

Abstract
Gain-of-function (GOF) mutations in the TP53 gene lead to acquisition of new functions by the mutated tumor suppressor p53 protein. A number of the over-represented ‘hot spot’ mutations, including the ones in codons 175, 248 or 273, convey GOF phenotypes. Such phenotypes may include resistance to chemotherapeutics or changes in motility and invasiveness. Whereas the prevalent notion is that the acquisition of the p53 GOF phenotype translates into poorer prognosis for the patient, the analysis of a human somatic p53 mutations dataset demonstrated earlier tumor onset, but decreased frequency and altered location of metastases in patients with the p53-R248Q allele. Therefore, the GOF activities of p53-R248Q and p53-D281G were analyzed in triple negative breast cancer MDA-MB-231 and lung adenocarcinoma H1299 cell lines with regard to invasive and metastatic traits. The expression of p53-D281G increased the motility and invasiveness of the lung cancer cells, but not those of the breast cancer cells. In contrast, the expression of p53-R248Q decreased the motility and invasiveness of the breast and lung cancer cells in a p53 transactivation-dependent manner. The intravenous xenotransplantation of MDA-MB-231 cells expressing p53-R248Q into zebrafish embryos resulted in an alteration of the distribution of cancer cells in the body of the fish. In p53-R248Q-expressing H1299 cells a decrease in the expression of TCF8/ZEB1 and N-cadherin was observed, suggesting partial mesenchymal-to-epithelial transition. In the two cell lines expressing p53-R248Q a decrease was noted in the expression of myosin light chain 2, a protein involved in actomyosin-based motility. To the best of our knowledge, the present study is one of only few reports demonstrating the mutated p53 GOF activity resulting in a decrease of a malignant trait in human cancer.
Keywords: p53, gain-of-function, hot spot, invasiveness, metastasis
Introduction
The TP53 tumor suppressor gene encodes the p53 protein, a transcription factor that, in a homotetrameric form, binds its specific target sites and regulates a plethora of genes. The function of p53 is crucial for proper control of cell cycle progression, apoptosis, senescence, DNA repair and genome maintenance, to name a few of its major functions (1). The importance of p53 in preventing tumor development is underlined by the fact that it is altered in >50% of human tumors, more frequently than any other gene. The majority of the alterations found in the TP53 gene are missense mutations and are more frequently found at particular codon positions: R175, R248, R249, R273 and R282 (2). Notably, the same codons are most frequently mutated in patients with Li-Fraumeni syndrome bearing germline TP53 mutations and in patients with cancer with somatic mutations. These positions are termed ‘hot spots’ and the majority of them are located in the DNA binding domain of the p53 protein. Consequently, a number of the ‘hot spot’ mutations lead to the loss of the DNA-binding capability and, therefore, transcriptional activator (TA) function (3). Furthermore, given the tetrameric structure of the bioactive form of p53, a mutation in one allele may lead to the functional inactivation of the remaining wild-type (WT) allele via the formation of heterotetrameric, transcriptionally inactive complexes (4-6). This mode of action is referred to as dominant negative. The notion that a mutated p53 may exhibit oncogenic activity was formulated in 1993, based on in vitro and in vivo evidence (7); mutations causing such activity are referred to as gain-of-function (GOF).
A number of studies involving p53 GOF mutations have been conducted in an in vitro setting, utilizing primary cells from transgenic animals as well as immortalized cell lines. This approach allows for the separation of the functional components of what is observed as ‘tumor progression’ in an animal model, ‘hallmarks of cancer’ (8,9), and the investigation of the molecular mechanisms underlying the observed GOF phenotypes. The range of these phenotypes is broad, depending on the cell type, the position and type of mutation, and other co-occurring changes. The most consistently reported phenotypes entail accelerated growth due to sustained proliferative signaling, genomic instability, increased survival of cancer cells that may manifest as resistance to chemotherapy, and increased mobility/invasiveness that may translate to increased rate of invasion and metastasis in vivo. Since the focus of the present study is on migration and invasiveness, other p53 GOF mutation-induced phenotypes will not be elaborated upon; several excellent recent reviews on these topics have been published (10-13).
Progress in molecular biology techniques has enabled the creation of transgenic mice with NULL-p53 and mutant (mut)-p53 genotypes (14-16). Initially, the mutations most commonly observed in human patients were introduced into the murine genome. In the p53 R172H model (equivalent to human R175H) no decrease in overall survival was revealed, compared with the NULL genotype, but a marked increase in the frequency of metastasis was observed in the p53+/R172H heterozygotes compared with the p53+/− animals (14). Notably, no such increase was observed in otherwise genetically identical mice with the p53+/R172P genotype, which reflects a less common mutation observed in humans (17). In the independently created set of mice featuring the R172H and R270H (equivalent to human R273H) heterozygotic alleles in WT and NULL backgrounds, no significant difference in survival time was noted, but the spectrum of tumors differed depending on the mutation. Also, an increased incidence of metastases was observed in the p53+/R172H heterozygotes compared with the p53+/− animals (16). In other studies, transgenic mice were created using humanized p53 knock-in (HUPKI) alleles (18,19). These animals bore homozygous R175H, R248W and R273H alleles. The mice expressing all mutated p53 variants exhibited serious impairment of DNA repair mechanisms, resulting in numerous interchromosomal translocations, compared with p53−/− mice. Whereas the p53 R175H and p53 R248W mice presented with a more complex tumor spectrum than their p53−/− counterparts, their overall survival time was not impacted. Using the same HUPKI technology, another study reported on the construction of p53 R248Q and G245S mice. The p53 G245S/- mice revealed no significant phenotype changes, yet the p53 R248Q mice (R248Q/- and R248Q/R248Q) suffered from accelerated tumor onset and decreased survival times in comparison with the p53−/− animals (20). In summary, whereas the majority of in vivo studies utilizing transgenic mice have reported changes in the tumor spectrum and onset time, evidence for decreased survival time and increased metastasis is limited. A number of studies conducted with cell lines, including xenotransplantations into experimental animals, addressed changes in the migratory and metastatic behavior of cells resulting from the expression of mutated p53 with the range of proposed explanatory mechanisms being very broad. Certain studies identified the sequestration of tumor proteins p63 and/or p73 by mut-p53 (21-23), resulting in the derepression of molecular pathways that support metastasis. The target molecules and cellular processes include Rab coupling receptor-dependent recycling of integrins and growth factor receptors (24), SMAD-mediated interference with the transforming growth factor β pathway (25), nuclear transcription factor Y (NF-Y)-mediated regulation of platelet-derived growth factor receptor β (26) and tumor suppressor microRNA (miRNA) let-7i (27). Other reported mechanisms involve direct interactions between mut-p53 and a variety of transcription factors, to include the co-recruitment of NF-Y and histone acetyltransferase p300 with mut-p53 to the Ephrin-B2 promoter, resulting in the enhancement of the epithelial-to-mesenchymal transition (EMT) (28), transcription factor ETS2-mediated upregulation of nucleotide biosynthesis required for increased invasiveness (29), or early growth response 1-mediated upregulation of myosin X, resulting in an increased invasiveness in breast cancer (30). In addition to the studies identifying molecular mediators of mut-p53 GOF-induced phenotypes, a number of studies exist that describe broad changes in transcription profiles, constituting a base for further research into the putative roles of differentially expressed genes in evoking the GOF-induced phenotypes (31-33); notably, even for a particular GOF mut-p53 (D281G) certain mut-p53-induced changes in expression profiles are dependent on the intactness of the p53 transactivation domain (31), whereas other are not (34). In addition, given the important role that chemokines and their receptors serve in the dissemination of tumor cells (35-37), it is not surprising that a number of studies have reported changes in the bioactivity of chemokines and/or their receptors in cells expressing mut-p53 (38-40) [reviewed in (41)].
Whereas the majority of the published studies demonstrate an increase in invasiveness following mut-p53 expression, certain studies have demonstrated a lack of change (42) or even a decrease (43) in this characteristic in mut-p53-expressing cells. Therefore, the present study tested whether the expression of two tumor-derived mut-p53 variants in breast cancer MDA-MB-231 and lung cancer H1299 cells leads to changes in their migratory behavior and whether these changes are dependent on p53 transactivation activity.
Materials and methods
Cell culture and genetic modification
H1299 (cat. no. CRL-5803), H358 (cat. no CRL-5807) and MDA-MB-231 (cat. no. HTB-26) cells were purchased from the American Type Culture Collection (Manassas, VA, USA). All cell culture media components were from Merck KGaA (Sigma-Aldrich; Merck KGaA, Darmstadt, Germany). The H1299 and H358 cells were cultured in RPMI base medium and the MDA-MB-231 cells were cultured in Dulbecco’s modified Eagle’s medium. Both base media were supplemented with 10% heat-inactivated fetal bovine serum (FBS), 100 U/ml penicillin and 0.1 mg/ml streptomycin. When the cells were 90% confluent, they were harvested by trypsinization and seeded at 10% confluence. Cells were incubated in 5% CO2 atmosphere at 37°C. For the stable expression of constructs in the cells, the following plasmids (44) were a gift from Eric Campeau and Paul Kaufman (Program in Gene Function and Expression, University of Massachusetts Medical School, Worcester, MA, USA): pENTR1A-no-ccDB, pENTR/pTER+, pLenti CMV TetR Blast, pLenti CMV/TO Puro DEST, pLenti CMV/TO Hygro DEST, pLenti X2 Hygro DEST and pLenti CMV Neo DEST. pMD2.G and psPAX2 were a gift from Didier Trono (School of Life Sciences, Lausanne Federal Institute of Technology, Lausanne, Switzerland). All the plasmids were deposited by their respective researchers to Addgene, Inc. (Cambridge, MA, USA) and obtained from there. The tetracycline-responsive regulatory element (TetR) utilized in this system is a repressor that, in the absence of tetracycline, occupies the CMV/TO promoter, blocking transcription. Upon addition of tetracycline or doxycycline (DOX), it dissociates from the promoter, allowing gene expression. This results in a Tet-ON system that includes a repressor rather than activator. A dsDNA fragment encoding a short hairpin RNA (shRNA) directed against p53 (CCCCGACTCCAGTGGTAATCTACTTCAAGAGAGTA GATTACCACTGGAGTCTTTTT; hairpin sequences in bold) was created by annealing complementary oligonucleotides according to the protocol of the BLOCK-iT™ U6 RNAi Entry Vector kit (Thermo Fisher Scientific, Inc., Waltham, MA, USA). The resulting dsDNA fragment was amplified by PCR using oligonucleotides (5′-GCAGATCTCCCCGACTCCAG TGG-3′ and 5′-CGAAGCTTAAAAAGACTCCAGTG-3′; restriction sites for PCR digestion marked in bold) and Phusion high-fidelity DNA polymerase (Thermo Fisher Scientific, Inc.). The resulting product was column-purified (Clean-up kit; A&A Biotechnology, Gdynia, Poland). Subsequently, the PCR product and pENTR/pTER+ plasmid were digested with BglII and HindIII restriction enzymes, gel-purified (Gel-Out kit; A&A Biotechnology), and ligated with T4 ligase. This resulted in the creation of pENTR/pTER-shp53 cloning vector, which was then recombined with pLenti X2 Hygro DEST using Gateway LR Clonase II (Thermo Fisher Scientific, Inc.), as per the manufacturer’s protocols, resulting in the pLenti X2 Hygro pTER-shp53 lentiviral vector.
The open reading frames (ORFs) of FLAG-p53, enhanced green fluorescent protein (EGFP) and tdTomato were PCR-cloned into pENTR1A-no-ccDB and then moved to pLenti CMV/TO Puro, pLenti CMV/TO Hygro DEST and pLenti CMV Neo DEST, respectively, using Gateway cloning, as described above. Silent mutations were introduced into the ORF of p53, making it resistant to silencing by the designed shRNA. Point mutations causing resistance to shRNA directed against endogenous p53 as well as amino acid changes L22S, W23Q, R248Q and D281G were introduced by PCR amplification of the pENTR-1A-FLAG-p53 plasmid using Phusion high-fidelity DNA polymerase (Thermo Fisher Scientific, Inc.) and mutagenic oligonucleotides, followed by digestion of the non-mutated template DNA with DpnI restriction endonuclease. The sequence targeted by the shRNA within the endogenous p53 ORF was 775-GACTCCAGTGGTAATCTACT-794 (numbers denote nucleotide positions in the p53 ORF without FLAG) with the following silent mutations introduced into all p53 ORFs used for regulated expression (bold and underlined): 775-GA(CTCGAG)CGGCAATCTACT-794. The amino acid residue changes L22S-W23Q, R248Q and D281G were obtained by introducing the following point mutations: 61-GACCTATGG-69 to 61-GATCAATCG-69 (L22S-W23Q), 742-CGG-744 to 742-CAG-744 (R248Q), and 832-CCTGGGAGAGAC-843 to 832-(CCCGGG)AGAGGC-843 (D281G). Introduction of additional silent mutations in the shRNA and D281G mutageneses allowed for restriction-based identification of mutated p53 ORFs by XhoI and SmaI digestion, respectively. The location of these newly created diagnostic restriction sites is marked by the parentheses. The full sequences of the oligonucleotides used for shRNA, L22S-W23Q, R248Q and D281G mutageneses were 5′-ACAC TGGAAGACTCGA GCGGCAATCTACTGGGACG-3′, 5′-GGAAACATTTTCA GATCAATCGAAACTACTTCCT-3′, 5′-GCATGGGCGGCA TGAACCAGAGGCCCATCCTCAC-3′ and 5′-GCCTGTCCCGGGAGAGGCCGGCGCACAGAGG-3′, respectively. The reaction mix included 50 ng template (WT) plasmid, 0.2 mM dNTP mix, 0.2 µM each of the forward and reverse oligonucleotides and 2 units Phusion polymerase in 25 µl. Following an initial denaturation at 98°C for 30 sec, 20 cycles of 98°C for 10 sec, 54°C for 45 sec and 72°C for 75 sec, were completed, followed by a final extension at 72°C for 10 min. The entire reaction was digested overnight with DpnI to remove template DNA and 2 µl were transformed into competent TOP10F’ bacteria (Thermo Fisher Scientific, Inc.).
DNA minipreps of all the constructs were prepared (Plasmid Mini AX kit; A&A Biotechnology) and commercially sequenced (Genomed S.A., Warsaw, Poland) to verify the integrity of the reading frames. The lentiviral particle preparation and target cell transduction were performed according to a protocol developed by Campeau et al (44).
Western blotting
Cells were harvested by scraping or trypsinization, rinsed in PBS and lysed in radioimmunoprecipitation assay buffer (Thermo Fisher Scientific, Inc.). The protein concentration was measured by micro BCA (Thermo Fisher Scientific, Inc.) and 10-40 µg total protein per lane was separated in 8-12% SDS-PAGE. The procedures for SDS-PAGE and blotting were performed according to established protocols (45), with nitrocellulose membranes (Pall Life Sciences, Port Washington, NY, USA), and 3% skim milk (Bio-Rad Laboratories, Inc., Hercules, CA, USA) in TBS-0.1% Tween as blocking and primary antibody incubation solutions. The blots were incubated with the primary antibodies at 4°C overnight and with the secondary antibodies at room temperature for 1 h. The antibodies were used at dilutions of 1:500-1,000 unless otherwise indicated. Primary antibodies against the following proteins were used: p53 (cat. no. sc-6243; 1:200 dilution), focal adhesion kinase (FAK; cat. no. sc-558; 1:200 dilution) (both from Santa Cruz Biotechnology, Inc., Dallas, TX, USA), GAPDH (cat. no. 2118), phosphoinositide 3-kinase (PI3K; cat. no. 4292), myosin light chain 2 (MLC2; cat. no. 8505), phosphorylated (p-)MLC2 (S19; cat. no. 3671), Ras homolog family member A (RhoA; cat. no. 2117), zinc finger protein SNAI1 (cat. no. 3879), zinc finger protein SNAI2 (SLUG; cat. no. 9585), Vimentin (cat. no. 5741), p-protein kinase B (AKT; S473; cat. no. 4060), β-catenin (cat. no. 8480), E-cadherin (cat. no. 3195), N-cadherin (cat. no. 13116), Rho-associated protein kinase 1 (ROCK1; cat. no. 4035), zinc finger E-box-binding homeobox 1 (TCF8/ZEB1; cat. no. 3396), p-mitogen-activated protein kinases 3/1 (ERK1/2; T202/Y204; cat. no. 4370) (all from Cell Signaling Technology, Inc., Danvers, MA, USA), AKT (cat. no. ADI-KAP-PK004), ERK1/2 (cat. no. ADI-KAP-MA001) (both Enzo Life Sciences, Inc., Farmingdale, NY, USA) and C-X-C chemokine receptor type 4 (cat. no. SAB3500383; Sigma-Aldrich; Merck KGaA). The secondary antibodies used were IRDye 680LT donkey anti-mouse IgG, IRDye 800CW donkey anti-mouse IgG, IRDye 680LT donkey anti-rabbit IgG and IRDye 800CW donkey anti-rabbit IgG (cat. nos. 926-68072, 926-32212, 926-68023 and 926-32213, respectively; LI-COR Biosciences, Lincoln, NE, USA); antibodies were diluted at 1:10,000 in 3% skimmed milk in TBS-0.1% Tween. The blots were scanned in an Odyssey infrared scanner (LI-COR Biosciences).
Flow cytometry
The EGFP expression in cells was assayed by analyzing cell suspensions in a BD FACSCalibur flow cytometer (BD Biosciences, San Jose, CA, USA). The data analysis was performed using FCS Express 4 Flow Cytometry software version 4.07.0005 (De Novo Software, Glendale, CA, USA).
Migration and invasion assays
For the 2-dimension (2D) migration assay (wound healing) an ORIS kit (Platypus Technologies, LLC, Madison, WI, USA) was used. The cells were seeded into 96-well plates with ORIS silicone stoppers inserted. The stoppers adhere tightly to the bottom of the well, creating a 2-mm diameter circular cell-free zone. The H1299 and MDA-MB-231 cells were seeded at 1×105 and 1.5×105 cells/cm2, respectively. At the time of seeding the cells were treated with 30 ng/ml DOX or vehicle (control). At 24 h (H1299) or 48 h (MDA-MB-231) after seeding, when the cells reached confluence, the pre-migration images were captured. The stoppers were then removed, free-floating cells were washed away, and the cells were allowed to migrate into the free space in the center of the well. After 24 h the cells were fixed and the post-migration images were captured. Based on the difference in diameter between the pre- and post-migration opening, the migration speed was calculated and for each cell line the relative difference in speed attributable to DOX treatment was calculated. For the extracellular matrix (ECM)-free migration, the xCELLigence Real-Time Cell Analyzer (Acea Biosciences, Inc., San Diego, CA, USA) was used with cell invasion/migration plates (46). The device measures the number of migrated cells based on changes in electric resistance, thus obviating the need for microscopic analysis and enabling live monitoring of migration. H1299 and MDA-MB-231 cells were seeded at 60,000 and 90,000 per well, respectively. Alternatively, for the Matrigel invasion assay, the ice-cold chambers were assembled with 10% FBS in culture medium as a chemoattractant in the lower chamber and 35 µl Matrigel diluted to 0.33 mg/ml in ice-cold culture medium with 0.2% FBS was added. The chambers were incubated for ≥4 h at 37°C to allow for Matrigel solidification and then the H1299 or MDA-MB-231 cells, pretreated with 30 ng/ml DOX for 48 h, were added on top of the gel. The chambers were placed in the xCELLigence Real-Time Cell Analyzer and scanned every 15 min for ≤96 h. The invasiveness was defined as the ratio of the number of cells migrating through the Matrigel to those migrating without Matrigel.
Microsphere matrix invasion assay
For the microsphere collagen I invasion assay, U-bottom 96-well plates were coated with poly-hydroxyethyl methacrylate (HEMA; Sigma-Aldrich; Merck KGaA). Briefly, 50 µl poly-HEMA (5 mg/ml) in 96% ethanol was added to each U-bottom well and the plates were left to dry in a 37°C incubator. When dry, the plates were sterilized under ultraviolet light and the H1299 or MDA-MB-231 cells (5,000 cells per well), with or without 30 ng/ml DOX, were seeded. The plates were immediately centrifuged for 3 min at 1,000 × g at room temperature to concentrate the cells in the center of each well. After 48 h the cells formed compact spheres. The plates were then placed on ice, 50 µl culture medium was removed, and 50 µl ice-cold neutralized 1 mg/ml rat tail collagen I (Corning Inc., Corning, NY, USA) in regular culture medium was added, at a final concentration of 0.5 mg/ml. The plate was gently vortexed to mix the collagen with medium, and centrifuged for 15 min at 200 × g at room temperature in order to re-center the spheres and prevent sphere flotation. Following the centrifugation the collagen had formed a gel. The plates were placed in the 37°C incubator for 1 h to complete the gel formation and 100 µl culture medium was added on top of the gel. At this point the plates were imaged in an automated wide-field fluorescence Olympus ScanR microscope (Olympus Corporation, Tokyo, Japan) using 5× objective and intrinsic cell tdTomato fluorescence. Subsequently, the spheres were imaged every 24 h for the following 3 days. The image set was manually curated to remove out-of-focus and otherwise unsuitable images and then processed using ImageJ software version 1.51U (National Institutes of Health, Bethesda, MD, USA). All the images were auto-thresholded and the objects were counted and measured. For the 1 day post-embedding (dpe) circularity assay the circularity parameter was calculated for the largest object. The relative circularity was obtained by dividing the circularity of the DOX-treated cells by that of the non-treated cells and normalizing to the reference cell line level (H1299-TR or MDA-MB-231-SH). Therefore, a relative circularity <1 indicates less round microspheres (and a more invasive phenotype) than in the reference cell line. Similarly, the relative number of cells (NOC) separated from the microsphere was obtaining from dividing the number of the small objects (cells) that separated from the sphere at 2 dpe in the DOX-treated group by the same parameter in non-treated cells and relating all the ratios to the reference cell line ratio. Therefore, a relative NOC >1 indicates a more invasive phenotype than in the reference cell line.
Adhesion assay
A culture plate-based assay was performed by aliquoting 50 µl culture medium containing 20,000 H1299 or 30,000 MDA-MB-231 cells with variants of p53, with or without pretreatment with 30 ng/ml DOX, into the wells of a 96-well plate. All the media and plastics were pre-warmed to 37°C. Following incubation in the cell culture incubator for 15, 30, 60 min or 20 h, the wells were gently rinsed 3 times with pre-warmed PBS, and the remaining adhered cells were fixed with ice-cold 3.7% formaldehyde in PBS. After 15 min the formaldehyde was replaced by PBS and the plates were stored in the fridge. When all time-points had been completed, the cells were stained with 1:10,000 GelGreen (Biotium, Inc., Freemont, CA, USA) in PBS with 0.05% Triton X-100 for 1 h at room temperature (Sigma-Aldrich; Merck KGaA), and imaged using an automated Olympus ScanR microscope with a 5× objective. The nuclei were counted and the number of nuclei for each cell line/DOX treatment combination for each time-point was normalized to the equivalent 20 h time-point that reflected 100% adhesion. This normalization accounted for the variability of input cell suspensions. For each cell line and time-point, the fraction of adhered cells in the DOX-treated population was then divided by that in the non-treated population and the fold-change resulting from DOX treatment was calculated.
Zebrafish (Danio rerio) xenotransplantation
MDA-MB-231-SH, -R248Q and -R248Q-TA cells were treated with 30 ng/ml DOX for 48 h, harvested by trypsinization, washed twice with PBS and resuspended in PBS with 2% polyvinylpyrrolidone 40 (Sigma-Aldrich; Merck KGaA) to prevent clumping. The cell suspension was then loaded into a home-pulled glass needle and microinjected into the common cardinal vein of a 2-days post fertilization (dpf) dechorionated Casper Tg(fli1:EGFP) embryo according to a published protocol (47), using a PicoPump PV820 microinjector (World Precision Instruments, Sarasota, FL, USA) at ca. 20 PSI and a 100-200 msec injection time. Following the injection, the fish larvae were kept at 32°C for the remainder of the experiment. Since the fish have green fluorescent vasculature and the cells express the red fluorescent tdTomato protein, they could be imaged directly under a fluorescence stereoscope for ≤3 days post-injection, following which the embryos were euthanized.
Statistical analysis
The data regarding the age of tumor onset in relation to p53 mutations were analyzed with a Mann-Whitney test due to non-normal distribution. The data regarding metastasis and primary tumor site characteristics were analyzed by pairwise χ2 analysis. The P-values obtained from these analyses were corrected for a false discovery ratio of 0.1 with the Benjamini-Hochberg method. In the comparison of patient groups with statistically significant differences in metastasis frequency with the NULL group (MIS and R248Q), the odds ratios and 95% confidence intervals were calculated using pairwise χ2 analysis. The tumor-free survival rate analysis was performed with a log-rank test. The cell migration, invasion and circularity results were analyzed with one-way analysis of variance (ANOVA) followed by a Sidak post-hoc test. The results of the adhesion assay were analyzed by two-way ANOVA with Dunnett’s post-hoc test for comparisons against the reference cell line and with a Sidak post-hoc test for the remaining comparisons. The xenotransplantation data were analyzed by pairwise χ2 analysis. The P-values obtained from this analysis were corrected for a false discovery ratio of 0.1 with the Benjamini-Hochberg method. The Benjamini-Hochberg corrections were performed in R statistical software (version 3.5.0; www.R-project.org) with the use of the p.adjust function. All other analyses were performed using GraphPad Prism (version 6.07; GraphPad Software, Inc., La Jolla, CA, USA). P<0.05 and Q<0.1 (Benjamini-Hochberg method) were considered to indicate statistical significance.
Results
Missense mutations in TP53 change tumor onset, spectrum and metastatic behavior
An analysis of clinical phenotypes concerning tumor onset, primary location and metastasis frequency and tropism, based on the latest release of the International Agency for Research on Cancer (IARC) p53 database (Release 18; http://p53.iarc.fr/), containing data on >30,000 patients with tumors with somatic or germline mutations in their TP53 genes (48), revealed that patients bearing the R248Q, but no other ‘hot-spot’ mutation, experienced significantly earlier onset of their first tumors than those bearing a NULL allele (Fig. 1A-C; Table I). The distribution of the primary tumor sites between patients with various somatic mutations and certain germline mutations also differed significantly from that in patients with the NULL allele (Tables II-IV). Notably, in patients with somatic missense TP53 mutations, metastasis was observed less frequently than in patients with NULL alleles (Table V), with an even more pronounced decrease in patients with R248Q. Amongst the patients with metastases from two large datasets, IARC and the Memorial Sloan Kettering Cancer Centre (MSKCC) (49), differences were observed in the distribution of metastatic sites in patients with certain GOF mutations (Tables VI and VII). When patients bearing the mutations R175H, R248Q, R248W and R273H were pooled, the difference between them and the NULL group were marked (IARC dataset, P=0.0016; MSKCC dataset, P=0.0071).
Figure 1.

GOF mutations in p53 accelerate tumor onset. (A and B) Tumor-free survival rates compared by log-rank analysis, and (C) the median age of first tumor onset in patients bearing germline NULL and MIS GOF mutations in TP53 gene. *P=0.0171 and Q=0.0855 vs. NULL group. GOF, gain-of-function; p53, tumor suppressor p53; MIS, any missense mutation.
Table I.
Median patient age at first tumor onset.
| p53 mutation | No. patients | Median age, years | P-value | Q |
|---|---|---|---|---|
| NULL | 519 | 30.0 | - | - |
| MIS | 1276 | 30.0 | 0.3801 | 0.3801 |
| R248Q | 45 | 23.0a | 0.0171 | 0.0855 |
| R248W | 69 | 24.0 | 0.0915 | 0.1525 |
| R175H | 73 | 26.0 | 0.2468 | 0.3085 |
| R273H | 54 | 27.5 | 0.0717 | 0.1525 |
P<0.05 and Q<0.1 vs. NULL group. p53, tumor suppressor p53; MIS, any missense mutation.
Table II.
Distribution of primary tumor sites in patients with somatic TP53 mutations.
| p53 mutation | Colon, n | Lung, n | Breast, n | Ovary, n | Brain, n | Esophagus, n | Bladder, n | Other, n | P-valuea | Q |
|---|---|---|---|---|---|---|---|---|---|---|
| NULL | 548 | 686 | 749 | 596 | 221 | 464 | 242 | 2333 | - | - |
| MIS | 2326 | 2307 | 2304 | 1632 | 1542 | 1423 | 1159 | 9114 | < 0.0001 | 0.0001 |
| R248Q | 159 | 37 | 113 | 54 | 75 | 49 | 53 | 415 | < 0.0001 | 0.0001 |
| R248W | 147 | 47 | 84 | 58 | 57 | 60 | 25 | 292 | < 0.0001 | 0.0001 |
| R175H | 290 | 37 | 131 | 107 | 107 | 99 | 38 | 412 | < 0.0001 | 0.0001 |
| R273H | 165 | 54 | 98 | 94 | 80 | 47 | 21 | 304 | < 0.0001 | 0.0001 |
Versus NULL group. p53, tumor suppressor p53; MIS, any missense mutation.
Table III.
Distribution of primary tumor sites in patients with germline TP53 mutations.
| p53 mutation | Breast, n | Brain, n | Soft tissues, n | Adrenal gland, n | Bone, n | Other, n | P-value | Q |
|---|---|---|---|---|---|---|---|---|
| NULL | 225 | 70 | 115 | 31 | 70 | 255 | - | - |
| MISa | 492 | 290 | 187 | 185 | 137 | 719 | <0.0001 | 0.0003 |
| R248Qa | 18 | 18 | 10 | 2 | 8 | 27 | 0.0161 | 0.0268 |
| R248Wa | 33 | 29 | 7 | 7 | 6 | 30 | <0.0001 | 0.0003 |
| R175H | 29 | 22 | 15 | 5 | 9 | 38 | 0.0654 | 0.0818 |
| R273H | 22 | 10 | 17 | 5 | 9 | 24 | 0.6412 | 0.6412 |
P<0.05 and Q<0.1 vs. NULL group for primary tumor site. p53, tumor suppressor p53; MIS, any missense mutation.
Table IV.
Distribution of primary tumor types in patients with germline TP53 mutations.
| p53 mutation | Sarcoma, n | Carcinoma, n | Other, n | P-value | Q |
|---|---|---|---|---|---|
| NULL | 190 | 97 | 479 | - | - |
| MISa | 364 | 380 | 1266 | <0.0001 | 0.0005 |
| R248Q | 21 | 7 | 55 | 0.5308 | 0.6635 |
| R248Wa | 16 | 19 | 77 | 0.0374 | 0.0623 |
| R175H | 27 | 14 | 77 | 0.8494 | 0.8494 |
| R273Ha | 29 | 17 | 41 | 0.0181 | 0.0453 |
P<0.05 and Q<0.1 vs. NULL group for primary tumor type. p53, tumor suppressor p53; MIS, any missense mutation.
Table V.
Frequency of metastasis in patients with somatic TP53 mutations.
| p53 mutation | M1 | M0 | P-value | Q | OR (95% CI) | % M1 |
|---|---|---|---|---|---|---|
| NULL | 200 | 3649 | - | - | - | 5.2 |
| MISa | 499 | 13242 | <0.0001 | 0.0005 | 0.6875 (0.5811-0.8134) | 3.6 |
| R248Qa | 18 | 561 | 0.0393 | 0.0983 | 0.5854 (0.3584-0.9561) | 3.1 |
| R248W | 23 | 484 | 0.5987 | 0.9267 | - | 4.5 |
| R175H | 41 | 772 | 0.9267 | 0.9267 | - | 5.0 |
| R273H | 29 | 487 | 0.7638 | 0.9267 | - | 5.6 |
P<0.05 and Q<0.1 vs. NULL group. M1/M0, number of patients with/without metastasis; % M1, percentage of patients with metastasis; MIS, any missense mutation; OR, odds ratio; CI, confidence interval.
Table VI.
Distribution of metastasis location sites in patients with somatic TP53 mutations from the International Agency for Research on Cancer database.
| p53 mutation | Liver, n | Lymph node, n | Other, n | P-value | Q |
|---|---|---|---|---|---|
| NULL | 82 | 36 | 82 | - | - |
| MISa | 226 | 114 | 159 | 0.0603 | 0.0905 |
| R248Q | 9 | 3 | 6 | 0.7493 | 0.7493 |
| R248W | 11 | 6 | 6 | 0.3483 | 0.4180 |
| R175Ha | 27 | 3 | 11 | 0.0122 | 0.0244 |
| R273Ha | 22 | 2 | 5 | 0.002 | 0.0060 |
| 3-GENEa | 69 | 14 | 28 | 0.0016 | 0.0060 |
P<0.05 and Q<0.1 vs. NULL group. p53, tumor suppressor p53; MIS, any missense mutation; 3-GENE, any one of R175H, R248Q, R248W or R273H mutations.
Table VII.
Distribution of metastasis location sites in patients with somatic TP53 mutations from the Memorial Sloan Kettering Cancer Center database.
| p53 mutation | Liver, n | Lymph node, n | Lung, n | Bone, n | Brain, n | Other, n | Versus NULL group
|
Versus WT group
|
||
|---|---|---|---|---|---|---|---|---|---|---|
| P-value | Q | P-value | Q | |||||||
| WTa | 482 | 506 | 350 | 213 | 76 | 975 | <0.0001 | 0.0007 | - | - |
| MISSb | 389 | 264 | 141 | 75 | 57 | 465 | 0.7006 | 0.7006 | <0.0001 | 0.0001 |
| NULLb | 231 | 158 | 97 | 41 | 43 | 266 | - | - | <0.0001 | 0.0001 |
| R248Qb | 18 | 11 | 2 | 4 | 3 | 31 | 0.1411 | 0.1768 | 0.0728 | 0.0849 |
| R248Wb | 22 | 5 | 3 | 1 | 3 | 13 | 0.0899 | 0.1573 | <0.0001 | 0.0001 |
| R175Hb | 36 | 10 | 5 | 3 | 4 | 28 | 0.0633 | 0.1477 | <0.0001 | 0.0001 |
| R273H | 13 | 11 | 5 | 0 | 1 | 26 | 0.1515 | 0.1768 | 0.1871 | 0.1871 |
| 3-GENEa,b | 89 | 37 | 15 | 8 | 11 | 98 | 0.0071 | 0.0249 | <0.0001 | 0.0001 |
P<0.05 and Q<0.1 vs. NULL group;
P<0.05 and Q<0.1 vs. WT group. p53, tumor suppressor p53; WT, wild-type; MIS, any missense mutation; 3-GENE, R175H, R248Q, R248W and R273H mutations.
Breast cancer is one of the most prevalent types of cancer, and the presence of metastasis either at the initial presentation of the patient or following the initial round of therapy is a poor prognostic marker. The present findings suggest that certain classes of mutations in the TP53 gene are associated with lower metastasis rates in patients. Therefore, the influence of the expression of certain mutated p53 variants was investigated in MDA-MB-231, a cell line commonly used as a model in breast cancer metastasis studies, and in lung adenocarcinoma H1299 cells, which do not express p53 and are an established model in studies on p53.
Construction and validation of cell lines with inducible p53 expression
In order to test the effects of short-term expression of mutant p53 on cellular migration and invasion, cell lines with inducible expression of the transgenes were constructed. H1299 and MDA-MB-231 cells were transduced with lentivirus encoding the tetracycline repressor (TR) and selected with blasticidin. To facilitate observation in downstream applications, the cells were also transduced with lentivirus encoding the tdTomato protein and selected by G418 until the population exhibited uniform red fluorescence. These cell lines were called TR and used as the common parent for all subsequently created cell lines. The TR cell lines were subsequently transduced with lentivirus encoding tetracycline-inducible GFP and selected with hygromycin. At this stage only single GFP-expressing cells could be observed in the absence of DOX. In order to test the inducibility and dynamic range of the Tet-ON system, the cells were seeded into a 96-well plate and, after 24 h of culture, induced with a range of DOX concentrations. After 24 h, the total cellular GFP fluorescence was measured using a multiwell spectrofluorimeter (Fig. 2A). The half maximal effective concentration (EC50) for 24-h DOX induction of GFP expression was estimated at 11.4 and 17.8 ng/ml in H1299 and MDA-MB-231 cells, respectively, with negligible p53 expression in the absence of DOX. In order to characterize the populations in more detail, the cells were seeded into a 12-well plate for 24 h and induced with a range of DOX concentrations. After 24 and 48 h, the green fluorescence was measured in arbitrary fluorescence units (AFU) using a flow cytometer. The parameters measured were the percentage of cell population that was GFP-positive and the median fluorescence intensity of the GFP-positive cells (Fig. 2B and C). At a moderately low DOX concentration of 25-35 ng/ml nearly 100% of cells were GFP-positive after 24 h with almost no further increase after 48 h (Fig. 2B). Meanwhile, the median intensity of fluorescence of GFP-positive cells continued to grow and during the following 24 h increased almost 2-fold in the MDA-MB-231 cells (348-633 AFU), and >2-fold in the H1299 cells (492-1,219 AFU) (Fig. 2C). The DOX EC50 for all the FACS-measured parameters was in the range of 5.5-9.5 ng/ml.
Figure 2.

DOX-induced EGFP and p53 expression in MDA-MB-231 and H1299 cells. (A) The EGFP expression in MDA-TR and H-TR cells induced with 30 ng/ml DOX for 24 h was measured by multiwell spectrofluorimetry. Total fluorescence from 96-well plate wells was measured, obtained data were fitted to a sigmoidal four-parameter curve and the calculated half maximal effective concentrations for DOX were 17.8 ng/ml for MDA and 11.4 ng/ml for H cells. (B and C) The EGFP expression in MDA-TR and H-TR cells induced with 30 ng/ml DOX for 24 and 48 h was also measured by flow cytometry and (B) the percentage of cells that were EGFP-positive and (C) the median EGFP intensity were plotted. The vertical dotted line indicates the 30 ng/ml DOX concentration used in subsequent experiments. The dashed lines indicate 50% of the normalized phenotype intensity. (D) Western blot analysis of the p53 expression in indicated MDA and H cells treated with 30 ng/ml DOX for 48 and 24 h, respectively. The protein expression visualized using anti-p53 and anti-FLAG antibodies in parallel, which were detected in separate fluorescence channels. Faint bands in the MDA-TR lanes and other non-DOX-induced lanes represent endogenous p53 R280K variants. DOX, doxycycline; EGFP, enhanced green fluorescent protein; p53, tumor suppressor p53; MDA, MDA-MB-231; H, H1299; TR, cells with Tet repressor only; SH, cells with silenced endogenous p53; TA, transactivatory domain disruption (L22S/W23Q) in addition to indicated mutation (R248Q or D281G).
As the tightness and inducibility of the TR system were evaluated, further genetic modifications were performed on the cells. The H1299-TR cells were transduced with a range of lentiviral vectors encoding for the following variants of N-terminally FLAG-tagged p53: WT, R248Q, L22S-W23Q-R248Q, D281G and L22S-W23Q-D281G. The cells were selected with puromycin. The MDA-MB-231 cells that endogenously expressed R280K were first transduced with a lentivirus that expressed p53-directed shRNA under a Tet-inducible promoter. Following selection with hygromycin, this cell line was called SH and was used as a parent cell line for the construction of all subsequent p53-expressing MDA-MB-231-based cell lines. The MDA-MB-231-SH cells were transduced with a range of lentiviral vectors encoding the following variants of N-terminally FLAG-tagged p53: WT, R248Q, L22S-W23Q-R248Q, D281G and L22S-W23Q-D281G. The mutations L22S and W23Q are located within the transactivation domain of p53 and are known to abolish its function as a TA (50,51). Therefore, cell lines expressing p53 with disruption in this domain are referenced to as TA (i.e. L22S-W23Q-R248Q as R248Q-TA, and L22S-W23Q-D281G as D281G-TA). In addition, a number of silent mutations were introduced into the p53 ORF in order to make the transgene resistant to the shRNA silencing of the endogenous TP53 gene in the MDA-MB-231 cells. The cells were selected with puromycin. The tightness and inducibility of the system with regard to exogenous p53 were tested by western blotting following treatment with 30 ng/ml DOX. The results confirmed that there was no p53 expression in the absence of induction, and that the endogenous p53 in the MDA-MB-231 cells was effectively silenced (Fig. 2D).
Cell lines expressing WT p53 were constructed in order to test the functionality of downstream response pathways, but were not intended for use in the migration studies. Indeed, the induction of WT p53 expression led to significant toxicity in the cells, exerting cytostatic and cytotoxic effects after 24 and 48 h of expression, respectively (data not shown). Based on the p53 mutation prevalence in breast and lung cancer, as well as on the phenotypes reported in the literature (33,44), a set of cell lines was selected for use in further migration studies. These included cells with R248Q, R248Q-TA, D281G and D281G-TA. In the MDA-MB-231 cells, the SH-only cell line was used as a NULL variant, enabling the identification of GOF p53 phenotypes.
Expression of mutated p53 influences the migration, invasion and adhesion properties of cancer cells
In order to test whether the selected p53 mutations exhibit a GOF phenotype with regard to cellular migration in H1299 and MDA-MB-231 cells, the migration speed was calculated and the data are presented in Fig. 3A. In the H1299 cells, the expression of the R248Q mutant significantly decreased the migration speed, in a transactivation-dependent manner (R248Q-TA). The expression of the p53 D281G variant significantly increased the migration speed. This increase was significantly, but not completely, countered by a mutation in the transactivation domain (D281G-TA). Since GOF p53 phenotypes were investigated, the MDA-MB-231-SH cell line (with silenced endogenous p53 R280K) was used as a reference (Fig. 2D). The expression of the R248Q variant significantly decreased the migration speed, in a transactivation-dependent manner. The expression of the D281G or D281G-TA variants resulted in no significant changes in the migration speed compared with SH cells. These results demonstrated that in the tested cell lines the expression of the p53 R248Q mutant led to decreased migration speed, and that this decrease depended on the functionality of the transactivation domain. Furthermore, the expression of the D281G variant increased the migration speed, but only in the H1299 cells, and this increase was partly dependent on the functionality of the p53 transactivation domain.
Figure 3.

Expression of mutated p53 influences the migration and invasiveness of cancer cells. (A) MDA-MB-231 and H1299 cells with indicated p53 variants were seeded into wells with ORIS stoppers and, where applicable, induced with 30 ng/ml DOX. After 48 h (MDA-MB-231) or 24 h (H1299) the stoppers were removed (t=0). The relative migration speed for each cell type was calculated by dividing the speed of the DOX-induced cells by the speed of non-induced cells and normalizing to the value obtained for the SH or TR cells, respectively. The migration speed with and without Matrigel was measured by xCELLigence for (B) MDA-MB-231 and (C) H1299 cells expressing the indicated p53 mutants. The migration speed values are normalized to the speed of MDA-MB-231-SH or H1299-TR cells without Matrigel. (D) The invasiveness (relative migration with vs. without Matrigel) was measured for MDA-MB-231 or H1299 cells expressing various p53 mutants at 16 or 48 h, respectively. The data are presented as the mean ± standard error of the mean. *P<0.05, **P<0.01, ***P<0.001 and ****P<0.0001. p53, tumor suppressor p53; DOX, doxycycline; ns, not significant; TR, cells with Tet repressor only; SH, cells with silenced endogenous p53; TA, transactivatory domain disruption (L22S/W23Q) in addition to indicated mutation (R248Q or D281G).
Since the migration speed of the MDA-MB-231 and H1299 cells was only significantly affected by the expression of p53-R248Q and p53-R248Q-TA, these cell lines were selected for further analysis concerning ECM invasiveness. Based on the migration curves, migration half-times were calculated and their reciprocals are presented in Fig. 3B and C. Regardless of the presence of Matrigel, the expression of p53-R248Q in MDA-MB-231 cells significantly decreased the migration speed. In the p53-R248Q-TA-expressing MDA-MB-231 cells, the migration speed in the absence of Matrigel did not significantly differ from either SH cells or cells with R248Q. When the R248Q-TA cells were migrating through Matrigel their ability to do so was significantly higher than that of cells with R248Q, and no significant difference was observed compared with that of the SH cells (Fig. 3B). For the H1299 cells (Fig. 3C) in the absence of Matrigel, the expression of R248Q decreased the migration speed significantly in a transactivation-dependent manner. When the cells were migrating through the Matrigel no significant difference was observed between the H1299-TR and H1299-R248Q cells. In order to present the data in a manner compatible with the traditional Transwell assay, the‘invasiveness’ parameter was calculated; it was defined as the ratio of the NOC that migrated through the Matrigel to that migrated without Matrigel. The read-out time-points (48 h for H1299 and 16 h for MDA-MB-231 cells) were selected at the time when the cells without Matrigel were completing their migration. The data are presented in Fig. 3D. The MDA-MB-231 and H1299 cells were less invasive upon expression of the R248Q mutant and this decrease was transactivation-dependent.
In order to analyze the ECM invasion ability in a qualitative and quantitative manner, a model was used whereby cellular microspheres were formed and embedded in collagen I. The microsphere formation was conducted concurrently with the induction of p53 for 48 h. Subsequently, the cells were embedded in 0.5 mg/ml collagen I, imaged immediately and every 24 h. The parameters measured include the microsphere circularity and NOC that separated from the main microsphere. The circularity decreases when cells make local infiltrations of the matrix without separating from the microsphere; the NOC is a reflection of a metastatic phenotype. For each cell line, any change in these parameters resulting from the addition of DOX was calculated and normalized to the change in the reference line: SH and TR for MDA-MB-231 and H1299, respectively (Fig. 4A). Notably, the expression of the R248Q variant led to a significant decrease in NOC, which was dependent on p53 transactivation activity. Even though no difference in microsphere circularity was observed between the reference and R248Q cells, a visible, yet difficult to quantify, change in cellular phenotype was observed in MDA-MB-231-R248Q (but not in MDA-MB-231-R248Q-TA) cells following treatment with DOX (Fig. 4B and C; SH vs. 248 at 2 dpe). These cells acquired an elongated, spindle-like phenotype, and infiltrated the matrix locally, but did not separate from the microsphere as readily as the MDA-MB-231-SH cells. In summary, following the expression of p53-R248Q in H1299 and MDA-MB-231 cells, a recurring phenotype of decreased transactivation-dependent motility and invasiveness was observed. In addition, a visible morphological difference was observed in MDA-MB-231 cells expressing p53-R248Q.
Figure 4.

Collagen I local infiltration and invasiveness are altered by the expression of mutated p53. (A) Microspheres were prepared from MDA or H cells conditionally expressing the indicated p53 variants and induced or not induced with 30 ng/ml DOX. The spheres were embedded in 0.5 mg/ml collagen I matrix and imaged at 0, 1 and 2 dpe. Changes in the main micro-sphere circularity and the NOC separated from the main microsphere were calculated in relation to the microspheres composed of cells not induced with DOX. A decrease in circularity reflects an irregular microsphere shape resulting from increased local invasiveness. (B) A representative set of microsphere images. Depicted are microspheres composed of MDA-MB-231 cells expressing the indicated p53 variants. The cells were DOX-induced 48 h prior to embedding. The images were captured directly post-embedding and at 48 h. Scale bar, 1 mm. (C) Magnification of the 2 dpe images. Scale bar, 0.5 mm. The data are presented as the mean ± standard error of the mean. *P<0.05 and **P<0.01, respectively. p53, tumor suppressor p53; MDA, MDA-MB-231; H, H1299; DOX, doxycycline; dpe, days post-embedding; NOC, number of cells; ns, not significant; TR, cells with Tet repressor only; SH, cells with silenced endogenous p53; TA, transactivatory domain disruption (L22S/W23Q) in addition to R248Q mutation.
Cellular migration in 2D or 3D is influenced by numerous factors, including microenvironment sensing, cytoskeletal remodeling and matrix degradation. One such factor is the ability to efficiently assemble and disassemble the matrix adhesion points. Therefore, the adhesion capability of MDA-MB-231 and H1299 cells expressing mutated p53 variants was assayed. The results of these analyses are presented in Fig. 5. The findings revealed that MDA-MB-231 cells expressing p53-R248Q, but not p53-R248Q-TA, adhered to the culture vessel faster than the MDA-MB-231-SH cells (Fig. 5A). In contrast, the adhesive properties of the H1299 cells did not change following the expression of either p53-R248Q or p53-R248Q-TA (Fig. 5B).
Figure 5.
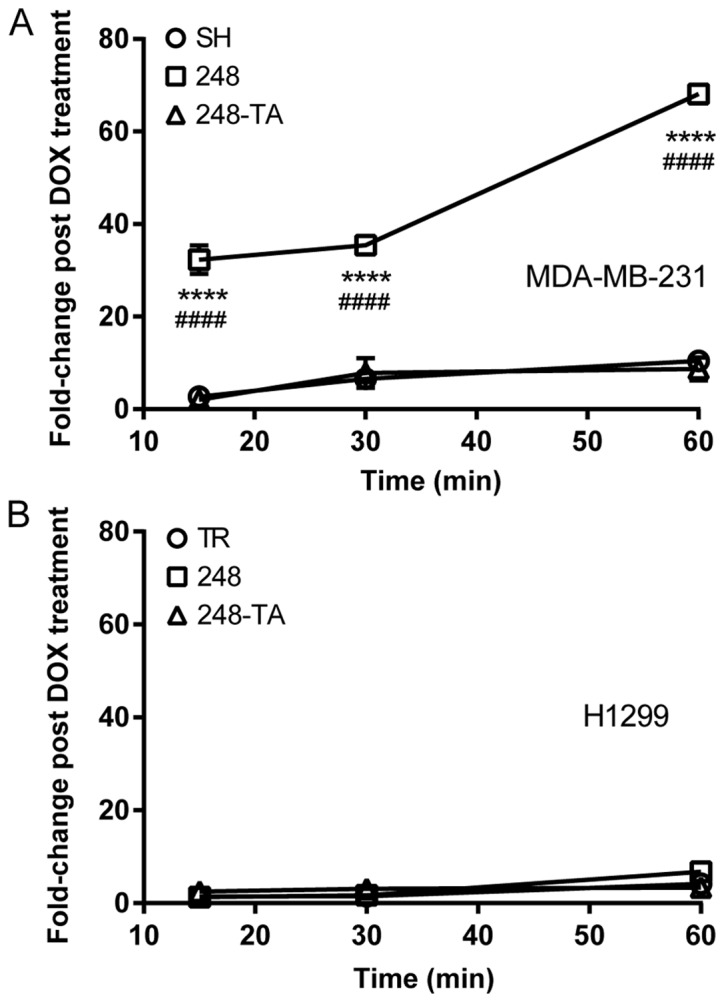
Mutant p53-R248Q results in pro-adhesive properties in a cell type- and transactivation-dependent manner. The adhesion of (A) MDA-MB-231 and (B) H1299 cells conditionally expressing indicated p53 variants to a tissue culture plate was measured. The cells were seeded and then rinsed off the plate at the indicated time-points. The remaining cells were counted and the number of adhered DOX-treated cells relative to the number of adhered non-treated cells (fold-change post DOX treatment) was plotted. The data are presented as the mean ± standard error of the mean. ****P<0.0001 vs. control group, two-way ANOVA with Dunnett’s post-hoc test; ####P<0.0001 R248Q vs. R248Q-TA group, two-way ANOVA with Sidak post-hoc test. p53, tumor suppressor p53; DOX, doxycycline; ANOVA, analysis of variance; TR, cells with Tet repressor only; SH, cells with silenced endogenous p53; TA, transactivatory domain disruption (L22S/W23Q) in addition to R248Q mutation.
In summary, following the expression of the p53 R248Q variant in MDA-MB-231 cells, they displayed a phenotype of decreasing invasiveness and increased adhesiveness, which was transactivation-dependent. In addition, a visible morphological change was observed in the MDA-MB-231 cells expressing p53-R248Q. In the H1299 cells, the motility and invasiveness, but not the adhesive properties, changed following the expression of p53-R248Q.
Expression of the p53 R248Q mutant alters the distribution of xenotransplanted cancer cells in zebrafish
In order to verify whether the differences observed in the in vitro assays in cancer cells expressing various p53 mutants would result in a changed phenotype in vivo, the MDA-MB-231-SH, -R248Q and -R248Q-TA cells were microinjected into the common cardinal vein of zebrafish embryos. The approximate injection site is indicated in Fig. 6A (red arrow). At 5 dpf images of the zebrafish were captured under a fluorescent stereoscope (Fig. 6A-C; the cell types used for injection are indicated). The area corresponding to the posterior 2 mm of the embryo was analyzed. The intensity profiles were analyzed to determine the locations of the transplanted cells along the fish trunk and tail. For analysis, this 2 mm section of the fish was divided into 10 sections of 200 µm and each cell was ascribed to its corresponding subsection. The relative cancer cell frequencies in the most posterior 2- and 0.5-mm sections of the embryo are plotted in Fig. 6D and E, respectively. Subsequently, pairwise contingency analysis was performed, and the difference between the distributions of the SH and R248Q cells was revealed to be significant (P<0.0001), whereas no significant difference was observed between the SH and R248Q-TA cells.
Figure 6.

Expression of the p53 R248Q mutant alters the distribution of breast cancer cells xenotransplanted into zebrafish. (A) MDA-MB-231-SH, (B) -R248Q and (C) -R248Q-TA cells were pretreated with 30 ng/ml doxycycline for 48 h and injected into the common cardinal vein of transgenic Tg(fli1:EGFP) zebrafish embryos with green fluorescent vasculature. Representative images of the embryos 3 days post injection are displayed. The distribution of the cells was analyzed in the posterior 2-mm section of ≥50 embryos for each cell type (box with white outline), based on the fluorescence intensity profiles (white profiles below the boxes). Based on fluorescence intensity profiles, the position of each cell within the measurement box was calculated and the distribution of cells in subsections covering the posterior (D) 2 or (E) 0.5 mm of the embryo was analyzed. The plots present the mean relative cancer cell frequencies in these sections for the analyzed embryo populations. ****P<0.0001 and Q<0.0003. p53, tumor suppressor p53; EGFP, enhanced green fluorescent protein; SH, cells with silenced endogenous p53; TA, transactivatory domain disruption (L22S/W23Q) in addition to R248Q mutation.
Expression of mutated p53 alters the expression of proteins involved in the EMT as well as the expression and phosphorylation of MLC2
In order to investigate the molecular mechanism responsible for the phenotypic changes observed in H1299 and MDA-MB-231 cells expressing mutated p53, lysates from cells treated with 30 ng/ml DOX for 2 or 3 days, respectively, were analyzed by western blotting (Fig. 7). The lysate from epithelial lung cancer H358 cells, which are p53-NULL, exhibit compact, islet-like growth and are essentially non-migratory, was used for comparison. As observed in Figs. 2D and 7A, the H1299 and MDA-MB-231 cells expressed p53 protein variants upon stimulation with 30 ng/ml DOX. In addition, efficient silencing of endogenous p53 was achieved in the MDA-MB-231-SH cells. In order to establish whether any major mitogen-response pathways are affected by the expression of the p53 variants, the expression and phosphorylation levels of PI3K, AKT and ERK1/2 were investigated (Fig. 7B); however, no apparent differences were observed. Since the p53 mutations are linked to the EMT (35,48,49) and this process is often accompanied by changes in migratory behavior, the levels of several EMT-associated proteins were analyzed (Fig. 7C). The cell lines used in the present study are of mesenchymal characteristic, therefore a partial shift of the molecular phenotype from mesenchymal towards epithelial was expected in the case of cells with decreased migration and invasiveness (i.e. cells with p53-R248Q). In the H1299-R248Q cells, a decrease was observed in TCF8/ZEB1 and N-cadherin, resulting in expression levels similar to those of the H358 cells (Fig. 7C). The other EMT markers, including Vimentin, E-cadherin and β-catenin, however, remained unchanged, suggesting only a partial phenotypic change. This is in agreement with the observation that, whereas H1299-R248Q cells migrate slower than H1299-TR cells, they are more mobile than the H358 cells (data not shown). Whereas no increase in the epithelial marker E-cadherin expression was observed (reference band clearly visible in the H358 lane), the expression of mesenchymal markers N-cadherin and β-catenin appeared lower in MDA-MB-231-R248Q cells than in MDA-MB-SH cells, suggesting a partial loss of mesenchymal characteristic following p53-R248Q expression. The multiple bands detected by the N-cadherin antibody represent the maturation stages of the protein. A small decrease in the expression of SLUG, an EMT regulator, was observed in the MDA-MB-231 cells expressing p53-R248Q, but not p53-R248Q-TA. Given that the difference in migration speed was the most consistently observed phenotype, and that p53 serves an important role in the functional regulation of cytoskeletal proteins (52), the expression and phosphorylation level of several proteins involved in the cytoskeletal regulation of migration were analyzed (Fig. 7D). No differences were observed in the expression levels of FAK and ROCK-1 following the induction of the mutated p53. However, in the MDA-MB-231-R248Q cells, but not the H1299-R248Q cells, a decrease in the expression of RhoA, a myosin regulator, was noted. Additionally, in the cells expressing p53-R248Q (but not p53-R248Q-TA) there was a decrease in total and S19-phosphorylated MLC2, which is involved in cellular migration. Notably, in the non-mobile H358 cells MLC2 was detected, but was phosphorylated at a relatively low level.
Figure 7.

Western blot analysis of protein expression in MDA-MB-231 cells expressing mutant p53. Cells were induced with 30 ng/ml doxycycline for 48 h, lysed and analyzed by western blotting with 20-40 µg total protein per lane, using fluorescent secondary antibodies and an Odyssey scanner for the detection of bands. (A) Protein expression of induced transgenes in H1299 and MDA-MB-231 cells, and the silencing of endogenous p53 in MDA-MB-231-SH cells. (B) Expression and phosphorylation levels of selected proteins associated with mitogenic signaling. (C) Expression levels of proteins associated with the epithelial-mesenchymal status. (D) Expression levels of proteins associated with the regulation of adhesion and actin-based migration. Wherever the identity of the band representing the marked protein was in doubt, it is marked with an asterisk (*). Apparent molecular weights, as assessed from the western blot markers, are indicated in the parentheses next to the protein identifiers. p53, tumor suppressor p53; p-, phosphorylated; ERK, mitogen-activated protein kinase; AKT, protein kinase B; PI3K, phosphoinositide 3-kinase; SLUG, zinc finger protein SNAI2; Snail, zinc finger protein SNAI1; TCF8/ZEB, zinc finger E-box-binding homeobox 1; Vim, Vimentin; β-CAT, β-catenin; CDH, cadherin; FAK, focal adhesion kinase 1; ROCK1, Rho-associated protein kinase 1; RhoA, Ras homolog family member A; MLC2, myosin light chain 2; TR, cells with Tet repressor only; SH, cells with silenced endogenous p53; TA, transactivatory domain disruption (L22S/W23Q) in addition to indicated mutation (R248Q or D281G).
Discussion
The key role of the p53 protein in tumor suppression has been recognized for many years. A vast body of evidence exists on its physiological role as tumor suppressor; for a number of years it has also been known that mutated p53 may serve an oncogenic role. Ever since the creation of the first transgenic mice bearing the ‘hot spot’ mutations in TP53 gene (15,16), it has been accepted that these mutations lead to an increase in the prevalence of tumors, change the tumor spectrum, and increase their invasiveness and metastatic potential.
The results of the analyses in the present study, performed using the largest publicly available database of p53 mutations in human patients (IARC, Release 18) (48), confirmed the changes in primary tumor onset and spectrum in patients bearing the ‘hot spot’ p53 mutations, for germline and somatic mutations. In addition, the locations of metastases were different in patients bearing the ‘hot spot’ p53 mutations within the IARC and MSKCC datasets. However, the analysis of the metastasis frequency revealed that, in patients with missense mutations in their TP53 gene, metastasis was less frequent than in patients with the NULL genotype; this was particularly accentuated in patients bearing R248Q mutation. These results seem contradictory to the reported phenotypes observed in mice (14-16), but one has to keep in mind that the variance in human phenotypes may result from the versatility of the genetic background in human cohorts, as opposed to isogenic murine strains.
Therefore, the present study reports on data describing changes in the migratory behavior and invasiveness of breast and lung cancer-derived cell lines upon the short-term expression of mutated forms of p53. The selected mutations are the one that, according to a previous study, leads to the largest increase in the migration speed of cancer H1299 cells (D281G) (38), and the one that, according to the present analyses, decreases the metastasis frequency most efficiently (R248Q).
The results of the present study demonstrated that the expression of the p53 D281G variant increased the wound-healing assay migration speed in a transactivation-dependent manner, but this effect was cancer type-specific and was only observed in lung cancer H1299 cells, as previously reported (38), but not in breast cancer MDA-MB-231 cells. To the best of our knowledge, this is the first attempt at testing the migration-associated GOF phenotype of p53-D281G in this popular breast cancer model. Notably, the expression of the R248Q mutant caused a significant decrease in the migration speed in the cell lines tested, in an exclusively transactivation-dependent manner. In experiments involving 3D culture in the presence of ECM, the expression of p53-R248Q significantly decreased the invasive phenotype in the tested cell types in a transactivation-dependent manner. The cells expressing the R248Q variant exhibited poorer invasiveness in experiments involving ECM, including the NOC that separated from microspheres. This could result from the overall lower mobility of the cells, alteration of interactions with the ECM mediated by adhesion molecules, or a differential regulation of the actin cytoskeleton. These mechanisms have been identified as relevant in tumor suppression by p53 (52-55). Indeed, MDA-MB-231-R248Q cells exhibited markedly accelerated adhesion in a transactivation-dependent manner. Notably, this phenotype was not observed in the lung cancer H1299 cells, suggesting that the underlying molecular mechanism is cancer type-specific. In conjunction with the 2D mobility assay results, this could be interpreted as accelerated assembly of focal adhesions in MDA-MB-231-R248Q cells. On the other hand, the inhibition of focal adhesions disassembly, possibly by interference with the function of FAK, may contribute to the decrease in migration of MDA-MB-231 and H1299 cancer cells expressing p53-R248Q. The role of FAK in cancer invasiveness, and the regulatory activity of p53 towards FAK have been demonstrated (56,57).
The zebrafish embryo has been successfully employed as a model for human cancer cell xenotransplantations (58), and it has been demonstrated that, not only do human cancer cells utilize their adhesion molecules for host endothelial attachment (59-61), but the distribution of the transplanted cells is non-random and may reflect the migratory properties and presumably a certain tropism exhibited by the cells (47). Accordingly, the alteration of the distribution of MDA-MB-231 cells that were injected intravenously into the zebrafish embryo upon expression of p53-R248Q, but not p53-R248Q-TA, was observed. This difference in distribution may reflect an altered tropism of the cancer cells, possibly resulting from the p53-induced changes in adhesive properties. In human patients this may translate to the differential distribution of metastatic sites (Tables VI and VII).
In H1299-R248Q cells, a decrease in TCF8/ZEB1 and N-cadherin protein levels was observed, compatible with the partial loss of the mesenchymal characteristic manifested by the decreased mobility and increased cell-cell coherence. TCF8/ZEB1 is a pleiotropic suppressor of epithelial features and inducer of EMT, whose importance in cancer metastasis is well recognized (62-64). Dong et al (65) reported a ZEB1 increase accompanied by an increase in cellular mobility upon mutant p53 expression, but different model cell lines were used than the ones employed in the present study, which might explain the difference in phenotype. In the MDA-MB-231-R248Q cells, a decrease in SLUG and RhoA expression was observed. SLUG is an EMT regulator and its decrease would be consistent with partial loss of the mesenchymal characteristic. RhoA is an important regulator of actomyosin contractile complexes and is involved in the regulation of cancer invasiveness (66,67). Furthermore, in H1299 and MDA-MB-231 cells expressing p53-R248Q, a decrease in the expression and phosphorylation of MLC2 was observed. The MLC2 protein is involved in the calcium-dependent regulation of actomyosin contractility and has recently been reported to have a positive impact on membrane mechanics and ECM remodeling (68); therefore, a decrease in its expression may result in a decrease in cellular mobility, particularly in the ECM environment. Notably, the immobile H358 cells express MLC2, but it is not phosphorylated, and thus inactive.
The molecular mechanisms of regulation of SLUG, RhoA or MLC2 expression by the p53 GOF mutant protein in a transactivation-dependent manner are not known. It could be hypothesized that a slightly misfolded p53 acquires affinity for a different set of promoters and activates the expression of negative regulators of the impacted proteins. Alternatively, it might activate the expression of miRNAs that limit the translation of the proteins in question. Chromatin immunoprecipitation/mRNA/miRNA sequencing would be good experimental approaches to address these questions.
In summary, the current study has presented data demonstrating that the mutated p53 GOF characteristics, including changes in adhesive properties, may be cancer type-specific. It may also translate to a cellular phenotype that seemingly decreases traits of malignancy. This should be a reminder that generalizations, even when they concern key regulators, including p53, should be made with caution. Further research into the molecular mechanisms of the decrease in invasiveness resulting from p53-R248Q expression is required. Furthermore, it would be interesting to see whether a larger meta-analysis of available clinical and molecular data confirms the lower occurrence of metastasis in patients bearing the p53-R248Q mutation.
Acknowledgments
Not applicable.
Funding
MO, MP, AZ and MZ were supported by the National Science Centre, Krakow, Poland, under MAESTRO grant no. 2012/06/A/NZ1/00089. The funding body had no influence over the design of the study, collection, analysis and interpretation of data.
Availability of data and materials
All data generated or analyzed during this study are included in this published article.
Authors’ contributions
MBO designed and performed experiments, analyzed the data and wrote the draft manuscript. MP performed the experiments and analyzed the data. ESJ contributed to the design and execution of the experiments involving zebrafish embryos. AZ and MZ contributed to the design of the experiments, data analysis and manuscript editing and improvement.
Ethics approval and consent to participate
Under Polish and EU guidelines, no specific ethics approval was required for this study, as all zebrafish embryos used were between 0 and 5 days old and therefore not considered animals. Fish embryos were obtained in-house, as the International Institute of Molecular and Cell Biology has an institutional license to house and breed fish. No patient-derived data or tissues were used in this study.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
References
- 1.Lane DP. Cancer. p53, guardian of the genome. Nature. 1992;358:15–16. doi: 10.1038/358015a0. [DOI] [PubMed] [Google Scholar]
- 2.Petitjean A, Achatz MI, Borresen-Dale AL, Hainaut P, Olivier M. TP53 mutations in human cancers: Functional selection and impact on cancer prognosis and outcomes. Oncogene. 2007;26:2157–2165. doi: 10.1038/sj.onc.1210302. [DOI] [PubMed] [Google Scholar]
- 3.Halevy O, Michalovitz D, Oren M. Different tumor-derived p53 mutants exhibit distinct biological activities. Science. 1990;250:113–116. doi: 10.1126/science.2218501. [DOI] [PubMed] [Google Scholar]
- 4.Milner J, Medcalf EA. Cotranslation of activated mutant p53 with wild-type drives the wild-type p53 protein into the mutant conformation. Cell. 1991;65:765–774. doi: 10.1016/0092-8674(91)90384-B. [DOI] [PubMed] [Google Scholar]
- 5.Chan WM, Siu WY, Lau A, Poon RY. How many mutant p53 molecules are needed to inactivate a tetramer? Mol Cell Biol. 2004;24:3536–3551. doi: 10.1128/MCB.24.8.3536-3551.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Willis A, Jung EJ, Wakefield T, Chen X. Mutant p53 exerts a dominant negative effect by preventing wild-type p53 from binding to the promoter of its target genes. Oncogene. 2004;23:2330–2338. doi: 10.1038/sj.onc.1207396. [DOI] [PubMed] [Google Scholar]
- 7.Dittmer D, Pati S, Zambetti G, Chu S, Teresky AK, Moore M, Finlay C, Levine AJ. Gain of function mutations in p53. Nat Genet. 1993;4:42–46. doi: 10.1038/ng0593-42. [DOI] [PubMed] [Google Scholar]
- 8.Hanahan D, Weinberg RA. The hallmarks of cancer. Cell. 2000;100:57–70. doi: 10.1016/S0092-8674(00)81683-9. [DOI] [PubMed] [Google Scholar]
- 9.Hanahan D, Weinberg RA. Hallmarks of cancer: The next generation. Cell. 2011;144:646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 10.Aschauer L, Muller PA. Novel targets and interaction partners of mutant p53 Gain-Of-Function. Biochem Soc Trans. 2016;44:460–466. doi: 10.1042/BST20150261. [DOI] [PubMed] [Google Scholar]
- 11.Shetzer Y, Molchadsky A, Rotter V. Oncogenic mutant p53 gain of function nourishes the vicious cycle of tumor development and cancer stem-cell formation. Cold Spring Harb Perspect Med. 2016;6:20. doi: 10.1101/cshperspect.a026203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Shetzer Y, Solomon H, Koifman G, Molchadsky A, Horesh S, Rotter V. The paradigm of mutant p53-expressing cancer stem cells and drug resistance. Carcinogenesis. 2014;35:1196–1208. doi: 10.1093/carcin/bgu073. [DOI] [PubMed] [Google Scholar]
- 13.Pfister NT, Prives C. Transcriptional regulation by wild-type and cancer-related mutant forms of p53. Cold Spring Harb Perspect Med. 2017;7:26. doi: 10.1101/cshperspect.a026054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Liu G, McDonnell TJ, Montes de Oca Luna R, Kapoor M, Mims B, El-Naggar AK, Lozano G. High metastatic potential in mice inheriting a targeted p53 missense mutation. Proc Natl Acad Sci USA. 2000;97:4174–4179. doi: 10.1073/pnas.97.8.4174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lang GA, Iwakuma T, Suh YA, Liu G, Rao VA, Parant JM, Valentin-Vega YA, Terzian T, Caldwell LC, Strong LC, et al. Gain of function of a p53 hot spot mutation in a mouse model of Li-Fraumeni syndrome. Cell. 2004;119:861–872. doi: 10.1016/j.cell.2004.11.006. [DOI] [PubMed] [Google Scholar]
- 16.Olive KP, Tuveson DA, Ruhe ZC, Yin B, Willis NA, Bronson RT, Crowley D, Jacks T. Mutant p53 gain of function in two mouse models of Li-Fraumeni syndrome. Cell. 2004;119:847–860. doi: 10.1016/j.cell.2004.11.004. [DOI] [PubMed] [Google Scholar]
- 17.Liu G, Parant JM, Lang G, Chau P, Chavez-Reyes A, El-Naggar AK, Multani A, Chang S, Lozano G. Chromosome stability, in the absence of apoptosis, is critical for suppression of tumorigenesis in Trp53 mutant mice. Nat Genet. 2004;36:63–68. doi: 10.1038/ng1282. [DOI] [PubMed] [Google Scholar]
- 18.Song H, Hollstein M, Xu Y. p53 gain-of-function cancer mutants induce genetic instability by inactivating ATM. Nat Cell Biol. 2007;9:573–580. doi: 10.1038/ncb1571. [DOI] [PubMed] [Google Scholar]
- 19.Liu DP, Song H, Xu Y. A common gain of function of p53 cancer mutants in inducing genetic instability. Oncogene. 2010;29:949–956. doi: 10.1038/onc.2009.376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hanel W, Marchenko N, Xu S, Yu SX, Weng W, Moll U. Two hot spot mutant p53 mouse models display differential gain of function in tumorigenesis. Cell Death Differ. 2013;20:898–909. doi: 10.1038/cdd.2013.17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gaiddon C, Lokshin M, Ahn J, Zhang T, Prives C. A subset of tumor-derived mutant forms of p53 down-regulate p63 and p73 through a direct interaction with the p53 core domain. Mol Cell Biol. 2001;21:1874–1887. doi: 10.1128/MCB.21.5.1874-1887.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Xu J, Reumers J, Couceiro JR, De Smet F, Gallardo R, Rudyak S, Cornelis A, Rozenski J, Zwolinska A, Marine JC, et al. Gain of function of mutant p53 by coaggregation with multiple tumor suppressors. Nat Chem Biol. 2011;7:285–295. doi: 10.1038/nchembio.546. [DOI] [PubMed] [Google Scholar]
- 23.Wiech M, Olszewski MB, Tracz-Gaszewska Z, Wawrzynow B, Zylicz M, Zylicz A. Molecular mechanism of mutant p53 stabilization: The role of HSP70 and MDM2. PLoS One. 2012;7:e51426. doi: 10.1371/journal.pone.0051426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Muller PA, Caswell PT, Doyle B, Iwanicki MP, Tan EH, Karim S, Lukashchuk N, Gillespie DA, Ludwig RL, Gosselin P, et al. Mutant p53 drives invasion by promoting integrin recycling. Cell. 2009;139:1327–1341. doi: 10.1016/j.cell.2009.11.026. [DOI] [PubMed] [Google Scholar]
- 25.Adorno M, Cordenonsi M, Montagner M, Dupont S, Wong C, Hann B, Solari A, Bobisse S, Rondina MB, Guzzardo V, et al. A Mutant-p53/Smad complex opposes p63 to empower TGFbeta-induced metastasis. Cell. 2009;137:87–98. doi: 10.1016/j.cell.2009.01.039. [DOI] [PubMed] [Google Scholar]
- 26.Weissmueller S, Manchado E, Saborowski M, Morris JP, IV, Wagenblast E, Davis CA, Moon SH, Pfister NT, Tschaharganeh DF, Kitzing T, et al. Mutant p53 drives pancreatic cancer metastasis through cell-autonomous PDGF receptor β signaling. Cell. 2014;157:382–394. doi: 10.1016/j.cell.2014.01.066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Subramanian M, Francis P, Bilke S, Li XL, Hara T, Lu X, Jones MF, Walker RL, Zhu Y, Pineda M, et al. A mutant p53/let-7i-axis-regulated gene network drives cell migration, invasion and metastasis. Oncogene. 2015;34:1094–1104. doi: 10.1038/onc.2014.46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Alam SK, Yadav VK, Bajaj S, Datta A, Dutta SK, Bhattacharyya M, Bhattacharya S, Debnath S, Roy S, Boardman LA, et al. DNA damage-induced ephrin-B2 reverse signaling promotes chemo-resistance and drives EMT in colorectal carcinoma harboring mutant p53. Cell Death Differ. 2016;23:707–722. doi: 10.1038/cdd.2015.133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kollareddy M, Dimitrova E, Vallabhaneni KC, Chan A, Le T, Chauhan KM, Carrero ZI, Ramakrishnan G, Watabe K, Haupt Y, et al. Regulation of nucleotide metabolism by mutant p53 contributes to its gain-of-function activities. Nat Commun. 2015;6:7389. doi: 10.1038/ncomms8389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Arjonen A, Kaukonen R, Mattila E, Rouhi P, Högnäs G, Sihto H, Miller BW, Morton JP, Bucher E, Taimen P, et al. Mutant p53-associated myosin-X upregulation promotes breast cancer invasion and metastasis. J Clin Invest. 2014;124:1069–1082. doi: 10.1172/JCI67280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Scian MJ, Stagliano KE, Ellis MA, Hassan S, Bowman M, Miles MF, Deb SP, Deb S. Modulation of gene expression by tumor-derived p53 mutants. Cancer Res. 2004;64:7447–7454. doi: 10.1158/0008-5472.CAN-04-1568. [DOI] [PubMed] [Google Scholar]
- 32.Vaughan CA, Singh S, Grossman SR, Windle B, Deb SP, Deb S. Gain-of-function p53 activates multiple signaling pathways to induce oncogenicity in lung cancer cells. Mol Oncol. 2017;11:696–711. doi: 10.1002/1878-0261.12068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kogan-Sakin I, Tabach Y, Buganim Y, Molchadsky A, Solomon H, Madar S, Kamer I, Stambolsky P, Shelly A, Goldfinger N, et al. Mutant p53(R175H) upregulates Twist1 expression and promotes epithelial-mesenchymal transition in immortalized prostate cells. Cell Death Differ. 2011;18:271–281. doi: 10.1038/cdd.2010.94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Vaughan CA, Singh S, Windle B, Yeudall WA, Frum R, Grossman SR, Deb SP, Deb S. Gain-of-Function Activity of Mutant p53 in Lung Cancer through Up-Regulation of Receptor Protein Tyrosine Kinase Axl. Genes Cancer. 2012;3:491–502. doi: 10.1177/1947601912462719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cavallaro S. CXCR4/CXCL12 in non-small-cell lung cancer metastasis to the brain. Int J Mol Sci. 2013;14:1713–1727. doi: 10.3390/ijms14011713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hinton CV, Avraham S, Avraham HK. Role of the CXCR4/CXCL12 signaling axis in breast cancer metastasis to the brain. Clin Exp Metastasis. 2010;27:97–105. doi: 10.1007/s10585-008-9210-2. [DOI] [PubMed] [Google Scholar]
- 37.Orimo A, Gupta PB, Sgroi DC, Arenzana-Seisdedos F, Delaunay T, Naeem R, Carey VJ, Richardson AL, Weinberg RA. Stromal fibroblasts present in invasive human breast carcinomas promote tumor growth and angiogenesis through elevated SDF-1/CXCL12 secretion. Cell. 2005;121:335–348. doi: 10.1016/j.cell.2005.02.034. [DOI] [PubMed] [Google Scholar]
- 38.Yeudall WA, Vaughan CA, Miyazaki H, Ramamoorthy M, Choi MY, Chapman CG, Wang H, Black E, Bulysheva AA, Deb SP, et al. Gain-of-function mutant p53 upregulates CXC chemokines and enhances cell migration. Carcinogenesis. 2012;33:442–451. doi: 10.1093/carcin/bgr270. [DOI] [PubMed] [Google Scholar]
- 39.Moskovits N, Kalinkovich A, Bar J, Lapidot T, Oren M. p53 Attenuates cancer cell migration and invasion through repression of SDF-1/CXCL12 expression in stromal fibroblasts. Cancer Res. 2006;66:10671–10676. doi: 10.1158/0008-5472.CAN-06-2323. [DOI] [PubMed] [Google Scholar]
- 40.Coradini D, Fornili M, Ambrogi F, Boracchi P, Biganzoli E. TP53 mutation, epithelial-mesenchymal transition, and stemlike features in breast cancer subtypes. J Biomed Biotechnol. 2012;2012;254085 doi: 10.1155/2012/254085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Cordani M, Pacchiana R, Butera G, D’Orazi G, Scarpa A, Donadelli M. Mutant p53 proteins alter cancer cell secretome and tumour microenvironment: Involvement in cancer invasion and metastasis. Cancer Lett. 2016;376:303–309. doi: 10.1016/j.canlet.2016.03.046. [DOI] [PubMed] [Google Scholar]
- 42.Dong P, Tada M, Hamada J, Nakamura A, Moriuchi T, Sakuragi N. p53 dominant-negative mutant R273H promotes invasion and migration of human endometrial cancer HHUA cells. Clin Exp Metastasis. 2007;24:471–483. doi: 10.1007/s10585-007-9084-8. [DOI] [PubMed] [Google Scholar]
- 43.Kalo E, Buganim Y, Shapira KE, Besserglick H, Goldfinger N, Weisz L, Stambolsky P, Henis YI, Rotter V. Mutant p53 attenuates the SMAD-dependent transforming growth factor beta1 (TGF-beta1) signaling pathway by repressing the expression of TGF-beta receptor type II. Mol Cell Biol. 2007;27:8228–8242. doi: 10.1128/MCB.00374-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Campeau E, Ruhl VE, Rodier F, Smith CL, Rahmberg BL, Fuss JO, Campisi J, Yaswen P, Cooper PK, Kaufman PD. A versatile viral system for expression and depletion of proteins in mammalian cells. PLoS One. 2009;4:e6529. doi: 10.1371/journal.pone.0006529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sambrook J, Russell DW. Molecular Cloning: A Laboratory Manual. Cold Spring Harbor Laboratory Press; Cold Spring Harbor, NY: 2001. [Google Scholar]
- 46.Limame R, Wouters A, Pauwels B, Fransen E, Peeters M, Lardon F, De Wever O, Pauwels P. Comparative analysis of dynamic cell viability, migration and invasion assessments by novel real-time technology and classic endpoint assays. PLoS One. 2012;7:e46536. doi: 10.1371/journal.pone.0046536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.He S, Lamers GE, Beenakker JW, Cui C, Ghotra VP, Danen EH, Meijer AH, Spaink HP, Snaar-Jagalska BE. Neutrophil-mediated experimental metastasis is enhanced by VEGFR inhibition in a zebrafish xenograft model. J Pathol. 2012;227:431–445. doi: 10.1002/path.4013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Petitjean A, Mathe E, Kato S, Ishioka C, Tavtigian SV, Hainaut P, Olivier M. Impact of mutant p53 functional properties on TP53 mutation patterns and tumor phenotype: Lessons from recent developments in the IARC TP53 database. Hum Mutat. 2007;28:622–629. doi: 10.1002/humu.20495. [DOI] [PubMed] [Google Scholar]
- 49.Zehir A, Benayed R, Shah RH, Syed A, Middha S, Kim HR, Srinivasan P, Gao J, Chakravarty D, Devlin SM, et al. Mutational landscape of metastatic cancer revealed from prospective clinical sequencing of 10,000 p atients. Nat Med. 2017;23:703–713. doi: 10.1038/nm.4333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lin J, Teresky AK, Levine AJ. Two critical hydrophobic amino acids in the N-terminal domain of the p53 protein are required for the gain of function phenotypes of human p53 mutants. Oncogene. 1995;10:2387–2390. [PubMed] [Google Scholar]
- 51.Lin J, Chen J, Elenbaas B, Levine AJ. Several hydrophobic amino acids in the p53 amino-terminal domain are required for transcriptional activation, binding to mdm-2 and the adenovirus 5 E1B 55-kD protein. Genes Dev. 1994;8:1235–1246. doi: 10.1101/gad.8.10.1235. [DOI] [PubMed] [Google Scholar]
- 52.Araki K, Ebata T, Guo AK, Tobiume K, Wolf SJ, Kawauchi K. p53 regulates cytoskeleton remodeling to suppress tumor progression. Cell Mol Life Sci. 2015;72:4077–4094. doi: 10.1007/s00018-015-1989-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Bon G, Di Carlo SE, Folgiero V, Avetrani P, Lazzari C, D’Orazi G, Brizzi MF, Sacchi A, Soddu S, Blandino G, et al. Negative regulation of beta4 integrin transcription by home-odomain-interacting protein kinase 2 and p53 impairs tumor progression. Cancer Res. 2009;69:5978–5986. doi: 10.1158/0008-5472.CAN-09-0244. [DOI] [PubMed] [Google Scholar]
- 54.Janouskova H, Ray AM, Noulet F, Lelong-Rebel I, Choulier L, Schaffner F, Lehmann M, Martin S, Teisinger J, Dontenwill M. Activation of p53 pathway by Nutlin-3a inhibits the expression of the therapeutic target α5 integrin in colon cancer cells. Cancer Lett. 2013;336:307–318. doi: 10.1016/j.canlet.2013.03.018. [DOI] [PubMed] [Google Scholar]
- 55.Guo AK, Hou YY, Hirata H, Yamauchi S, Yip AK, Chiam KH, Tanaka N, Sawada Y, Kawauchi K. Loss of p53 enhances NF-κB-dependent lamellipodia formation. J Cell Physiol. 2014;229:696–704. doi: 10.1002/jcp.24505. [DOI] [PubMed] [Google Scholar]
- 56.Sulzmaier FJ, Jean C, Schlaepfer DD. FAK in cancer: Mechanistic findings and clinical applications. Nat Rev Cancer. 2014;14:598–610. doi: 10.1038/nrc3792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Golubovskaya V, Kaur A, Cance W. Cloning and characterization of the promoter region of human focal adhesion kinase gene: Nuclear factor kappa B and p53 binding sites. Biochim Biophys Acta. 2004;1678:111–125. doi: 10.1016/j.bbaexp.2004.03.002. [DOI] [PubMed] [Google Scholar]
- 58.Tulotta C, Stefanescu C, Beletkaia E, Bussmann J, Tarbashevich K, Schmidt T, Snaar-Jagalska BE. Inhibition of signaling between human CXCR4 and zebrafish ligands by the small molecule IT1t impairs the formation of triple-negative breast cancer early metastases in a zebrafish xenograft model. Dis Model Mech. 2016;9:141–153. doi: 10.1242/dmm.023275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Stoletov K, Kato H, Zardouzian E, Kelber J, Yang J, Shattil S, Klemke R. Visualizing extravasation dynamics of metastatic tumor cells. J Cell Sci. 2010;123:2332–2341. doi: 10.1242/jcs.069443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Li Y, Drabsch Y, Pujuguet P, Ren J, van Laar T, Zhang L, van Dam H, Clément-Lacroix P, Ten Dijke P. Genetic depletion and pharmacological targeting of αv integrin in breast cancer cells impairs metastasis in zebrafish and mouse xenograft models. Breast Cancer Res. 2015;17:28. doi: 10.1186/s13058-015-0537-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Truong HH, Xiong J, Ghotra VP, Nirmala E, Haazen L, Le Dévédec SE, Balcioğlu HE, He S, Snaar-Jagalska BE, Vreugdenhil E, et al. β1 integrin inhibition elicits a prometastatic switch through the TGFβ-miR-200-ZEB network in E-cadherin-positive triple-negative breast cancer. Sci Signal. 2014;7:ra15. doi: 10.1126/scisignal.2004751. [DOI] [PubMed] [Google Scholar]
- 62.Spaderna S, Schmalhofer O, Wahlbuhl M, Dimmler A, Bauer K, Sultan A, Hlubek F, Jung A, Strand D, Eger A, et al. The transcriptional repressor ZEB1 promotes metastasis and loss of cell polarity in cancer. Cancer Res. 2008;68:537–544. doi: 10.1158/0008-5472.CAN-07-5682. [DOI] [PubMed] [Google Scholar]
- 63.Wellner U, Schubert J, Burk UC, Schmalhofer O, Zhu F, Sonntag A, Waldvogel B, Vannier C, Darling D, zur Hausen A, et al. The EMT-activator ZEB1 promotes tumorigenicity by repressing stemness-inhibiting microRNAs. Nat Cell Biol. 2009;11:1487–1495. doi: 10.1038/ncb1998. [DOI] [PubMed] [Google Scholar]
- 64.Zhang P, Sun Y, Ma L. ZEB1: At the crossroads of epithelial-mesenchymal transition, metastasis and therapy resistance. Cell Cycle. 2015;14:481–487. doi: 10.1080/15384101.2015.1006048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Dong P, Karaayvaz M, Jia N, Kaneuchi M, Hamada J, Watari H, Sudo S, Ju J, Sakuragi N. Mutant p53 gain-of-function induces epithelial-mesenchymal transition through modulation of the miR-130b-ZEB1 axis. Oncogene. 2013;32:3286–3295. doi: 10.1038/onc.2012.334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Jeong D, Park S, Kim H, Kim CJ, Ahn TS, Bae SB, Kim HJ, Kim TH, Im J, Lee MS, et al. RhoA is associated with invasion and poor prognosis in colorectal cancer. Int J Oncol. 2016;48:714–722. doi: 10.3892/ijo.2015.3281. [DOI] [PubMed] [Google Scholar]
- 67.O’Connor K, Chen M. Dynamic functions of RhoA in tumor cell migration and invasion. Small GTPases. 2013;4:141–147. doi: 10.4161/sgtp.25131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Choi DS, Stark DJ, Raphael RM, Wen J, Su J, Zhou X, Chang CC, Zu Y. SDF-1α stiffens myeloma bone marrow mesenchymal stromal cells through the activation of RhoA-ROCK-Myosin II. Int J Cancer. 2015;136:E219–E229. doi: 10.1002/ijc.29145. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
All data generated or analyzed during this study are included in this published article.