT VSV or the rVSV-M51R strain in infected HFFs. In contrast to the significant reduction in WT VSV replication observed in IL-1β-treated cells, IL-1β had no effect on the already reduced replication of rVSV-M51R (Figure 4A). The antiviral activities of IL-1 are therefore most important to control immune-evasive strains of VSV.
Figure 4. IL-1 Cytokines Restrict the Replication of Immune Evasive Viruses.
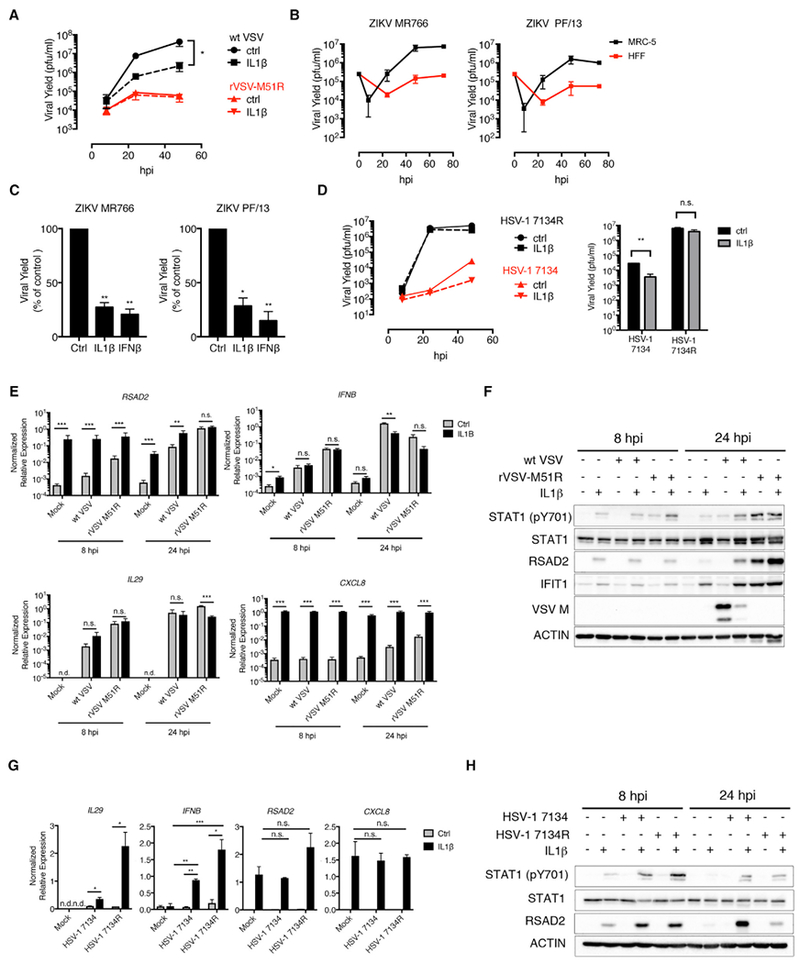
(A) HFFs were infected with WT VSV or rVSV-M51R (MOI 0.1) in the presence of IL-1β. Cell-free supernatants were collected at 8, 24, and 48 hpi, and viral yields were determined by plaque assay.
(B) HFF and MRC-5 fibroblasts were infected with ZIKV strains MR766 and PF/13 (MOI 1). Viral yields were determined as in (A) at 8, 24, 48, and 72 hpi.
(C) MRC-5 lung fibroblasts were infected with indicated ZIKV strains in the presence ofIL-1β or IFNβ. Viral titers from infected-cell supernatants were determined at 48 hpi by plaque assay. Data are presented as a percentage of PFUs in control (ctrl)-treated cells.
(D) HFFs were infected with HSV-1 7134 or 7134R (MOI 0.1). (Left panel) Virus was isolated from infected cells at 8, 24, and 48 hpi, and viral yields were determined by plaque assay on U2OS cells. (Right panel) Plaque assays were performed on infected-cell lysates isolated at 48 hpi.
(E and F) HFFs were infected as described in (A).
(E) Total RNA was isolated at 8 and 24 hpi and analyzed by qRT-PCR. Data are normalized to GAPDH.
(F) Whole-cell lysates were isolated at 8 and 24 hpi, separated by SDS-PAGE, and analyzed by immunoblot.
(G and H) HFFs were infected as described in (D).
(G) Total RNA was isolated at 8 hpi and analyzed by qRT-PCR. Data are normalized to 18S rRNA.
(H) Whole-cell lysates were isolated at 8 and 24 hpi, separated by SDS-PAGE, and analyzed by immunoblot.
(A–H) IL-1 and IFNβ cytokine stimulations were performed at 10 ng/mL or 10 U/mL, respectively. Data are the average of three independent experiments ± SEM.
(D, left panel) Data are representative of two independent experiments. Student’s t test: *p < 0.05; **p < 0.01; and ***p < 0.001.
To determine whether other viruses are susceptible to IL-1-mediated restriction, we examined Zika virus (ZIKV) and herpes simplex virus 1 (HSV-1). Like VSV, ZIKV and HSV-1 produce proteins that block PRR-dependent IFN expression (Kumar et al., 2016; Kurt-Jones et al., 2017). We observed that two strains of ZIKV, MR766 and PF/13, replicated poorly in HFFs but reached higher titers in MRC-5 cells (Figure 4B). We therefore compared the antiviral activities of IFN and IL-1 on ZIKV replication in MRC-5 cells. The replication of both ZIKV strains was suppressed to a similar extent by IL-1β or IFNβ (Figure 4C). We noted, however, that the reduction in ZIKV titer by IL-1β or IFNβ treatment was relatively modest and corresponded to a 3- to 4-fold decrease in viral titers (Figure 4C). Interestingly, HSV-1 was also insensitive to the antiviral activities of IL-1β (Figure 4D; HSV-1 7134R).
These observations, coupled with the high sensitivity of VSV to IFNβ and IL-1β, suggested a correlation between IFN sensitivity and the susceptibility of a particular virus to IL-1-mediated inhibition. We therefore sought to identify a strain of HSV-1 that may be similar to WT VSV, in that it would be sensitive to the actions of IFNs but capable of preventing their expression. HSV-1 strains lacking infected-cell protein 0 (ICP0) fulfill these criteria (Mossman et al., 2000). Consistent with our hypothesis, we found that IL-1β treatment reduced the replication of an ICP0-null virus at 48 hpi by ~1-log (Figure 4D; 7134). Overall, these results suggest that IL-1 family cytokines are most important during encounters with viruses that block IFN expression but are sensitive to IFNs.
IL-1 Overcomes the Immune-Evasive Activities of VSV and HSV-1
We monitored antiviral transcriptional responses in VSV- or HSV-1-infected HFFs treated with IL-1β. As expected, WT VSV infection weakly induced RSAD2 expression in infected cells at 8 hr, whereas the rVSV-M51R strain strongly induced RSAD2 expression (Figure 4E). IL-1β treatment resulted in an increase in RSAD2 expression in infected cells (Figure 4E). IL-1β also induced an increase in RSAD2 and IFIT1 protein in cells infected with WT VSV (Figure 4F; 24 hpi). VSV M protein abundance was also decreased in lysates from infected cells treated with IL-1β (Figure 4F). Confocal microscopic analysis revealed that the increased RSAD2 production observed following IL-1β treatment was at the level of individual cells and was inversely correlated with the number of VSV-M-positive cells (Figures S2A and S2B). Thus, IL-1 overcomes the immune-evasive activities of VSV at the population level and at the level of individual cells. We did note, however, that not all antiviral responses were enhanced by IL-1β in VSV-infected cells. Indeed, IL-1β inhibited IFNB expression in WT VSV-infected HFFs at 24 hr (Figure 4E). This decrease was not observed at the earlier 8 hr time point (Figure 4E), and we observed enhanced STAT1 phosphorylation in virus-infected lysates of IL-1-stimulated cells at 24 hr (Figure 4F).
Similar experiments were performed during HSV-1 infections. In the absence of IL-1β, WT HSV-1 (7134R) and ICP0-null (7134) infections induced minimal expression of IL29, IFNB, or RSAD2 (Figure 4G). By contrast, IL-1β treatment during infections strongly enhanced the expression of these antiviral factors (Figure 4G). This augmented transcriptional response corresponded to increased STAT1 phosphorylation and RSAD2 protein in HSV-1-infected-cell lysates, neither of which were detectable following viral infection in the absence of IL-1β (Figure 4H). The striking increase in IL29 and IFNB expression in HSV-1-infected cells was surprising, given the inability of IL-1 to enhance these responses following VSV infection (Figures 4E and 4F), but suggests the potential for unique inhibitory mechanisms of IL-1 action against disparate viruses. Taken together, these data indicate that IL-1 can overcome the PRR-evasive activities of multiple pathogenic viruses.
IRF1 Promotes IL-1-Mediated Antiviral Responses
To identify regulators of IL-1-mediated antiviral responses, we considered transcription factors that regulate PRR-mediated antiviral gene expression. The transcription factor IRF3 regulates antiviral gene expression downstream of several TLRs, the RIG-I-like receptors (RLRs), and cGAS (Chen et al., 2016; Iwasaki, 2012). To determine whether IRF3 is involved in IL-1-mediated antiviral responses, we determined whether IRF3 was activated in IL-1-stimulated HFFs. We did not detect IRF3 phosphorylation in whole-cell lysates from IL-1-stimulated HFFs (Figure 5A), nor did we observe IRF3 accumulation in the nuclei of IL-1-stimulated HFFs by confocal microscopy using an antibody that preferentially detects IRF3 dimers (Melroe et al., 2004; Figure 5B). By contrast, infection with Sendai virus (SeV) or treatment with poly(I:C) resulted in IRF3 phosphorylation or increased nuclear IRF3 detection, respectively (Figures 5A and 5B). We confirmed that cells were successfully stimulated by IL-1α, IL-1β, or poly(I:C) by co-staining for IRF1, which was induced by all three stimuli (Figure 5B). These data indicate that IRF3 is not activated in response to IL-1 treatment.
Figure 5. IRF1 Is a Critical Regulator of IL-1-Mediated Antiviral Responses.
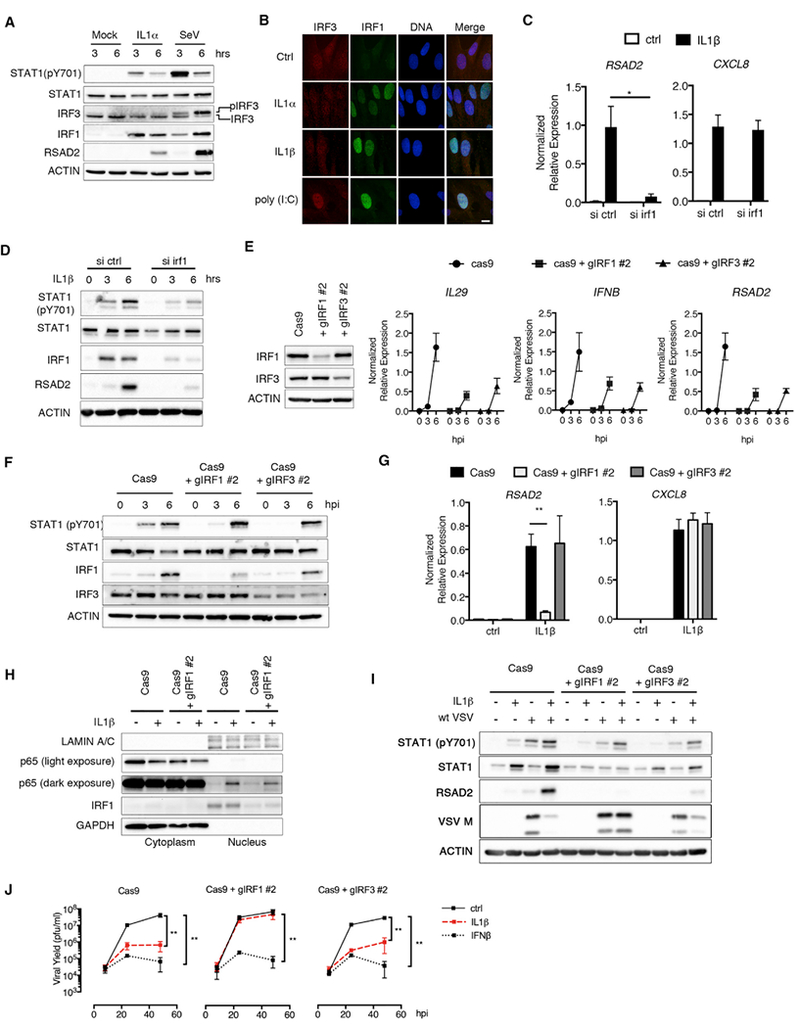
(A) HFFs were stimulated with IL-1α (100 pg/mL) or infected with SeV (0.001 hemagglutinating units [HAUs]/cell). Whole-cell lysates were harvested at 3 and 6 hpi, separated by SDS-PAGE, and analyzed by immunoblot.
(B) HFFs were stimulated with IL-1α (10ng/mL), IL-1β (10ng/mL), or poly(I:C) (1 μg/mL) for 6hr. Cells were fixed and stained with IRF3 and IRF1 antibodies. Nuclei were counterstained with DRAQ5. Data are representative of two independent experiments. The scale bar represents 10 μm.
(C and D) HFFs were transfected with ctrl siRNAs or siRNAs targeting IRF1 for 72 hr. siRNA-transfected cells were then treated with IL-1β (10 ng/mL), and total RNA or protein were harvested at 3 and 6 hr or 6 hr, respectively.
(C) Indicated transcripts were quantified by qRT-PCR and normalized to GAPDH.
(D) Whole-cell lysates were separated by SDS-PAGE and analyzed by immunoblot.
(E) (Left panel) Whole-cell lysates from HFFs transduced with lentiviruses expressing Cas9 alone or in combination with IRF1-or IRF3-targeted guide RNAs were separated by SDS-PAGE and analyzed by immunoblot. (Right panel) IRF1-and IRF3-deficient cells were infected SeV (0.001 HAU/cell). Total RNA was isolated from infected cells at 0, 3, and 6 hpi, and IL29, IFNB, and RSAD2 expressions were quantified by qRT-PCR. Samples were normalized to GAPDH.
(F) Whole-cell lysates from SeV-infected Cas9 and IRF1- and IRF3-deficient cells were separated by SDS-PAGE and analyzed by immunoblot.
(G) Cas9, IRF1-deficient, or IRF3-deficient HFFs were stimulated with IL-1β (10 ng/mL). Total RNA was harvested from cells at 6 hr and analyzed by qRT-PCR.
(H) Cas9 or IRF1-deficient HFFs were stimulated with IL-1β (10 ng/mL). Cellular cytoplasmic and nuclear fractions were isolated at 1 hr post-stimulation and analyzed by immunoblot.
(I and J) Indicated cell lines were infected with WT VSV (MOI 0.1) in the presence or absence of IL-1β (I and J) or IFNβ (J).
(I) Whole-cell lysates were harvested at 24 hpi, separated by SDS-PAGE, and analyzed by immunoblot.
(J) Infected-cell supernatants were harvested at indicated time points, and viral yields were determined by plaque assay.
Data are the average of at least three independent experiments ± SEM. Student’s t test: *p < 0.05, **p < 0.01
In addition to IRF3, IRF1 regulates antiviral responses, in particular in human cells (Schoggins et al., 2011). We found that small interfering RNA (siRNA)-mediated depletion of IRF1 in HFFs reduced IL-1β-induced RSAD2, but not CXCL8, expression, suggesting a role for this transcription factor in IL-1-mediated antiviral responses (Figure 5C). This decrease in ISG expression corresponded to a reduction in STAT1 phosphorylation and RSAD2 protein abundance within IL-1β-stimulated cells (Figure 5D). These data suggest that IRF1 is necessary for IL-1-mediated STAT1 phosphorylation and ISG expression.
We constructed HFFs genetically deficient in IRF1 or IRF3 by utilizing a lentivirus-based CRISPR/Cas9 approach. We observed a specific reduction in IRF1 and IRF3 protein in HFFs transduced with lentiviruses expressing Cas9 and specific guide RNAs (gRNAs) targeting either the IRF1 or IRF3 genes (Figure 5E). Functionally, IRF1- and IRF3-deficient cells were validated, as they exhibited defects in SeV-induced expression of IL29, IFNB, and RSAD2 (Figure 5E). In addition, we observed delayed STAT1 phosphorylation in SeV-infected cells in the absence of IRF1 or IRF3 (Figure 5F).
We next used these cells to examine the role of IRF1 and IRF3 in IL-1-mediated responses. IL-1β stimulation of Cas9-expressing cells induced RSAD2 expression, which was reduced in IRF1-deficient, but not IRF3-deficient, cells (Figure 5G). Neither IRF1 nor IRF3 deficiency influenced the expression of CXCL8 (Figure 5G). Consistent with this observation, IRF1 deficiency had no effect on the accumulation of the NF-κB p65 subunit in the nucleus of IL-1-stimulated cells (Figure 5H). To determine the function of IRF1 and IRF3 in IL-1-mediated control of viral infection, we examined VSV replication in our genetically deficient HFFs. IL-1 retained the ability to restrict VSV M protein expression and replication in IRF3-deficient cells (Figures 5I and 5J). By contrast, the ability of IL-1 to restrict VSV replication was abolished in IRF1-deficient cells (Figure 5J). Whereas IRF1 was specifically required for IL-1-mediated restriction of VSV replication, IRF1 and IRF3 promoted RSAD2 induction in IL-1-stimulated cells infected with WT VSV (Figure 5I). However, consistent with our viral replication data (Figure 5J), only IRF1 deficiency rescued VSV M protein levels in IL-1-stimulated cells (Figure 5I). Neither IRF1 nor IRF3 were necessary for the antiviral actions of IFNβ (Figure 5J). Taken together, these data identify IRF1 as a regulator of the IL-1R signaling pathway that specifically controls antiviral responses.
IL-1 Stimulates the Secretion of an Antiviral Factor that Signals through a gp130/JAK/STAT1 Axis
The cytokine tumor necrosis factor alpha (TNF-α) activates IRF1-dependent transcriptional responses in macrophages and endothelial cells (Venkatesh et al., 2013; Yarilina et al., 2008). As IL-1 induces the expression of IFNs and TNF-α (Figure 2A), we determined the role for IFN or TNF-α in regulating IL-1-mediated ISG expression. We first blocked secretory pathway activity by treatment of cells with brefeldin A (BFA) (Lippincott-Schwartz et al., 1989). BFA potently blocked IL-1-induced RSAD2, but not CXCL8, expression (Figure 6A), suggesting protein secretion is required to induce ISG expression. The potential role of TNF-α was discounted, as siRNA-mediated depletion of TNFR1 or the downstream adaptor TRADD reduced TNF-α-induced, but not IL-1-induced, RSAD2 or CXCL8 expression (Figures S3A and S3B). In addition, we could detect CXCL8, but not TNF-α, protein in the supernatants of IL-1β-treated HFFs (Figure S3C). Furthermore, although TNFA transcripts were induced in IL-1β-treated HFFs, the amount of TNFA mRNA present in stimulated cells was comparable to the amount of CXCL8 transcripts present in resting cells (Figure S3D). These data suggest that the ability of IL-1 to induce ISG expression is unlikely to be regulated by a TNF-α-dependent intermediate step.
Figure 6. Gp130 and STAT1 Are Essential for IL-1-Mediated Antiviral Responses.
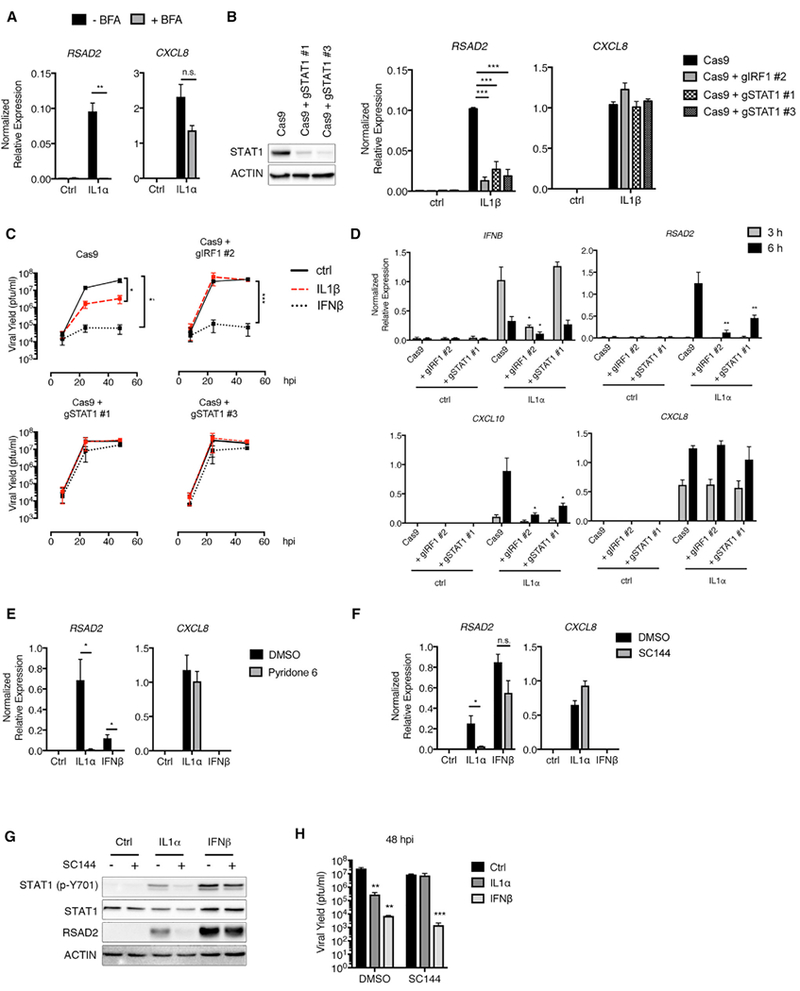
(A) HFFs were stimulated with IL-1α (100 pg/mL) in the presence or absence of brefeldin A (BFA) (1 μg/mL). Total RNA was isolated at 6 hr post-stimulation and analyzed by qRT-PCR.
(B) (Left panel) STAT1 deficiency was demonstrated by the absence of STAT1 protein by immunoblot. (Right panel) Cells were stimulated with IL-1β (10 ng/mL).Total RNA or whole-cell lysates were harvested from cells at 6 hr and analyzed by qRT-PCR.
(C) Cells were infected with WT VSV (MOI 0.1) in the presence or absence of IL-1β (10 ng/mL) or IFNβ (10 U/mL). Infected-cell supernatants were harvested at indicated time points, and viral yield was determined by plaque assay.
(D) Cells were stimulated with IL-1α (100 pg/mL). Total RNA was harvested at 3 and 6 hr and analyzed by qRT-PCR.
(E) HFFs pre-treated with DMSO or pyridone 6 (5 μg/mL) for 1 hr were stimulated with IL-1α (100 pg/mL) or IFNβ (1 U/mL). Total RNA was isolated at 6 hr and analyzed by qRT-PCR.
(F and G) HFFs were pre-treated with a gp130 inhibitor (SC144; 25 μM) for 1 hr, prior to dilution with an equal volume of media containing IL-1α (200 pg/mL) or IFNβ (20 U/mL). Total RNA or whole-cell lysates were harvested at 6 hr and analyzed by (F) qRT-PCR or (G) separated by SDS-PAGE and analyzed by immunoblot.
(H) SC144 (12.5 μM) alone or in combination with IL-1α (100 pg/mL) or IFNβ (10 U/mL) was added to cells 1 hr after infection with WT VSV (MOI 0.1). Infected-cell supernatants were isolated at 48 hpi and analyzed by plaque assay.
Data are the average of at least three independent experiments ± SEM. Student’s t test: *p < 0.05; **p < 0.01; and ***p < 0.001.
We next considered the role of IFNs in IL-1-mediated antiviral responses. We have observed that IL-1 treatment results in STAT1 phosphorylation (Figure 2D), which can be activated downstream of a number of cytokines, including IFNs. To determine whether STAT1 is required for IL-1-mediated responses, we examined antiviral activities in STAT1-deficient cells. These cells phenocopied IRF1 deficiency in terms of IL-1β-induced ISG expression and restriction of VSV replication (Figures 6B and 6C). In addition, IL-1-mediated ISG (RSAD2 and CXCL10), but not IFNB, expression was dependent on STAT1 (Figure 6D). STAT1 is activated by Janus kinase (JAK) family members, whose activities can be inhibited by pyridone 6. Pyridone 6 treatment specifically reduced IL-1-mediated RSAD2, but not CXCL8, expression (Figure 6E). These results together indicate that IL-1 stimulates a JAK/STAT1-dependent antiviral signaling pathway.
The IFNα/β receptor (IFNAR) utilizes a JAK/STAT1 signaling pathway to induce ISGs. To assess the role of type I IFN in the antiviral actions of IL-1, we inhibited IFNAR signaling with an IFNAR2 neutralizing antibody. Using concentrations of recombinant IL-1α and IFN β that induce comparable RSAD2 expression, IFNAR neutralization only prevented IFNβ activity (Figure S4A). The inhibition of IFNAR signaling was also evident when using an IFNβ dose that induced higher RSAD2 expression than IL-1α can achieve (Figure S4A). Similarly, IL-1α-induced RSAD2 expression was not reduced in fibroblasts from patients with a genetic mutation in IFNAR1 that reduces IFNβ responsiveness (Hoyos-Bachiloglu et al., 2017; Figure S4B). IFNAR1-deficient cells also retained the ability to restrict VSV replication upon IL-1α treatment when compared to control fibroblasts (Figure S4C). Thus, neither IFNAR1 nor IFNAR2 are necessary for IL-1-mediated antiviral responses. We therefore considered other cytokines that signal through JAK/STAT1. Certain members of the IL-6 family of cytokines can induce STAT1 phosphorylation (Hirahara et al., 2015), and our observation that IL-6 was upregulated in IL-1-stimulated HFFs (Figure 2A) prompted us to examine the role of IL-6 family cytokines in IL-1-mediated antiviral responses. All IL-6 family members signal through a receptor complex that contains a unique subunit receptor and the protein glycoprotein 130 (gp130). Chemical inhibition of gp130 greatly diminished IL-1-mediated STAT1 phosphorylation and RSAD2 expression, whereas CXCL8 expression was unaffected (Figures 6F and 6G). Importantly, inhibition of gp130 had no effect on IFNβ-induced RSAD2 transcripts or protein (Figures 6F and 6G). Consistent with these findings, the ability of IL-1 to prevent VSV replication was lost upon inhibition of gp130, and IFNβ retained the ability to restrict VSV replication (Figure 6H). Although it is possible that the chemical inhibitor used may influence pathways other than gp130, the fact that IFN signaling is intact in inhibitor-treated cells formally demonstrates that IFN signaling cannot explain the antiviral actions of IL-1. We therefore propose that IL-1 promotes the secretion of a factor that activates a gp130-JAK-STAT1-dependent pathway to restrict viral replication.
IRF2 Negatively Regulates IL-1-Mediated Antiviral Responses
The IAP family member cIAP2, encoded by the BIRC3 gene, was reported to activate IRF1 to promote an IL-1-mediated inflammatory response (Harikumar et al., 2014). In addition, the IRF2 transcription factor may act as a repressor of IRF1-mediated transcriptional responses (Harada et al., 1989). We therefore investigated the roles of cIAP2 and IRF2 in IRF1-dependent antiviral activities in IL-1-stimulated cells. Deletion of BIRC3 by CRISPR had no effect on IL-1-mediated gene expression or phosphorylation of STAT1 (Figures 7A and 7B). cIAP1 can compensate for loss of cIAP2 under certain conditions (Mahoney et al., 2008; Varfolomeev et al., 2008). To determine whether additional IAP family members compensate for cIAP2 deficiency, we depleted fibroblasts of IAP proteins through treatment with the BV6 SMAC mimetic. Although BV6 treatment sensitized HFFs to TNF-α-induced caspase 3 cleavage (Figure 7C), it had no effect on IL-1-induced ISG expression or STAT1 phosphorylation (Figures 7D and 7E). These data indicate that IAP family members do not regulate IRF1-mediated antiviral activities in IL-1-treated cells.
Figure 7. IRF2 Negatively Regulates IL-1-Mediated Antiviral Responses.
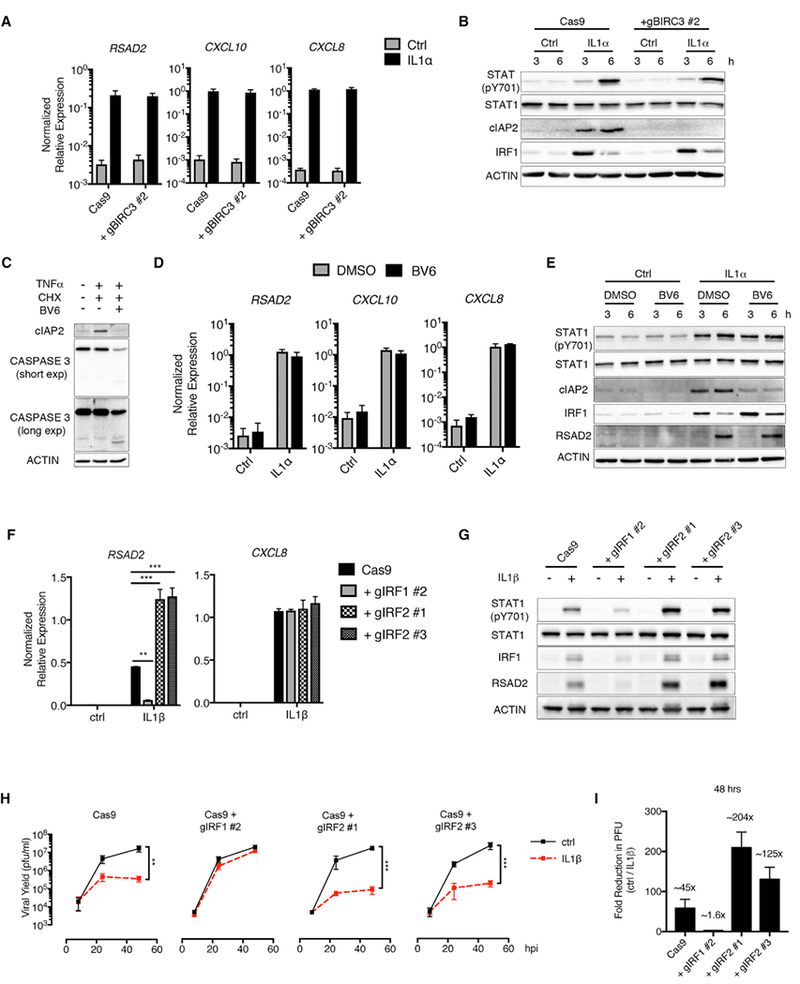
(A and B) Cas9 control or BIRC3-deficient fibroblasts were stimulated with IL-1α (100 pg/mL).
(A) Total RNA was isolated at 6 hr and analyzed by qRT-PCR.
(B) Whole-cell lysates were isolated at 3 and 6 hr post-stimulation, separated by SDS-PAGE, and analyzed by immunoblot.
(C) HFFs were treated with cycloheximide (CHX) (10 μg/mL) and BV6 (5 μM) for 1 hr, followed by stimulation with TNF (10 ng/mL). Whole-cell lysates were isolated at 6 hr post-stimulation, separated by SDS-PAGE, and analyzed by immunoblot.
(D and E) HFFs were pre-treated with BV6 as described in (C) and stimulated with IL-1α (100 pg/mL).
(D) Total RNA was isolated at 6 hr post-stimulation and analyzed by qRT-PCR.
(E) Whole-cell lysates were isolated at 3 and 6 hr post-stimulation, separated by SDS-PAGE, and analyzed by immunoblot.
(F–I) Cas9 and IRF1- and IRF2-deficient HFFs were stimulated with (F and G) IL-1β (10 ng/mL) alone or (H–I) infected with WT VSV (MOI 0.1) in the presence or absence of IL-1β (10 ng/mL).
(F) Total RNA was isolated at 6 hr post-stimulation and analyzed by qRT-PCR.
(G) Whole-cell lysates were isolated at 6 hr, separated by SDS-PAGE, and analyzed by immunoblot.
(H) Infected-cell supernatants were harvested at 8, 24, and 48 hpi, and viral yields were quantified by plaque assay.
(I) The fold reduction in viral PFUs in IL-1b-treated cells (H) was determined at 48 hpi.
Data are the average of at least three independent experiments ± SEM. Student’s t test: **p < 0.01; and ***p < 0.001.
We next examined a role for IRF2 in regulating IL-1-mediated antiviral immunity. Transduction of HFFs with lentiviruses expressing two different guide RNAs targeting IRF2 reduced IRF2 protein, as demonstrated by immunofluorescence and flow cytometry (Figures S5A and S5B). The guide RNAs targeting IRF2 were specific, as we did not observe a reduction in IRF1 protein abundance in IRF2-deficient cells (Figure 7G). IL-1 treatment of IRF2-deficient cells increased RSAD2 transcripts and protein to an extent that exceeded what was observed in WT Cas9-expressing cells (Figures 7F and 7G). Notably, neither IRF1 nor IRF2 regulated IL-1-induced CXCL8 expression (Figure 7F). Increased ISG expression corresponded to an increase in STAT1 phosphorylation in IRF2-deficient cells (Figure 7G). Consistent with IRF2 acting as a negative regulator of IL-1-induced ISG expression, we found that IL-1 treatment reduced VSV replication to a greater extent in IRF2-deficient cells than cells expressing Cas9 alone (Figure 7H). For example, at 48 hpi, we observed a ~2.5- to 4.5-fold greater reduction in VSV replication in the two IRF2-deficient cell lines treated with IL-1 when compared to the fold reduction observed in IL-1-treated Cas9 cells (Figure 7I). Based on these findings, we propose the existence of an IRF1/IRF2-regulated branch in the IL-1R pathway that is specifically dedicated to antiviral defense (Figure S6).
DISCUSSION
A core characteristic of virus-sensing PRRs is their ability to act within the cells that are infected and subsequently promote the upregulation and secretion of IFNs. This antiviral response system is therefore predicated on the need of the infected cell to induce a transcriptional response in order to protect the host. Consequently, pathogenic evasion strategies that disrupt transcriptional activities of the infected cell should render the host unable to express IFN and therefore unable to warn other cells of an infection. This study was designed to identify pathways that may serve as backup systems to protect the host during encounters with pathogens that block PRR-induced IFN responses.
We have identified IL-1 family members as proteins that exhibit hallmarks of antiviral signaling molecules. This finding expands the activities of DAMPs beyond their established role as a backup system that promotes inflammation during encounters with pathogens that block PRR-induced inflammatory gene expression. DAMPs, like IL-1, may now also be considered to provide backup antiviral activities under conditions where PRR-induced responses are prevented.
Several observations support our conclusion that IL-1 family cytokines exhibit antiviral activity to prevent pathogenic virus replication. First, IL-1 family cytokines differ from other cytokines in their lack of an N-terminal secretion signal (Auron et al., 1984). These cytokines can therefore be released from cells after lysis, as opposed to being released via the secretory pathway. The fact that IL-1 is released after membrane-damage allows these cytokines to be omnipresent at sites of infection. Second, as reported in this manuscript, IL-1α and IL-1β induce ISG expression. Third, we found that IL-1 family members can restrict the replication of PRR-evasive viruses, such as wild-type strains of VSV and ZIKV, and an ICP0-deficient strain of HSV-1. Importantly, this antiviral activity was not limited to fibroblasts, as our studies of primary human endothelial cells also revealed the ability of IL-1α to induce antiviral gene expression and restrict viral replication.
IL-1 cytokines may not be the only atypical source of antiviral activity, as TNF-α has also been reported to induce IRF1-dependent ISG expression (Venkatesh et al., 2013; Yarilina et al., 2008). TNF-α-mediated ISG expression is dependent on IFNAR signaling, and we report here that IL-1-mediated ISG expression relies on a JAK/STAT1-signaling axis that may involve gp130. The exact nature of the antiviral factors induced by IL-1 is undefined, but may consist of one or more IL-6 family members that are transcriptionally regulated by IRF1. The identification of this undefined cytokine(s) may reveal a novel class of proteins that can directly regulate ISG expression through a JAK/STAT1-signaling pathway. This idea, and the discoveries reported herein, provides a mandate to further explore the antiviral activities of IL-1 family cytokines and to re-examine the mechanisms of IL-1R-mediated signal transduction in health and disease.
STAR★METHODS
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Jonathan C. Kagan (jonathan.kagan@childrens.harvard.edu).
METHOD DETAILS
Cells and Viruses
Human foreskin fibroblasts, human lung fibroblasts, HEK293T cells, and Vero cells were cultured in DMEM supplemented with 10% Fetal Bovine Serum (FBS, GIBCO), 2mM glutamine (GIBCO), and pen/strep (GIBCO). MEFs were cultures in DMEM supplemented with 15% FBS, 2mM glutamine (GIBCO), and pen/strep (GIBCO). Normal oral keratinocytes (NOKs) immortalized with hTERT were cultured in Keratinocyte-SFM (GIBCO) media. HUVECs were cultured on collagen-coated plates in EGM media (Lonza). HUVECs were cultured in low serum (0.3% FBS)for24 hours prior to stimulations and infections. Human neutrophils and PBMCs were isolated from whole blood using Polymorphprep according to the manufacturer’s instructions (Broggi et al., 2017). For stimulation experiments, 1 × 106 neutrophils or PBMCs were stimulated with indicated cytokines. Fibroblasts from healthy controls or patients with loss-of-function mutations in IFNAR1 (Hoyos-Bachiloglu et al., 2017) were cultured like the human foreskin fibroblasts above.
Primary mouse lung and skin fibroblasts were isolated from C57BL/6J mice. Lungs and ears were isolated from euthanized mice, cut into ~3mm pieces, and incubated in 5 mL digestion buffer (0.1% collagenase A, 2.4U/ml dispase II, 150 mM NaCl, 10 mM HEPES, 2mM CaCl2) for 1 hr at 37°C with gentle agitation. Supernatants from digested lung tissues were filtered through a 70 μm filter and cells were isolated by centrifugation at 1500 rpm for 5 min. Cell pellets were washed with DMEM supplemented with 15% FBS and plated in a 100 mm dish. For isolation of skin fibroblasts, digested tissue samples were allowed to attach to 100 mm dishes for 30 min followed by addition of 10 mL DMEM supplemented with 15% FBS. Cells were passaged 1× prior to experimental use.
Wt VSV (Whelan et al., 1995) and rVSV-M51R (Kopecky et al., 2001) were propagated in BHK-21 cells (ATCC) and titrated on Vero cells (ATCC) as described previously (Cureton et al., 2009). HSV-17134 and 7134R (Cai and Schaffer, 1989) viruses were grown and titrated on U2OS cells. ZIKV stocks were propagated and titrated by plaque assay on Vero cells as described previously for West Nile virus (Brien et al., 2013).
Mice and in vivo infections
Ifnar1−/−, Il1r1−/−, and C57BL/6 mice were bred and housed at Yale University. Mice of both sexes were between 6–8 weeks of age for the initiation of all experiments conducted. No randomization protocol or blinding was used. All animal procedures were completed in compliance with approved Yale Institutional Animal Care and Use Committee protocols. Mice were infected with 5×106 PFU of VSV via intranasal injection or by epidermal scarification. Indicated tissue was harvested at specified time points, weighed, homogenized and stored at −80C for subsequent analysis via plaque assay. To determine viral titer, Vero cell monolayers were incubated with cell-free, homogenates of mouse tissues in 10-fold serial dilutions for 1 hour at 37°C and overlaid with a mixture of 2% agarose and 2× media. 1–2 days post infection, cells were fixed by 10% formalin, stained with crystal violet and plaque-forming units (PFU) were counted.
In Vitro Viral Infections and Plaque Assays
HFFs and human lung fibroblasts were plated at a concentration of 5×104 cells per well of a 24-well plate and incubated at 37°C for 72 hours to reach confluence. Viral stocks were diluted in DMEM supplemented with 1% FBS (DMEV), 2mM glutamine, and pen/strep and added to cell monolayers. After a 1-hour incubation at 37°C with gentle shaking at 10 min intervals, viral inocula were removed and replaced with 500 μL of DMEV in the presence or absence of indicated cytokines. Infected cells were incubated at 37°C for the remainder of the experiment.
To assess viral yield, supernatants from VSV- or ZikV-infected cells were harvested at indicated time points, centrifuged to remove cell debris, and frozen at −80°C until analyzed. For HSV-1-infected samples, an equal amount of DMEV supplemented with 30% (v/v) glycerol was added to wells containing infected cells and supernatants, and samples were freeze-thawed 2× at −80°C. Lysates were clarified by centrifugation. Vero or U2OS cells were plated at a concentration of 2.5×105 cells per well of 12-well plates 24 hours prior to infection. Serial 10-fold dilutions of supernatant (VSV) or clarified lysates (HSV-1) were added to Vero or U2OS cells, respectively, in duplicate and incubated at 37°C for 1 h as described above. Viral inocula were removed and infected cells were overlayed with 1× MEM containing 0.25% agarose (VSV, HSV-1) or 0.7% agarose (ZikV). Plaques were fixed at 18 hpi (VSV), 48 hpi (HSV-1), or 72 hpi (ZikV) with 10% buffered formalin for at least 1 hr at 25°C. Agarose overlays were removed and cells were stained with crystal violet in 10% methanol and air-dried overnight at 25°C.
Ex vivo Human Foreskin Infections
Normal human foreskin tissues were acquired with Partners Institutional Review Board approval from infants undergoing circumcisions and were provided by the Human Skin Disease Research Center (Brigham & Women’s Hospital). Tissues were stored in sterile normal saline solution (0.9% NaCl) prior to processing. Multiple 5mm diameter tissues were isolated from foreskin by punch biopsy (Tru-Punch, Sklar). Tissues were injected with 5 μL of PBS containing 2.5×103 pfu VSV in the presence or absence of 500 ng IL-1ra. Individual infected tissues were placed in the upper chambers of 24-well transwell plates (1 μm PET, Millipore) with the epidermis side up. Keratinocyte culture media (Carlson et al., 2008) supplemented with 20 μg/ml gentamicin and 1 μg/ml amphotericin B was added to the bottom chambers and tissues were incubated at 37°C for 48 hpi. Supernatants were isolated from the bottom chambers and viral titers were determined by plaque assay.
Cytokine and poly (I:C) stimulations.
Confluent cell monolayers were stimulated with indicated concentrations of recombinant human or mouse IL-1α, IL-1β, IL-18, IL-33, IL-36α, IFNβ or Poly (I:C)-HMW diluted in DMEM supplemented with 1% FBS.
Chemical Inhibitors
BV6 (5 μM), Pyridone 6 (5 μg/ml), Cycloheximide (10 μg/ml). Cells were pre-treated with SC144 (12.5 μM) for 1 h prior to and throughout stimulation with indicated cytokines. For infections, SC144 (12.5 μM) was added to media following 1 h virus adsorption period.
siRNA knockdown
HFFs were plated at a density of 5×104 cells per well of a 24-well dish 24 h pre-siRNA transfection. Cells were transfected with 50 nM of human control, IRF1, TNFR1, or TRADD pooled siRNAs using DharmaFECT 2 according to the manufacturer’s instructions. Transfection media was replaced at 24 hpt and depletion efficiency was determined at 72 hpt.
CRISPR/Cas9 gene targeting
Prior to the implementation of CRISPR, HFF were immortalized (HFF-hTERT) by transduction with the pWZL-hTERT retrovirus and selected for hygromycin resistance for 5 days. The pRRL-Cas9-Puro lentivirus construct used in this study was described previously (Eckard et al., 2014). Double stranded DNA oligos encoding specific gRNAs were inserted into the pRRL-Cas9-Puro vector by homologous recombination using the In-Fusion cloning kit. Lentivirus plasmids expressing Cas9 alone or co-expressing gRNAs were co-transfected with the psPAX2 packaging vector, and pVSV-G into 10 mm dishes of 50% confluent HEK293T cells using PEI. Media containing transfection complexes was replaced at 24 hpt, supernatants were harvested at 48 hpt and Altered through a 0.45 μm syringe Alter. Filtered supernatants were added to 1×105 HFF-hTERT cells plated in a 6-well plate and spinfected at 2000 rpm for 30 min. Media was replaced at 24 h post-transduction. At 3 days post-transduction, cells were selected for puromycin resistance. Cells were passaged for 1–2 weeks prior to experimental use. Targeting of the desired gene was evaluated by western blot or flow cytometry for loss of endogenous protein.
RNA isolation, qRT-PCR, and nCounter
Total RNA was isolated from cells using the PureLink RNA mini kit and eluted from columns with 150 μL nuclease-free H2O. For the detection of IFNB transcripts, an aliquot of 100 ng of RNA was DNase-treated for 1hr at 37°C and used for subsequent reverse transcription and detection steps. For other transcripts, 40 ng of RNA was used for detection steps. Reverse transcription and PCR reactions were performed on a CFX384 Touch Real-Time PCR detection system (Bio-Rad) using the Taqman RNA-to-CT 1-Step Kit following the manufacturer’s instructions. Nanostring analysis was carried out as described previously (Odendall et al., 2014). Briefly, 100 ng of purified RNA was hybridized with an nCounter CodeSet for 16 h. Samples were then loaded onto the nCounter prep station, followed by quantification with the nCounter Digital Analyzer. Data were processed using nSolver Analysis Software. Data were normalized to the expression of a control gene (GAPDH) and plotted as log 2-fold change compared to 0 h time point.
Western Blot and Antibodies
Cells were harvested in 1× Laemmli sample buffer and run on 10% Tris-Glycine SDS Gels. Proteins were transferred to PVDF membranes overnight at 4°C, and the membranes were blocked in 5% (w/v) milk suspended in Tris-buffered saline (TBS) prior to incubation in primary antibody diluted in TBS containing 0.05% (v/v) Tween-20 (TBST) and 2.5% (w/v) bovine serum albumin. Membranes were washed 3× with TBST, incubated in secondary antibody for 1 h at RT, washed 2× with TBST, and 1× TBS. Membranes were incubated in SuperSignal West Pico Chemiluminescent Substrate and detected using a ChemiDocXRS+ System (Bio-Rad). Secondary goat anti-mouse and goat anti-rabbit antibodies conjugated to HRP were used at a 1:5000 dilution.
ELISA
Supernatants from mock-infected or VSV-infected NOKs were clarified at 10,000 rpm for 10 min to remove cellular debris. Aliquots of 500 μL of supernatant were frozen at −80°C until analysis was performed. To detect intracellular IL-1 cytokines, 5×105 cells were lysed in 500 μL 10% (v/v) NP40 buffer (10% NP40, PBS). The concentrations of supernatant or intracellular IL-1 cytokines were determined using Human IL-1α and IL-1β ELISA MAX kits according to the manufacturer’s instructions. Extracellular IL29 concentrations were determined using the Human IL-29/IFN-lambda 1 DuoSet ELISA kit according to the manufacturer’s instructions
Immunofluorescence
Cells were fixed with 4% formaldehyde, permeabilized with 0.5% NP40, and blocked in 5% normal goat serum overnight at 4°C. Fixed cells were incubated with primary antibodies for 30 min at 37°C and washed two times with PBS containing 0.05% Tween 20 followed by one wash with PBS. Alexa Fluor 488- and 594-conjugated secondary antibodies (1:500) were incubated with cells for 2 hr at 25°C. The coverslips were washed as above and mounted on slides with ProLong Gold antifade reagent. Images were acquired using a Ziess Axiovert Spinning Disk Confocal Microscope.
QUANTIFICATION AND STATISTICAL ANALYSIS
Results are expressed as mean ± standard error of the mean. 2-tailed Student’s t tests were used to determine p values. A p value < 0.05 was considered statistically significant. *p < 0.05, **p <0.01, and ***p <0.001. All statistical analyses were performed using GraphPad Prism software.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| pSTAT1 (Y701) | BD biosciences | # 612132; RRID:AB_399503 |
| STAT1 | Cell Signaling | #9172; RRID:AB_2198289 |
| RSAD2 | Cell Signaling | # 13996; RRID:AB_2734772 |
| IRF1 | Cell Signaling | # 8478; RRID:AB_10949108 |
| IFIT1 | GeneTex | # GTX103452; RRID:AB_1950546 |
| VSV M | KeraFast | # EB0011; RRID:AB_2734773 |
| Beta-actin | Sigma | # A5441; RRID:AB_476744 |
| IRF3 | Santa Cruz | # sc-33641; RRID:AB_627826 |
| IRF3 | Cell Signaling | # 4302; RRID:AB_1904036 |
| c-IAP2 | Cell Signaling | # 3130; RRID:AB_10693298 |
| IRF2 | Biolegend | # 657502; RRID:AB_2562588 |
| IFNAR2 | ThermoFisher | # 213851; RRID:AB_387828 |
| Mouse IgG2a | ThermoFisher | # 02-6200; RRID:AB_2734774 |
| Goat anti-Mouse IgG Alexa Fluor 488 | ThermoFisher | # A-11001; RRID:AB_2534069 |
| Goat anti-Mouse IgG Alexa Fluor 594 | ThermoFisher | # A-11005; RRID:AB_141372 |
| Goat anti-rabbit IgG Alexa Fluor 488 | ThermoFisher | # R37116; RRID:AB_2734776 |
| Goat anti-rabbit IgG Alexa Fluor 594 | ThermoFisher | # R37117; RRID:AB_2734777 |
| Goat-anti-mouse HRP | Millipore | # 12-349; RRID:AB_390192 |
| Goat-anti-rabbit HRP | Millipore | # 12-348; RRID:AB_11214240 |
| Bacterial and Virus Strains | ||
| Wt VSV | Provided by Dr. Sean Whelan, (Whelan et al., 1995) | N/A |
| rVSV M51R | Provided by Dr. Sean Whelan, (Kopecky et al., 2001) | N/A |
| HSV-1 7134 | Provided by Dr. David Knipe, (Cai and Schaffer, 1989) | N/A |
| HSV-1 7134R | Provided by Dr. David Knipe, (Cai and Schaffer, 1989) | N/A |
| ZikaV MR766 | Provided by Dr. Michael Diamond | N/A |
| ZikaV PF/13 | Provided by Dr. Michael Diamond | N/A |
| Sendai Virus, Cantell Strain | Charles River Laboratory | #10100774 |
| Biological Samples | ||
| Normal human foreskin | Human Skin Disease Research Center (Brigham & Women’s Hospital) | N/A |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Draq5 | ThermoFisher | # 62251 |
| Recombinant human IL-1ra | Biolegend | # 553902 |
| Recombinant human IL-1α | Biolegend | # 570002 |
| Recombinant human IL-1β | Biolegend | # 579402 |
| Recombinant human IL-18 | Biolegend | #592102 |
| Recombinant human IL-33 | Biolegend | # 581802 |
| Recombinant human IL-36α | Biolegend | # 551602 |
| Recombinant human IFNβ | Millipore | # IF014 |
| Recombinant human TNFα | Biolegend | # 570102 |
| Poly (I:C)-HMW | Invivogen | # tlr-pic |
| Recombinant mouse IL-1α | Biolegend | # 575002 |
| Recombinant mouse IL-1β | Biolegend | # 575102 |
| Bv6 | Millipore | # 533965 |
| Pyridone 6 | Millipore | # 420099 |
| Cycloheximide | Sigma | # C7698–1G |
| SC144 | Millipore | # 506387 |
| Polyethylenimine (PEI) | Polysciences, Inc | # 23966-1 |
| Critical Commercial Assays | ||
| ProLong Gold antifade reagent | ThermoFisher | # P36930 |
| Polymorphprep | Axis Shield | # 1114683 |
| Dharmafect 2 | Dharmacon | # T-2002-01 |
| PureLink RNA mini kit | ThermoFisher | # 12183018A |
| In-Fusion HD cloning system | Clontech | # 639645 |
| Dnase-I | ThermoFisher | # EN0525 |
| Taqman RNA-to-CT 1-Step Kit | ThermoFisher | # 4392653 |
| SuperSignal West Pico Chemiluminescent Substrate | ThermoFisher | # 34577 |
| Human IL-1α ELISA MAX kit | Biolegend | # 433404 |
| Human IL-1β ELISA MAX kit | Biolegend | # 437004 |
| Human IL-29/IFN-lambda 1 DuoSet ELISA kit | R&D Systems | # DY7246 |
| Experimental Models: Cell Lines | ||
| Human Foreskin Fibroblasts, HFF | ATCC | # SCRC-1041 |
| Human Lung Fibroblasts, MRC-5 | ATCC | # CCL-171 |
| HEK293T | ATCC | # CRL-3216 |
| Vero | ATCC | # CCL-81 |
| BHK-21 | Provided by Dr. Sean Whelan, Harvard Medical School | N/A |
| U2OS | Provided by Dr. David Knipe, Harvard Medical School | N/A |
| Normal oral keratinocytes (NOK) | Provided by Dr. Karl Munger, Tufts University School of Medicine | N/A |
| HUVEC | Provided by Dr. Dezheng Zhao (Beth Israel Deaconess Medical Center) | N/A |
| Human control dermal fibroblasts (C1) | Provided by Dr. Raif Geha, Boston Children’s Hospital (Hoyos-Bachiloglu et al., 2017) | N/A |
| IFNAR1 loss of function mutant fibroblasts (P1) | Provided by Dr. Raif Geha, Boston Children’s Hospital (Hoyos-Bachiloglu et al., 2017) | N/A |
| HFF Cas9 | This paper | N/A |
| HFF gIRF1 #1 | This paper | N/A |
| HFF gIRF2 #1 | This paper | N/A |
| HFF gIRF2 #3 | This paper | N/A |
| HFF gIRF3 #2 | This paper | N/A |
| HFF gSTAT1 #1 | This paper | N/A |
| HFF gSTAT1 #3 | This paper | N/A |
| HFF gBIRC3 #2 | This paper | N/A |
| Experimental Models: Organisms/Strains | ||
| C57BL/6J mice (in vitro experiments) | The Jackson Laboratory | #000664 |
| Il1r1−/−(in vivo experiments) | The Jackson Laboratory | #003245 |
| Ifna1r−/−(in vivo experiments) | The Jackson Laboratory | #32045-JAX |
| C57BL/6 mice (in vivo experiments) | Charles River | # 027 |
| Oligonucleotides | ||
| Human IL1R1 Taqman primer/probe | ThermoFisher | Hs00991010_m1 |
| Human IL1A Taqman primer/probe | ThermoFisher | Hs00174092_m1 |
| Human IL1B Taqman primer/probe | ThermoFisher | Hs00174097_m1 |
| Human IL29 Taqman primer/probe | ThermoFisher | Hs00601677_g1 |
| Human IFNB Taqman primer/probe | ThermoFisher | Hs01077958_s1 |
| Human RSAD2 Taqman primer/probe | ThermoFisher | Hs00369813_m1 |
| Human CXCL8 Taqman primer/probe | ThermoFisher | Hs00174103_m1 |
| Human CXCL10 Taqman primer/probe | ThermoFisher | Hs01124252_g1 |
| Human GAPDH Taqman primer/probe | ThermoFisher | Hs02786624_g1 |
| Human 18S rRNA Taqman primer/probe | ThermoFisher | # 4333760F |
| Mouse Cxcl1 Taqman primer/probe | ThermoFisher | Mm04207460_m1 |
| Mouse Ifit2 Taqman primer/probe | ThermoFisher | Mm00492606_m1 |
| Mouse Rsad2 Taqman primer/probe | ThermoFisher | Mm00491265_m1 |
| Mouse Tbp Taqman primer/probe | ThermoFisher | Mm01277042_m1 |
| gIRF1 #2 GTCTCATGCGCATCCGAGTGA | This paper | N/A |
| gIRF2 #1 GAAAGGATGCGCATGCGCCCG | This paper | N/A |
| gIRF2 #3 GCTTGAGCCCCGGGATCGTGT | This paper | N/A |
| gIRF3 #2 GATGGTCCGGCCTGCGCGTAT | This paper | N/A |
| gSTAT1 #1 GCATGCCAACGCACCCTCAG | This paper | N/A |
| gSTAT1 #3 GGAGTCCACCAATGGCAGTC | This paper | N/A |
| gBIRC3 #2 GCGTGCTGGTTTCTATTACAC | This paper | N/A |
| OnTARGETplus IRF1 siRNAs | Dharmacon | L-011704-00-0005 |
| OnTARGETplus TRADD siRNAs | Dharmacon | L-004452-00-0005 |
| OnTARGETplus TNFR1 siRNAs | Dharmacon | L-005197-00-0005 |
| OnTARGETplus Control siRNAs | Dharmacon | D-001810-10-05 |
| Recombinant DNA | ||
| pWZL-hTERT | Provided by Dr. Steen Hansen, Boston Children’s Hospital | N/A |
| pCL-Eco | Addgene | # 12371 |
| pRRL-Cas9-Puro | Provided by Dr. Dan Stetson, University of Washington, (Eckard et al., 2014) | N/A |
| psPAX2 | Provided by Dr. Dan Stetson, University of Washington, (Eckard et al., 2014) | N/A |
| pVSV-G | Provided by Dr. Dan Stetson, University of Washington, (Eckard et al., 2014) | N/A |
Highlights.
Keratinocytes release IL-1 cytokines in response to PRR-evasive virus infection
IL-1 cytokines induce an antiviral state in human fibroblasts and endothelial cells
This antiviral response is important during encounters with immune-evasive viruses
IL-1 stimulates this antiviral response through an IRF1/gp130/STAT1 signaling axis
ACKNOWLEDGMENTS
We thank members of the Kagan and Garlick labs for helpful discussions. We thank David Knipe and Sean Whelan (Harvard Medical School) for assistance with HSV-1 and ZIKV infections, respectively. We thank Raif Geha for providing us with IFNAR1 mutant human fibroblasts. We thank the Human Skin Disease Resource Center of Brigham and Women’s Hospital and Harvard Medical School, which provided neonatal foreskin samples. The Human Skin Disease Resource Center is supported in part by NIAMS Resource-Based Center grant no. 1P30AR069625. This work was supported by the NIH grants AI133524, AI093589, AI116550, and P30DK34854 to J.C.K. J.C.K. holds an Investigators in the Pathogenesis of Infectious Disease Award from the Burroughs Wellcome Fund. M.H.O. was supported by NIH grants T32AI007512 and K99AI130258.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information includes six figures and can be found with this article online at https://doi.org/10.1016/j.molcel.2018.07.009.
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- Ahmed M, McKenzie MO, Puckett S, Hojnacki M, Poliquin L, and Lyles DS (2003). Ability of the matrix protein of vesicular stomatitis virus to suppress beta interferon gene expression is genetically correlated with the inhibition of host RNA and protein synthesis. J. Virol 77, 4646–4657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexopoulou L., Holt AC., Medzhitov R., and Flavell RA. (2001). Recognition of double-stranded RNA and activation of NF-kappaB by Toll-like receptor 3. Nature 413, 732–738. [DOI] [PubMed] [Google Scholar]
- Arpaia N, and Barton GM (2011). Toll-like receptors: key players in antiviral immunity. Curr. Opin. Virol 1, 447–454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Auron PE, Webb AC, Rosenwasser LJ., Mucci SF, Rich A, Wolff SM, and Dinarello CA (1984). Nucleotide sequence of human monocyte interleukin 1 precursor cDNA. Proc. Natl. Acad. Sci. USA 81, 7907–7911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brien JD., Lazear HM., and Diamond MS. (2013). Propagation, quantification, detection, and storage of West Nile virus. Curr. Protoc. Microbiol 31, 15D.3.1.–15D.3.18. [DOI] [PubMed] [Google Scholar]
- Broggi A., Tan Y., Granucci F., and Zanoni I. (2017). IFN-l suppresses intestinal inflammation by non-translational regulation of neutrophil function. Nat. Immunol 18, 1084–1093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai WZ, and Schaffer PA (1989). Herpes simplex virus type 1 ICP0 plays a critical role in the de novo synthesis of infectious virus following transfection of viral DNA. J. Virol 63, 4579–4589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlson MW., Alt-Holland A., Egles C., and Garlick JA. (2008). Three-dimensional tissue models of normal and diseased skin. Curr. Protoc. Cell Biol Chapter 19. Unit 19.9.book. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan YK, and Gack MU (2016). Viral evasion of intracellular DNA and RNA sensing. Nat. Rev. Microbiol 14, 360–373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Q, Sun L, and Chen ZJ (2016). Regulation and function of the cGAS-STING pathway of cytosolic DNA sensing. Nat. Immunol 17, 1142–1149 [DOI] [PubMed] [Google Scholar]
- Chow J, Franz KM, and Kagan JC (2015). PRRs are watching you: localization of innate sensing and signaling regulators. Virology 479–480, [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cureton DK, Massol RH, Saffarian S, Kirchhausen TL, and Whelan SP (2009). Vesicular stomatitis virus enters cells through vesicles incompletely coated with clathrin that depend upon actin for internalization. PLoS Pathog 5, e1000394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dinarello CA (2009). Immunological and inflammatory functions of the interleukin-1 family. Annu. Rev. Immunol 27, 519–550. [DOI] [PubMed] [Google Scholar]
- Eckard SC, Rice GI, Fabre A, Badens C, Gray EE, Hartley JL, Crow YJ, and Stetson DB (2014). The SKIV2L RNA exosome limits activation of the RIG-I-like receptors. Nat. Immunol 15, 839–845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fensterl V., Chattopadhyay S, and Sen GC. (2015). No love lost between viruses and interferons. Annu. Rev. Virol 2, 549–572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferran MC, and Lucas-Lenard JM (1997). The vesicular stomatitis virus matrix protein inhibits transcription from the human beta interferon promoter. J. Virol 71,371–377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Sastre A (2017). Ten strategies of interferon evasion by viruses. Cell Host Microbe 22, 176–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garlanda C., Dinarello CA, and Mantovani A. (2013). The interleukin-1 family: back to the future. Immunity 39, 1003–1018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gitlin L, Barchet W, Gilfillan S, Cella M, Beutler B, Flavell RA, Diamond MS, and Colonna M (2006). Essential role of mda-5 in type IIFN responses to polyriboinosinic:polyribocytidylic acid and encephalomyocarditis picorna-virus. Proc. Natl. Acad. Sci. USA 103, 8459–8464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harada H, Fujita T, Miyamoto M, Kimura Y, Maruyama M, Furia A, Miyata T, and Taniguchi T (1989). Structurally similar but functionally distinct factors, IRF-1 and IRF-2, bind to the same regulatory elements of IFN and IFN-inducible genes. Cell 58, 729–739. [DOI] [PubMed] [Google Scholar]
- Harikumar KB., Yester JW., Surace MJ., Oyeniran C., Price MM., Huang WC., Hait NC., Allegood JC., Yamada A., Kong X., et al. (2014). K63-linked polyubiquitination of transcription factor IRF1 is essential for IL-1-induced production of chemokines CXCL10 and CCL5. Nat. Immunol 15, 231–238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirahara K, Onodera A, Villarino AV, Bonelli M, Sciume G, Laurence A, Sun HW, Brooks SR, Vahedi G, Shih HY, et al. (2015). Asymmetric action of STAT transcription factors drives transcriptional outputs and cytokine specificity. Immunity 42, 877–889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoyos-Bachiloglu R, Chou J, Sodroski CN, Beano A, Bainter W, Angelova M, Al Idrissi E, Habazi MK, Alghamdi HA, Almanjomi F, et al. (2017). A digenic human immunodeficiency characterized by IFNAR1 and IFNGR2 mutations. J. Clin. Invest 127, 4415–4420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwasaki A (2012). A virological view of innate immune recognition. Annu. Rev. Microbiol 66, 177–196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwasaki A, Foxman EF., and Molony RD. (2017). Early local immune defences in the respiratory tract. Nat. Rev. Immunol 17, 7–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwata M., Vieira J., Byrne M., Horton H., and Torok-Storb B. (1999). Interleukin-1 (IL-1) inhibits growth of cytomegalovirus in human marrow stromal cells: inhibition is reversed upon removal of IL-1. Blood 94, 572–578. [PubMed] [Google Scholar]
- Kono H, and Rock KL (2008). How dying cells alert the immune system to danger. Nat. Rev. Immunol 8, 279–289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kopecky SA, Willingham MC, and Lyles DS (2001). Matrix protein and another viral component contribute to induction of apoptosis in cells infected with vesicular stomatitis virus. J. Virol 75, 12169–12181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar A, Hou S, Airo AM, Limonta D, Mancinelli V, Branton W, Power C, and Hobman TC (2016). Zika virus inhibits type-I interferon production and downstream signaling. EMBO Rep 17, 1766–1775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurt-Jones EA., Orzalli MH., and Knipe DM. (2017). Innate immune mechanisms and herpes simplex virus infection and disease. Adv. Anat. Embryol. Cell Biol 223, 49–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lippincott-Schwartz J, Yuan LC, Bonifacino JS, and Klausner RD (1989). Rapid redistribution of Golgi proteins into the ER in cells treated with brefeldin A: evidence for membrane cycling from Golgi to ER. Cell 56,801–813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma Z, and Damania B (2016). The cGAS-STING defense pathway and its counteraction by viruses. Cell Host Microbe 19, 150–158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahoney DJ, Cheung HH, Mrad RL, Plenchette S, Simard C, Enwere E, Arora V, Mak TW, Lacasse EC, Waring J, and Korneluk RG (2008). Both cIAP1 and cIAP2 regulate TNFalpha-mediated NF-kappaB activation. Proc. Natl. Acad. Sci. USA 105, 11778–11783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melroe GT, DeLuca NA, and Knipe DM (2004). Herpes simplex virus 1 has multiple mechanisms for blocking virus-induced interferon production. J. Virol 78, 8411–8420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mossman KL, Saffran HA, and Smiley JR (2000). Herpes simplex virus ICP0 mutants are hypersensitive to interferon. J. Virol 74, 2052–2056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nair S, Michaelsen-Preusse K, Finsterbusch K, Stegemann-Koniszewski S., Bruder D., Grashoff M., Korte M., Keister M., Kalinke U., Hauser H., and Kroger A. (2014). Interferon regulatory factor-1 protects from fatal neurotropic infection with vesicular stomatitis virus by specific inhibition of viral replication in neurons. PLoS Pathog 10, e1003999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Odendall C, and Kagan JC (2015). The unique regulation and functions of type III interferons in antiviral immunity. Curr. Opin. Virol 12, 47–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Odendall C., Dixit E., Stavru F., Bierne H., Franz KM., Durbin AF., Boulant S., Gehrke L., Cossart P., and Kagan JC. (2014). Diverse intracellular pathogens activate type III interferon expression from peroxisomes. Nat. Immunol 15, 717–726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Randolph-Habecker J., Iwata M., Geballe AP., Jarrahian S., and Torok-Storb B. (2002). Interleukin-1-mediated inhibition of cytomegalovirus replication is due to increased IFN-beta production. J. Interferon Cytokine Res 22, 765–772. [DOI] [PubMed] [Google Scholar]
- Schoggins JW, and Rice CM (2011). Interferon-stimulated genes and their antiviral effector functions. Curr. Opin. Virol 1, 519–525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schoggins JW, Wilson SJ, Panis M, Murphy MY, Jones CT, Bieniasz P, and Rice CM (2011). A diverse range of gene products are effectors of the type I interferon antiviral response. Nature 472, 481–485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sims JE, and Smith DE (2010). The IL-1 family: regulators of immunity. Nat. Rev. Immunol 10, 89–102. [DOI] [PubMed] [Google Scholar]
- Van Damme J, De Ley M, Opdenakker G, Billiau A, DeSomer P, and Van Beeumen J (1985). Homogeneous interferon-inducing 22Kfactor is related to endogenous pyrogen and interleukin-1. Nature 314, 266–268. [DOI] [PubMed] [Google Scholar]
- Varfolomeev E, Goncharov T, Fedorova AV, Dynek JN, Zobel K, Deshayes K, Fairbrother WJ, and Vucic D (2008). c-IAP1 and c-IAP2 are critical mediators of tumor necrosis factor alpha (TNFalpha)-induced NF-kappaB activation. J. Biol. Chem 283, 24295–24299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venkatesh D, Ernandez T, Rosetti F, Batal I, Cullere X, Luscinskas FW, Zhang Y, Stavrakis G, Garcia-Carderna G, Horwitz BH, and Mayadas TN (2013). Endothelial TNF receptor 2 induces IRF1 transcription factor-dependent interferon-β autocrine signaling to promote monocyte recruitment. Immunity 38, 1025–1037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whelan SP, Ball LA, Barr JN, and Wertz GT (1995). Efficient recovery of infectious vesicular stomatitis virus entirely from cDNA clones. Proc. Natl. Acad. Sci. USA 92, 8388–8392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yarilina A, Park-Min KH, Antoniv T, Hu X, and Ivashkiv LB (2008). TNF activates an IRF1-dependent autocrine loop leading to sustained expression of chemokines and STAT1-dependent type I interferon-response genes. Nat. Immunol 9, 378–387. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.