

Abstract
Successive adaptation of the bone marrow (BM) from homeostatic hematopoietic microenvironment to a self-reinforcing niche is an integral aspect of leukemogenesis. Yet, the cellular mechanisms underlying these functional alterations remain to be defined. Here, we found that AML incursion precipitates compartmental endoplasmic reticulum (ER) stress and an unfolded protein response (UPR) in both leukemia and stromal cells. We observed that extracellular vesicles (EV) transmit ER stress in vivo from the AML xenograft to BM stroma, whereby the upregulation of core UPR components drives subsequent osteolineage differentiation of mesenchymal stem cells (MSC). Finally, we show that the underlying mechanism involves quantitative incorporation and cell-cell transfer of Bone Morphogenic Protein 2 (BMP2), a potent osteogenic signal, by AML-EVs. Corroborative studies in AML patient samples support the translational relevance of AML-EVs as a platform for BMP trafficking and source of compartmental crosstalk. Transmissible ER stress was previously identified as a source of chemoresistance in solid tumor models, and this work reveals a role in remodeling the BM niche in AML.
INTRODUCTION:
Hematopoiesis occurs in operationally defined niches in the bone marrow (BM) and is regulated through reciprocal signaling between hematopoietic and stromal tissue components (1–6). Leukemia cells, including Acute Myeloid Leukemia (AML), actively compete with hematopoietic stem cells (HSC) for niche occupancy. The successive tumor growth in turn affects stromal cell function, and results in reduced chemotherapeutic efficacy as well as impaired blood formation (7–10). These observations do not appear to be AML subtype-specific, and in fact similar defects have been described in murine models of CML (11, 12). Evidence from several groups indicates that remodeling and secretory conversion of the microenvironment accounts for the role of the BM as a sanctuary site for residual, drug-resistant disease and relapse (13, 14). This notion is further consistent with observations that AML patient-derived mesenchymal stem cells (MSCs) exhibit an altered secretion of cytokines with reduced hematopoietic support and a more chemoprotective phenotype (15–17). While inherently translational, snapshot analysis of patient samples at diagnosis limits our ability to model the dynamic crosstalk that reconfigures the BM microenvironment, and risks artifacts due to propagation in tissue culture. Murine xenograft studies have been widely used for the study of leukemia-stroma cell interactions, providing an opportunity for prospective in vivo modeling while benefitting from validated strategies for immunophenotypic isolation of distinct stromal populations for study (1, 8, 12, 18, 19).
To better understand how leukemia induces changes in the composition of the BM compartment, we focused on the two mesenchymal populations central to AML leukemogenesis: MSCs, which maintain the potential to differentiate along adipo-, chondro-, and osteolineages; and Osteoblastic Progenitor Cells (OPCs), a population of osteolineage committed progeny that will mature into osteoblasts (12). Both populations contribute to hematopoietic homeostasis or, conversely, their functional disruption can lead to myelodysplastic growth and clonal evolution (20). We were particularly interested in understanding the reciprocal crosstalk in the AML niche that would spur osteogenic differentiation bias, previously implicated during AML expansion (11, 12, 21, 22), and known to alter the release of soluble factors that regulate growth and niche adhesion (23).
The studies herein identify significant compositional changes in the niche of AML xenograft animals associated with osteogenic MSC differentiation. We show that the underlying mechanism relies on transmissible ER stress (TERS) (24, 25), and identify AML derived extracellular vesicles (EVs) as a contributory factor in promoting the unfolded protein response (UPR) in stromal cells, a known stimulus for altering secretion and inducing osteogenic MSC differentiation (26–28). We show that EVs accomplish these changes in part by trafficking Bone Morphogenic Protein 2 (BMP2), a known regulator of osteogenesis and inflammation.
MATERIALS AND METHODS:
Mice and xenografts
NOD-scid IL2R γ null mice were purchased from The Jackson Laboratory (Bar Harbor, ME, USA). Male and female animals, 6–8 weeks old, were used in the experiments. Molm-14 cells (1 × 105 per animal), HL-60 cells (5 × 106 per animal), or U937 (2 × 105 per animal) were engrafted into non-irradiated animals by tail vein injection. No randomization process was used. Chimerism was determined by flow cytometry using a human CD45 antibody. We used a chimerism cutoff of >%60 for use in xenograft experiments. Animals were sacrificed at indicated time points and bone marrow from femurs and tibias was collected from each animal. Husbandry and experimental procedures were performed in accordance with federal guidelines and protocols approved by the Institutional Animal Care and Use Committee at Oregon Health & Science University.
MSC and OPC isolation
Long bones were isolated and marrow plugs flushed as previously described (29). Bones were then broken into small pieces with surgical scissors and incubated in Collagenase II (Sigma Aldrich) buffer (DMEM, 2% FBS, 1% Penicillin/Streptomycin, 2mg/mL Collagenase II) for one hour at 37°C and 200 RPM. The solution was then filtered through a 70μm filter, and then washed with Hemolytic Buffer. Hemolyzed cells were then filtered into Cell-Strainer tubes (Corning, Corning, NY, USA) and resuspended in FACS Wash (DPBS, 2%FBS), stained with antibodies, and then sorted/analyzed using flow cytometry. In vitro cultured cells were propagated in MSC media (MEMα, 15% FBS, 1X penicillin/streptomycin) at 37°C, 5% CO2, and >95% humidity.
Antibodies
Detailed information on antibodies used is available in Supplemental Table 1.
Intrafemoral injections of cell line and serum-derived EVs
Animals were anesthetized using isoflurane (1.5–2.0%) and carefully shaved at the injection site using an electric razor. The shaved area was disinfected and sterilized by alternately scrubbing with 1% betadine and 70% ethanol three times. The injected leg was bent at the knee at a 90˚ angle keeping the femur vertical. The injected femur was accessed by gentle twist, using a 25-gauge needle at the patellar groove into the femoral cavity. The coring needle was carefully removed, a 27-gauge injection needle was inserted into the femoral cavity and 50 μl of sample suspended in PBS was slowly injected.
Statistical analysis
All experiments were replicated at least three times with the exception of the intrafemoral injections of patient-derived plasma, where available material was limiting (Figure 5g). Statistical analyses were performed using the PRISM software for Windows produced by Graphpad Software Inc. (La Jolla, CA, USA). Comparisons between two groups were performed with a two-tailed Student’s t-test. One-way analysis of variance (ANOVA) was utilized when comparing more than two groups. T-tests with Bonferroni correction were used to determine p-values. An N of at least three animals or biological replicates was used in all analyses. Error bars represent ± Standard Error of the Mean. For qRT-PCR analyses, a pairwise comparison of fold change (2ˆ-ΔΔCT) between control and xenograft-derived cells, or between vehicle-treated and EV-treated cells was performed. An N of at least three was used in these analyses. For all experiments, statistical significance was set at *= p <0.05, **=p <0.01, and ***=p <0.001. No statistical test was used a priori to determine the sample size. No randomization was used to allocate animals to particular groups; age and sex-matched recipients were used for transplantation experiments. The investigators were not blinded to experimental groups during analysis.
Figure 5: AML cells exhibit an UPR in vivo.
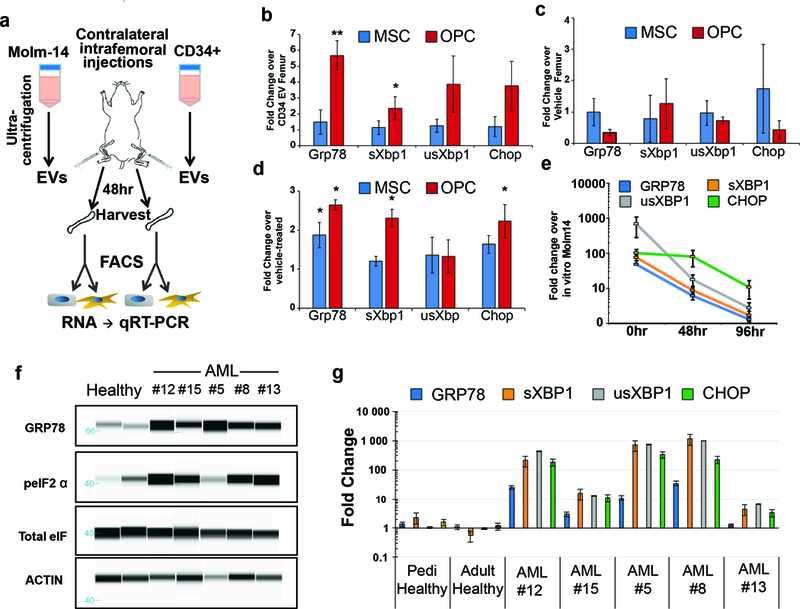
(a) Experimental outline for intrafemoral injections. EVs from Molm-14-mGFP cells and healthy CD34+ cells were injected contralaterally into femurs of recipient mice. Femurs were harvested 48hrs later, MSCs and OPCs were sorted into RNA extraction buffer for gene expression analysis. (b) Expression analysis of UPR genes from MSCs and OPCs from Molm-14-mGFP EV injected femurs. Fold change was determined by 2-ΔΔCt against respective cells from CD34+ EV injected femurs. Error bars are standard error of the mean from three animals per condition. (c) Expression analysis of UPR genes from MSCs and OPCs from CD34+-derived EV injected femurs. Fold change was determined by 2-ΔΔCt against respective cells from vehicle injected femurs. Error bars are standard error of the mean from three animals per condition. (d) Expression analysis of UPR genes from in vitro cultured MSCs and OPCs exposed to Molm-14-mGFP EVs. Fold change determined by 2-ΔΔCt against vehicle-treated cells. Error bars are standard error of the mean from three separate experiments. Significance in (b) and (c) was determined by Student’s t-test, *P<0.05, **P<0.01. (e) Timecourse of UPR gene expression in explanted Molm-14-mGFP. Molm-14-mGFP cells were sorted out of xenograft bone marrow based on human CD45 expression and cultured ex vivo. RNA was extracted from cells directly from the sort (0hr) or 48 and 72hrs in normal culture media in vitro. Fold change determined by 2-ΔΔCt against in vitro cultured Molm-14-mGFP cells. Error bars are standard error of the mean from three separate experiments. (f) UPR status was determined by protein levels of GRP78, and phosphorylated eIF2α in AML-patient samples and healthy pediatric and adult controls. (g) Expression analysis of UPR genes from blasts from AML patient samples. Fold change determined by 2-ΔΔCt in pairwise analysis against control samples. Error bars are standard error of the mean.
Additional experimental methods are provided in the Supplementary Experimental Methods.
RESULTS:
AML remodels the bone marrow
To examine BM niche composition and function in vivo, we used NOD-scid IL2R γ null (NSG) xenografts (8–10) with tail vein grafting and without conditioning irradiation to ascertain undisturbed BM niche function (29, 30). To avoid expansion artifacts during in vitro cell culture of MSCs and OPCs, we isolated endosteal MSC and OPC populations directly from mice using fluorescence activated cell sorting (FACS) (31). Specifically, we excluded hematopoietic and endothelial cells (CD45 and TER119, and CD31, respectively) to sort two immunophenotypically distinct mesenchymal populations: SCA-1+/CD51+ MSCs and SCA-1-/CD51+ OPCs (12, 32) (Figure 1a and Supplemental Figure 1). We analyzed cells sorted from long bones for morphological differences, clonogenic growth, and extracted RNA to transcriptionally validate differential expression of genes characteristically expressed in either population (Supplemental Figure 2). With this strategy in place, we generated several xenograft cohorts via intravenous tail vein injection of three human AML cell lines: Molm-14, U937, and HL-60. Chimerism was tracked by the percentage of human CD45+ cells over time in the peripheral blood and at time of harvest in the bone marrow. We included xenografts with a marrow chimerism > 60% in this study to simulate niche remodeling effects during advanced disease, reflective of AML patients at diagnosis (33). Functionally, we observed reduced fibroblastic colony forming (CFU-F) potential in both MSCs and OPCs from all three AML xenograft cohorts (Figure 1b). Strikingly, we also observed a significant shift in the proportion of the two populations (Figure 1c), with increased MSC/OPC ratios, signifying a compositional change within the BM niche.
Figure 1: AML negatively impacts the endosteal niche.
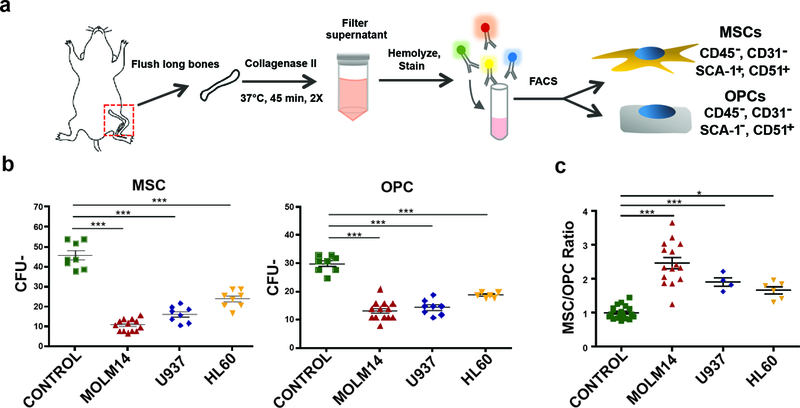
(a) MSC and OPC harvest workflow. Long bones from control and xenografted mice are removed, flushed, crushed, and incubated in Collagenase II. Disadhered cells are then filtered, stained with antibodies, and sorted via FACS. (b) CFU-F assay of MSCs and OPCs derived control and AML xenografted animals. Error bars are standard error of the mean (n= 8, 13, 8, 8 animals per condition). (c) The ratio of MSCs / OPCs in control and AML xenograft animals. Error bars are standard error of the mean (n= 22, 14, 4, 6 animals per condition). Significance in (b) and (c) was determined using ANOVA and Bonferroni correction. *P<0.05, ***P<0.001.
MSCs and OPCs exhibit differential fates in the AML bone marrow
As Osterix-expressing progenitor cells have been previously shown to be sensitive to AML-induced apoptosis (19, 34), we hypothesized that the altered ratio of MSCs over OPCs may be due to increased apoptotic turnover of OPCs. Indeed, isolated OPCs exhibited increased apoptosis within xenografts as measured by increases in Annexin V positivity and Ser-15 phosphorylated p53, but not overall p53 (Figure 2a). These OPC differences were significant in Molm-14 and U937 xenografts, and did not reach statistical significance for HL-60 xenografts. Xenograft-derived MSCs on the other hand did not show evidence of apoptosis or p53 engagement under these conditions (Figure 2b). In MSCs from AML xenografts, we observed significant induction of osteolineage differentiation. Specifically, MSCs in all three xenograft models showed increased expression of Runx2, Osterix, and Dkk3, whereas markers of late osteoblastic development, Col1α1 and Spp1, were significantly reduced when compared to MSCs from control animals (Figure 2c). This was further consistent with increased in vitro osteogenic- and reduced adipolineage differentiation of Molm-14 xenograft-derived MSCs, using Alizarin Red S (binds to calcium) and Oil Red O (lipophilic) stains, respectively (Supplemental Figure 3). Together, the data suggest that leukemia cells provide extrinsic cues that promote osteogenic MSC differentiation and increased apoptosis in OPCs, resulting in aggregate changes in the overall composition of the BM compartment in vivo.
Figure 2: MSCs and OPCs exhibit differential fates in the leukemic bone marrow.
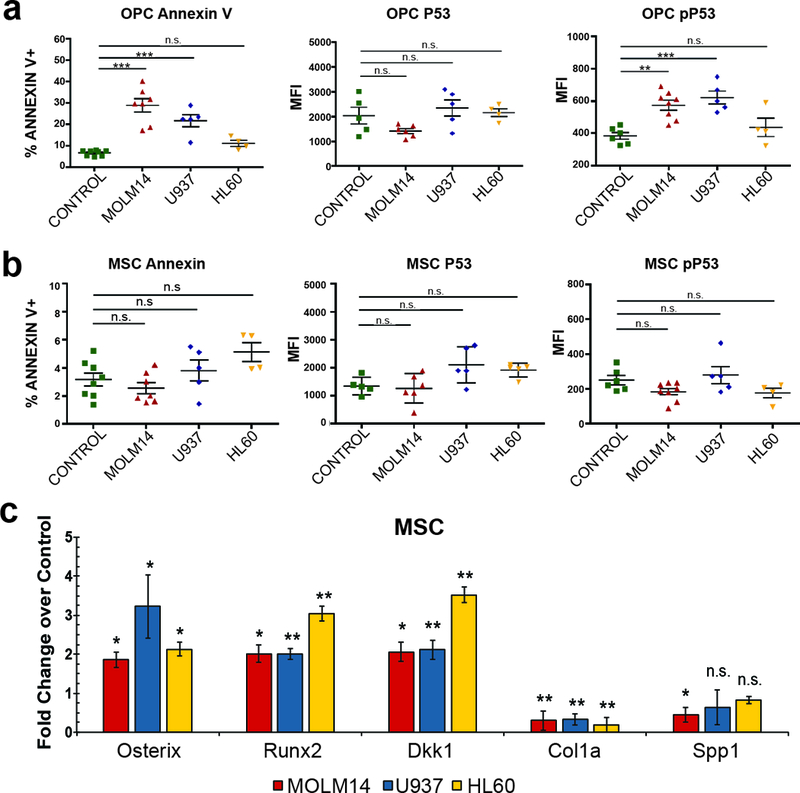
(a-b) Analysis of apoptosis in stromal populations. The percentage of Annexin V+ cells, and the MFI of P53 and Ser-15 phosphorylated P53 in control and xenografted animals within OPC (a) and MSC (b) gates. Error bars are standard error of the mean from (n= 8, 7, 5, 4) animals for Annexin V dataset and (n= 5, 6, 5, 4) for the pP53 and P53 experiments. Significance in (a) and (b) was determined using ANOVA and Bonferroni correction. **P<0.01, ***P<0.001. (c) Expression analysis of genes involved in early and late osteogenesis of xenograft-derived MSCs compared to control MSCs. Fold change determined by 2-ΔΔCt in pairwise analysis against control MSCs. Error bars are standard error of the mean from four animals per condition. Significance was determined by ANOVA and Student’s t-test. *P<0.05, **P<0.01, ***P<0.001.
Stromal UPR induction in AML xenografts
Both the increasing translational burden within the ER during osteogenic differentiation (35) and reports of the XBP1-dependent upregulation of Osterix, a master regulator of osteogenesis (26), led us to consider the involvement of the UPR in inducing AML-mediated MSC differentiation. We therefore assembled an RT-PCR survey panel to screen for UPR induction, observing broad engagement in xenograft-derived MSC and OPC populations compared to those from control mice (Figure 3a-b). With Molm-14 xenografts, both populations exhibited 2–30 -fold upregulation of Glucose regulated protein 78kDa (Grp78), a core regulatory component of the UPR and spliced X-box binding protein 1 (Xpb1), a transcription factor involved in upregulating chaperones and ER stress sensors. There were more subtle increases in the unspliced isoform of Xbp1 (usXbp1) and upregulation of Chop. Promotion of osteogenesis by the UPR (36) under these ER stress conditions was supported by increased Runt Related Transcription Factor 2 (Runx2) and Osterix gene expression after exposure of MSCs to the UPR inducer thapsigargin (37) (Figure 3c). By contrast, hematopoietic progenitor cells, identified by the expression of c-Kit, Sca-1, and the absence of lineage markers (i.e. KSL) did not exhibit induction of the UPR, suggesting a stroma-restricted response (Figure 3d). To further implicate the UPR in the increase in apoptosis within the OPC population, we sorted for Annexin V+ OPCs and Annexin- populations from both Molm-14 xenografts and controls. Specifically, we reasoned that the UPR should preferentially be induced in Annexin V+ cells, and indeed we observed significant upregulation of Grp78, Chop, Xbp1 and sXbp1 genes in Molm14 xenograft derived Annexin V+ cells compared with those from controls (Supplemental Figure 4). Altogether, the data suggest that ER stress in the AML niche contributes to the adaptive changes in BM stromal fate and composition.
Figure 3: MSCs and OPCs exhibit increased ER stress.
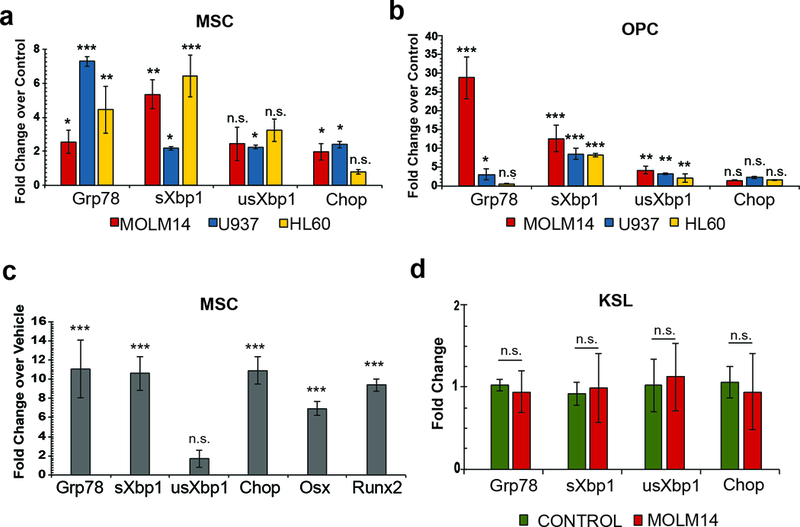
(a-b). Expression analysis of genes involved in the UPR from xenograft-derived MSCs (a) and OPCs (b). Fold change determined by 2-ΔΔCt in pairwise analysis against control MSC and OPCs, respectively. Error bars are standard error of the mean from four animals per condition. Significance was determined by ANOVA and Student’s t-test. *P<0.05, **P<0.01, ***P<0.001. (c) Expression analysis of genes involved in the UPR and osteogenesis in MSCs cultured in 1ng/mL thapsigargin compared to vehicle-treated MSCs. Fold change determined by 2-ΔΔCt against control cells. Error bars are standard error of the mean from three biological replicates. Significance was determined by Student’s t-test. ***P<0.001. (d) Expression analysis of UPR genes in hematopoietic stem cells (c-KIT+, SCA-1+, lin−; KSL) from Molm-14 xenografts. Fold change determined by 2-ΔΔCt against KSL from control animals. Error bars are standard error of the mean from three animals per condition. Significance was determined by Student’s t-test. *P<0.05, **P<0.01, ***P<0.001.
Uptake of AML-derived EV into endosteal cells in vivo and in vitro
We and others previously demonstrated that AML-derived EVs enter into stromal cells (CD45- / plastic-adherent) in vitro (9, 30). To more specifically visualize MSC and OPC uptake of AML-derived EVs in vivo, we generated Molm-14 cells stably expressing a myristoylated GFP transgene (Molm-14-mGFP), which functions as a continuously produced lipid membrane label (29). The Molm-14-mGFP cells release brightly labeled EVs into the extracellular space and enable us to detect EV dissemination in the BM and within the peripheral blood for the lifespan of the xenograft animals (Figure 4a). Here, we used this approach to map the cellular uptake of xenograft-derived EVs in vivo and performed live-cell microscopy of FACS-purified MSC and OPC. At animal sacrifice, the sorted cells are transferred to culture dishes and labeled with Hoechst nuclear stain and Cellmask, a lipophilic dye used to mark the cytoplasm. Results reveal that both MSCs and OPCs from Molm-14-mGFP xenografts, but not control animals, contained discrete mGFP+ vesicles (Figure 4b). For quantification of mGFP+ vesicle uptake, we next scored mGFP+ foci in both cell types. The MSCs and OPCs from xenografted animals (day 21 sacrifice) were found to contain GFP+ vesicles in 55% and 34% of cells respectively (Figure 4c). In both cell types the total number of internalized foci ranged from one to thirty EVs with no difference between mean the number of EVs in MSC vs. OPC (Figure 4d). We further confirmed vesicle uptake using confocal live-cell microscopy of MSCs and OPCs exposed to Molm-14-mGFP EVs in vitro (Figure 4e). Next, in determining the spatial distribution inside recipient cells, we found that most Molm-14 EVs traffic to the ER in both MSCs and OPCs, as shown by ER Tracker co-staining (Figure 4e-f). This intracellular localization at the ER is consistent with previous observations of EV fate (38). Finally, because UPR induction generally leads to an increase in the size of the ER compartment (37, 39), we visualized ER size and morphology of MSCs and OPCs exposed to Molm-14-mGFP-derived EVs in vitro using ER-Tracker (40). The images show that EV-treated cells display a dilated ER like cells treated with thapsigargin, when qualitatively compared to vehicle-treated cells (Supplemental Figure 5).
Figure 4: AML EVs traffic to the ER of MSCs and OPCs.
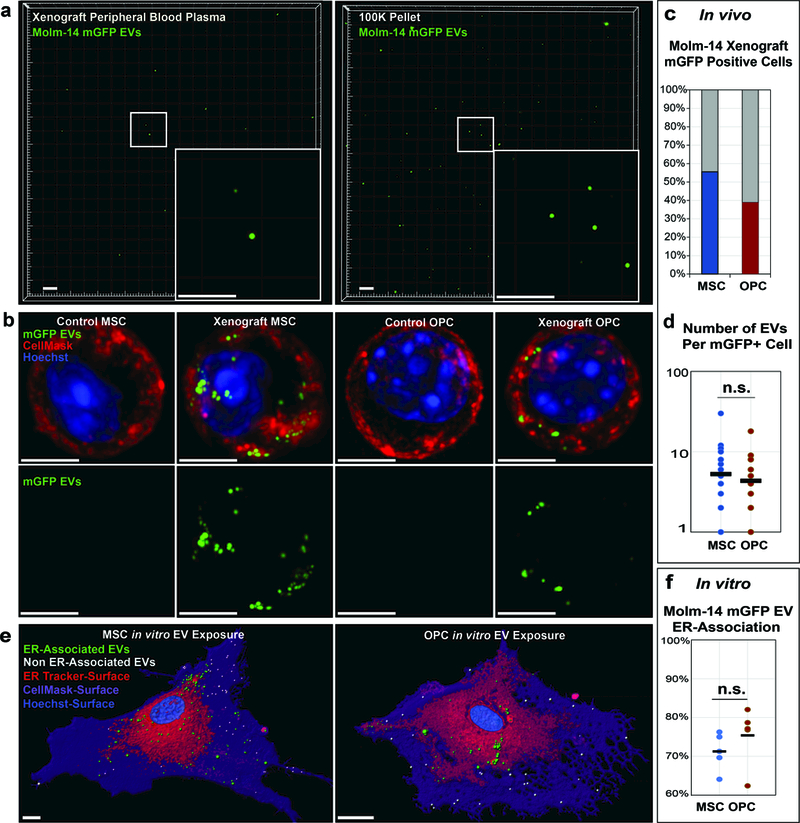
(a) Solid capture imaging of mGFP+ EVs from peripheral blood of Molm-14-mGFP xenografts (left) and from in vitro Molm-14-mGFP cells (right). Scale bars are 5μm. (b) Live-cell imaging of MSCs and OPCs derived from control and Molm-14-mGFPxenografts. mGFP: green, Cellmask: red, Hoechst: blue. Scale bars are 5μm. (c) Quantification of Molm-14-mGFP xenograft-derived MSCs and OPCs containing mGFP+ vesicles. 50 cells per cell type analyzed from 2 animals. (d) Quantification of mGFP+ vesicles per positive MSC and OPC derived from Molm-14-mGFP xenografts. 50 cells per cell type analyzed from 2 animals, significance determined by Student’s t-test *P<0.05. (e) Representative images of live-cell, confocal microscopy of in vitro expanded MSCs (left) and OPCs (right) exposed to EVs harvested from Molm-14-mGFP cells. Green: ER-localized mGFP+ vesicles, white: cytosol-localized mGFP+ vesicles, red: ER surface, purple: plasma membrane surface, blue: Hoechst. Scale bars are 5μm. (f) Quantification of ER-localization of internalized mGFP+ vesicles in in vitro expanded MSCs and OPCs. 5 cells per cell type analyzed, significance determined by student’s T-test *P<0.05
AML EVs induce the UPR in MSCs and OPC in vivo
The observation that EVs traffic to the ER in MSC and OPC prompted us to systematically test the possibility that EVs transmit ER stress, and may account for UPR activation and altered MSC and OPC fates (41–43). To test this hypothesis, we performed intrafemoral injections, delivering dose-matched, purified EVs from in vitro cultured healthy human bone marrow-derived CD34+ cells versus either vehicle or Molm-14-mGFP cells (Figure 5a), an approach we validated previously (29). Animals were sacrificed 48 hours later, and MSCs and OPCs were sorted directly into RNA extraction buffer for subsequent examination of UPR induction by qRT-PCR. Molm-14-mGFP-derived EVs induced a UPR in OPCs when compared to femurs receiving CD34+ derived EVs (Figure 5b). Furthermore, EVs derived from the CD34+ cell cultures did not promote a UPR in MSC and OPC cells when compared to vehicle-treated femurs, indicating that the observed ER stress is not a cross-species artifact (Figure 5c). Consistent with their apoptotic fate, OPCs exhibited a more substantial increase in Grp78 and spliced Xbp1 expression than MSCs when exposed to AML-derived, but not healthy CD34+ cell-derived, EVs (Figure 5d). For further confirmation of EV-mediated UPR induction we expanded MSCs in vitro and again observed a persistent, but more modest increase of the UPR in both cell types 48 hours post EV-exposure, where CD34+-derived EVs had no effect. (Supplemental Figure 6). Recent reports also indicate TERS crosstalk between cancer and bystander cells in pancreatic and breast tumor microenvironments (24, 25). To test the possibility of TERS from AML cells to stroma, we first measured UPR induction in FACS-purified Molm-14-mGFP cells explanted from xenografts at sacrifice and at 48 and 96 hours during subsequent in vitro propagation. Remarkably, when compared against Molm-14-mGFP cells grown in vitro, we found sharply increased, but rapidly diminishing transcriptional activity of several key UPR components over time in cell culture (Figure 5e). Further consistent with a model whereby AML EVs elicit UPR in MSCs and the loss of GRP78 response following pharmacological inhibition using the UPR inhibitor GSK2606414, (Supplemental Figure 7). Next, we had the opportunity to test blasts enriched from AML patients at diagnosis and compared their UPR status to healthy controls. In a set of 38 AML samples, we found that a subset of 8 exhibited a robust UPR, illustrated by the upregulation of GRP78 and phosphorylated Elongation initiation factor 2α (peIF2α) protein (Supplemental Figure 8). Material was available from five of those patients to perform repeat experiments that confirmed elevation of both GRP78 and peIF2α protein levels (Figure 5f). To better correlate the magnitude of UPR induction in patients with our xenograft results, we also performed an additional transcriptional analysis of GRP78, s/usXBP1 and CHOP expression in these patient samples and found highly significant differences from healthy control bone marrows (Figure 5g). Similarly, we observed induction of phosphorylated Inositol-requiring enzyme 1α (pIRE1α) at the protein level in the U937 leukemia cells, with rapid kinetics that peak at 3–6 hours, consistent with the subsequent transcriptional upregulation of GRP78 (Supplemental Figure 9). Consistent with the UPR induction in three AML cell lines representing different subtypes in our xenograft studies, UPR activity in AML patient samples did not segregate by AML subtype or cytogenetic lesion (Supplemental Table 2). With the availability of peripheral blood plasma from the UPR+ AML patients, we purified plasma-derived EVs by serial ultracentrifugation and performed intrafemoral injections into recipient mice (29), with contralateral vehicle controls and harvested of MSCs and OPCs 48hrs later. Even though modest, and likely suffering from both dilution effects and single dosing, serum EVs from AML patients appeared to induce UPR in bone marrow MSC and OPC, whereas serum-EVs from healthy donors failed to do so (Supplemental Figure 10). Our experiments suggest that ER stress in the AML niche is transmitted by a leukemia-derived, transmissible, EV-bound factor.
UPR induction enhances EV release and cargo
To further test the observation that AML cells experiencing UPR transfer ER stress and induce UPR in stromal cells, we harvested and quantified EVs from thapsigargin-treated Molm-14-mGFP cells. Consistent with prior observations (44), we found that Molm-14-mGFP also produce more EVs when experiencing ER stress, without compromise to cell viability or proliferation (Supplemental Figure 11). The dose-matched EVs derived from thapsigargin-treated Molm-14-mGFP in turn strongly induced a UPR in MSCs and OPCs when compared to the cells exposed to dose-matched, vehicle-treated Molm-14-mGFP EVs (Supplemental Figure 12). To understand the EV component responsible for UPR induction we turned to a recent study of AML patients and murine models that revealed an osteogenic differentiation bias in BM MSCs elicited through BMP signaling (21). We tested the hypothesis that AML elicits stromal UPR and osteogenic differentiation via BMP cargo trafficked via EV. We observed that ER stress via thapsigargin treatment significantly induced the expression of several BMP family genes in Molm-14 cells (Figure 6a). In striking similarity to loss of UPR during tissue culture (Figure 5d), ex vivo analysis of the BMP family gene expression revealed significant upregulation in vivo, and rapid decline during four days propagation in culture (Figure 6b). This BMP gene set was also upregulated in our patient samples (Figure 6c). Since AML patients have been previously reported to have elevated circulating BMP2 levels (45), we determined BMP2 levels by ELISA on EVs harvested from Molm-14-mGFP cells and found a significant increase in BMP2 protein associated with EVs obtained from thapsigargin-treated cells compared to vehicle treated cells (Figure 6d). Intriguingly, there was no detectable change in vesicle free BMP2 (that is, not contained in EVs), suggesting that EV trafficking accounts for increased BMP signaling under these circumstances. Due to the increased EV output of thapsigargin-treated cells, we repeated the BMP2 ELISA following normalization of EV counts, and confirmed that Molm-14-mGFP both increase their EV output, and the amount of BMP2 protein packaged in EVs (Supplemental Figure 13). Altogether, these data suggest a model whereby AML blasts utilize the UPR to adapt to metabolic ER stress in the BM niche. In aggregate, we demonstrate that the trafficking of EVs from AML cells to stroma leads to ER stress transmission and promotes the subsequent changes in stromal at least in part through the action of EV-associated BMP2 (Figure 6e).
Figure 6: AML cells alter their EV cargo upon UPR induction.
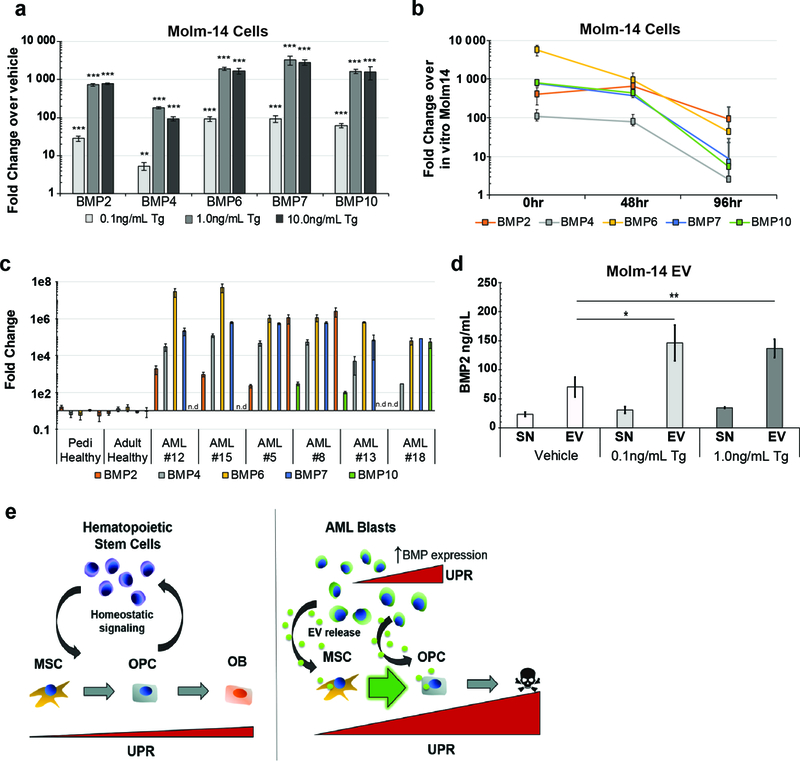
(a) Expression analysis of BMP genes in Molm-14-mGFP cells cultured in thapsigargin. Fold change determined by 2-ΔΔCt against vehicle-treated cells. Error bars are standard error of the mean from (n=4, two separate experiments). Significance was determined using ANOVA and Bonferroni correction. *P<0.05, **P<0.01. (b) Timecourse of BMP gene expression in explanted Molm-14-mGFP. Molm-14-mGFP cells were sorted out of xenograft bone marrow based on human CD45 expression and cultured ex vivo. RNA was extracted from cells directly from the sort (0hr) or 48 and 72hrs in normal culture media in vitro. Fold change determined by 2-ΔΔCt against in vitro cultured Molm-14-mGFP cells. Error bars are standard error of the mean from (n=7, two separate experiments). (c) Expression analysis of BMP genes from AML patient samples. Fold change determined by 2-ΔΔCt in pairwise analysis against control samples. Error bars are standard error of the mean. (d) Concentration of BMP2 protein in the supernatant (SN) and pellet (EV) from EV harvest of Molm-14-mGFP cells cultured in thapsigargin. ELISA was used to determine protein concentration. Error bars are standard error of the mean from three separate experiments. Significance was determined by ANOVA and Student’s t-test. *P<0.05, **P<0.01. (e) Model for AML-mediated remodeling of the endosteal niche. Niche homeostasis is maintained by reciprocal crosstalk between stromal and hematopoietic components (left). AML proliferation results in an intrinsic UPR that is transferred to stromal cells in part by increased EV output and BMP2 incorporation into EVs. This signaling axis promotes ER stress in recipient stroma and contribute to the differential fates of MSCs and OPCs (right).
DISCUSSION:
While circulating blasts are rapidly eliminated by conventional chemotherapy, at least 40% of AML patients relapse with drug-resistant disease that persists in the BM. The adaptive changes that foster the survival of AML clones in the BM niche not only coincide with disease progression, but may play a causative role in leukemic drug resistance and hematopoietic suppression (14, 46). Accordingly, the crosstalk between tumor cells and the microenvironment that adapts stromal function represents a critical gap in our understanding of AML leukemogenesis. We and others previously demonstrated the trafficking of AML derived EVs to BM stroma (9, 30, 47, 48), and this work demonstrates EV involvement in the transmission of ER stress, whereby AML cells actively shape the BM composition and alter the MSC phenotype.
To better understand the cell-cell signaling that alters and coopts the BM during AML invasion, we relied on a xenograft model that does not require host irradiation, and we prioritized the direct ex vivo analysis of immunophenotypically defined cell populations without tissue culture propagation in an unmanipulated microenvironment. First, we set out to undertake an unbiased survey of stroma composition with emphasis on the two key populations involved in endosteal niche function, where AML preferentially localizes (14, 49). Using a validated immunophenotyping strategy, we observed a systematic shift in the proportion of MSCs and OPC that proved to be highly reproducible across different AML xenograft cell lines and correlated directly with suppression of clonal fibroblast expansion (CFU-F) and a p53-mediated pro-apoptotic response in OPC. Intriguingly, MSC simultaneously experienced a broad and significant induction of osteogenesis with a reduction in adipogenesis.
The UPR represents a strong stimulus for osteogenic differentiation (27). Here, we reasoned that MSC differentiation toward a more secretory phenotype might be provoked by ER stress and engage one of three branches of the UPR as an adaptive mechanism to adjust protein folding and secretory load (26, 28, 50). Others previously showed that AML blasts similarly undergo a UPR to adjust to ER stress (51, 52). Our observations not only confirm those studies, but demonstrate that AML cells can transfer ER stress to elicit these changes in the BM. The transmission of UPR responses from AML cells to both MSC and OPC is entirely consistent with recent evidence that ER stress can be transferred between cells in a tissue compartment, a known mechanism of drug resistance in pancreatic and breast cancer (24, 25). Remarkably, some of the key events, namely UPR induction and BMP expression in AML cells rely on conditions encountered in vivo. Tissue culture propagation on the other hand prompted a rapid loss in expression of central UPR components GRP78, spliced XBP1 and BMP. From a clinical perspective, leukemia cell UPR has already been implicated in resistance to standard-of-care agents such as cytarabine (53). Along with the microenvironmental signaling promoting the UPR in AML blasts, this may provide a target to ameliorate compartmental resistance. Furthermore, the effects of these potential therapeutics on the composition and function of stromal cells should also be considered, as they could potentially reduce or exacerbate microenvironmental remodeling.
We previously showed that AML cells produce EVs that enter bystander cells in the AML niche and deliver protein and RNA regulatory cargo (9, 29). While often viewed as effectors of broad phenotypic changes, EV trafficking can be a narrowly specific signaling paradigm, as in the case of suppressed colony formation via EV miRNA targeting of the c-Myb transcription factor that is highly expressed in hematopoietic progenitors, but not in HSC, MSC or OPC (29). In other words, EVs are multicomponent signaling devices and the outcomes in target cells differs based on target cell identity. The data certainly do not exclude mechanisms other than BMP trafficking in EV-mediated stromal UPR induction (54), and additional EV cargo likely modulates ER stress responses and subsequent phenotypic changes in BM stroma further. It is tempting to speculate that intracellular localization and cargo deposition at ER membranes (38) may also be involved in the translational suppression via miRNA by ER resident components of the miRNA processing machinery (55, 56).
Among reports of osteogenic differentiation in AML patient-derived MSCs (19, 22, 34), one study directly linked BMP2 release by AML blasts with MSC differentiation and leukemia promotion (21). Our data now indicate that AML-derived EVs are carriers of BMP2, and elicit a UPR response and osteogenic differentiation in MSC. Whether the UPR results directly from BMP signaling, or as a consequence of osteogenic differentiation, remains unclear (28). Importantly, just like BMP2 expression and UPR responses, upregulated EV production by AML cells requires specific environmental conditions found in vivo. Indeed, we demonstrate significant increases in gene expression of several BMPs other than BMP2 under in vivo conditions, and it is conceivable that several BMPs contribute to UPR response. Studies in CML have revealed a self-reinforcing BMP loop with autocrine and paracrine BMP2/4 signaling as a source of inflammation and chemoresistance, and our observations leave open the possibility that similar events may operate in the AML niche (57).
Reciprocal signaling in the BM microenvironment contributes to AML pathogenesis, and the successive emergence of a more leukemia-permissive microenvironment can be viewed as a part of leukemogenesis (14, 19, 22, 58–61). Our patient sample numbers are too small to conclusively comment on AML subtype specific differences, or on disease outcome by UPR status (Supplemental Table 3). However, it is worthwhile to note that BM stroma effects in our studies seemed remarkably similar among different AML subtypes, an observation that echoes studies by others (9). The intensity of the response however, may fluctuate across AML subtypes, evidenced by the reduced propensity for HL-60 to induce apoptosis in OPCs (Figure 2a). This should provide strong motivation to uncover the durability of the observed changes, determine their impact on drug resistance, and develop adjuvant therapies that increase treatment efficacy without further escalating toxicity.
In aggregate, we demonstrate that AML EVs contribute to changes in BM stromal composition through ER stress transfer. We believe that these studies tie together several disparate observations in patients and murine models to support a model whereby AML EV trafficking of BMP2, transmits a stromal UPR and induces osteogenic differentiation of MSC.
Supplementary Material
ACKNOWLEDGEMENTS
We are indebted to staff in the Department of Comparative Medicine for excellent veterinary care, the technicians at the OHSU Flow Cytometry core, and acknowledge contributions by all members of the labs. These studies were funded in part by: Hyundai Hope on Wheels (PK), Max Blue Butterfly Campaign (PK), R01-CA164024 (TMH) and Hyundai Hope on Wheels grant (TMH).
Footnotes
AUTHOR CONTRIBUTIONS:
BD conceived the study, designed and performed the experiments, analyzed data, interpreted the results, and wrote the paper; SA performed xenograft studies, intrafemoral injections, and helped with experiments; JTB performed microscopy and in vitro experiments, analyzed the data, generated figures and edited the manuscript; SKH organized, generated and interpreted patient data, and edited the manuscript; TMH consented patients, supervised patient data collection and experiments, interpreted patient data, and edited the manuscript; PK conceived the study, designed the experiments, interpreted the results, wrote the paper, and oversaw the research project.
Conflict of Interest: The authors have no conflicts of interest to disclose
REFERENCES
- 1.Mendez-Ferrer S, Michurina TV, Ferraro F, Mazloom AR, Macarthur BD, Lira SA, et al. Mesenchymal and haematopoietic stem cells form a unique bone marrow niche. Nature. 2010;466(7308):829–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chitteti BR, Cheng YH, Poteat B, Rodriguez-Rodriguez S, Goebel WS, Carlesso N, et al. Impact of interactions of cellular components of the bone marrow microenvironment on hematopoietic stem and progenitor cell function. Blood. 2010;115(16):3239–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Jung Y, Song J, Shiozawa Y, Wang J, Wang Z, Williams B, et al. Hematopoietic stem cells regulate mesenchymal stromal cell induction into osteoblasts thereby participating in the formation of the stem cell niche. Stem Cells. 2008;26(8):2042–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.El-Badri NS, Wang BY, Cherry Good RA. Osteoblasts promote engraftment of allogeneic hematopoietic stem cells. Exp Hematol. 1998;26(2):110–6. [PubMed] [Google Scholar]
- 5.Mendez-Ferrer S, Chow A, Merad M, Frenette PS. Circadian rhythms influence hematopoietic stem cells. Curr Opin Hematol. 2009;16(4):235–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Morrison SJ, Scadden DT. The bone marrow niche for haematopoietic stem cells. Nature. 2014;505(7483):327–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Colmone A, Amorim M, Pontier AL, Wang S, Jablonski E, Sipkins DA. Leukemic cells create bone marrow niches that disrupt the behavior of normal hematopoietic progenitor cells. Science. 2008;322(5909):1861–5. [DOI] [PubMed] [Google Scholar]
- 8.Boyd AL, Reid JC, Salci KR, Aslostovar L, Benoit YD, Shapovalova Z, et al. Acute myeloid leukaemia disrupts endogenous myelo-erythropoiesis by compromising the adipocyte bone marrow niche. Nat Cell Biol. 2017;19(11):1336–47. [DOI] [PubMed] [Google Scholar]
- 9.Kumar B, Garcia M, Weng L, Jung X, Murakami JL, Hu X, et al. Acute myeloid leukemia transforms the bone marrow niche into a leukemia-permissive microenvironment through exosome secretion. Leukemia. 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Miraki-Moud F, Anjos-Afonso F, Hodby KA, Griessinger E, Rosignoli G, Lillington D, et al. Acute myeloid leukemia does not deplete normal hematopoietic stem cells but induces cytopenias by impeding their differentiation. Proc Natl Acad Sci U S A. 2013;110(33):13576–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhang B, Ho YW, Huang Q, Maeda T, Lin A, Lee SU, et al. Altered microenvironmental regulation of leukemic and normal stem cells in chronic myelogenous leukemia. Cancer Cell. 2012;21(4):577–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Schepers K, Pietras EM, Reynaud D, Flach J, Binnewies M, Garg T, et al. Myeloproliferative neoplasia remodels the endosteal bone marrow niche into a self-reinforcing leukemic niche. Cell Stem Cell. 2013;13(3):285–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Duan CW, Shi J, Chen J, Wang B, Yu YH, Qin X, et al. Leukemia propagating cells rebuild an evolving niche in response to therapy. Cancer Cell. 2014;25(6):778–93. [DOI] [PubMed] [Google Scholar]
- 14.Ishikawa F, Yoshida S, Saito Y, Hijikata A, Kitamura H, Tanaka S, et al. Chemotherapy-resistant human AML stem cells home to and engraft within the bone-marrow endosteal region. Nat Biotechnol. 2007;25(11):1315–21. [DOI] [PubMed] [Google Scholar]
- 15.Chandran P, Le Y, Li Y, Sabloff M, Mehic J, Rosu-Myles M, et al. Mesenchymal stromal cells from patients with acute myeloid leukemia have altered capacity to expand differentiated hematopoietic progenitors. Leuk Res. 2015;39(4):486–93. [DOI] [PubMed] [Google Scholar]
- 16.Reikvam H, Brenner AK, Hagen KM, Liseth K, Skrede S, Hatfield KJ, et al. The cytokine-mediated crosstalk between primary human acute myeloid cells and mesenchymal stem cells alters the local cytokine network and the global gene expression profile of the mesenchymal cells. Stem Cell Res. 2015;15(3):530–41. [DOI] [PubMed] [Google Scholar]
- 17.Huang JC, Basu SK, Zhao X, Chien S, Fang M, Oehler VG, et al. Mesenchymal stromal cells derived from acute myeloid leukemia bone marrow exhibit aberrant cytogenetics and cytokine elaboration. Blood Cancer J. 2015;5:e302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Passaro D, Di Tullio A, Abarrategi A, Rouault-Pierre K, Foster K, Ariza-McNaughton L, et al. Increased Vascular Permeability in the Bone Marrow Microenvironment Contributes to Disease Progression and Drug Response in Acute Myeloid Leukemia. Cancer Cell. 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Frisch BJ, Ashton JM, Xing L, Becker MW, Jordan CT, Calvi LM. Functional inhibition of osteoblastic cells in an in vivo mouse model of myeloid leukemia. Blood. 2012;119(2):540–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Raaijmakers MH, Mukherjee S, Guo S, Zhang S, Kobayashi T, Schoonmaker JA, et al. Bone progenitor dysfunction induces myelodysplasia and secondary leukaemia. Nature. 2010;464(7290):852–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Battula VL, Le PM, Sun JC, Nguyen K, Yuan B, Zhou X, et al. AML-induced osteogenic differentiation in mesenchymal stromal cells supports leukemia growth. JCI Insight. 2017;2(13). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lim M, Pang Y, Ma S, Hao S, Shi H, Zheng Y, et al. Altered mesenchymal niche cells impede generation of normal hematopoietic progenitor cells in leukemic bone marrow. Leukemia. 2016;30(1):154–62. [DOI] [PubMed] [Google Scholar]
- 23.Mirantes C, Passegue E, Pietras EM. Pro-inflammatory cytokines: emerging players regulating HSC function in normal and diseased hematopoiesis. Exp Cell Res. 2014;329(2):248–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rodvold JJ, Chiu KT, Hiramatsu N, Nussbacher JK, Galimberti V, Mahadevan NR, et al. Intercellular transmission of the unfolded protein response promotes survival and drug resistance in cancer cells. Sci Signal. 2017;10(482). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rodvold JJ, Mahadevan NR, Zanetti M. Immune modulation by ER stress and inflammation in the tumor microenvironment. Cancer Lett. 2016;380(1):227–36. [DOI] [PubMed] [Google Scholar]
- 26.Tohmonda T, Miyauchi Y, Ghosh R, Yoda M, Uchikawa S, Takito J, et al. The IRE1alpha-XBP1 pathway is essential for osteoblast differentiation through promoting transcription of Osterix. EMBO Rep. 2011;12(5):451–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Horiuchi K, Tohmonda T, Morioka H. The unfolded protein response in skeletal development and homeostasis. Cell Mol Life Sci. 2016;73(15):2851–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Moore KA, Hollien J. The unfolded protein response in secretory cell function. Annu Rev Genet. 2012;46:165–83. [DOI] [PubMed] [Google Scholar]
- 29.Hornick NI, Doron B, Abdelhamed S, Huan J, Harrington CA, Shen R, et al. AML suppresses hematopoiesis by releasing exosomes that contain microRNAs targeting c-MYB. Sci Signal. 2016;9(444):ra88. [DOI] [PubMed] [Google Scholar]
- 30.Huan J, Hornick NI, Goloviznina NA, Kamimae-Lanning AN, David LL, Wilmarth PA, et al. Coordinate regulation of residual bone marrow function by paracrine trafficking of AML exosomes. Leukemia. 2015;29(12):2285–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Doron B, Handu M, Kurre P. Concise Review: Adaptation of the Bone Marrow Stroma in Hematopoietic Malignancies: Current Concepts and Models. Stem Cells. 2017. [DOI] [PubMed] [Google Scholar]
- 32.Su X, Yu M, Qiu G, Zheng Y, Chen Y, Wen R, et al. Evaluation of nestin or osterix promoter-driven cre/loxp system in studying the biological functions of murine osteoblastic cells. Am J Transl Res. 2016;8(3):1447–59. [PMC free article] [PubMed] [Google Scholar]
- 33.Hornick NI, Huan J, Doron B, Goloviznina NA, Lapidus J, Chang BH, et al. Serum Exosome MicroRNA as a Minimally-Invasive Early Biomarker of AML. Sci Rep. 2015;5:11295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hawkins ED, Duarte D, Akinduro O, Khorshed RA, Passaro D, Nowicka M, et al. T-cell acute leukaemia exhibits dynamic interactions with bone marrow microenvironments. Nature. 2016;538(7626):518–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Boot-Handford RP, Briggs MD. The unfolded protein response and its relevance to connective tissue diseases. Cell Tissue Res. 2010;339(1):197–211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hamamura K, Liu Y, Yokota H. Microarray analysis of thapsigargin-induced stress to the endoplasmic reticulum of mouse osteoblasts. J Bone Miner Metab. 2008;26(3):231–40. [DOI] [PubMed] [Google Scholar]
- 37.Oslowski CM, Urano F. Measuring ER stress and the unfolded protein response using mammalian tissue culture system. Methods Enzymol. 2011;490:71–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Heusermann W, Hean J, Trojer D, Steib E, von Bueren S, Graff-Meyer A, et al. Exosomes surf on filopodia to enter cells at endocytic hot spots, traffic within endosomes, and are targeted to the ER. J Cell Biol. 2016;213(2):173–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Riggs AC, Bernal-Mizrachi E, Ohsugi M, Wasson J, Fatrai S, Welling C, et al. Mice conditionally lacking the Wolfram gene in pancreatic islet beta cells exhibit diabetes as a result of enhanced endoplasmic reticulum stress and apoptosis. Diabetologia. 2005;48(11):2313–21. [DOI] [PubMed] [Google Scholar]
- 40.Shim SH, Xia C, Zhong G, Babcock HP, Vaughan JC, Huang B, et al. Super-resolution fluorescence imaging of organelles in live cells with photoswitchable membrane probes. Proc Natl Acad Sci U S A. 2012;109(35):13978–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hoshino A, Costa-Silva B, Shen TL, Rodrigues G, Hashimoto A, Tesic Mark M, et al. Tumour exosome integrins determine organotropic metastasis. Nature. 2015;527(7578):329–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mulcahy LA, Pink RC, Carter DR. Routes and mechanisms of extracellular vesicle uptake. J Extracell Vesicles. 2014;3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zanetti M, Rodvold JJ, Mahadevan NR. The evolving paradigm of cell-nonautonomous UPR-based regulation of immunity by cancer cells. Oncogene. 2016;35(3):269–78. [DOI] [PubMed] [Google Scholar]
- 44.Kanemoto S, Nitani R, Murakami T, Kaneko M, Asada R, Matsuhisa K, et al. Multivesicular body formation enhancement and exosome release during endoplasmic reticulum stress. Biochem Biophys Res Commun. 2016;480(2):166–72. [DOI] [PubMed] [Google Scholar]
- 45.Hara Y, Yamato G, Shiba N, Ohki K, Park M-J, Tomizawa D, et al. High BMP2 Expression Is a Poor Prognostic Factor and a Good Candidate to Identify CBFA2T3-GLIS2-like High-Risk Subgroup in Pediatric Acute Myeloid Leukemia. Blood. 2015;126(23):2583–. [Google Scholar]
- 46.Krause DS, Scadden DT. A hostel for the hostile: the bone marrow niche in hematologic neoplasms. Haematologica. 2015;100(11):1376–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Huan J, Hornick NI, Shurtleff MJ, Skinner AM, Goloviznina NA, Roberts CT Jr., et al. RNA trafficking by acute myelogenous leukemia exosomes. Cancer Res. 2013;73(2):918–29. [DOI] [PubMed] [Google Scholar]
- 48.Valadi H, Ekstrom K, Bossios A, Sjostrand M, Lee JJ, Lotvall JO. Exosome-mediated transfer of mRNAs and microRNAs is a novel mechanism of genetic exchange between cells. Nat Cell Biol. 2007;9(6):654–9. [DOI] [PubMed] [Google Scholar]
- 49.Duarte D, Hawkins ED, Akinduro O, Ang H, De Filippo K, Kong IY, et al. Inhibition of Endosteal Vascular Niche Remodeling Rescues Hematopoietic Stem Cell Loss in AML. Cell Stem Cell. 2018;22(1):64–77 e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Yadav RK, Chae SW, Kim HR, Chae HJ. Endoplasmic reticulum stress and cancer. J Cancer Prev. 2014;19(2):75–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Schardt JA, Mueller BU, Pabst T. Activation of the unfolded protein response in human acute myeloid leukemia. Methods Enzymol. 2011;489:227–43. [DOI] [PubMed] [Google Scholar]
- 52.Sun H, Lin DC, Guo X, Kharabi Masouleh B, Gery S, Cao Q, et al. Inhibition of IRE1alpha-driven pro-survival pathways is a promising therapeutic application in acute myeloid leukemia. Oncotarget. 2016;7(14):18736–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wey S, Luo B, Tseng CC, Ni M, Zhou H, Fu Y, et al. Inducible knockout of GRP78/BiP in the hematopoietic system suppresses Pten-null leukemogenesis and AKT oncogenic signaling. Blood. 2012;119(3):817–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Murakami T, Saito A, Hino S, Kondo S, Kanemoto S, Chihara K, et al. Signalling mediated by the endoplasmic reticulum stress transducer OASIS is involved in bone formation. Nat Cell Biol. 2009;11(10):1205–11. [DOI] [PubMed] [Google Scholar]
- 55.Montgomery TA, Ruvkun G. MicroRNAs visit the ER. Cell. 2013;153(3):511–2. [DOI] [PubMed] [Google Scholar]
- 56.Stalder L, Heusermann W, Sokol L, Trojer D, Wirz J, Hean J, et al. The rough endoplasmatic reticulum is a central nucleation site of siRNA-mediated RNA silencing. EMBO J. 2013;32(8):1115–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Zylbersztejn F, Flores-Violante M, Voeltzel T, Nicolini FE, Lefort S, Maguer-Satta V. The BMP pathway: a unique tool to decode the origin and progression of leukemia. Exp Hematol. 2018. [DOI] [PubMed] [Google Scholar]
- 58.Geyh S, Rodriguez-Paredes M, Jager P, Khandanpour C, Cadeddu RP, Gutekunst J, et al. Functional inhibition of mesenchymal stromal cells in acute myeloid leukemia. Leukemia. 2016;30(3):683–91. [DOI] [PubMed] [Google Scholar]
- 59.Krause DS, Fulzele K, Catic A, Sun CC, Dombkowski D, Hurley MP, et al. Differential regulation of myeloid leukemias by the bone marrow microenvironment. Nat Med. 2013;19(11):1513–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Raaijmakers MH. Niche contributions to oncogenesis: emerging concepts and implications for the hematopoietic system. Haematologica. 2011;96(7):1041–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Viola S, Traer E, Huan J, Hornick NI, Tyner JW, Agarwal A, et al. Alterations in acute myeloid leukaemia bone marrow stromal cell exosome content coincide with gains in tyrosine kinase inhibitor resistance. Br J Haematol. 2016;172(6):983–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.