

Abstract
Inhibition of ubiquitin ligases with small molecule remains a very challenging task, given the lack of catalytic activity of the target and the requirement of disruption of its interactions with other proteins. Siah1/2, which are E3 ubiquitin ligases, are implicated in melanoma and prostate cancer and represent high-value drug targets. We utilized three independent screening approaches in our efforts to identify small-molecule Siah1/2 inhibitors: Affinity Selection-Mass Spectrometry, a protein thermal shift-based assay and an in silico based screen. Inhibitors were assessed for their effect on viability of melanoma and prostate cancer cultures, colony formation, prolyl-hydroxylase-HIF1α signaling, expression of selected Siah2-related transcripts, and Siah2 ubiquitin ligase activity. Several analogues were further characterized, demonstrating improved efficacy. Combination of the top hits identified in the different assays demonstrated an additive effect, pointing to complementing mechanisms that underlie each of these Siah1/2 inhibitors.
Keywords: Siah1, Siah2, ubiquitin ligase, melanoma, prostate cancer
1. Introduction
Ubiquitin ligases limit the stability and activity of substrates by targeting them for proteasomal-dependent degradation. Siah is a two-member family of ubiquitin ligases implicated in control of key cellular processes. Among those, Siah ligases control prolyl hydroxylases, which regulate HIF1α and ATF4 stability [1–3]. Inhibition of Siah1/2 attenuates expression of HIF1α, resulting in attenuated tumor development and metastasis in melanoma [4] and inhibits neuroendocrine (NE) prostate tumor development [4, 5]. Through its regulation of the co-repressor NCOR1, Siah2 controls transcription of select androgen receptor-regulated genes that contribute to development of hormone refractory prostate cancers (PCa) [6]. Siah2 also determines ATF4 levels with a concomitant effect on cell death programs following stress, as occurs in ischemic conditions [3]. Moreover, Siah2 is implicated in control of mitochondrial fission, which impacts cell death under stress conditions, such as those associated with heart infarction [7]. In all cases, blocking Siah activity appears to alleviate pathological conditions – from ischemia to tumor development and metastasis – making it a high value drug target.
There have been numerous attempts to inhibit Siah2 ubiquitin ligase activity, including application of a meso-scale-based screening that led to identification of menadione [8]. A structure-based approach enabled identification of short peptides that covalently bind Siah2 and inhibit its ubiquitin ligase activity [9]. The crystal structure of Siah2-bound peptides has been solved, revealing a newly defined structural pocket. Siah2 inhibitory peptides were found to significantly suppress Siah2 ubiquitin ligase activity in cell culture and in vitro [9]. Yet, the utilization of such peptides has been largely limited to cultures, limiting further characterization in vivo.
We undertook three independent screening approaches to identify prospective Siah1/2 inhibitors, on the assumption that each strategy might reveal different classes of compounds. Using these approaches, we succeeded in identifying small molecule Siah1/2 inhibitors that could provide the foundation for further development of small molecules that disrupt the ubiquitin ligase activities of Siah1/2.
2. Materials and Methods
Purification of GST-Siah2 and full length Siah2
GST-Siah2 proteins were produced in bacteria and purified from using Glutathione-Sepharose (Amersham Bioscience) as described before [8].
Screen of Siah2 inhibitors by Affinity Selection-Mass Spectrometry (AS-MS)
The affinity selection-mass spectrometry (AS-MS) ligand binding screen was optimized following Cloutier, et. al. [10]. Briefly, an ultrafiltration (UF) plate (Pall Acroprep Advance 96 filter plate, 3k MW cutoff) was incubated with 150 µL of protein buffer, placed on top of a receiver plate (Greiner-Bio 96 well microplate) and centrifuged (Eppendorf centrifuge 5810R) at 2,000g. The eluent was discarded and replaced with a fresh receiver plate. Next, 100 µL of a 20 µM protein solution, 3.2 µL of 400 compounds mixture in DMSO with each at 125 µM, and 100 µL of protein buffer was added to one well of the UF plate. This step was repeated for each of the 80 wells for a total of 32,000 compounds. The final concentration is 10 µM of protein, 2 µM of each compound and 1.6 % D MSO. The plate was incubated for 60 minutes at room temperature in the dark and subsequently centrifuged for 45 mins. The eluent was discarded, 150 µL of buffer was added to each well and centrifuged for 45 mins. This washing step was repeated three times while discarding the eluent after each wash. After the final wash step, 150 µL of buffer was added, mixed, and 50 µL aliquot was transferred to a fresh UF plate. 150 µL of methanol was added to each well, thoroughly mixed and centrifuged to collect the compounds in the eluent. The compounds were dried with nitrogen stream (Argonaut SPE DRY 96) and reconstituted in a mixture of methanol:H2O (25:75). A 2 µL injection of the eluent was analyzed on an Agilent Technologies (Wilmington, DE) 1200 HPLC system with 1290 Affinity HTS autosampler and 6224A TOF-MS using a gradient profile on an Agilent Zorbax SB-C18 R.R. HT, 2.1 × 50mm, 1.8 µm HPLC column. Agilent MassHunter Data acquisition and MassHunter Qualitative Analysis softwares were used to collect the data and identify compounds with protein affinity, respectively.
Purification of Siah1 for PTS screening
Full length Siah1 was produced in the baculovirus/insect cell system. The full-length gene was cloned into the pVL intracellular insect cell expression vector. 8 × 1 L of Sf9 cells at 3 × 106 cells/ml were infected with ~10 ml of freshly-produced virus P2 stock. Cells were infected for 72 h at 27°C, shaken at 120 rpm, then harvested by centrifugation at 3000g for 10 min. Pellets were resuspended in 500 mM NaCl, 20 mM Tris pH 8.0, 10 mM imidazole, EDTA-free protease inhibitor cocktail, and 10 mM DTT (final volume was 2–3x cell pellet). Cells were lysed gently on ice using a glass dounce homogenizer (6 times). Cell debris was pelleted by centrifugation at 19,000 RPM at 4°C for 1 h. Supernatants were bound to cobalt Talon resin for four hours at 4°C. Protein was eluted on an FPLC column using a gradient of 10 mM to 1 M imidazole, 500 mM NaCl, 20 mM HEPES pH 7.4, 10 mM DTT. Peak fractions containing Siah1 were collected and run over a Superdex S200 gel filtration column equilibrated in 150 mM NaCl, 20 mM HEPES at pH 7.4, and 1 mM TCEP (tris(2-carboxyethyl) phosphine)). Peak fractions were concentrated to 1–2 mg/ml, stored at 4°C and used within 2 weeks. Truncated Siah1 (residues 90–282), comprising the ligand binding domain and 2 N-terminal Zinc finger domains, but lacking the RING domain, was produced as a HIS-tag protein in E. coli as previously described [11].
Screen of Siah2 inhibitors by Protein Thermal Shift assay (PTS)
Compounds dissolved in DMSO were dispensed into 384-well PCR plates by Echo 555 acoustic dispenser. Pre-chilled 1.7 µM full-length Siah2 protein, prepared in assay buffer containing 20 mM Tris pH 8, 150 mM NaCl, and 1 mM TCEP, was dispensed by Multi-Drop Combi. After 15-minute pre-incubation, equal volume of 10X Sypro Orange dye in room temperature assay buffer was added. The plate was sealed and spun for 2 minutes at 1000 RPM. The plate was read on ViiA7 RT-PCR instrument with a ramping speed of 0.15 °C/min using X1-M3 setting. Transition temperature (Tm) values were determined using Protein Thermal Shift™ software from ThermoFisher. Hit compounds were selected using hit criteria Z-score ≥ 3 or DTm ≥ 1 °C and reconfirmed in triplicate-well single-con centration and duplicate-well concentration-dependent assays.
In silico screening for small molecule Siah2 inhibitors
The virtual screening pipeline involving numerous methods of computer-aided drug discovery and successfully used in many previous studies [12, 13] has been employed to Siah2 hit discovery. The crystal structure of Siah2 SBD domain (pdb code 5H9M) was used for docking. We identified a possible small molecule binding pocket using the SiteFinder module of MOE suite of software [14]. When superimpose the crystal structure 5H9M of Siah2 and the co-crystal structure of Siah1 and a bound peptide (see Fig 4a), we found that the identified small molecule binding pocket is located at exactly where Siah1 peptide inhibitor (pink) binds.
Figure 4. In Silico screen and characterization of Siah small molecule inhibitors.
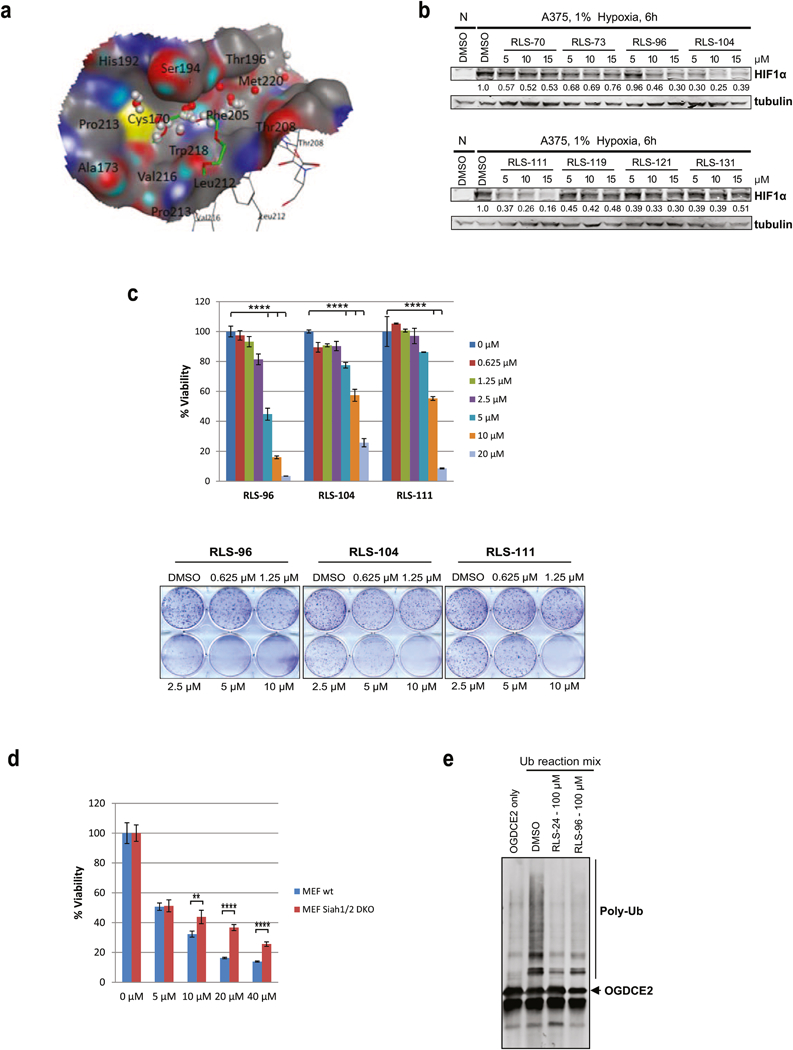
(a) The SiteFinder module of MOE software predicted a small molecule binding pocket using Siah2 crystal structure 5H9M at where the co-crystalized pentaethylene glycol PEG400 in green is. The grey spheres are the hydrophobic probes and the red spheres are the electrostatic probes. (b) Melanoma cells were incubated with 5 µM, 10 µM and 15 µM of selected compounds for 6 h under hypoxia. Cells were harvested and whole cell lysates were immunoblotted with indicated antibodies. Quantification of immunoblots was performed using BioRad densitometer, relative to loading controls, noted under the blots (c) Viability and colony formation assays of RLS-96, RLS-104, and RLS-111 in A375 cells. Each bar represents the mean ± standard deviation of three measurements. Representative data is shown. ****p < 0.0001 was calculated based on comparison with the control (one-way ANOVA with Dunnett’s test). (d) MEFs and Siah1/2 double knock out MEFs were treated with indicated concentrations of RLS-96, and cell viability was assessed by ATPlite after 72 h. Each bar represents the mean ± standard deviation of three measurements. **p < 0.01, ***p < 0.001, were calculated using Student’s t test (e) Compounds RLS-24 and RLS-96 were incubated with the Siah2 substrate OGDCE2 and subjected to an in-vitro ubiquitination assay, as described in Figure 3g.
Approximately 5M ZINC15 commercially available lead-like compounds was first docked to the identified Siah2 pocket using Glide [15] with default settings. Subsequently, 235103 compounds that have the Glide docking score better than −6.0 were selected for a second round of docking using eHiTS program [16] with default parameter settings. Only compounds of RMSD between the predicted binding poses from the Glide and eHITS less than 2.0 Å, which are 16402 structures, were subsequently subjected to consensus ranking using glide-docking score, eHITS-score, and Dock-pKi (calculated using MOE’s scoring.svl). After the final visual check, 133 compounds were selected as hits, of which 94 compounds were purchased for Siah2 assays.
Experimental procedures for the synthesis of RLS-7 and derivatives
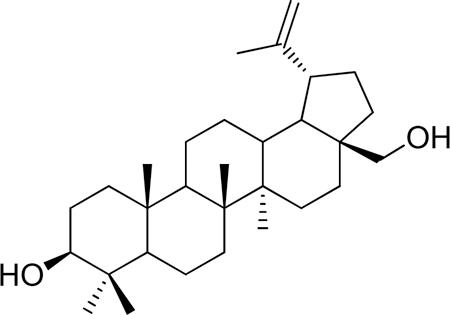
Betulin was purchased commercially and purified on silica gel (hexane/EtOAc gradient) to give pure material. 1H-NMR (500 MHz-CDCl3) δ 4.68 (s, 1H), 4.58 (s, 1H), 3.79 (d, J = 10.6 Hz, 1H), 3.33 (d, J = 10.8 Hz, 1H), 3.18 (dd, J = 11.4, 4.8 Hz, 1H), 2.38 (m, 1H), 2.08 – 0.53 (m, >10H), 1.68 (s, 3H), 1.02 (s, 3H), 0.98 (s, 3H), 0.96 (s, 3H), 0.82 (s, 3H), 0.76 (s, 3H). 13C-NMR (125 MHz-CDCl3) δ 150.68, 109.89, 79.19, 60.75, 55.50, 50.61, 48.97, 48.00, 42.93, 41.13, 39.07, 38.92, 37.52, 37.37, 34.45, 34.18, 29.97, 29.39, 28.20, 27.60, 27.26, 25.42, 21.05, 19.30, 18.52, 16.32, 16.20, 15.58, 14.98. HRMS: calculated [M+H]: 443.3884, found [M+H]: 443.3868.
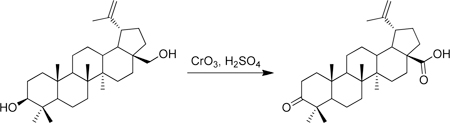
A round bottom flask was charged with betulin (750 mg, 1.69 mmol) and acetone (50 mL) and cooled to 0 °C in an ice water bath. Separatel y, a solution of Jones reagent was prepared by combining CrO3 (1 g), concentrated H2SO4 (1 mL) and water (3 mL) at 0 °C. This Jones reagent was then slowly added to the betulin solution which caused the initially white suspension to become green. As the addition is continued, the orange color of the Jones reagent is maintained in the reaction mixture. The combined mixture was then stirred at 0 °C for 2 h, after which methanol was slowly added until the solution became a dark green color and the solution was warmed to room temperature. The mixture was then partially concentrated in vacuo and then partitioned between EtOAc and water. The aqueous portion was thrice extracted with EtOAc and the combined organic portions were washed with brine and dried over sodium sulfate. Purification on silica gel (25 g, preabsorbed onto silica gel, 5–25% hexane/EtOAc gradient) gave betulonic acid (426 mg, 55% yield) as a white solid. 1H-NMR (500 MHz-CDCl3) δ 4.75 (s, 1H), 4.62 (s, 1H), 3.00 (m, 1H), 2.57 – 2.34 (m, 2H), 2.33 – 2.18 (m, 2H), 2.05 – 1.85 (m, 2H), 1.83 – 0.75 (m, >12H), 1.69 (s, 3H), 1.07 (s, 3H), 1.02 (s, 3H), 0.99 (s, 3H), 0.98 (s, 3H), 0.93 (s, 3H). 13C-NMR (125 MHz-CDCl3) δ 218.41, 182.46, 150.51, 109.97, 56.60, 55.13, 50.05, 49.40, 47.54, 47.11, 42.70, 40.84, 39.81, 38.72, 37.25, 37.12, 34.33, 33.81, 32.31, 30.77, 29.89, 26.85, 25.70, 21.58, 21.20, 19.83, 19.58, 16.16, 16.03, 14.83. HRMS: calculated [M+H]: 455.3520, found [M+H]: 455.3519.
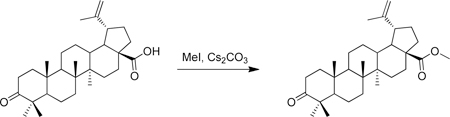
A vial was charged with betulonic acid (50 mg, 0.11 mmol), cesium carbonate (54 mg, 0.16 mmol) and DMF (1 mL) at room temperature. Iodomethane (22 µL, 0.33 mmol) was added and the resulting solution was stirred for 17 h at room temperature. Water (~ 5 mL) was then added to the solution which initiated the precipitation of a white solid. This solid was collected via filtration and washed with water. Purification of the solid on silica gel (10 g biotage column, material pre-adsorbed onto silica gel) with a hexane/EtOAc gradient (2–25%) resulted in methyl betulonate (37 mg, 70% yield) as a white solid. 1H-NMR (500 MHz-CDCl3) δ 4.74 (bs, 1H), 4.60 (bs, 1H), 3.67 (s, 3H), 3.00 (m, 1H), 2.54 – 2.33 (m, 2H), 2.31 – 2.19 (m, 2H), 1.89 (m, 3H), 1.69 (s, 3H), 1. 81 – 1.12 (m, 16H), 1.07 (s, 3H), 1.02 (s, 3H), 0.97 (s, 3H), 0.95 (s, 3H), 0.92 (s, 3H), 0.88 (m, 1H). 13C-NMR (125 MHz-CDCl3) δ 218.33, 176.85, 150.73, 109.86, 56.77, 55.23, 51.52, 50.15, 49.63, 47.57, 47.17, 42.68, 40.85, 39.86, 38.58, 37.18, 37.15, 34.38, 33.86, 32.34, 30.82, 29.89, 26.84, 25.76, 21.64, 21.27, 19.88, 19.61, 16.17, 15.99, 14.86. HRMS: calculated [M+H]: 469.3676, found [M+H]: 469.3664.
A vial was charged with betulonic acid (50 mg, 0.11 mmol), triethylamine (31 µL, 0.22 mmol), HATU (63 mg, 0.16 mmol) and DMF (1 mL) at room temperature. 2-aminoethanol (13 µL, 0.22 mmol) was added and the resulting yellow solution was stirred for 18 h at room temperature. Water (~ 5 mL) was then added to the solution which initiated the precipitation of a white solid. This solid was collected via filtration and washed with water. Purification of the solid on silica gel (10 g biotage column, material preabsorbed onto silica gel) with a hexane/EtOAc gradient (2–100%) resulted in (1R,3aS,5aR,5bR,11aR)-N-(2-hydroxyethyl)-5a,5b,8,8,11a-pentamethyl-9-oxo-1-(prop-1-en-2-yl)icosahydro-1H-cyclopenta[a]chrysene-3a-carboxamide (36 mg, 65% yield) as a white solid. 1H-NMR (500 MHz-CDCl3) δ 6.03 (t, J= 5.7 Hz, 1H), 4.74 (bs, 1H), 4.60 (bs, 1H), 3.73 (t, J = 4.9 Hz, 2H), 3.55 – 3.31 (m, 2H), 3.11 (td, J = 11.1, 4.5 Hz, 1H), 2.49 (m, 2H), 2.40 (m, 1H), 2.05 – 1.85 (m, 2H), 1.81 – 1.65 (m, 2H), 1.69 (3H, s), 1.65 – 1.14 (m, >10H), 1.06 (s, 3H), 1.02 (s, 3H), 0.98 (s, 6H), 0.92 (s, 3H). 13C-NMR (125 MHz-CDCl3) δ 218.32, 178.17, 150.96, 109.71, 63.56, 55.92, 55.29, 50.30, 50.23, 47.59, 46.95, 42.78, 42.63, 40.94, 39.88, 38.64, 38.07, 37.17, 34.40, 34.05, 33.92, 31.09, 29.66, 26.80, 25.85, 21.67, 21.27, 19.86, 19.75, 16.16, 16.10, 14.81. HRMS: calculated [M+H]: 498.3942, found [M+H]: 498.3949.
A microwave vial was charged with betulonic acid (50 mg, 0.11 mmol), hydroxylamine hydrochloride (53 mg, 0.77 mmol) and pyridine (1 mL) at room temperature. The solution was then heated in a microwave at 120 °C for 4h. Water (~ 8 mL) was then added to the solution which initiated the precipitation of a white solid. This solid was collected via filtration and washed with water to give (1R,3aS,5aR,5bR,11aR,E)-9-(hydroxyimino)-5a,5b,8,8,11a-pentamethyl-1-(prop-1-en-2-yl)icosahydro-1H-cyclopenta[a]chrysene-3a-carboxylic acid as a white solid (49 mg, 94%). 1H-NMR (500 MHz-CDCl3) δ 12.07 (s, 1H), 10.25 (s, 1H), 4.70 (s, 1H), 4.57 (s, 1H), 3.04 – 2.90 (m, 1H), 2.81 (dt, J = 15.4, 4.9 Hz, 1H), 2.13 (m, 2H), 1.94 – 0.75 (m, >12H), 1.66 (s, 3H), 1.08 (s, 3H), 0.96 (s, 3H), 0.94 (s, 3H), 0.91 (s, 3H), 0.87 (s, 3H).13 C-NMR (125 MHz-CDCl3) δ 177.22, 163.41, 150.28, 109.64, 30.08, 29.17, 27.56, 25.10, 23.01, 20.74, 18.94, 18.62, 16.54, 15.63, 15.42, 14.26. HRMS: calculated [M+H]: 470.3629, found [M+H]: 470.3623.
A round bottom flask was charged with 10% Pd/C (293 mg, 0.275 mmol) and flushed with nitrogen. Anhydrous THF (20 mL) and methanol (20 mL) were sequentially added, followed by betulonic acid (5.00 g). A hydrogen-filled balloon was then added and the flask was evacuated and back-filled with hydrogen. This evacuation and fill procedure was repeated two additional times. The mixture was then vigorously stirred at room temperature until the reduction was complete (~ 22 h). The mixture was then filtered through celite, washing with methanol, and concentrated under vacuum to give (1S,3aS,5aR,5bR,11aR)-1-isopropyl-5a,5b,8,8,11a-pentamethyl-9-oxoicosahydro-1H-cyclopenta[a]chrysene-3a-carboxylic acid (4.92 g, 98% yield) as a white solid. 1H-NMR (500 MHz-CDCl3) δ 2.58 – 2.34 (m, 2H), 2.25 (m, 2H), 2.02 – 1.12 (m, 22H), 1.08 (s, 3H), 1.02 ( s, 3H), 0.98 (s, 3H), 0.97 (s, 3H), 0.94 (s, 3H), 0.86 (d, J = 6.9 Hz, 3H), 0.76 (d, J = 6.7 Hz, 3H). 13C-NMR (125 MHz-CDCl3) δ 218.30, 179.80, 56.91, 55.18, 49.87, 48.91, 47.57, 44.31, 42.88, 40.89, 39.83, 38.53, 37.57, 37.12, 34.37, 33.93, 32.22, 29.95, 29.91, 27.13, 26.86, 23.19, 22.93, 21.64, 21.27, 19.87, 16.15, 16.10, 14.90, 14.76. HRMS: calculated [M-H]: 455.3531, found [M-H]: 455.3525.
A round bottom flask containing 4 angstrom molecular sieves was flame dried under an atmosphere of nitrogen. To this flask was then added betulonic acid (43 mg, 0.095 mmol), anhydrous THF (2 mL) and LaCl3.LiCl (1.57 mL, 0.6 M in THF, 0.95 mmol) at room temperature. After 10 min, MeMgBr (316 µL, 3 M in Et2O, 0.95 mmol) was added and the solution was stirred at room temperature for 3 h. The solution was then quenched with NH4Cl and twice extracted with CH2Cl2. The product was adsorbed onto SiO2 and purified via flash chromatography (10 g SiO2, 2–40% hexane/EtOAc gradient) to give (1R,3aS,5aR,5bR,11aR)-9-hydroxy-5a,5b,8,8,9,11a-hexamethyl-1-(prop-1-en-2-yl)icosahydro-1H-cyclopenta[a]chrysene-3a-carboxylic acid (14 mg) as a white solid. 1H-NMR (500 MHz-CDCl3) δ 4.74 (s, 1H), 4.61 (s, 1H), 3.49 (s, 1H), 3.00 (td, J = 10.7, 4.7 Hz, 1H), 2.27 (dd, J = 13.0, 3.6 Hz, 1H), 2.24 – 2.13 ( m, 1H), 2.08 – 0.65 (m, >18H), 1.69 (s, 3H), 1.20 (s, 3H), 0.99 (s, 3H), 0.93 (s, 3H), 0.85 (s, 3H), 0.84 (s, 6H). 13C-NMR (125 MHz-CDCl3) δ 181.55, 150.62, 109.89, 75.56, 56.58, 53.65, 51.18, 51.10, 49.51, 47.13, 42.64, 41.12, 40.95, 38.64, 38.12, 37.73, 37.27, 34.71, 34.36, 32.41, 30.81, 29.95, 25.72, 24.71, 23.44, 21.05, 19.61, 19.10, 16.59, 16.30, 15.03. HRMS: calculated [M+H]: 469.3687, found [M+H]: 469.3681.
Cell culture
All human and mouse melanoma cell lines, liver cancer cell lines and pancreatic cell lines were cultured in high-glucose Dulbecco’s modified Eagle’s medium supplemented with 10% fetal bovine serum and 1% penicillin-streptomycin. All prostate cancer cell lines are maintained in RPMI medium supplemented with 10% fetal bovine serum and 1% penicillin-streptomycin. Cell cultures were maintained in cell culture incubator at 37°C in 5% CO2. For hypoxia treatment, cell cultures were maintained in hypoxia cell incubator at 37°C, 5% CO2 and 1% oxygen.
Reagents and drug treatment protocol
RLS-7 and derivatives were synthesized and purified at the CPCCG of SBP Medical Discovery Institute as described above. Other compounds were purchased from MolPort (Latvia) and Enamine Chemicals (New Jersey, USA). All compounds were maintained as 10 mM DMSO stocks. To determine the effect of selected compounds and derivatives, cells were plated at 50% confluency and grown overnight in 6-well plates. Test compounds at different concentrations were added to the plates, cells were exposed to hypoxia (1%) for 6 hours and lysed immediately for Western blot analysis.
Western blot analysis and antibodies
Cells were rinsed with PBS and lysed in lysis buffer (50 mM Tris-HCl, pH 7.4, 150 mM NaCl, 1% NP-40, 1 mM EDTA, 10 µg/ml aprotinin, 1 µg/ml pepstatin A, 10 µg/ml leupeptin, 2 mM phenylmethylsulfonyl fluoride, 2.5 mM sodium orthovanadate). Protein concentration was determined using Coomassie Plus Protein Assay Reagent (Thermo Scientific). Equal amounts of cell lysate proteins (50 μg) were separated on SDS-PAGE and transferred to PVDF membrane. Membranes were blocked with 3% BSA/TBST, incubated with primary antibodies and secondary antibodies. Detection and quantifications were made using Odyssey Infrared Imaging System (LiCor Biosciences). Antibodies against HIF1α were kindly provided by Dr. Gary Chiang (1:1000). Antibodies α-tubulin, GAPDH, HA and Myc were obtained from Santa Cruz Biotechnology. Anti-Flag antibody was obtained from Sigma-Aldrich. Secondary antibodies were goat anti-rabbit Alexa-680 F(ab’)2 (Molecular Probes) and goat anti-mouse IRDye 800 F(ab’)2 (Rockland Immunochemicals). All antibodies were used according to the suppliers’ recommendations.
Proliferation assay
All cell lines were seeded (4,000 cells in 100 µL per well) in 96-well plates. Cells were allowed to attach overnight. Tested compounds and derivatives were serially diluted using media from stock solutions and were added to cells. Tests were performed in triplicate, and each microplate included media and DMSO control wells. Cell viability was assessed using ATPlite after 72 h according to manufacturer’s protocol. Cell growth inhibition was calculated as a percentage of DMSO-treated controls and plotted against the compound concentration.
Colony formation assay
For colony formation assays, cells were plated in triplicate (500 cells/well) in 6-well plates or 100 cells/well in 24-well plates and grown overnight before compounds were added. After 1– 2 weeks, depending on cell line, colonies were stained with Accustain Crystal Violet solution (Sigma-Aldrich) for 30 min. Plates were rinsed with water and images were acquired by scanning. Colony number and size were analyzed using Image J software. Colony formation efficiency was calculated relative to the number of colonies in control (DMSO)-treated wells.
Soft agar assay
Soft agar culture was performed as described before. Briefly, 50,000 melanoma cells mixed in top agar (0.35% in 1× DMEM) were plated over a second layer of agar (0.7% in 1× DMEM). After 24 h, medium containing indicated compounds was added over the top agar and replaced every 5 days. Colonies were visualized by crystal violet staining (0.005% crystal violet in 4% paraformaldehyde/PBS). Images were acquired with a microscope (×40) or scanned (HP ScanJet G4010). Colony number and size were analyzed using Image J software (NIH).
Silencing Siah2 expression by shRNA
The shRNA vectors for silencing of Siah2 were previously described [3]. The sequences used for scramble and Siah2 shRNA were respectively: caacaagatgaagagcaccaa (scramble), ccttggaatcaatgtcacgat (Siah2). Melanoma cells were transfected with scramble or shSiah2 using the jetPRIME DNA transfection reagent kit (Polyplus, France) according to the manufacture’s protocol.
Reverse Transcription and real-time qPCR.
Total RNA was extracted using a total RNA miniprep kit (Sigma) with the On-column DNase I digestion step included. cDNA was synthesized using cDNA kit from Applied Biosystems. Real-time PCR was performed on a Bio-Rad CFX Connect Real-Time System using FastStart Universal SYBR Green Master (Rox) from Roche. H3.3A was used as an internal control. Quantitative PCR reactions were performed in triplicates. PCR primers were designed using PrimerBank (http://pga.mgh.harvard.edu/primerbank).
In vitro ubiquitination assay
GST-Siah2 was purified from the bacteria using glutathione-Sepharose (Amersham Biosciences). Siah substrates were obtained from 293T cells transfected with FLAG-OGDCE2, myc-Sprouty 2 or V5-ASPP2. After 24 h, indicated Siah substrate was immunoprecipitated with agarose A/G beads. Purified GST-Siah2 was incubated with test compounds for 30 min at room temperature before adding to Siah substrate beads and ubiquitination buffer (50 mM Tris-HCl, pH 8.0, 5 mM MgCl2, 0.5 mM dithiothreitol, 2 mM NaF) supplemented with purified ubiquitin (2 µg), 2 mM ATP, E1 (50 ng) (Boston Biochem, Cambridge, MA), purified E2 (UbcH5b) (250 ng) for 45 min at 37 °C. These reactions were washed four times with buffer containing (50 mM Tris-HCl, pH 8.0, 150 mM NaCl, 1% Triton X-100, 1 mM EDTA). Samples were loaded on SDS-PAGE and followed by Western blot analysis using anti-ubiquitin antibody.
Luciferase assay
A375 cells (50% confluency) were transiently transfected with the firefly luciferase reporter plasmid HRE-Luc and renilla luciferase plasmid pRL-TK (20:1) using jetPRIME according to the manufacturer’s protocol. After 24 h, cells were plated (1×105 cells/well in 24-well plate) and allowed to attach overnight. Cells were then treated with different concentration of selected compounds with or without 10 µM of FG4592. Luciferase assay was performed using the dual luciferase kit (Promega, Madison, WI) according to the manufacturer’s protocol after 24 h. Firefly luciferase activity was normalized to renilla luciferase activity as a control for transfection efficiency.
Neuroendocrine differentiation
RV1 cells were plate into 6-well plate at low density (2000 cells per well) and treated with different concentrations of tested compounds. Cells were maintained under hypoxia (1% oxygen) at 37°C, 5% CO2 for one week before images were taken using bright field microscopy.
Animal studies
Male beige/SCID mice (Envigo, 8–10 weeks old) were orthotopically grafted as previously described [17]. In accordance with institutional animal care and use committee approval, 2×105 RV1 cells and 6×105 human primary prostatic CAF were suspended in 30 µL type I collagen for grafting into mice supplemented with testosterone (30 mg pellet implanted subcutaneously). After allowing the tumors to expand for four weeks, the testosterone pellets were removed, the mice were castrated, and the mice were treated 3 times weekly with enzalutamide (1 mg/mouse by oral gavage) and/or RLS-12 (20 mg/kg i.v.). The mice were sacrificed 14 days after castration. The tumor tissues were fixed in 4% para-formaldehyde and embedded in paraffin for sectioning and immunohistochemical staining.
3. Results
AS-MS screen for Siah inhibitors
To identify potential inhibitors of Siah1/2 ubiquitin ligases we performed an Affinity Selection-Mass Spectrometry (AS-MS)-based screen, which is based on a compound’s propensity to exhibit tight association with the target protein (Fig S1a). A screen of over 30,000 compounds revealed 150 compounds that bound with high affinity to Siah2 but not to RNF5, a different RING finger ubiquitin ligase used in a counter-assay (Table S1). Assessment of hits identified a subset of compounds with similar structures. Of those, seven were selected for further analysis in a secondary assay to evaluate their effect on expression levels of HIF1α. Since Siah2 regulates stability of prolyl hydroxylases 1 and 3 which are negative regulators of HIF1α, it was expected that inhibition of Siah2 should upregulate PHD1/3 levels with concomitant reduction in HIF1α levels [1, 5].
In further referencing, compounds identified by different screening campaigns were designated Ronai Lab Siah (RLS) inhibitors and numbered sequentially. Of the seven compounds selected by primary screening, four (RLS-2, RLS-5, RLS-6, RLS-7) reduced HIF1α protein expression, with RLS-7 the most active (Fig 1a). Notably, RLS-7, also known as betulonic acid (Fig 1b), was found to effectively inhibit HIF1α expression. We then further assessed the effect of the two top hits (RLS-6, RLS-7) on HIF1α expression, using purchased betulinic acid (BA, RLS-7 is the oxidized form of betulinic acid) and two its close analogs - lupeol and oleanolic acid (Fig S1b). Notably, RLS-7 and BA were most efficient in inhibiting HIF1α levels in a dose-dependent manner. Lupeol was active in inhibiting HIF1α level, albeit to lesser degree. Although oleanolic acid caused inhibition of HIF1α, such inhibition was not dose-dependent (Fig 1c). As an independent assessment, we measured HIF1α transcriptional activity using HRE-luc reporter assay. Notably, RLS-7 treatment of HEK293T cells harboring the HRE-Luc reporter resulted in a dose-dependent inhibition of transcriptional activity (Fig 1d).
Figure 1. AS-MS screen and characterization of RLS-7 as a Siah small molecule inhibitor.
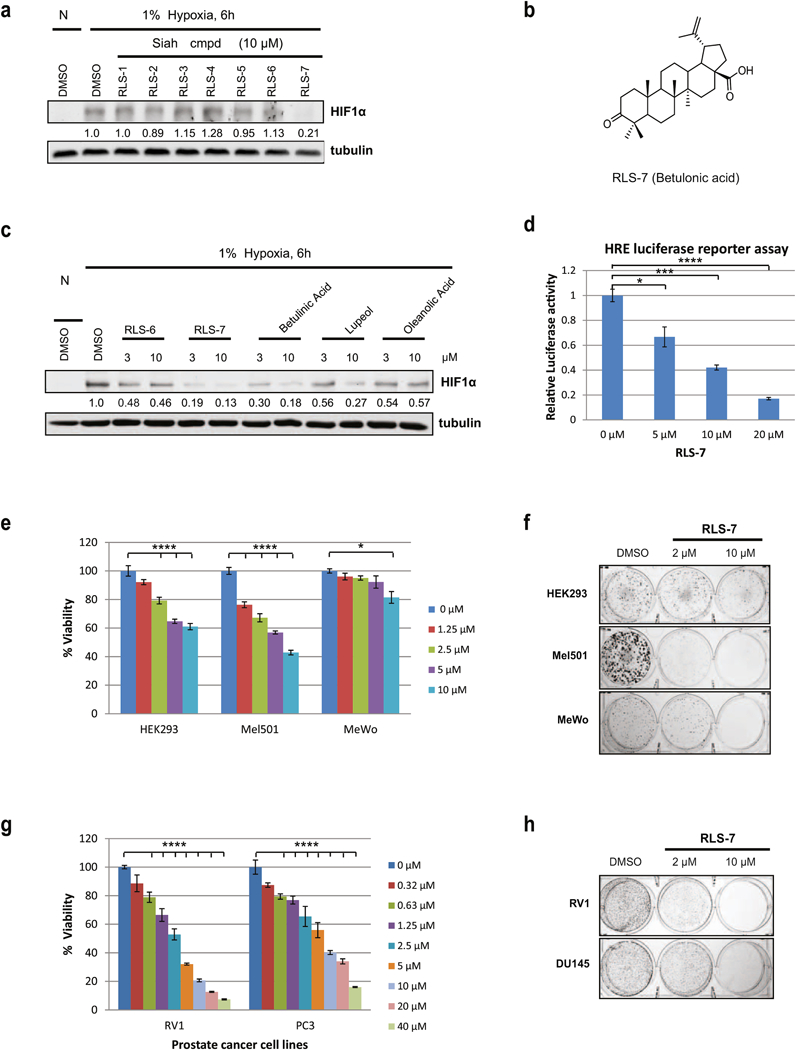
(a) Inhibition of hypoxia-induced HIF1α induction by seven representative compounds from 3 scaffolds. HEK293 cells were incubated with either vehicle (DMSO) or 10 µM of indicated compounds for 6 h under hypoxia. Cells were harvested and whole cell lysates were immunoblotted with the indicated antibodies. (N – normoxia; H - hypoxia). Quantification of the proteins in immunoblots was performed with the aid of BioRad densitometer, relative to loading control, noted under the respective gels. (b) RLS-7 structure. (c) Comparison of RLS-6 with RLS-7 and close analogs. Cells were treated with different compounds at 3 µM and 10 µM and subjected to Wester n Blot, as described above. Quantification of immunoblots was performed using BioRad densitometer, relative to loading controls, noted under the westerns (d) Inhibition of HRE luciferase activity by RLS-7. 293T cells were transfected with HRE-firefly and renilla luciferase plasmids for 24 hours and treated with indicated concentrations of RLS-7 under hypoxia. After 24 hours, cells were lysed and subjected to a luciferase assay. Data were calculated relative to DMSO-treated cells. Means ± standard errors calculated from three independent experiments. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001 compared to controls (one-way ANOVA with Dunnett’s test). (e) Inhibition of HEK293 and melanoma cell viability by RLS-7. HEK293 and different melanoma cells were plated in 96-well plates and incubated with indicated concentrations of RLS-7. Cell viability was assessed by ATPlite after 72 h. Growth inhibition was calculated as percentage of DMSO-treated controls. Each bar represents the mean ± standard deviation of three experiments. Shown is a representative experiment. ****p < 0.0001 as compared to the control (one-way ANOVA with Dunnett’s test). (f) Colony formation assay. Indicated cultures were plated at low density (500 cells/well in 6-well plates) and grown in medium containing vehicle, or 2 µM or 10 µM RLS-7. The number of colonies formed after 10 days in culture was determined by crystal violet staining. (g) Prostate cancer cell lines RV1 and PC3 were treated with indicated concentrations of RLS-7. Cell viability was assessed 72 h later. ****p < 0.0001 as compared with the control, as described above (one-way ANOVA with Dunnett’s test). (h) RV1 and Du145 cells were plated at low density and grown in medium containing indicated concentrations of RLS-7. The number of colonies formed after 2 weeks was determined by crystal violet staining.
We further confirmed the effect of RLS-7 on Siah2 by assessing viability of melanoma cells transfected with control shRNA or shRNA targeting Siah2 for 24h followed by RLS-7 treatment. Melanoma cell viability following RLS-7 treatment was less pronounced in cells that were subjected to knockdown of Siah2, supporting Siah2 as RLS-7 target. Along these lines, inhibition of Siah2 expression by shSiah2 abolished cell death seen upon RLS-7 treatment (10 µM), compared with 80% cell viability in cells subjected to control shRNA. At a higher dose of RLS-7 (20 µM) the degree of cell viability was 71% (shSiah2) compared with 42% (control shRNA; Fig S1c). These findings provide independent confirmation of Siah2 being the target of RLS-7.
Next, we determined the effect of RLS-7 on viability of various melanoma cell lines in comparison with HEK293 cells. Notably, RLS-7 exhibited a pronounced inhibition of growth in Mel501 melanoma line, to degree seen in HEK293 cells (Fig 1e, Fig S1d). Then we asked whether RLS-7 treatment could alter the efficiency of colony formation (CFE) by melanoma cells that were plated at low density and allowed to grow and form colonies over 10-day period. Notably, inhibition of CFE by RLS-7 was not limited to cells where it was found to affect the viability (HEK293 and Mel501 cells, Fig 1f), as evident from its effect on CFE of WM793, MeWo and WM35 lines (Fig S1e). These observations suggest that RLS-7 effectiveness is mediated by a number of pathways, not limited to its ability to induce cell death programs.
Given these effects on established melanoma cell lines, we next determined whether RLS-7 treatment had any effect on primary melanoma cells, which had been cultured for a short time after collection from patient tumors. RLS-7 inhibited melanoma cell growth in 3 of 6 primary cell lines, albeit to different degrees, again raising the possibility that effective Siah2 inhibition of melanoma viability is determined by select genetic and possibly epigenetic characteristics, seen in specific tumor types or genetic backgrounds (Fig S1f).
Siah activity was also shown to play an important role in the development of aggressive forms of prostate cancer [18]. Thus, we extended our analysis of RLS-7 to include prostate cancer cell lines. We found that the effect of RLS-7 on viability of PCa cell lines CW22RV1 (RV1) and PC3 was more robust than on melanoma cells tested so far (Fig 1g). Also, RLS-7 treatment very effectively inhibited colony formation by PCa lines (Fig 1h). Siah2’s effect on transformation of PCa cells is reportedly mediated, in part, by its regulation of transcription of AR-dependent genes [5, 6]. To this end we evaluated whether RLS-7 could block transcription of Siah2-regulated, AR-dependent genes, such as PSA, PMEPA1 and NKX3.1. Indeed, the expression of these three genes was inhibited in PCa cells treated with RLS-7, while expression of the Siah-independent AR target gene FKBP5 was not affected (Fig S1g).
Identification of RLS-7 analogs
Our initial analysis of RLS-7 activity led us to synthesize 6 analogues modifying either the carboxylic acid, the ketone or the isopropene group of RLS-7 (RLS-8 to RLS-13) (Fig 2a) to assess potential structure/activity relationships within the series. These compounds were all tested for inhibition of HIF1α expression under hypoxia. Modifications to the ketone (RLS-11 and RLS-13) or isopropene (RLS-12) demonstrate similar inhibition compared to RLS-7 (10 µM; Fig 2b). These analogs were further tested for their efficacy in causing cell death of cultured A375 melanoma cells using the ATPlite protocol. Comparing the IC50 for RLS-7, and RLS-12, the latter exhibited somewhat greater efficacy in killing cells (Fig 2c). Viability assay of a series of human melanoma cell lines treated with RLS-12 showed it is more potent in inhibiting multiple cell lines growth compared with RLS-7 (Fig 2d and Fig S1d). We also analyzed the efficacy of RLS-12 against the human prostate lines RV1 and PC3 and observed killing efficacy in both (Fig 2e, and S2a). Dose-response analysis of RLS-7 and RLS-12 effects using Lu1205 melanoma cells revealed that RLS-12 was more potent in inhibiting HIF1α protein expression (Fig S2b). We also monitored effects of RLS-7 and RLS-12 on melanoma ability to form colonies (Fig 2f), where RLS-12 was shown to be more potent in inhibiting colony formation. The inhibition of RLS-12 on melanoma and prostate cancer cell growth on soft agar was shown in Fig S2c.
Figure 2. Characterization of RLS-7 derivatives.
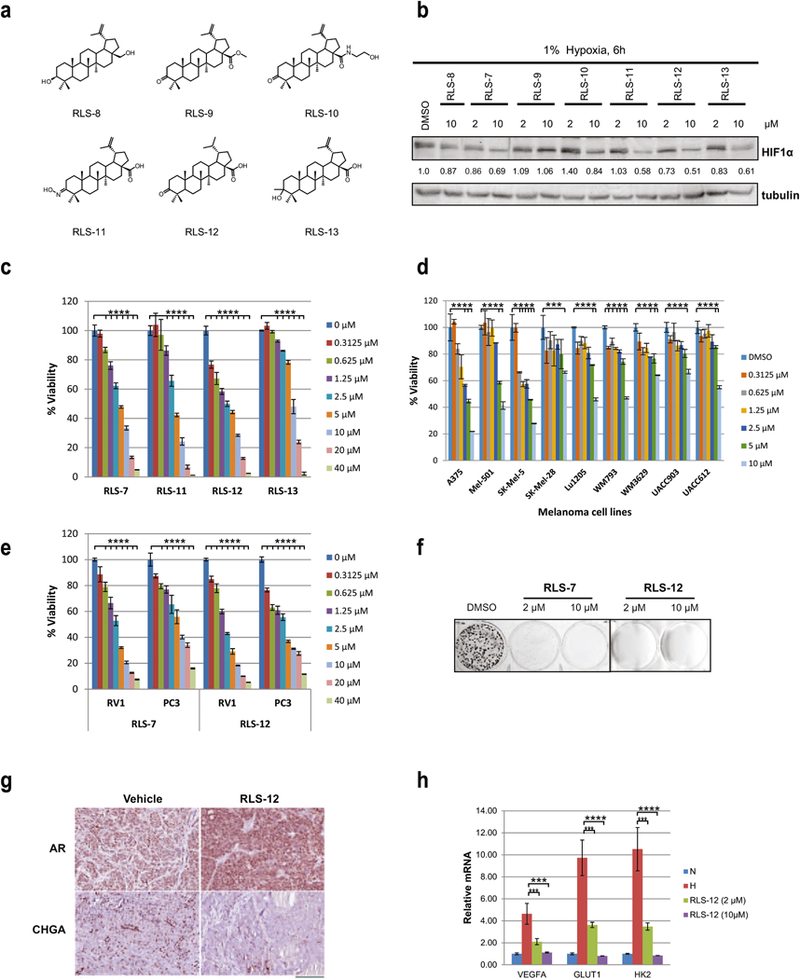
(a) Structures of RLS-7 derivatives. (b) A375 melanoma cells were treated with RLS-7 and indicated derivatives for 6 h under hypoxia. Whole cell lysates were immunoblotted with indicated antibodies. Quantification of immunoblots was performed using BioRad densitometer, relative to loading controls, noted under the blots (c) A375 cells were treated with different concentrations of indicated compounds and cell viability was assessed by ATPlite after 72 h. Each bar represents the mean ± standard deviation from three experiments. Representative data is shown. ****p < 0.0001 compared to the control (one-way ANOVA with Dunnett’s test). (d) Different melanoma cells were treated with RLS-12 and cell viability was assessed by ATPlite after 72 h. ****p < 0.0001 based on comparison with control using one-way ANOVA, as described above. (e) Prostate cancer cells RV1 and PC3 were treated with different concentrations of RLS-7 or RLS-12, and cell viability was assessed by ATPlite after 72 h. ****p < 0.0001 based on comparison with control using (one-way ANOVA with Dunnett’s test). (f) A375 cells were plated at low density and grown in medium containing vehicle, 2 µM or 10 µM of RLS-7, or RLS-12. The num ber of colonies formed after 10 days in culture was determined by crystal violet staining. (g) Immunohistochemistry for the expression of AR and chromogranin A (CHGA) of PCa tumors from castrated mice treated with RLS-12. Scale bar indicates 100 µm. (h) A375 cells treated with vehicle, 2 µM or 10 µM of RLS-12 for 24 h under hypoxia. mRNA was extra cted and subjected to qPCR analysis of indicated HIF target genes. Means ± standard errors were calculated based on two independent experiments. ***p < 0.001, ****p < 0.0001 were calculated based on comparison with DMSO-treated control using Student’s t test.
Siah inhibition has a marked effect on the ability of RV1 cells to exhibit neuroendocrine (NE) phenotypes in vitro and in vivo [5]. Thus, we assessed a potential effect of RLS-12 treatment of RV1 cells on NE phenotypes, such as neurite extension. RLS-12 treatment of RV1 cells, maintained under hypoxic conditions required to induce NE phenotypes, markedly inhibited neurite extension (Fig S2d), consistent with the role of Siah in their formation. We have also tested RLS-12 in a tissue recombination orthotopic grafts having RV1 prostatic epithelia with human carcinoma associated fibroblasts (CAF). In this study all mice were castrated after allowing the tumor to expand to initiate a neuroendocrine response by the tumor.
Subsequently, vehicle or RLS-12 was administered for two weeks prior to sacrifice. While no appreciable differences in tumor volume were found among the treatment groups, there was a marked change in the differentiation state of the tumors. We found that the neuroendocrine differentiation marker, chromogranin A (CHGA), reproducibly expressed in the vehicle treated castrated mice, was largely absent in the mice treated with RLS-12 (Fig 2g). Analogously, the observed down regulation of androgen receptor (AR) potentiated by castration, seemed to be restored in the tumors when RLS-12 was administered. However, there were minimal differences in proliferation or cell death in the treatment and control mice (data not shown).
RLS-12 also elicited effective growth inhibition of melanoma cells resistant to vemurafenib (A375R), suggesting that Siah2 inhibition could also address some melanoma drug sensitivity problems (Fig S2e). Consistent with previously observed effects of the developed compounds on HIF1α, we further established a dose-dependent decrease in transcript levels of HIF1α target genes VEGF, GLUT1, and HK2 (Fig 2h), following the treatment of human melanoma A375 cells with RLS-12.
Use of a Protein Thermal Shift assay to identify Siah inhibitors
An independent approach to identify small molecule inhibitors of Siah - is a Protein Thermal Shift assay (PTS), which is based on the principle that protein melting temperature is potentially altered by binding of a small molecule [19]. As this screen requires large quantities of purified proteins, we produced full-length Siah1 in baculovirus as well as truncated Siah1 (residues 90–282; i.e., lacking the N-terminal Ring domain) in E. coli, and purified them to homogeneity. Siah1 protein that was produced and purified from insect cultures was used for the final screen. Of 30,000 small molecules used in the previously described high throughput screening, 80 elicited a change in Siah1 melting temperature (Fig S3a), and of those, 15 (RLS-14 to RLS-28) were selected for the initial assessment, based on structure and biophysical properties. Among them, RLS-24 (adapalene, Fig 3a) elicited a more potent effect, as assayed by the degree of inhibition of HIF1α protein expression in melanoma cells (Fig 3b). Correspondingly, we also observed parallel dose-dependent inhibition of HRE-Luc reporter activity by RLS-24 (Fig 3c). Moreover, the effect on cell viability in melanoma cells treated with RLS-24 was more potent than that seen with other selected small molecule inhibitors in this group (Fig S3b).
Figure 3. Protein thermal shift assay screen and characterization of RLS-24 (adapalene) as a Siah small molecule inhibitor.
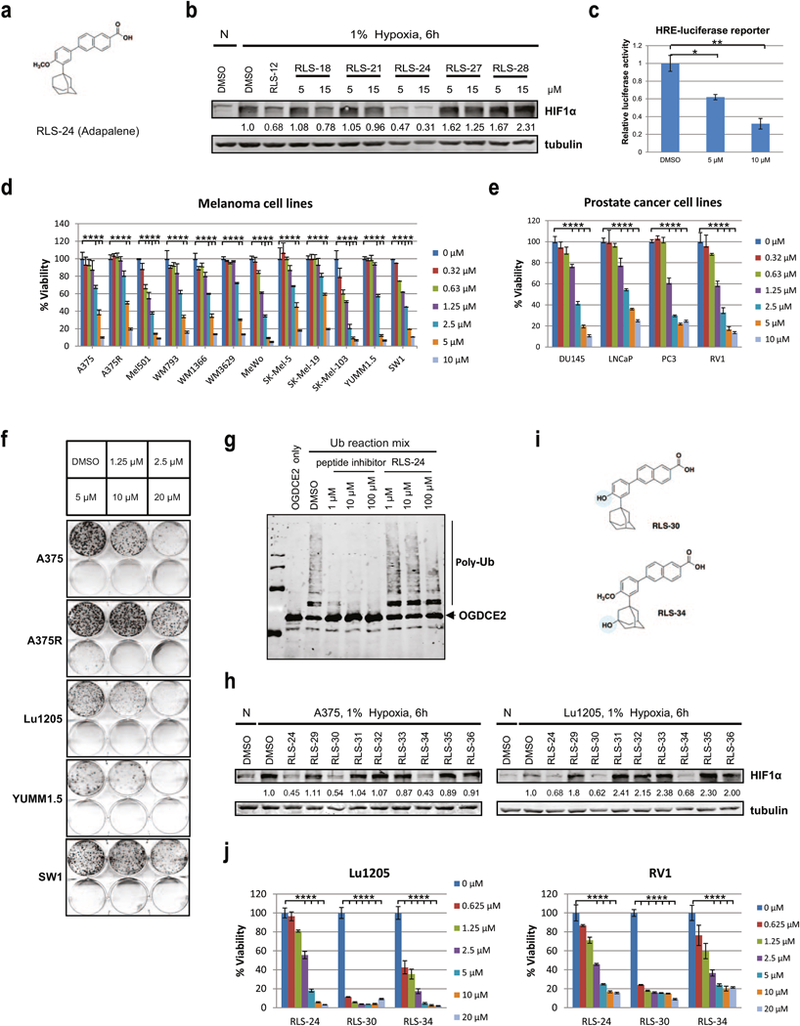
(a) Structure of RLS-24/adapalene. (b) Different compounds at 5 µM or 15 µM concentrations were added to A375 melanoma cells, which were cultured in 1% oxygen for 6 h. HIF1α levels were examined by Western Blot analysis. Quantification of immunoblots was performed using BioRad densitometer, relative to loading controls, noted under the blots. (c) A375 cells were transfected with HRE-firefly and renilla luciferase plasmids for 24 hours and treated with 2 µM or 10 µM of adapalene (RLS-24) under hypoxia. After 24 hours, cells were lysed and subjected to a luciferase assay. Data were calculated relative to DMSO-treated cells. Means ± standard errors calculated from three experiments. *p <0.05, **p < 0.01, compared to controls (one-way ANOVA). (d) Different human and mouse melanoma cells were treated with indicated concentrations of RLS-24, and cell viability was assessed by ATPlite after 72 h. Each bar represents the mean ± standard deviation of three measurements. Representative data was shown. ****p < 0.0001 was calculated based on comparison with the control (one-way ANOVA with Dunnett’s test). (e) Different human prostate cancer cells were treated with indicated concentrations of RLS-24, and cell viability was assessed by ATPlite after 72 h. ****p < 0.0001, compared to control (one-way ANOVA with Dunnett’s test). (f) Indicated human and mouse melanoma cells were plated at low density and grown in medium containing different concentrations of RLS-24. The number of colonies formed after 10 days in culture was determined by crystal violet staining. (g) Indicated concentrations of RLS-24 and a Siah peptide inhibitor (as a positive control) were incubated with purified Siah2 for 30 min, followed by addition of Siah2 substrate OGDCE2 and ubiquitination reagents (E1, E2, Ub). Mixtures were then incubated at 37°C for 45 min and subjected to Western Blot an alysis. (h) A375 and Lu1205 melanoma cells were treated with RLS-24 and derivatives for 6 h under hypoxia. Whole cell lysates were immunoblotted with indicated antibodies. Quantification of immunoblots was performed using BioRad densitometer, relative to loading controls, noted under the blots (i) Structures of RLS-24 derivatives RLS-30 and RLS-34. (j) Cells of the melanoma line Lu1205 and the prostate cancer line RV1 were treated with different concentrations of RLS-24, RLS-30 or RLS-34. Cell viability was assessed by ATPlite after 72 h. ****p < 0.0001 was calculated based on comparison with the control (one-way ANOVA with Dunnett’s test).
RLS-24 has been shown by others to possess anti-proliferative and pro-apoptotic effects in cultured colon carcinoma (CC-531, HT-29 and LoVo) and hepatoma (HepG2, Hep1B) lines [20, 21], largely by altering Caspase 3, Bcl2 and Bax activities [22]. We used RLS-24 to treat 12 human and murine melanoma lines of different genetic backgrounds and observed effective growth inhibition in all of them, with Mel501, SW1, and SK-MEL103 showing greatest sensitivity (~40% inhibition at 0.625 µM; Fig 3d). Similar analysis indicated a loss of viability in different PCa lines (Fig 3e). Likewise, colony formation culture by several melanoma and PCa lines (Fig 3f, S3c), including the vemurafenib-resistant melanoma line A375R (Fig 3f), was suppressed by RLS-24. The degree of the colony formation inhibition varied, with the less profound effect seen for vemurafenib-resistant melanoma, and SW1, a mouse melanoma line which was relatively sensitive to RLS-24-induced effective cell death (Fig 3d, 3f). However, overall results indicate that RLS-24 is generally effective in decreasing viability and growth of both melanoma and PCa lines.
We next investigated whether RLS-24 blocked Siah E3 ligase activity by performing ubiquitination assays and monitoring the degree of in vitro ubiquitination of Siah2 substrates in the presence and absence of the inhibitor. To achieve this, we examined in vitro ubiquitination of the Siah2 substrates ASPP2, Sprouty2 and OGDCE2 using bacterially-produced and then purified Siah2 plus substrates immuno-purified from HEK293 cells. Addition of RLS-24 to these reactions effectively decreased the degree of ubiquitination (Fig S3d). We also performed dose-response analysis of RLS-24’s effect on OGDCE2 ubiquitination, which confirmed inhibition of ubiquitination in vitro (Fig 3g).
We next assessed several RLS-24 analogs which one of them has been previously reported [23] and tested the effect of 8 compounds (RLS-29 to RLS-36) on HIF1α protein expression levels in both A375 and Lu1205 cells. It has been established that two analogs, demethylated Adapalene (RLS-30) and the hydroxylated analog RLS-34 were as potent as RLS-24 itself (Fig 3h). Notably, treatment of melanoma cells with an analogue RLS-35, which has a modified retinoic acid moiety, did not reduce HIF1α expression, pointing to the importance of this moiety for inhibiting Siah. Structures of the two most active analogues, RLS-30 and RLS-34, are shown in Fig 3i, and the comparison of both of them with RLS-24 in a viability assay revealed their greater potency (Fig 3j, Fig S3e; data not shown). Their propensity to induce apoptosis, was previously reported (15), and was confirmed for RLS-30 using Annexin V-PI FACS analyses (data not shown), which may be independent of their effect on Siah ubiquitin ligases (15).
In silico screen identifies new Siah inhibitors
A third, independent approach to identify Siah2 inhibitors relied on in silico screening of a published three dimensional structure of Siah [24]. We focused on the Siah substrate binding domain characterized in earlier studies [9, 25]. This domain binds a peptide designated as PHYL derived from the Drosophila protein phyllopod [26–28], which in flies serves as an adaptor for Siah2 substrates. Although PHYL homologues were not identified in mammalian systems, they are as potent in inhibiting Siah binding and ubiquitination of its substrates [29]. The 19aa PHYL peptide used in our earlier studies potently inhibited Siah ubiquitin ligase activities and has served as the basis to design other short Siah2 inhibitory peptides [9]. We evaluated 5 million drug-like compounds in a virtual screen (Fig 4a, S4a) and identified ~100 putative inhibitors of Siah2 (RLS-37 to RLS-135), which we then purchased and tested for Siah ubiquitin ligase inhibition by monitoring changes of HIF1α expression in melanoma cultured cells (Fig S4b). Of those, we confirmed dose-dependent HIF1α inhibition by RLS-96, RLS-104, and RLS-111 (Fig 4b). Cell viability assays revealed RLS-96 to be the most effective, both in promoting cell death and in CFE assays (Fig 4c; S4c). Moreover, degree of cell death seen in WT mouse embryo fibroblasts that were treated with RLS-96 was attenuated in Siah1/2 DKO cells (1–7) (Fig 4d), supporting the notion that cell death can partially be attributed to Siah inhibition. In vitro ubiquitination studies also confirmed that RLS-96 antagonizes Siah-dependent OGDCE2 ubiquitination (Fig 4e), although to a lesser degree than RLS-24.
Parallel comparison of top Siah1/2 small molecule inhibitors.
To evaluate the efficacy of the top inhibitor identified in each of the three screening campaigns (RLS-12, RLS-24, RLS-96) we performed parallel assessments for key Siah1/2 related indications. Effect on viability of 9 melanoma cell lines confirmed RLS-24 as the most potent compound (2.5μM causing 50% inhibition in about half of the lines; Fig S5a), which was also reflected in effective inhibition of CFE (Fig 5a). In both cases, RLS-12 scored second in its effects on melanoma cells (Fig S5a, 5a). To assess the toxicity of these compounds on non-transformed cells, we compared the effect RLS-12, RLS-24 and RLS-96 on human melanocytes (Hermes H3A) with their effect on the melanoma cell line A375.
Figure 5. Parallel comparison of top Siah small molecule inhibitors.
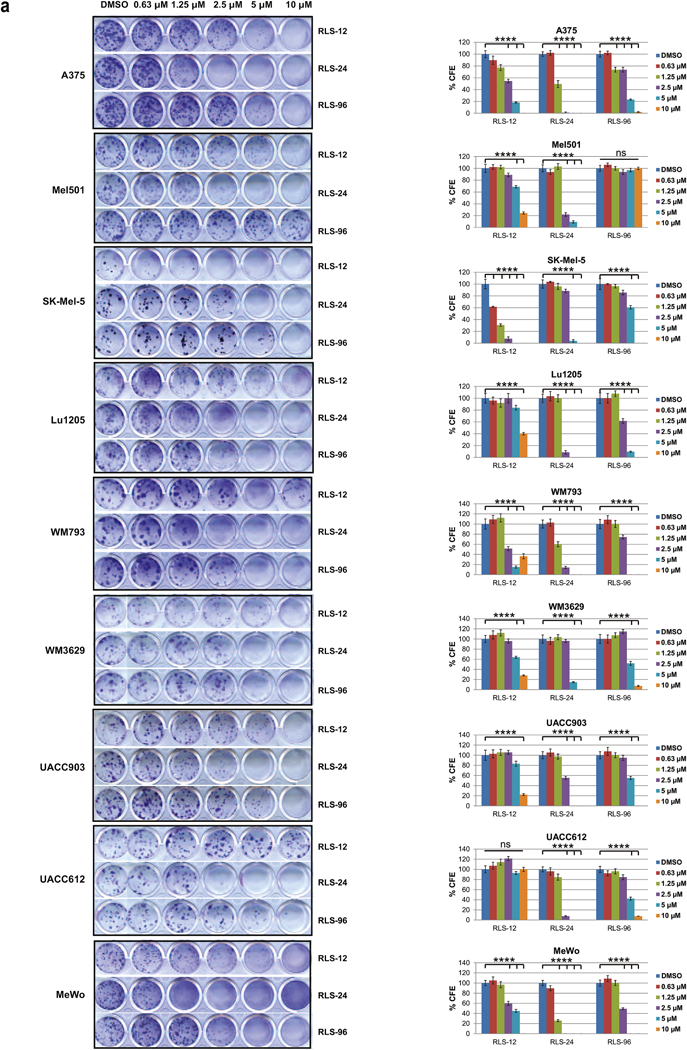
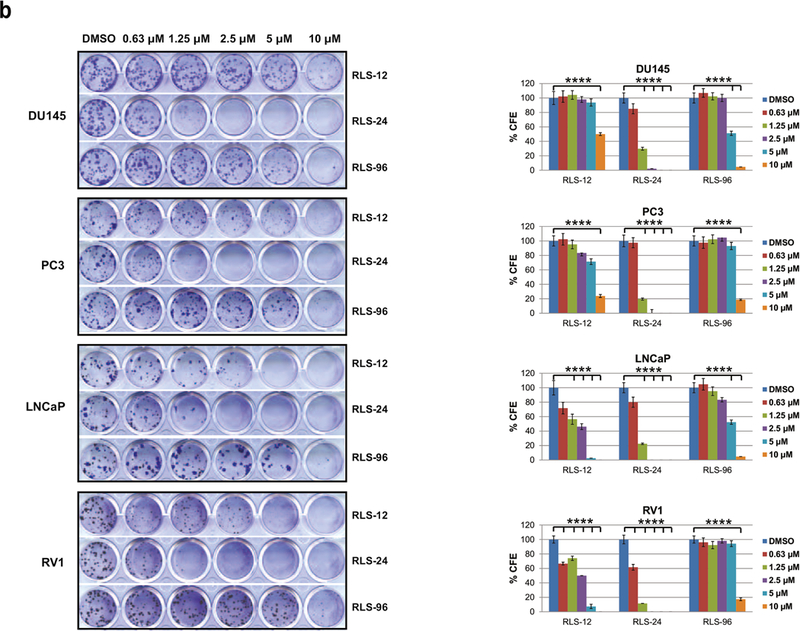
(a) Colony formation assay. Nine human melanoma cell lines were plated at low density (100 cells/well in 24-well plates) and grown in medium containing DMSO or indicated concentration of RLS-12, RLS-24 and RLS-96. The number of colonies formed after 7–14 days in culture was determined by crystal violet staining. Right panel, quantification of colonies shown on the left panel. Each bar represents the mean ± standard deviation of three measurements. ****p < 0.0001 was calculated based on comparison with control (one-way ANOVA with Dunnett’s test). (b) Four prostate cancer cell lines were subjected to colony formation assay as described in 5a. Each bar represents the mean ± standard deviation of three measurements. ****p < 0.0001 computed based on comparison with the control (one-way ANOVA with Dunnett’s test). (c) Inhibition of melanoma cell growth by RLS-12 and RLS-24 is partially rescued by CRISPR/CAS9 deletion of Siah2. Mouse melanoma cell line YUMM1.7 and CRISPR deletion of Siah2 YUMM1.7 were treated with indicated compounds and cell viability was assessed 72 h later using ATPlite. Viability of YUMM1.7 parental cells was set at 1 for each indicated compound at each indicated concentration. Each bar represents the mean ± standard deviation of two measurements. *p < 0.05, **p < 0.01, ***p < 0.001, were calculated using Student’s t test. (d) Inhibition of MEF cell growth by RLS-12 and RLS-24 is partially rescued in Siah1/2 double knockout MEF cells. MEF WT and Siah1/2 double knock out cells were treated with indicated compounds and cell viability was assessed 72 h later using ATPlite. Each bar represents the mean ± standard deviation of two measurements. *p < 0.05, **p < 0.01, were calculated using Student’s t test. (e) A375 cells were transfected with HRE-firefly and renilla luciferase plasmids for 24 h and treated with different compounds at indicated concentrations, with or without 10 µM of the PHD inhibitor FG4592. Cells were then lysed 24 hours later and subjected to a luciferase assay. Data are expressed relative to DMSO-treated cells. Means ± standard error were calculated based on two experiments. *p < 0.05, **p < 0.01, ***p < 0.001, were calculated based on comparison with the control (one-way ANOVA with Dunnett’s test). (f) A375 cells were incubated with either vehicle (DMSO) or different concentrations of indicated compounds under hypoxia overnight. Cells were harvested and whole cell lysates were analyzed by immunoblot using the indicated antibodies. Quantification of immunoblots was performed using BioRad densitometer, relative to loading controls, noted under the blots (g) Indicated concentrations of RLS-12, RLS-24 and RLS-96 and a Siah peptide inhibitor (as a positive control) were subjected to in-vitro ubiquitination assay as described in Fig 3g.
Although at the higher concentrations, all three compounds elicited toxicity on normal melanocytes, 80% of the melanocytes retained their viability at the dose of EC50 for melanoma cells (Fig. S5a, lower panel).
Assessment of these compounds in 4 prostate cancer cell lines confirmed RLS-24 to be superior over RLS-12 and RLS-96, respectively for both viability and inhibition of CFE (Fig S5b, 5b). Likewise, the ability to attenuate formation of neurites in culture was best seen following RLS-24 treatment (Fig S5c). Assessment of 4 liver and 2 pancreatic cancer cell lines also identified RLS-24 to be most effective, compared with RLS-12 and RLS-96, on viability and CFE (Fig. S5d, S5e, S5f, S5g; efficiency of Siah2 KD is shown in representative Fig. S5c).
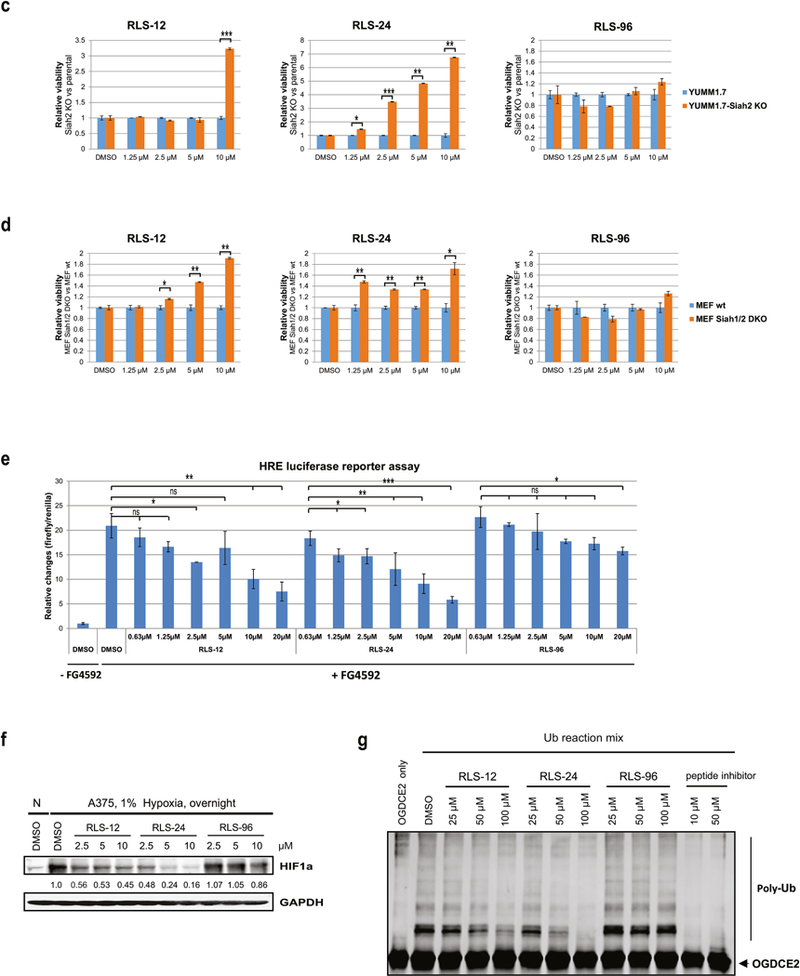
To verify that the effect of these compounds on cell viability is Siah1/2 dependent we used the melanoma A375 cells that were subjected to Siah2 KD using corresponding shRNA (Fig S5h; S5c), and then to treatment with each of these inhibitors. In the absence of Siah2, each of these inhibitors was less effective in killing melanoma cells, in particular at the higher concentration used (Fig S5i). Further, using murine melanoma cells in which Siah2 was genetically inactivated by the CRISPR/CAS9 technology (confirmed by genomic sequencing; not shown) and MEF cells with Siah1 and Siah2 double knockout (3,7), confirmed greater resistance to RLS-12 and RLS-24, compared with RLS-96 (Fig. 5c & 5d). These findings suggest RLS-96 may be more specific for Siah1, whereas RLS-12 and RLS-24 are more Siah2-directed.
Siah inhibition can also be assessed by monitoring activity or availability of prolyl hydroxylases, which are Siah substrates that hydroxylate on proline residues and hence destabilize HIF1α. Since Siah2 targets PHD1/3 for degradation, resulting in elevated HIF1α levels, Siah2 inhibition would increase PHD1/3 levels and promote robust HIF1α protein degradation. Moreover, PHD inhibitors are available, allowing us to test their ability to override Siah2 inhibition, as treatment with PHD inhibitors should increase HIF1α levels and activity. Thus, we combined the pharmacological PHD inhibitor FG4592 with Siah2 inhibitors and monitored changes in HIF1α transcriptional activities, by assessing HRE-Luc that was transfected into the indicated melanoma cells. While FG4592 treatment increased HRE-Luc activity (due to PHD inhibition and increased HIF1α activity), the combined treatment of FG4592 with the indicated Siah inhibitor dose-dependently decreased HRE-Luc activity (Fig 5e; S4d as proof of concept, using PHYL as positive control). Overall, comparison of all the inhibitors assessed here suggest that RLS-24 may be the most effective in antagonizing HRE-Luc activity, with RLS-12 exhibiting similar effectiveness (Fig. 5e). The latter corresponds with the effect of these compounds on the level of HIF1α expression (Fig. 5f).
Next, we compared the effect of the three inhibitors on in vitro ubiquitination of OGDCE2, one of Siah substrates. Here again, RLS-24 was most effective, compared with the other two inhibitors (Fig. 5g).
RNAseq analysis in murine melanoma subjected to CRISPR/CAS9 deletion of Siah2 led us to determine whether Siah1/2 inhibitors will also attenuate their expression. Analysis of four genes, Afap1/2, Neb, Fbn2, Casd1, that were upregulated in the Siah2 CRISPR’d YUMM1.7 murine melanoma cells (Fig 6a) revealed that they were also upregulated following treatment by each of the Siah1/2 inhibitors (Fig. 6b). These findings further support the selectivity of Siah1/2 inhibitors, which phenocopy effect of genetic Siah2 inactivation.
Figure 6. Siah1/2 inhibitors phenocopy gene expression pattern seen in genetically inactivated Siah2 and their combination elicits additive effect.
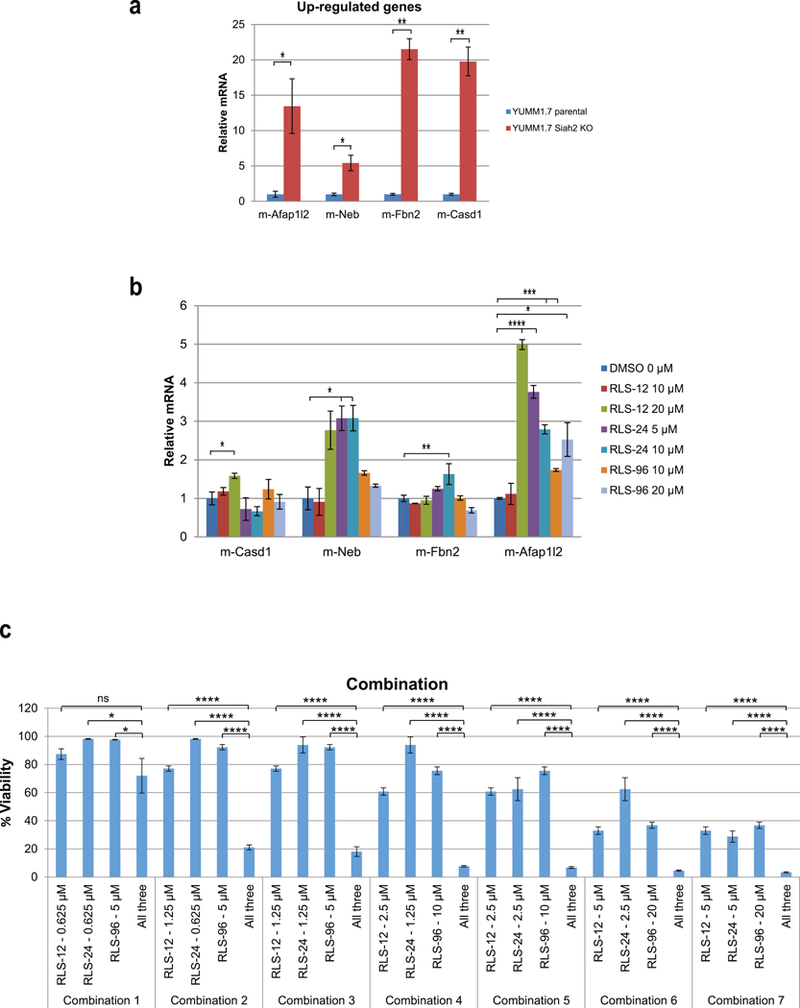
(a) Level of transcripts for selected genes upregulated in YUMM1.7, in which Siah2 was subjected to CRISPR-CAS9 KO compared with control YUMM1.7 Siah2 WT cells. Each bar represents the means ± standard error from two experiments. *p < 0.05, **p < 0.01, were calculated using Student’s t test. (b) RNAs were extracted from YUMM1.7 cells following treatment with indicated concentration of RLS-12, RLS-24 and RLS-96 for 24 h, followed by qPCR analysis. Each bar represents the means ± standard error from two experiments. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001 were calculated using Student’s t test. (c) A375 cells were treated with single or combination of three compounds at indicated concentrations. Cell viability was assessed 72 h later using ATPlite. Each bar represents the mean ± standard deviation of three measurements. *p < 0.05, ****p < 0.0001 were calculated using Student’s t test.
Lastly, we assessed the possible combination of Siah1/2 inhibitors, on viability of melanoma cells. Parallel assessment of the 3 inhibitors allowed us to identify the IC50 for each, a concentration that was used in assessing their possible additive or synergistic effect on melanoma cell viability (Fig S6a). Notably, the combination of the three inhibitors was superior, to their effect when tested individually (Fig. 6c). Likewise, combination of only two out of the three compounds resulted in additive effect on melanoma cell viability (Fig. S6b, S6c, S6d).
4. Discussion
Ubiquitin ligases, which regulate signaling in numerous cell contexts, are highly desirable targets in medicine, as their deregulation results in disease-related phenotypes [30, 31]. Yet, ubiquitin ligases are also challenging drug targets, given that with the exception of HECT domain family of ligases, RING finger ubiquitin ligases lack catalytic activity_and their inhibition likely requires the disruption of specific protein-protein interactions.
Among two members of the Siah ubiquitin ligases family, Siah2 has been linked to oncogenic activities and shown by independent laboratories to function in melanoma, breast, and prostate cancers [4, 5, 18, 32, 33]. Siah2 inhibition reportedly attenuates melanoma growth, development, and metastatic capacity and antagonizes formation of neuroendocrine prostate cancer and evolution of hormone-refractory prostate tumors, albeit by distinct mechanisms. Thus, inhibiting Siah2 is desirable in these conditions. Our earlier attempts to inhibit Siah ubiquitin ligases have included a Meso-scale based analysis (Meso Scale Discovery) that identified menadione as Siah1/2 inhibitory small molecule. Later, using a structure-based approach, we designed small peptides that effectively inhibited Siah by covalent binding, with proven effect in vitro and in cultured cells.
Here we employed three independent platforms to identify putative Siah inhibitors. Each resulted in identification of distinct small molecule inhibitors that inhibit Siah activity, measured by a number of independent parameters. The effect on HIF1α protein levels was used as first surrogate marker, given Siah1/2 inhibition of its regulator prolyl-hydroxylase 3. Along those lines, a more rigorous assessment for Siah1/2 putative inhibitors was their ability to reverse the effect of FG4592 – PHD inhibitors, which was measured at the level of HIF1α transcriptional activities. Further, assessment of Siah2 ubiquitin ligase activity in vitro, confirmed the ability of inhibitors identified here, to attenuate, albeit to different degrees, its ability to ubiquitinate one of its select substrates. Independent assessment for Siah1/2 inhibitors also relied on their effectiveness on cells lacking Siah2, confirming greater resistance, varying among the inhibitors tested. Since changes in gene expression were mapped for melanoma cells lacking Siah2, we were able to confirm that such changes can be phenocopied by the inhibitors characterized here. Collectively, these independent assessments establish the impact of Siah1/2 inhibitors we have identified in independent screening campaigns, on Siah1/2 related cellular functions as on its own activity as ubiquitin ligases. The select inhibitors were effective in inhibition of melanoma, but also prostate and to lesser degree liver and pancreatic cancer viability and colony formation, attesting to their overall impact on these tumor cells.
Each inhibitor was also capable of decreasing viability of both melanoma and to a greater extent prostate cancer cultures and decreasing CFE, indicative of a positive anti-tumor effect. The most effective compound identified in the 3 campaigns was adapalene/RLS-24 and its analogue (RLS-30), both of which promoted effective cell death at nanomolar concentrations, effectively inhibited Siah2 ubiquitin ligase activity in vitro, and altered HIF1α activity in a PHD-dependent manner. At times, inhibitors were more potent in prostate cancer cells compared with melanoma, and others, melanoma better then prostate cancer cells. Among the plausible explanations are the differential activity of Siah1/2 genes in the different tumor cells, and correspondingly, different affinity of RLS’s to Siah1 vs. Siah2. The latter is supported by the observation that certain RLS are more effective in Siah1 KO compared with Siah2 KO MEFs and vice versa, requiring further assessment of RLS to each of the Siah proteins.
Since initial characterization of these inhibitors took place individually, we have repeated key assessments by comparing these inhibitors in parallel. These assessments clearly identified RLS-24 as the most potent inhibitor, in each of the assays performed in this study, with RLS-12 and RLS-96 following, albeit with lower potency and effectiveness. Clearly, combination of RLS-12 and RLS-96 using IC20-IC50 for each inhibitor, revealed a clear additive effect. Likewise, combination of all 3 inhibitors, each at low concentrations that had limited impact on tumor cells viability, confirmed their significant additive effect. These observations point to the possible evaluation of such combination for in vivo assessment of tumor models where Siah1/2 were demonstrated to play critical role, including melanoma and prostate cancer. Initial assessments in xenografts for prostate and melanoma tumors revealed that further development of these compounds properties and formulations is needed, to improve solubility and stability.
Among lessons learned from our exhaustive approach to identify Siah1/2 inhibitors are: (i) the need to use diverse assay platforms; (ii) the importance of assessing Siah2 inhibition using a cell-based screen; (iii) the consideration for studying larger molecules, including natural products, to target large protein-protein complexes, characteristic of ubiquitin ligase complexes; and (iv) the consideration of using additional approaches, including cereblon-tethered structures, to develop Siah2-specific inhibitors. Future studies should also consider the possible need to distinguish Siah2 inhibition from the inhibition of Siah1, given the notion that these two family members, although shares some of the substrates identified so far, may also have distinct targets and biological properties, which may be cell type dependent.
In all, our screening campaigns identified inhibitor prototypes for the ubiquitin ligases Siah1/2 which provide the basis for further development to improve their overall potency and to optimize their drug-like properties and formulation.
Supplementary Material
Figure S1. (a) Basic workflow of the affinity selection-mass spectrometry (AS-MS) ligand binding screen. A detailed description of the screen is found at Materials and Methods. (b) Structures of lupeol and oleanolic acid. (c) Siah2 knockdown is partially rescued inhibition of cell growth by RLS-7. HEK293 cells were transfected with either empty vector pLKO or shSiah2 for 24 h and treated with RLS-7 at indicated concentrations. Cell viability was assessed 48 h later using ATPlite. Each bar represents the mean ± standard deviation of three measurements. *p < 0.05, **p < 0.01, were calculated using Student’s t test. (d) Different melanoma cells and HEK293 cells were plated in 96-well plates and incubated with indicated concentrations of RLS-7. Cell viability was assessed 72 h later. Each bar represents the mean ± standard deviation of three measurements. ****p < 0.0001 were computed based on comparison to the control (one-way ANOVA with Dunnett’s test). ns- not significant. (e) Indicated cultures were plated at low density (500 cells/well in 6-well plates) and grown in medium containing DMSO, 2 μM or 10 μM RLS-7. The number of colonies formed after 10 days in culture was determined by crystal violet staining. (f) Different patient-derived primary melanoma cells were plated in 96-well plates and incubated with indicated concentrations of RLS-7. Cell viability was assessed 72 h later. Each bar represents the mean ± standard deviation of three measurements. ****p < 0.0001 was computed based on comparison with the control (one-way ANOVA with Dunnett’s test). (g) RV1 cells were treated with RLS-7 at indicated concentrations for 12 h and 24 h. RNAs were then isolated from cells and subjected to RT-qPCR analysis for indicated AR target genes. Each bar represents the mean ± standard deviation of three measurements. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001, were calculated based on comparison with the control using Student’s t test. ns- not significant.
Figure S2. (a) Prostate cancer cell lines RV1 and PC3 were treated with indicated concentrations of RLS-7 derivatives. Cell viability was assessed 72 h later. Each bar represents the mean ± standard deviation of three measurements. ****p < 0.0001 were calculated based on comparison with the control (one-way ANOVA with Dunnett’s test). (b) Lu1205 melanoma cells were treated with RLS-7 or RLS-12 at the indicated concentrations under hypoxia. Whole cell lysates were immunoblotted with indicated antibodies. Quantification of immunoblots was performed using BioRad densitometer, relative to loading controls, noted under the blots (c) A375, RV1 and PC3 cells were plated and grown in soft agar with medium containing vehicle, 2 μM or 10 μM of RLS-12. The number of colonies formed after 2-3 weeks in culture was determined by crystal violet staining. (d) RV1 cells were plated at low density and treated with 5 μM of RLS-12. Cells were kept in 1% hypoxia for one week before images were taken using bright field microscopy. (e) A375 melanoma cells and Vemurafenib-resistant cells A375R were treated with indicated concentrations of RLS-12. Cell viability was assessed 72 h later. Each bar represents the mean ± standard deviation of three measurements. ****p < 0.0001 was calculated based on comparison to the control (one-way ANOVA with Dunnett’s test).
Figure S3. (a) Representative melting curve plot with PHYL (positive control) and representative compound. (b) Melanoma cells A375 were treated with different concentrations of compounds selected from the protein thermal shift assay, and cell viability was assessed by ATPlite after 72 h. Each bar represents the mean ± standard deviation of three measurements. ****p < 0.0001 was calculated based on comparison with the control (one-way ANOVA with Dunnett’s test). (c) Different human prostate cancer cells were plated at low density and grown in medium containing different concentrations of RLS-24. The number of colonies formed after 10 days in culture was determined by crystal violet staining. (d) RLS-24 was incubated with the purified Siah2 for 30 min followed by addition of ubiquitination reagents (E1, E2, Ub) and Siah2 substrates ASPP2, Sprouty 2 or OGDCE2. Mixtures were then incubated at 37°C for 45 min and subjected to Western Blot analysis. (e) Human melanoma A375 and mouse melanoma SW1 cells were treated with different concentrations of RLS-24, RLS-30 or RLS-34. Cell viability was assessed by ATPlite after 72 h. Each bar represents the mean ± standard deviation of three measurements. ****p < 0.0001 based on comparison with the control (one-way ANOVA with Dunnett’s test).
Figure S4. (a) Model of compound RLS-96 binding to Siah 2. (b) Melanoma cells were incubated with 5 μM, 10 μM of selected compounds for 6 h under hypoxia. Cells were harvested and whole cell lysates were immunoblotted with indicated antibodies. Quantification of immunoblots was performed using BioRad densitometer, relative to loading controls, noted under the blots (c) Viability assay of A375 cells in the presence of indicated compounds. Each bar represents the mean ± standard deviation of three measurements. ****p < 0.0001 based on comparison with the control (one-way ANOVA with Dunnett’s test).
Figure S5. (a) Nine different melanoma cells were plated in 96-well plates and incubated with indicated concentrations of RLS-12, RLS-24 and RLS-96. Cell viability was assessed 72 h later. Each bar represents the mean ± standard deviation of three measurements. ****p < 0.0001 was calculated based on comparison with the control (one-way ANOVA with Dunnett’s test). Lower panel, A375 and human melanocytes Hermes 3A cells were treated with RLS-12, RLS-24 and RLS-96 followed 72 h later by a viability assay. Each point represents the means ± standard error from two experiments. **p < 0.01, ***p < 0.001, were calculated using Student’s t test. (b) Four prostate cancer cell lines were subjected to the viability assay as described in (a). (c) RV1 cells were plated at low density and treated with 1.25 μM of RLS-12, RLS-24 and RLS-96. Cells were kept in 1% hypoxia for one week before images were taken using bright field microscopy. (d) Viability assay of four human liver cancer cell lines as described in Figure S5a. (e) Colony formation assay of four human liver cancer cell lines. Each bar represents the mean ± standard deviation of three measurements. ****p < 0.0001 was calculated based on comparison with the control (oneway ANOVA with Dunnett’s test). (f) Viability assay of two human pancreatic cancer cell lines as described in Figure S5a. (g) Colony formation assay of two human pancreatic cancer cell lines as described in Figure S5e. (h) A375 cells were transfected with either shCtrl or shSiah2 for 24 h. RNA was extracted from cells and subjected to qPCR analysis. Each bar represents the means ± standard error from two experiments. ***p < 0.001, calculated using Student’s t test. (i) A375 cells were transfected with shCtrl or shSiah2 for 24 h and treated with RLS-12, RLS-24 and RLA-96 at indicated concentrations. Cell viability was assessed 48 h later using ATPlite. Each bar represents the means ± standard deviation of three measurements. *p < 0.05, **p < 0.01, ***p < 0.001, were calculated using Student’s t test. (j) A375 cells were transfected with HRE-luciferase plasmid and pcDNA3-PHYL for 24 h and treated with or without 10 μM of the PHD inhibitor FG4592 under normoxia or hypoxia. Cells were lysed and subjected to a luciferase assay after 24 h. Each bar represents the means ± standard error from two experiments. *p < 0.05, **p < 0.01, were calculated using Student’s t test.
Figure S6. (a) A375 cells were treated with RLS-12, RLS-24 and RLS-96 at indicated concentrations. Cell viability was assessed 72 h later using ATPlite. Each bar represents the mean ± standard deviation of three measurements. ****p < 0.0001 was calculated based on comparison with the control (one-way ANOVA). (b) A375 cells were treated with both RLS-12 and RLS-24 at indicated concentrations. Cell viability was assessed 72 h later using ATPlite. Each bar represents the mean ± standard deviation of three measurements. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001, calculated using Student’s t test. (c) A375 cells were treated with both RLS-12 and RLS-96 at indicated concentrations. Cell viability was assessed 72 h later using ATPlite as described in Figure S6b. (d) A375 cells were treated with both RLS-24 and RLS-96. Cell viability was assessed 72 h later using ATPlite as described in Figure S6b.
Highlights.
Three screening campaigns identify small molecule inhibitors to Siah1/2 ubiquitin ligases
Each screening campaign identifies distinct small molecule Siah1/2 inhibitors
Growth of melanoma and prostate cancer is inhibited by Siah1/2 inhibitors
Adapalene and related analogs identified as potent Siah1/2 inhibitors
5. Acknowledgements
This work is dedicated to the memory of Dr. Gregory Roth, who has contributed to the AS-MS screen and synthesis of related analogs, guiding our first steps in the quest to identify and study novel Siah1/2 small molecule inhibitors. We also thank Drs. Shiri Ashkenazi for generating the CRISPR Siah2 melanoma cells and to Stephanie Meyers for their characterization, Brian James for help with their RNAseq, Hyungsoo Kim for critical review of this manuscript, and members of the Ronai lab for continued discussions.
Funding: Support by DOD grant W81XWH-14-1-0551 and P01 grant P01 CA128814-08 (to ZAR) is gratefully acknowledged.
Abbreviations
- HIF1α
hypoxia inducible factor 1 alpha
- NCOR1
Nuclear co-repressor 1
- ATF4
activating transcription factor 4
- PHYL
phyllopod
- RLS
Ronai Lab Siah inhibitor
- CFE
colony forming efficiency
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of Interest
Authors declare they do not have any conflict of interest pertaining to the studies performed in this manuscript.
References:
- [1].Nakayama K, Frew IJ, Hagensen M, Skals M, Habelhah H, Bhoumik A, Kadoya T, Erdjument-Bromage H, Tempst P, Frappell PB, Bowtell DD, Ronai Z, Siah2 regulates stability of prolyl-hydroxylases, controls HIF1alpha abundance, and modulates physiological responses to hypoxia, Cell, 117 (2004) 941–952. [DOI] [PubMed] [Google Scholar]
- [2].Qi J, Kim H, Scortegagna M, Ronai ZA, Regulators and effectors of Siah ubiquitin ligases, Cell Biochem Biophys, 67 (2013) 15–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Scortegagna M, Kim H, Li JL, Yao H, Brill LM, Han J, Lau E, Bowtell D, Haddad G, Kaufman RJ, Ronai ZA, Fine tuning of the UPR by the ubiquitin ligases Siah1/2, PLoS Genet, 10 (2014) e1004348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Qi J, Nakayama K, Gaitonde S, Goydos JS, Krajewski S, Eroshkin A, Bar-Sagi D, Bowtell D, Ronai Z, The ubiquitin ligase Siah2 regulates tumorigenesis and metastasis by HIF-dependent and -independent pathways, Proc Natl Acad Sci U S A, 105 (2008) 16713–16718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Qi J, Nakayama K, Cardiff RD, Borowsky AD, Kaul K, Williams R, Krajewski S, Mercola D, Carpenter PM, Bowtell D, Ronai ZA, Siah2-dependent concerted activity of HIF and FoxA2 regulates formation of neuroendocrine phenotype and neuroendocrine prostate tumors, Cancer Cell, 18 (2010) 23–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Qi J, Tripathi M, Mishra R, Sahgal N, Fazli L, Ettinger S, Placzek WJ, Claps G, Chung LW, Bowtell D, Gleave M, Bhowmick N, Ronai ZA, The E3 ubiquitin ligase Siah2 contributes to castration-resistant prostate cancer by regulation of androgen receptor transcriptional activity, Cancer Cell, 23 (2013) 332–346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Kim H, Claps G, Moller A, Bowtell D, Lu X, Ronai ZA, Siah2 regulates tight junction integrity and cell polarity through control of ASPP2 stability, Oncogene, 33 (2014) 2004–2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Shah M, Stebbins JL, Dewing A, Qi J, Pellecchia M, Ronai ZA, Inhibition of Siah2 ubiquitin ligase by vitamin K3 (menadione) attenuates hypoxia and MAPK signaling and blocks melanoma tumorigenesis, Pigment Cell Melanoma Res, 22 (2009) 799–808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Stebbins JL, Santelli E, Feng Y, De SK, Purves A, Motamedchaboki K, Wu B, Ronai ZA, Liddington RC, Pellecchia M, Structure-based design of covalent Siah inhibitors, Chem Biol, 20 (2013) 973–982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Cloutier P, Lavallee-Adam M, Faubert D, Blanchette M, Coulombe B, A newly uncovered group of distantly related lysine methyltransferases preferentially interact with molecular chaperones to regulate their activity, PLoS Genet, 9 (2013) e1003210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Santelli E, Leone M, Li C, Fukushima T, Preece NE, Olson AJ, Ely KR, Reed JC, Pellecchia M, Liddington RC, Matsuzawa S, Structural analysis of Siah1-Siah-interacting protein interactions and insights into the assembly of an E3 ligase multiprotein complex, J Biol Chem, 280 (2005) 34278–34287. [DOI] [PubMed] [Google Scholar]
- [12].Lack NA, Axerio-Cilies P, Tavassoli P, Han FQ, Chan KH, Feau C, LeBlanc E, Guns ET, Guy RK, Rennie PS, Cherkasov A, Targeting the binding function 3 (BF3) site of the human androgen receptor through virtual screening, J Med Chem, 54 (2011) 8563–8573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Ban F, Dalal K, Li H, LeBlanc E, Rennie PS, Cherkasov A, Best Practices of Computer-Aided Drug Discovery: Lessons Learned from the Development of a Preclinical Candidate for Prostate Cancer with a New Mechanism of Action, J Chem Inf Model, 57 (2017) 1018–1028. [DOI] [PubMed] [Google Scholar]
- [14].MolecularOperatingEnvironment, https://www.chemcomp.com/, Molecular Operating Environment (MOE), Chemical Computing Group ULC, 1010 Sherbooke St. West, Suite #910, Montreal, QC, Canada, H3A 2R7, 2017, (2016). [Google Scholar]
- [15].Halgren TA, Murphy RB, Friesner RA, Beard HS, Frye LL, Pollard WT, Banks JL, Glide: a new approach for rapid, accurate docking and scoring. 2. Enrichment factors in database screening, J Med Chem, 47 (2004) 1750–1759. [DOI] [PubMed] [Google Scholar]
- [16].Zsoldos Z, Reid D, Simon A, Sadjad SB, Johnson AP, eHiTS: a new fast, exhaustive flexible ligand docking system, J Mol Graph Model, 26 (2007) 198–212. [DOI] [PubMed] [Google Scholar]
- [17].Banerjee J, Mishra R, Li X, Jackson RS 2nd, Sharma A, Bhowmick NA, A reciprocal role of prostate cancer on stromal DNA damage, Oncogene, 33 (2014) 4924–4931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Qi J, Pellecchia M, Ronai ZA, The Siah2-HIF-FoxA2 axis in prostate cancer - new markers and therapeutic opportunities, Oncotarget, 1 (2010) 379–385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Axelsson H, Almqvist H, Seashore-Ludlow B, Lundback T, Screening for Target Engagement using the Cellular Thermal Shift Assay - CETSA, in: Sittampalam GS, Coussens NP, Brimacombe K, Grossman A, Arkin M, Auld D, Austin C, Baell J, Bejcek B, Chung TDY, Dahlin JL, Devanaryan V, Foley TL, Glicksman M, Hall MD, Hass JV, Inglese J, Iversen PW, Kahl SD, Kales SC, Lal-Nag M, Li Z, McGee J, McManus O, Riss T, Trask OJ Jr., Weidner JR, Xia M, Xu X (Eds.) Assay Guidance Manual, Bethesda (MD), 2004. [PubMed] [Google Scholar]
- [20].Ocker M, Herold C, Ganslmayer M, Hahn EG, Schuppan D, The synthetic retinoid adapalene inhibits proliferation and induces apoptosis in colorectal cancer cells in vitro, Int J Cancer, 107 (2003) 453–459. [DOI] [PubMed] [Google Scholar]
- [21].Shi XN, Li H, Yao H, Liu X, Li L, Leung KS, Kung HF, Lin MC, Adapalene inhibits the activity of cyclin-dependent kinase 2 in colorectal carcinoma, Mol Med Rep, 12 (2015) 6501–6508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Ocker M, Herold C, Ganslmayer M, Zopf S, Hahn EG, Schuppan D, Potentiated anticancer effects on hepatoma cells by the retinoid adapalene, Cancer Lett, 208 (2004) 51–58. [DOI] [PubMed] [Google Scholar]
- [23].Han T, Goralski M, Capota E, Padrick SB, Kim J, Xie Y, Nijhawan D, The antitumor toxin CD437 is a direct inhibitor of DNA polymerase alpha, Nat Chem Biol, 12 (2016) 511–515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Polekhina G, House CM, Traficante N, Mackay JP, Relaix F, Sassoon DA, Parker MW, Bowtell DD, Siah ubiquitin ligase is structurally related to TRAF and modulates TNF-alpha signaling, Nat Struct Biol, 9 (2002) 68–75. [DOI] [PubMed] [Google Scholar]
- [25].Zhang Q, Wang Z, Hou F, Harding R, Huang X, Dong A, Walker JR, Tong Y, The substrate binding domains of human SIAH E3 ubiquitin ligases are now crystal clear, Biochim Biophys Acta, 1861 (2017) 3095–3105. [DOI] [PubMed] [Google Scholar]
- [26].Tang AH, Neufeld TP, Kwan E, Rubin GM, PHYL acts to down-regulate TTK88, a transcriptional repressor of neuronal cell fates, by a SINA-dependent mechanism, Cell, 90 (1997) 459–467. [DOI] [PubMed] [Google Scholar]
- [27].Dong X, Tsuda L, Zavitz KH, Lin M, Li S, Carthew RW, Zipursky SL, ebi regulates epidermal growth factor receptor signaling pathways in Drosophila, Genes Dev, 13 (1999) 954–965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Boulton SJ, Brook A, Staehling-Hampton K, Heitzler P, Dyson N, A role for Ebi in neuronal cell cycle control, EMBO J, 19 (2000) 5376–5386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].House CM, Frew IJ, Huang HL, Wiche G, Traficante N, Nice E, Catimel B, Bowtell DD, A binding motif for Siah ubiquitin ligase, Proc Natl Acad Sci U S A, 100 (2003) 3101–3106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Lipkowitz S, Weissman AM, RINGs of good and evil: RING finger ubiquitin ligases at the crossroads of tumour suppression and oncogenesis, Nat Rev Cancer, 11 (2011) 629–643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Wong CS, Moller A, Siah: a promising anticancer target, Cancer Res, 73 (2013) 2400–2406. [DOI] [PubMed] [Google Scholar]
- [32].Sarkar TR, Sharan S, Wang J, Pawar SA, Cantwell CA, Johnson PF, Morrison DK, Wang JM, Sterneck E, Identification of a Src tyrosine kinase/SIAH2 E3 ubiquitin ligase pathway that regulates C/EBPdelta expression and contributes to transformation of breast tumor cells, Mol Cell Biol, 32 (2012) 320–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Wong CS, Sceneay J, House CM, Halse HM, Liu MC, George J, Hunnam TC, Parker BS, Haviv I, Ronai Z, Cullinane C, Bowtell DD, Moller A, Vascular normalization by loss of Siah2 results in increased chemotherapeutic efficacy, Cancer Res, 72 (2012) 1694–1704. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. (a) Basic workflow of the affinity selection-mass spectrometry (AS-MS) ligand binding screen. A detailed description of the screen is found at Materials and Methods. (b) Structures of lupeol and oleanolic acid. (c) Siah2 knockdown is partially rescued inhibition of cell growth by RLS-7. HEK293 cells were transfected with either empty vector pLKO or shSiah2 for 24 h and treated with RLS-7 at indicated concentrations. Cell viability was assessed 48 h later using ATPlite. Each bar represents the mean ± standard deviation of three measurements. *p < 0.05, **p < 0.01, were calculated using Student’s t test. (d) Different melanoma cells and HEK293 cells were plated in 96-well plates and incubated with indicated concentrations of RLS-7. Cell viability was assessed 72 h later. Each bar represents the mean ± standard deviation of three measurements. ****p < 0.0001 were computed based on comparison to the control (one-way ANOVA with Dunnett’s test). ns- not significant. (e) Indicated cultures were plated at low density (500 cells/well in 6-well plates) and grown in medium containing DMSO, 2 μM or 10 μM RLS-7. The number of colonies formed after 10 days in culture was determined by crystal violet staining. (f) Different patient-derived primary melanoma cells were plated in 96-well plates and incubated with indicated concentrations of RLS-7. Cell viability was assessed 72 h later. Each bar represents the mean ± standard deviation of three measurements. ****p < 0.0001 was computed based on comparison with the control (one-way ANOVA with Dunnett’s test). (g) RV1 cells were treated with RLS-7 at indicated concentrations for 12 h and 24 h. RNAs were then isolated from cells and subjected to RT-qPCR analysis for indicated AR target genes. Each bar represents the mean ± standard deviation of three measurements. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001, were calculated based on comparison with the control using Student’s t test. ns- not significant.
Figure S2. (a) Prostate cancer cell lines RV1 and PC3 were treated with indicated concentrations of RLS-7 derivatives. Cell viability was assessed 72 h later. Each bar represents the mean ± standard deviation of three measurements. ****p < 0.0001 were calculated based on comparison with the control (one-way ANOVA with Dunnett’s test). (b) Lu1205 melanoma cells were treated with RLS-7 or RLS-12 at the indicated concentrations under hypoxia. Whole cell lysates were immunoblotted with indicated antibodies. Quantification of immunoblots was performed using BioRad densitometer, relative to loading controls, noted under the blots (c) A375, RV1 and PC3 cells were plated and grown in soft agar with medium containing vehicle, 2 μM or 10 μM of RLS-12. The number of colonies formed after 2-3 weeks in culture was determined by crystal violet staining. (d) RV1 cells were plated at low density and treated with 5 μM of RLS-12. Cells were kept in 1% hypoxia for one week before images were taken using bright field microscopy. (e) A375 melanoma cells and Vemurafenib-resistant cells A375R were treated with indicated concentrations of RLS-12. Cell viability was assessed 72 h later. Each bar represents the mean ± standard deviation of three measurements. ****p < 0.0001 was calculated based on comparison to the control (one-way ANOVA with Dunnett’s test).
Figure S3. (a) Representative melting curve plot with PHYL (positive control) and representative compound. (b) Melanoma cells A375 were treated with different concentrations of compounds selected from the protein thermal shift assay, and cell viability was assessed by ATPlite after 72 h. Each bar represents the mean ± standard deviation of three measurements. ****p < 0.0001 was calculated based on comparison with the control (one-way ANOVA with Dunnett’s test). (c) Different human prostate cancer cells were plated at low density and grown in medium containing different concentrations of RLS-24. The number of colonies formed after 10 days in culture was determined by crystal violet staining. (d) RLS-24 was incubated with the purified Siah2 for 30 min followed by addition of ubiquitination reagents (E1, E2, Ub) and Siah2 substrates ASPP2, Sprouty 2 or OGDCE2. Mixtures were then incubated at 37°C for 45 min and subjected to Western Blot analysis. (e) Human melanoma A375 and mouse melanoma SW1 cells were treated with different concentrations of RLS-24, RLS-30 or RLS-34. Cell viability was assessed by ATPlite after 72 h. Each bar represents the mean ± standard deviation of three measurements. ****p < 0.0001 based on comparison with the control (one-way ANOVA with Dunnett’s test).
Figure S4. (a) Model of compound RLS-96 binding to Siah 2. (b) Melanoma cells were incubated with 5 μM, 10 μM of selected compounds for 6 h under hypoxia. Cells were harvested and whole cell lysates were immunoblotted with indicated antibodies. Quantification of immunoblots was performed using BioRad densitometer, relative to loading controls, noted under the blots (c) Viability assay of A375 cells in the presence of indicated compounds. Each bar represents the mean ± standard deviation of three measurements. ****p < 0.0001 based on comparison with the control (one-way ANOVA with Dunnett’s test).
Figure S5. (a) Nine different melanoma cells were plated in 96-well plates and incubated with indicated concentrations of RLS-12, RLS-24 and RLS-96. Cell viability was assessed 72 h later. Each bar represents the mean ± standard deviation of three measurements. ****p < 0.0001 was calculated based on comparison with the control (one-way ANOVA with Dunnett’s test). Lower panel, A375 and human melanocytes Hermes 3A cells were treated with RLS-12, RLS-24 and RLS-96 followed 72 h later by a viability assay. Each point represents the means ± standard error from two experiments. **p < 0.01, ***p < 0.001, were calculated using Student’s t test. (b) Four prostate cancer cell lines were subjected to the viability assay as described in (a). (c) RV1 cells were plated at low density and treated with 1.25 μM of RLS-12, RLS-24 and RLS-96. Cells were kept in 1% hypoxia for one week before images were taken using bright field microscopy. (d) Viability assay of four human liver cancer cell lines as described in Figure S5a. (e) Colony formation assay of four human liver cancer cell lines. Each bar represents the mean ± standard deviation of three measurements. ****p < 0.0001 was calculated based on comparison with the control (oneway ANOVA with Dunnett’s test). (f) Viability assay of two human pancreatic cancer cell lines as described in Figure S5a. (g) Colony formation assay of two human pancreatic cancer cell lines as described in Figure S5e. (h) A375 cells were transfected with either shCtrl or shSiah2 for 24 h. RNA was extracted from cells and subjected to qPCR analysis. Each bar represents the means ± standard error from two experiments. ***p < 0.001, calculated using Student’s t test. (i) A375 cells were transfected with shCtrl or shSiah2 for 24 h and treated with RLS-12, RLS-24 and RLA-96 at indicated concentrations. Cell viability was assessed 48 h later using ATPlite. Each bar represents the means ± standard deviation of three measurements. *p < 0.05, **p < 0.01, ***p < 0.001, were calculated using Student’s t test. (j) A375 cells were transfected with HRE-luciferase plasmid and pcDNA3-PHYL for 24 h and treated with or without 10 μM of the PHD inhibitor FG4592 under normoxia or hypoxia. Cells were lysed and subjected to a luciferase assay after 24 h. Each bar represents the means ± standard error from two experiments. *p < 0.05, **p < 0.01, were calculated using Student’s t test.
Figure S6. (a) A375 cells were treated with RLS-12, RLS-24 and RLS-96 at indicated concentrations. Cell viability was assessed 72 h later using ATPlite. Each bar represents the mean ± standard deviation of three measurements. ****p < 0.0001 was calculated based on comparison with the control (one-way ANOVA). (b) A375 cells were treated with both RLS-12 and RLS-24 at indicated concentrations. Cell viability was assessed 72 h later using ATPlite. Each bar represents the mean ± standard deviation of three measurements. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001, calculated using Student’s t test. (c) A375 cells were treated with both RLS-12 and RLS-96 at indicated concentrations. Cell viability was assessed 72 h later using ATPlite as described in Figure S6b. (d) A375 cells were treated with both RLS-24 and RLS-96. Cell viability was assessed 72 h later using ATPlite as described in Figure S6b.