

Abstract
Solid organ transplantation disrupts virus-host relationships, potentially resulting in viral transfer from donor to recipient, reactivation of latent viruses, and new viral infections. Viral transfer, colonization, and reactivation are typically monitored using assays for specific viruses, leaving the behavior of full viral populations (the “virome”) understudied. Here we sought to investigate the temporal behavior of viruses from donor lungs and transplant recipients comprehensively. We interrogated the bronchoalveolar lavage (BAL) and blood viromes during the peri-transplant period and 6–16 months post-transplant in 13 donor-recipient pairs using shotgun metagenomic sequencing. Anelloviridae, ubiquitous human commensal viruses, were the most abundant human viruses identified. Herpesviruses, parvoviruses, polyomaviruses and bacteriophages were also detected. Anelloviridae populations were complex, with some donor organs and hosts harboring multiple contemporaneous lineages. We identified transfer of Anelloviridae lineages from donor organ to recipient serum in 4 of 7 cases that could be queried, and immigration of lineages from recipient serum into the allograft in 6 of 10 such cases. Thus, metagenomic analyses revealed that viral populations move between graft and host in both directions, showing that organ transplantation involves implantation of both the allograft and commensal viral communities.
Introduction
Solid organ transplantation exposes recipients to viruses present in donor tissues, and also requires immunosuppression, which facilitates virus reactivation and de novo infection. Organ donors are routinely screened for viruses of known clinical concern (1) and viral infections are common in lung transplant recipients (LTxRs) (2). Viruses may also contribute indirectly to acute cellular rejection (ACR) and chronic lung allograft dysfunction (3, 4).
Advances in metagenomic DNA sequencing methods now enable queries of whole viral populations (the “virome”), including viruses not monitored by current clinical assays. The virome of various human body sites, including the respiratory tract (5–8), is only beginning to be characterized. Recent studies have shown that Anelloviridae are ubiquitous in the human eukaryotic virome (9). Anelloviridae is a family of highly diverse, non-enveloped, small circular single-stranded DNA (ssDNA) viruses that infect humans and other mammals (9). Human Anelloviridae have not been associated with any diseases, although other small circular ssDNA viruses are important veterinary pathogens (10–12). Anelloviridae are likely under chronic immune control given that levels in blood increase in immune-suppressed states (13–15). Lower Anelloviridae burden in blood has been associated with solid organ rejection, suggesting that Anelloviridae DNA copy numbers may be a useful “functional” indicator of immune status (14, 16, 17). Whether lung allograft rejection might be associated with low Anelloviridae levels within the lung itself, perhaps as an indicator of local immune function, has not been investigated.
We previously investigated the virome in bronchoalveolar lavage (BAL) from healthy subjects and a cross-sectional sample of LTxRs (18) and found that lung allografts had markedly elevated Anelloviridae DNA levels compared to healthy adults. We also observed unexpectedly high levels of Anelloviridae DNA in the lungs of donors prior to organ recovery, and found an association between perioperative Anelloviridae dynamics and primary graft dysfunction (19). In these studies, it was unclear whether Anelloviridae genomes in recipients’ allografts were derived from the donated organ or by entry of circulating viruses present in the recipient before transplantation. It was also unknown whether Anelloviridae from the donor organ could disseminate and establish infection outside the lung. We hypothesized that the abundance of Anelloviridae in the donor lungs would result in virus transfer to immunosuppressed LTxRs and that viruses already present in the recipient might also transfer into the allograft. To test this, we investigated the dynamics of whole viral populations from donor lungs before procurement, and in lung allografts and blood during the first 6–16 months post-transplant.
Materials and Methods
Study population
Samples and clinical data were obtained from subjects who had been prospectively enrolled in the multicenter Clinical Trials in Organ Transplantation-03 (CTOT-03) study (NCT00531921). We also collected longitudinal samples of post-transplantation BAL and serum from the recipient (average: 290 days, median: 304 days). BAL samples were obtained from healthy volunteers, as reported previously (Table S1) (20, 21). Serum from a separate group of 21 healthy adult volunteers was also collected. All subjects provided written informed consent under protocols approved by Institutional Review Boards at their respective institutions.
Collection and processing of biological samples
BAL was collected in the operating room from donors immediately before organ procurement, and BAL of the allograft and serum were obtained from LTxRs 1 hour after organ reperfusion as described previously (22). Additional BAL and serum were obtained during routine surveillance bronchoscopy and when indications for further testing arose. Details of virus-like-particle preparation and nucleic acid extraction are in Supplemental Methods. Paired environmental controls were not available from CTOT-03 subjects, so we analyzed a panel of unrelated bronchoscopy prewash specimens and extraction blanks to approximate sequences originating from environmental sources (23–25).
Quantitative PCR
Anelloviridae quantitative PCR (qPCR) was performed on serum and BAL as described previously (18) and in Supplemental Methods.
Shotgun metagenomic sequencing and bioinformatics pipeline
Library preparation and Illumina sequencing are described in Supplemental Methods. To account for contaminating environmental and/or artefactual sequences introduced during acquisition, processing and library preparation of low biomass samples (23, 24, 26, 27), bronchoscope prewashes, buffer and sterile water blanks were analyzed using the same workflow. High quality non-human reads were classified by Kraken (28) against a database containing complete fungal, bacterial, archaeal, and viral genomes from RefSeq and the human genome (GRCH38). Hits to viruses known to be reagent contaminants or otherwise spurious (details in Supplemental Methods) were removed. As previously described (19), a threshold of 10 read pairs assigned to a given viral family per sample was used to call a positive detection, based upon distinguishing clinical samples from environmental controls. Additionally, to validate samples positive for human viruses by the k-mer based approach, reads were aligned to reference viral genomes using Bowtie2 (29) and visually inspected using Integrative Genomics Viewer (30) to ensure broad and even coverage.
Contigs were assembled from reads within each sample using IDBA-UD (31) followed by CAP3 (32). Partial viral genomes were identified by performing nucleotide BLAST of contigs to the NCBI viral genomes database retaining a single target genome hit with an E-value <10−10. This analysis was performed using the Sunbeam pipeline (https://github.com/sunbeam-labs/sunbeam).
Read pairs were aligned to Anelloviridae contigs using Bowtie2 with settings as described in Supplemental Methods (33) and the fraction of the genome covered was generated using BEDtools (34). Alignments were visually inspected as described above.
Statistical analyses
To determine the relatedness of Anelloviridae sequences in longitudinal post-transplant lung and blood samples to Anelloviridae present in the initial donor lung and recipient serum samples, we generated Anelloviridae contigs from donor BAL and peri-transplant recipient serum, and aligned shotgun sequence reads from post-transplantation time points to those contigs. The fold coverage at each base was calculated using python and R scripts (https://github.com/sherrillmix/dnapy, https://github.com/sherrillmix/dnar). We then used a modification of the Gini index, a measure of inequality in a distribution of values (35), to evaluate the evenness of read mapping to target genomes with the premise that authentic detections would result in deep and even coverage. In contrast, uneven coverage represents reads aligning to conserved genomic regions or cross-contamination between samples. Differences in viral lineage detections, quantities, and homology between groups were tested using Wilcoxon Rank Sum Test and ANOVA. The relationship between Anelloviridae levels and clinical outcomes was tested using Student’s t-test. Further details are in Supplemental Methods.
Data availability
Sequence data is collected in BioProject record PRJNA419524.
Results
Study subjects
A total of 114 samples (13 donor BAL, 49 recipient BAL, 52 recipient serum) from 13 organ donors and their respective LTxRs were available for metagenomic sequence analysis (Table 1). These included perioperative samples (donor BAL, and recipient BAL and serum taken immediately following organ implantation and reperfusion) and samples taken during routine post-transplant surveillance (approximately 1, 6, 12, 24, and 48 weeks post transplantation) and for clinical indications. The sampling schedule and relevant clinical events are summarized in Figure 1, Table 1 and Table S1.
Table 1:
Clinical Features of Lung Transplant Recipients
| Subject ID | Age (Years) | Sex | Preoperative Diagnosis | Donor Age (Years) | Transplant Type | Infectious Clinical Events (Organisms) | Non-Infectious Events |
|---|---|---|---|---|---|---|---|
| 11-03 | 28 | M | Cystic Fibrosis | 36 | BLT | Pseudomonas, Haemophilus influenzae, MRSA | ACR |
| 11-09 | 64 | M | Pulmonary Fibrosis | 40 | SLT | Staphylococcus epidermidis | Death |
| 11-15 | 66 | F | Pulmonary Fibrosis | 28 | SLT | Aspergillus terreus | ACR |
| 11-21 | 44 | F | Cystic Fibrosis | 50 | BLT | None | None |
| 11-27 | 22 | M | Cystic Fibrosis | 32 | BLT | None | ACR, death |
| 12-02 | 62 | F | Emphysema | 43 | SLT | None | None |
| 12-09 | 53 | F | Bronchiectasis | 20 | BLT | Streptococcus | PGD |
| 12-12 | 36 | M | Cystic Fibrosis | 52 | BLT | None | None |
| 13-17 | 36 | F | Cystic Fibrosis | 25 | BLT | Pseudomonas, Stenotrophomonas maltophila, Haemophilus influenzae, Candida glabrata, Parainfluenza | None |
| 13-19 | 26 | F | Cystic Fibrosis | 42 | BLT | Pseudomonas, MRSA | None |
| 13-20 | 21 | M | Cystic Fibrosis | 13 | BLT | Escherichia coli, Pseudomonas | ACR, death |
| 13-28 | 54 | M | Pulmonary Fibrosis | 19 | SLT | None | None |
| 13-31 | 45 | M | Pulmonary Fibrosis | 34 | SLT | None | ACR, death |
Subjects were derived from the multicenter Clinical Trials in Organ Transplantation-03 study. All subjects received maintenance immunosuppression post lung transplantation and all but one (12-12) received induction immunosuppression (Simulect/Basilixomab). Infectious events reported were for the lung only and indicate any and all events in the duration of subject enrollment. The virome of peri-transplant BAL samples from three subjects (12-2, 12-9, 13-17) was previously examined in a cross-sectional study of primary graft dysfunction (19).
BLT, bilateral lung transplant ; SLT, single lung transplant; F, female; M, male; MRSA, Methicillin-resistant Staphylococcus aureus; ACR, acute cellular rejection; PGD, primary graft dysfunction
Figure 1: Sample Collection and Clinical Events.
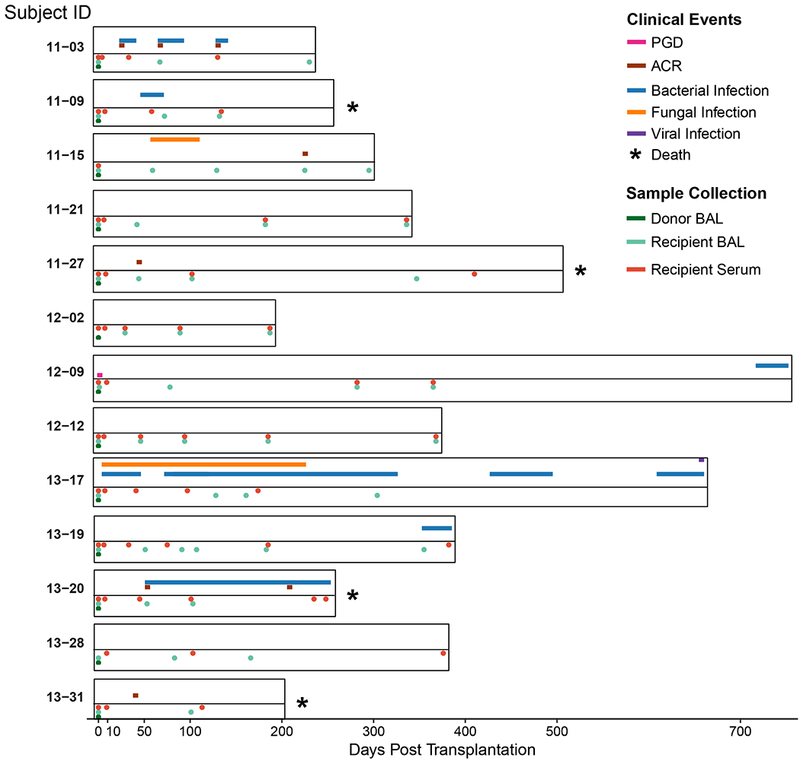
The duration of each subject’s enrollment in the study is shown. Sample collection (Bronchoalveolar lavage (BAL) and blood specimens) time points are displayed below the timeline and indicated by the color code. The timing and duration of adverse clinical events for each subject are displayed above the time line and similarly annotated. For subject 12–09, primary graft dysfunction (PGD) grade 3 occurred within the first 72 h following lung transplantation (55). Five subjects that experienced acute cellular rejection (ACR) had a maximum grade of A2 (56).
Metagenomic shotgun sequencing of donor and recipient samples
To analyze the virome, virus-like particles were isolated from acellular BAL and serum. Total nucleic acid was extracted, whole-genome amplified and then analyzed by metagenomic sequencing. Initial analysis for RNA viruses on a subset of 51 samples yielded no authentic RNA virus detection as determined by the limited extent of genome coverage when sequence reads were mapped onto reference genomes. Therefore, subsequent analysis focused on DNA viruses.
Human sequences and technical artifacts were removed as described previously (27). Further filtering removed artefactual calls due to mis-annotation of human reads (19), database errors, barcode hopping during Illumina sequencing (36) and contamination from environmental sources (23, 26). We acquired over 5.25×1010 base pairs (bp) of DNA sequence from 114 clinical samples, 6 environmental controls (Table S2) and 24 bronchoscope prewashes (27). Samples differed considerably in the number of reads remaining after filtration of human and artifactual sequences (Table S2), likely reflecting both the abundance of the filtered sequences, and differences in the authentic content of viruses.
We assessed the representation of known viruses by analyzing sequence reads using a k-mer based classification scheme (28), revealing a range of abundances (0–52% in BAL and 0.06–33% in serum). We identified viruses from four families known to infect humans: Anelloviridae, Herpesviridae, Polyomaviridae, and Parvoviridae (Fig. 2 and Table 2). The most frequently identified human-cell virus was Anelloviridae, consistent with previous reports (14, 18, 19). On average, 8.7% of all classifiable reads in BAL and 12.7% in serum were derived from Anelloviridae species.
Figure 2: The Viral Microbiome before and after Transplantation.
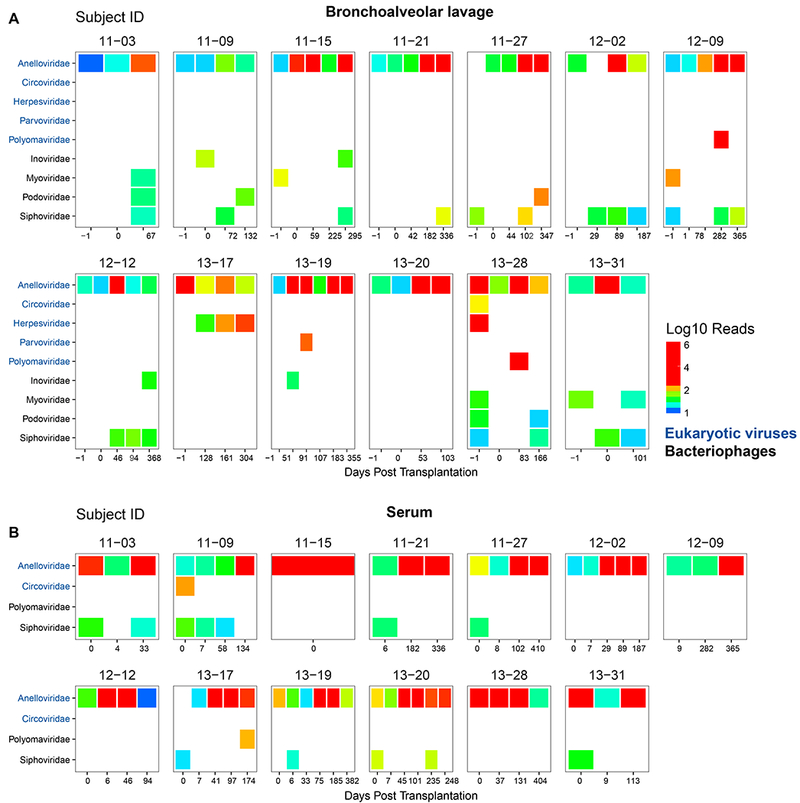
Displayed are the distribution and number of read pair assignments (on a log10 scale) from shotgun metagenomic sequencing of BAL (A) and serum (B). Shown are results from DNA sequences that match known viruses, filtered to remove spurious hits. Each box represents a different donor-recipient pair and each column a different BAL (A) or serum (B) sample. BAL of the donor lung prior to procurement and transplantation is the first column and indicated as “−1” days post-transplantation. Post transplantation BAL samples were taken during routine surveillance bronchoscopy or for other indications. Reads that could not be classified at the species level are not displayed. Only hits with a minimum of 10 reads assigned to a viral family per sample are included. Hits believed to be spurious or derived from environmental contamination are not displayed. Viral families are grouped according to target host. Sequencing, pre-processing of reads, classification and quality control was carried out as described in Supplemental Methods.
Table 2:
Detection of Human Viruses in BAL and Serum
| Subject | Time Point | Species | Mapped Reads | % Coverage |
|---|---|---|---|---|
| 12-09 | Visit 5 BAL | Merkel cell polyomavirus | 5296 | 100 |
| 13-17 | Visit 4 BAL | CMV | 24 | 1 |
| 13-17 | Visit 5 BAL | CMV | 184 | 7 |
| 13-17 | Visit 5 Serum | Merkel cell polyomavirus | 82 | 72 |
| 13-17 | Visit 6 Serum | CMV | 348 | 11 |
| 13-19 | Visit 4 BAL | Human bocavirus | 262 | 87 |
| 13-19 | Visit 5 BAL | Parvovirus B19 | 58 | 48 |
| 13-19 | Visit 6 Serum | Parvovirus B19 | 22 | 27 |
| 13-28 | Donor BAL | EBV | 1848 | 45 |
| 13-28 | Visit 4 BAL | KI polyomavirus | 702 | 100 |
Viral hits discovered through the k-mer based approach (28) were validated by aligning read pairs to reference viral genomes. The number of mapped reads and coverage of the reference genome were calculated as described in Supplemental Methods.
BAL, bronchoalveolar lavage; CMV, cytomegalovirus; EBV, Epstein-Bar virus
To validate assignments from the k-mer analysis, reads from virus-positive samples were aligned to reference viral genomes and inspected for depth and evenness of coverage (Table 2, Fig. S1). In donor BAL 13–28, Epstein-Barr virus (EBV) was detected (1800 reads covering 45% of the genome). The organ donor had positive serology for EBV (Table S3); however, EBV was not detected in subsequent recipient BAL or serum samples. A complete genome of KI polyomavirus was also detected in BAL of this subject at 83 days post transplantation (Fig. S1A). Merkel cell polyomavirus was detected once each in BAL of subject 12–09 and serum of subject 13–17 (Fig. S1A). Cytomegalovirus (CMV) was also detected in the lung of subject 13–17 at two consecutive time points and in serum, even though they were receiving CMV prophylaxis. A near-complete human bocavirus genome was detected in BAL 91 days post-transplantation in subject 13–19. Two subsequent samples from this subject (one BAL and one serum) were positive for parvovirus B19 (Fig. S1B).
We also identified reads that matched to regions of several non-human eukaryotic viruses (Table S4). These included two species within the Circoviridae family and micromonas pusilla virus, which infects green algae. The significance of sequences aligning to these viruses is uncertain.
In addition to eukaryotic viruses, we identified sequences from four bacteriophage families in BAL (Inoviridae, Myoviridae, Podoviridae, Siphoviridae; Fig. 2A). These bacteriophages have diverse bacterial hosts, including members of common oropharyngeal flora and respiratory pathogens (37). In contrast, in serum there were only a few hits to bacteriophages, which mainly overlapped with species or families found in environmental controls (Fig. 2B and Fig. S2). Thus, we lack convincing evidence for authentic detection of bacteriophage in blood.
Most viral hits in background samples matched bacteriophages (Fig. S2), predominantly bacteriophages of Propionibacterium, a skin inhabitant and common reagent contaminant (23, 27). Several controls yielded Anelloviridae reads, but at far lower levels than in clinical samples, likely reflecting “barcode hopping” during sequence acquisition (24). Additional hits in controls were to nonhuman eukaryotic-cell viruses reported previously in metagenomic analyses of bronchoscope prewash controls such as Genomoviridae (27), which we did not study further.
Quantitative analysis of Anelloviridae dynamics in BAL and serum
Anelloviridae were the most prevalent viruses, and were present in donor lungs prior to transplantation. Therefore, we focused on investigating their temporal dynamics and relationship to clinical outcomes.
We first quantified absolute genome copy numbers using qPCR (Fig. 3). Consistent with our previous work (19), Anelloviridae genome copies in BAL from donor lungs prior to transplantation were higher than those in healthy adults (p=0.006; Wilcoxon Rank Sum Test, Table S5). Levels in BAL generally remained elevated compared to healthy adults both early and late post-transplantation (p<0.05 for visits 1, 3, 4, and 6; Wilcoxon Rank Sum Test, Table S5), extending prior observations from our cross-sectional study of LTxRs (18)
Figure 3: Anelloviridae Dynamics by Body Site.
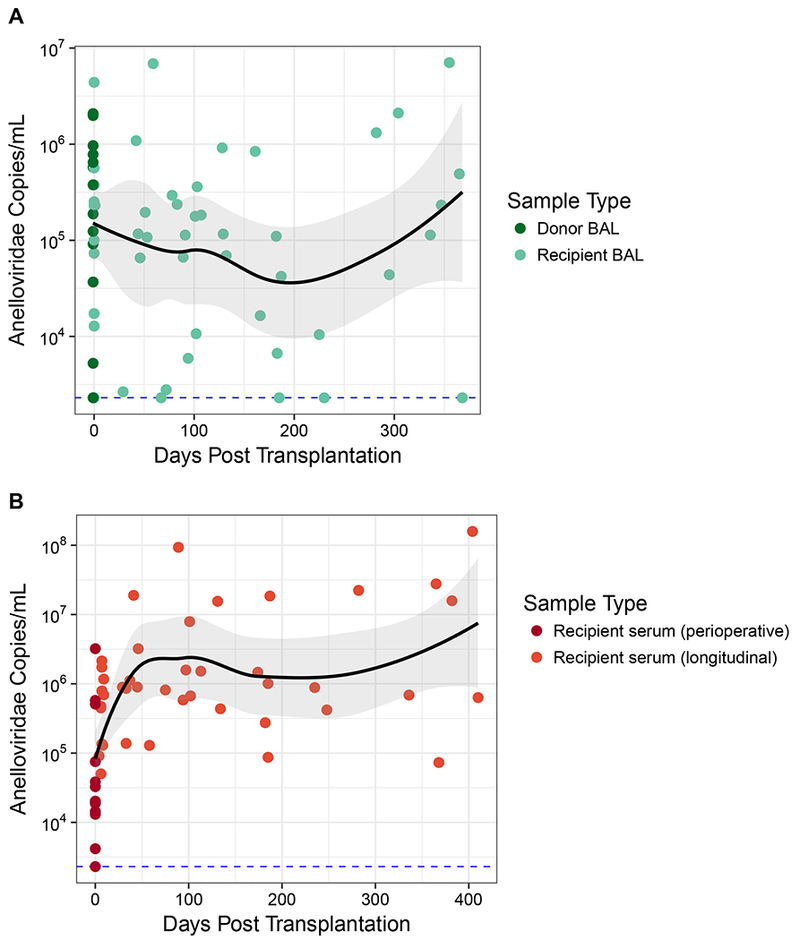
Anelloviridae species were quantified in BAL and serum by qPCR that targets torque teno viruses, torque teno midi viruses and torque teno mini viruses (Anelloviridae family members). The black line represents the local regression curve and standard error for all longitudinal samples. The dotted line represents the limit of detection for the qPCR assay (38 target copies/reaction). Samples at or below this limit were assigned this minimal value. Dots represent individual samples. (A) Longitudinal analysis of Anelloviridae genome copies in BAL. Donor BAL was taken prior to organ procurement. Recipient BAL was first obtained an hour after organ reperfusion and at various time points post transplantation. (B) Longitudinal analysis of Anelloviridae genome copies in serum from lung transplant recipients. Perioperative serum from the transplant recipients was obtained an hour after organ reperfusion.
In contrast to BAL levels, Anelloviridae genome copy numbers in serum of LTxRs immediately after organ reperfusion were lower than levels in healthy adults (p=0.01; Wilcoxon Rank Sum Test, Table S5), consistent with a previous report (38). Serum Anelloviridae levels then increased above the levels of healthy subjects (p<0.05 for all time points greater than 60 days post transplantation; Wilcoxon Rank Sum Test), as previously shown with iatrogenic immunosuppression (15, 39, 40).
Blood Anelloviridae levels have been shown to correlate with ACR in pediatric lung transplantation (41), but no reports have addressed levels in lung. Therefore, we asked if there was a relationship between ACR and Anelloviridae levels in BAL, as well as serum. Consistent with prior reports, we found lower Anelloviridae levels in serum during the month preceding episodes of ACR compared to all non-ACR sera (average 105.5 copies/mL vs average of 106.9 copies/mL; p=0.039; Student’s two-tailed t-test), consistent with previous reports (41). In addition, we also found lower Anelloviridae levels in BAL during the 30 days preceding a diagnosis of ACR (average 104.8 copies/mL vs average of 105.8 copies/mL; p=0.014; Student’s two-tailed t-test).
In contrast, neither levels of Anelloviridae in the donor organ prior to procurement nor diversity of viral lineages were associated with ACR (data not shown). We previously reported that Anelloviridae dynamics during the peri-transplant period were also associated with primary graft dysfunction (19).
Assessing transfer of Anelloviridae populations between allograft and recipient
Given the presence of Anelloviridae in both donor lungs and recipients at the time of transplantation, we investigated transfer of Anelloviridae lineages between the graft and transplant recipient. To do this, we assembled sequence reads from the donor lung and initial recipient serum samples into contigs and asked whether these contigs were represented by reads appearing in later samples. Contigs greater than 2000 bp were chosen to allow sufficient length to query the presence of each Anelloviridae lineage against the background of other variants. Such contigs could be assembled in 7 donor BAL samples, and in 10 post-reperfusion LTxR serum samples (used to represent viral lineages present in recipients at the time of transplantation).
We first analyzed the Anelloviridae ORF1 amino acid sequences to understand the baseline similarity of Anelloviridae swarms within and between subjects (Fig. S3), and determine whether there would be adequate diversity to enable lineage-specific tracking. Across all 140 perioperative contigs, ORF1 sequences exhibited 36% amino acid identity between subjects, and 37% identity within subjects. The low within- and between-subject identity emphasizes the extreme diversity of Anelloviridae, and allows tracking of initial lung and blood lineages at later time points.
We aligned reads from subsequent BAL and blood to these initially present donor lung and blood contigs to track their appearance post-transplantation. Representation at later time points was calculated using the Gini index (35). The Gini index scores evenness of contig coverage and is sensitive to regions of genomic divergence while accommodating different sequence depths in each sample and for each Anelloviridae genome. The Gini index has been used previously for analyses of metagenomic data (42, 43).
To validate this approach, we first investigated whether sharing of Anelloviridae lineages based on Gini index values was more likely in cognate donor-recipient pairs than in unrelated pairs. Indeed, Anelloviridae lineages present in initial donor lung or recipient serum were more likely to be found in later BAL or blood specimens from cognate recipients than in samples from unrelated subjects (p<0.001; Wilcoxon Rank Sum Test on Gini values, Fig. 4–5).
Figure 4: Longitudinal Monitoring of Donor Lung Anelloviridae in Transplant Recipients’ Subsequent Lung and Serum Samples.

Contigs >2000 bp that could be assembled from 7 organ donor BAL were aligned with reads found in post-transplantation BAL (A) and serum (B) samples. Each row is a contig representing an Anelloviridae partial genome in donor BAL. Rows are grouped according to the donor organ in which the contig was found, shown on the left. Columns represent individual samples of BAL (A) or serum (B) arranged chronologically, grouped by recipient ID and annotated based on the color key. Recipient perioperative serum in B was sampled one hour after organ reperfusion. The color in each block represents the Gini index of each comparison of initial sample contig to subsequent samples’ reads. A value of 1 is highly uneven coverage suggesting lack of detection, while 0 is even coverage across the genome suggesting highly confident detection. Alignments of samples to contigs in cognate donor-recipient pairs (donor lung and recipient of that specific donor organ) are outlined in black for ease of visualization. As a group, Anelloviridae lineages present in initial donor lungs were significantly more likely to be found in BAL and blood specimens from cognate recipients than in samples from unrelated subjects (p<0.001; Wilcoxon Rank Sum Test). Individual donor-recipient pairs where there was significant detection of donor lineages in either BAL (A) or serum (B) are indicated by the asterisk (p<0.05; Wilcoxon Rank Sum Test).
Figure 5: Longitudinal Monitoring of Initial Recipient Serum Anelloviridae in Transplant Recipients’ Subsequent Serum and Lung Samples.
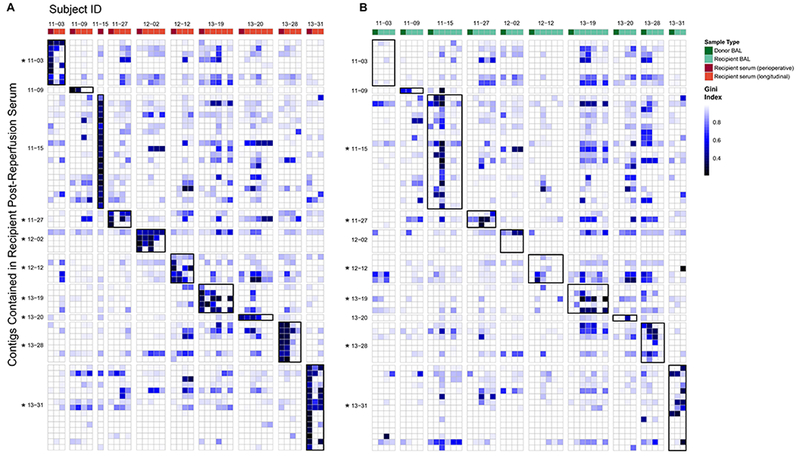
Contigs >2000 bp that could be assembled from 10 perioperative serum samples (obtained within an hour after transplantation and taken to represent recipient Anelloviridae at the time of transplantation) were aligned with reads found in all post-transplantation serum (A) and BAL (B) samples. Each row is a contig representing an Anelloviridae partial genome present in perioperative serum, grouped and annotated by subject. Columns represent individual serum (A) or BAL (B) samples arranged chronologically and grouped by subject (subject 11–15 only had serum from the perioperative time point available). In B, BAL of the donor organ is the first column. The color in each block represents the Gini index of each genome in each sample. A value of 1 is highly uneven coverage suggesting lack of detection while 0 is even coverage across the genome suggesting highly confident detection. Alignment results of only the 10 subjects with contigs from perioperative serum are shown here. Alignments of samples to contigs in cognate donor-recipient pairs are outlined in black for ease of visualization. As a group, Anelloviridae lineages present in initial recipient serum were significantly more likely to be found in BAL and blood specimens at later time points from that subject than in samples from unrelated subjects (p<0.001; Wilcoxon Rank Sum Test). Individual recipients where there was significant detection of these perioperative lineages in either BAL (A) or serum (B) are indicated by the asterisk (p<0.05; Wilcoxon Rank Sum Test).
We also compared samples using several additional approaches. First, we compared lineage representation in each sample by calculating reads per kilobase of alignment target per million reads sequenced (RPKM), using reads remaining after filtration to remove human reads and low quality reads. This normalizes for variable recovery of reads after filtration (Table S6). Second, we calculated the total fraction of the contig covered by reads in subsequent samples (Table S6). We also observed that full and even coverage of a contig by sequences from an unrelated individual was rare (Tables S7–8). Finally, we assessed temporal dynamics of specific Anelloviridae contigs within a donor-recipient pair by visually inspecting alignments (Fig. S4). These approaches yielded similar conclusions as the analysis using the Gini index.
Detection of donor lung Anelloviridae lineages in recipient allograft and serum
To assess transfer of Anelloviridae lineages from donor lungs to recipients, we focused on the 7 LTxRs for whom donor BAL samples yielded contigs >2000 bp (range 1–28 contigs per donor). Figure 4A illustrates the persistence of Anelloviridae lineages from donor lung in longitudinal BAL of recipients, and Figure 4B illustrates the appearance of donor lung lineages in serum of recipients. As a group, donor lung contigs were more similar to sequences in subsequent cognate BAL and serum samples than samples from other transplant recipients (p<0.001; Wilcoxon Rank Sum Test on Gini index). Tracking individual donor viruses revealed that they were more evenly covered in BAL specimens from their cognate recipients than in random comparisons in 4/7 pairs (Fig. 4A). The same analysis comparing donor lung contigs and longitudinal recipient serum reads revealed the appearance of donor lung Anelloviridae lineages in recipient blood in 4/7 pairs (Fig. 4B). Thus, post-transplantation Anelloviridae persistence within the lung and dissemination to serum was detectable in the majority of cases where this could be evaluated.
Detection of recipient Anelloviridae lineages in lung allografts and recipient serum over time
We next asked whether Anelloviridae lineages present in recipients’ blood at the time of transplant appeared in the allograft after transplantation, and whether these lineages persisted in serum. Post-reperfusion serum contigs were used as a representation of recipient-derived lineages. Contigs >2000 bp were available for 10 LTxRs (range 1–20 contigs per subject).
Recipient serum Anelloviridae lineages were more closely related to both cognate serum and BAL after transplantation, compared with unrelated subjects (p<0.001; Wilcoxon Rank Sum test on Gini index). Recipient Anelloviridae lineages present at time of transplantation were then tracked individually, and could be detected in serum at later time points in 8/10 pairs (Fig. 5A), indicating persistence in blood. Entry of recipient blood Anelloviridae lineages into the allograft was investigated by comparing contigs from recipient post-reperfusion serum samples to longitudinal recipient BAL samples. As expected, in most cases there was no significant coverage of recipient serum lineages in donor BAL pre-transplant (i.e. before exposure to recipient lineages) (Table S8). After transplantation, recipient Anelloviridae lineages were detectable in subsequent BAL samples in 6/10 subjects (Fig. 5B). Thus, Anelloviridae lineages present systemically in recipients at the time of transplantation commonly entered and populated the graft.
Discussion
In this study, we investigated dynamics of the lung and blood virome in lung transplant donors and recipients. Both human viruses and bacteriophages were identified. One viral family, Anelloviridae, was ubiquitous and abundant. Anelloviridae have not been reported to cause disease in humans, but they appear to be under immune control and monitoring their abundance and diversity provides a window on viral interactions with the host immune system.
Anelloviridae blood levels increase in states of immune deficiency such as AIDS and decrease with immune reconstitution (13, 15, 39, 44). Transplant recipients with episodes of organ rejection have been shown to have lower Anelloviridae levels than those without rejection, consistent with inadequate immune suppression (14, 41) . Thus, while the specific immune mechanisms responsible for regulating Anelloviridae in vivo are unknown, it has been proposed that monitoring levels in blood might serve as a “functional” measure of immune activity to manage organ transplant immunosuppression (14, 16). Our data are consistent with previous findings in blood. In addition, a novel observation here is that Anelloviridae within the lung allograft are also relatively decreased prior to ACR. Since controlling immune activity within the allograft is the key goal of organ transplantation management, further studies appear warranted to determine whether lung Anelloviridae levels might reflect local compartmentalized immune function, and/or could offer actionable information useful in immunosuppression management.
The extensive intra-subject Anelloviridae sequence diversity observed here is consistent with previous reports of diverse lineages coexisting within individuals (18, 45–49). The origin of multiple independent lineages within individuals is unclear and could arise from intra-host evolution, initial infection by multiple lineages, or episodes of superinfection (9). Additionally, certain individuals had, by chance, some Anelloviridae sequences that were highly similar to those in unrelated individuals. Tracking populations of related yet distinct viral genomes within subjects is challenging, so we combined breadth and depth of genome coverage using the Gini index and compared representation of specific Anelloviridae lineages within and between individuals. We show that some Anelloviridae lineages contained within the donor organ emigrate from the allograft and circulate systemically in recipients. Conversely, some Anelloviridae species from the recipient entered and populated the allograft. Donor-recipient transmission of clinically important viruses during organ transplantation is well-described (50), but engraftment of an entire viral population along with the organ, with bidirectional flow from the allograft to the recipient and vice-versa, is novel.
We found that some Anelloviridae lineages present either in the donor organ or the recipients’ systemic circulation persisted for months, while others disappeared, possibly being replaced by new variants. The consequences of donor- or recipient-derived Anelloviridae within the graft are unknown. It is plausible that introduction of novel Anelloviridae lineages may have subtle deleterious consequences such as by triggering localized inflammation within the allograft. Conversely, Anelloviridae have been implicated in suppressing NFkB-mediated cellular activation (51). Unfortunately we did not have sufficient power to ask what outcomes might be linked to donor- versus recipient-derived lineages within the graft, which should be a topic of future studies.
Only one donor BAL revealed a known human virus other than Anelloviridae, which was EBV. In recipients, Herpesviruses (CMV, EBV), parvoviruses (B19, human bocavirus) and polyomaviruses (KI, Merkel cell) were identified in lung and blood. Detection of Merkel cell polyomavirus and KI polyomavirus in whole BAL or tissue of lung transplantation patients has been described (52), although DNA in acellular BAL is a novel finding that suggests production of extracellular viral particles, consistent with active viral replication. Subject 13–17 yielded CMV in one serum and two BAL specimens despite being on CMV suppressive therapy, and another subject’s BAL revealed human bocavirus. Although infection with traditional community-acquired respiratory viruses is common after lung transplantation, none of our subjects had clinically-recognized viral infection at the time of sample collection. Thus, our limited detection of known pathogenic viruses in acellular BAL and serum suggest that asymptomatic or subclinical infection in lung transplant recipients is uncommon, or that these metagenomic methods have limited sensitivity for their detection.
Our study has several limitations. The use of acellular BAL is appropriate for analysis of the extracellular (replicating) virome, but cannot detect intracellular viral nucleic acids of non-replicating or latent forms. Due to geographic heterogeneity within the lung (53), any single sample may report only part of the virome. Because replicate samples were not available, the effect of sampling stochasticity could not be quantified. Long term storage of BAL and our methods of sample processing may have reduced our sensitivity to detect low abundance enveloped DNA and RNA viruses. Our small sample size limited our ability to compare the magnitude of association of Anelloviridae and ACR in lung versus blood, and the number of subjects in whom we could track individual lineages precluded an analysis of donor versus recipient strain-specific relationship to outcomes. Finally, our findings report on viruses present in reference databases. Indeed, between 12–94% of metagenomic reads in each sample could not be classified into any known kingdom, similar to other metagenomic studies (25, 54). Illuminating this viral dark matter may reveal uncharacterized viruses that may play a role in lung health and disease.
In summary, our study shows that lung transplantation is associated with engraftment not just of a donor organ, but of its endogenous population of Anelloviridae as well. Future studies will be needed to determine factors that regulate viral transfer between graft and host, and further define the relationship between compartmentalized lung Anelloviridae and transplantation outcomes.
Supplementary Material
Acknowledgments
We are grateful to subjects and volunteers for providing specimens, to the CTOT investigators, the staff participating in the CTOT-03 and Lung HIV Microbiome Project studies, and to members of the Bushman and Collman laboratories for help and suggestions.
This work was supported by NIH grants R01-HL113252, R61-HL137063, U01-HL098957, R01-HL087115, K24-HL115354, and received assistance from the Penn Center for AIDS Research (P30-AI045008) and the PennCHOP Microbiome Program.
A.A.A was supported by NSF grant DGE-1321851 and J.M.D was supported by K23-HL121406.
Abbreviations:
- ACR
acute cellular rejection
- BAL
bronchoalveolar lavage
- bp
base pairs
- CMV
cytomegalovirus
- EBV
Epstein-Bar virus
- LTxR
lung transplant recipient
- ORF
open reading frame
- ssDNA
single-stranded DNA
Footnotes
Disclosure
The authors of this manuscript have no conflicts of interest to disclose as described by the American Journal of Transplantation.
Supporting Information
Additional Supporting Information may be found online in the supporting information tab for this article.
References
- 1.Grossi PA, Fishman JA, of Practice AST. Donor-Derived Infections in Solid Organ Transplant Recipients. American Journal of Transplantation. 2009;9(s4). [DOI] [PubMed] [Google Scholar]
- 2.Burguete SR, Elli DJ, Feandez JF, Levine SM. Lung transplant infection. Respirology. 2013;18(1):22–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Peghin M, Hirsch HH, Len Ó, Codina G, Berastegui C, Sáez B, et al. Epidemiology and Immediate Indirect Effects of Respiratory Viruses in Lung Transplant Recipients: A 5-Year Prospective Study. American journal of transplantation : official journal of the American Society of Transplantation and the American Society of Transplant Surgeons. 2017;17(5):1304–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Vu DLL, Bridevaux POO, Aubert JDD, Soccal PM, Kaiser L. Respiratory viruses in lung transplant recipients: a critical review and pooled analysis of clinical studies. American journal of transplantation : official journal of the American Society of Transplantation and the American Society of Transplant Surgeons. 2011;11(5):1071–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lewandowska DW, Schreiber PW, Schuurmans MMM, Ruehe B, Zagordi O, Bayard C, et al. Metagenomic sequencing complements routine diagnostics in identifying viral pathogens in lung transplant recipients with unknown etiology of respiratory infection. PloS one. 2017;12(5). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wylie KM, Mihindukulasuriya KA, Sodergren E, Weinstock GM, Storch GA. Sequence analysis of the human virome in febrile and afebrile children. PloS one. 2012;7(6). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Taboada B, Espinoza MA, Isa P, Aponte FE, Arias-Ortiz MA, Monge-Martínez J, et al. Is there still room for novel viral pathogens in pediatric respiratory tract infections? PloS one. 2014;9(11). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Willner D, Furlan M, Haynes M, Schmieder R, Angly FE, Silva J, et al. Metagenomic analysis of respiratory tract DNA viral communities in cystic fibrosis and non-cystic fibrosis individuals. PloS one. 2009;4(10). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Spandole S, Cimponeriu D, Berca LM, Mihăescu G. Human anelloviruses: an update of molecular, epidemiological and clinical aspects. Archives of virology. 2015;160(4):893–908. [DOI] [PubMed] [Google Scholar]
- 10.Meng XJ. Emerging and re-emerging swine viruses. Transboundary and emerging diseases. 2012;59 Suppl 1: 85–102. [DOI] [PubMed] [Google Scholar]
- 11.Ellis J Porcine circovirus: a historical perspective. Veterinary pathology. 2014;51(2):315–27. [DOI] [PubMed] [Google Scholar]
- 12.Todd D Avian circovirus diseases: lessons for the study of PMWS. Veterinary microbiology. 2004;98(2):169–74. [DOI] [PubMed] [Google Scholar]
- 13.Thom K, Petrik J. Progression towards AIDS leads to increased torque teno virus and torque teno minivirus titers in tissues of HIV infected individuals. Journal of Medical Virology. 2007;79(1):1–7. [DOI] [PubMed] [Google Scholar]
- 14.De Vlaminck I, Khush KK, Strehl C, Kohli B, Luikart H, Neff NF, et al. Temporal response of the human virome to immunosuppression and antiviral therapy. Cell. 2013;155(5):1178–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Görzer I, Haloschan M, Jaksch P, Klepetko W, Puchhammer-Stöckl E. Plasma DNA levels of Torque teno virus and immunosuppression after lung transplantation. The Journal of Heart and Lung Transplantation. 2014;33(3):320–3. [DOI] [PubMed] [Google Scholar]
- 16.Focosi D, Antonelli G, Pistello M, Maggi F. Torquetenovirus: the human virome from bench to bedside. Clinical microbiology and infection : the official publication of the European Society of Clinical Microbiology and Infectious Diseases. 2016;22(7):589–93. [DOI] [PubMed] [Google Scholar]
- 17.Masouridi-Levrat S, Pradier A, Simonetta F, Kaiser L, Chalandon Y, Roosnek E. Torque teno virus in patients undergoing allogeneic hematopoietic stem cell transplantation for hematological malignancies. Bone marrow transplantation. 2016;51(3):440–2. [DOI] [PubMed] [Google Scholar]
- 18.Young JC, Chehoud C, Bittinger K, Bailey A, Diamond JM, Cantu E, et al. Viral metagenomics reveal blooms of anelloviruses in the respiratory tract of lung transplant recipients. American journal of transplantation : official journal of the American Society of Transplantation and the American Society of Transplant Surgeons. 2015;15(1):200–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Abbas AA, Diamond JM, Chehoud C, Chang B, Kotzin JJ, Young JC, et al. The Perioperative Lung Transplant Virome: Torque Teno Viruses Are Elevated in Donor Lungs and Show Divergent Dynamics in Primary Graft Dysfunction. Am J Transplant. 2017;17(5):1313–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Charlson ES, Bittinger K, Haas AR, Fitzgerald AS, Frank I, Yadav A, et al. Topographical Continuity of Bacterial Populations in the Healthy Human Respiratory Tract. American Journal of Respiratory and Critical Care Medicine. 2011;184(8):957–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Beck JM, Schloss PD, Venkataraman A, Twigg H, Jablonski KA, Bushman FD, et al. Multicenter Comparison of Lung and Oral Microbiomes of HIV-infected and HIV-uninfected Individuals. American journal of respiratory and critical care medicine. 2015;192(11):1335–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cantu E, Lederer DJ, Meyer K, Milewski K, Suzuki Y, Shah RJ, et al. Gene set enrichment analysis identifies key innate immune pathways in primary graft dysfunction after lung transplantation. American journal of transplantation : official journal of the American Society of Transplantation and the American Society of Transplant Surgeons. 2013;13(7):1898–904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Salter SJ, Cox MJ, Turek EM, Calus ST, Cookson WO, Moffatt MF, et al. Reagent and laboratory contamination can critically impact sequence-based microbiome analyses. BMC Biology. 2014;12(1):1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kim D, Hofstaedter CE, Zhao C, Mattei L, Tanes C, Clarke E, et al. Optimizing methods and dodging pitfalls in microbiome research. Microbiome. 2017;5(1):52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Quince C, Walker AW, Simpson JT, Loman NJ, Segata N. Shotgun metagenomics, from sampling to analysis. Nature biotechnology. 2017;35(9):833–44. [DOI] [PubMed] [Google Scholar]
- 26.Naccache SN, Greninger AL, Lee D, Coffey LL, Phan T, Rein-Weston A, et al. The perils of pathogen discovery: origin of a novel parvovirus-like hybrid genome traced to nucleic acid extraction spin columns. Journal of virology. 2013;87(22):11966–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Clarke EL, Lauder AP, Hofstaedter CE, Hwang Y, Fitzgerald AS, Imai I, et al. Microbial Lineages in Sarcoidosis: A Metagenomic Analysis Tailored for Low Microbial Content Samples. American journal of respiratory and critical care medicine. 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wood DE, Salzberg SL. Kraken: ultrafast metagenomic sequence classification using exact alignments. Genome biology. 2014;15(3). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Langmead B, Salzberg SL. Fast gapped-read alignment with Bowtie 2. Nature methods. 2012;9(4):357–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Robinson JT, Thorvaldsdóttir H, Winckler W, Guttman M, Lander ES, Getz G, et al. Integrative genomics viewer. Nature biotechnology. 2011;29(1):24–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Peng Y, Leung HCM, Yiu SM, Chin FYL. IDBA-UD: a de novo assembler for single-cell and metagenomic sequencing data with highly uneven depth. Bioinformatics. 2012;28(11):1420–8. [DOI] [PubMed] [Google Scholar]
- 32.Huang X, Madan A. CAP3: A DNA sequence assembly program. Genome research. 1999;9(9):868–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics (Oxford, England). 2009;25(16):2078–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Quinlan AR, Hall IM. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics (Oxford, England). 2010;26(6):841–2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gini C Variabilità e mutabilità. Contributi allo studio dele relazioni e delle distribuzioni statistiche. Studi Economico-Giuridici della Università di Cagliari. 1912. [Google Scholar]
- 36.Lauder AP, Roche AM, Sherrill-Mix S, Bailey A, Laughlin AL, Bittinger K, et al. Comparison of placenta samples with contamination controls does not provide evidence for a distinct placenta microbiota. Microbiome. 2016;4(1):29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Edlund A, Santiago-Rodriguez TM, Boehm TK, Pride DT. Bacteriophage and their potential roles in the human oral cavity. Journal of oral microbiology. 2015;7:27423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Focosi D, Macera L, Boggi U, Nelli LC, Maggi F. Short-term kinetics of torque teno virus viraemia after induction immunosuppression confirm T lymphocytes as the main replication-competent cells. The Journal of general virology. 2015;96(Pt 1):115–7. [DOI] [PubMed] [Google Scholar]
- 39.Görzer I, Jaksch P, Kundi M, Seitz T, Klepetko W, Puchhammer-Stöckl E. Pre-transplant plasma Torque Teno virus load and increase dynamics after lung transplantation. PloS one. 2015;10(3). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Moen EM, Sagedal S, Bjøro K, Degré M, Opstad PK, Grinde B. Effect of immune modulation on TT virus (TTV) and TTV-like-mini-virus (TLMV) viremia. Journal of medical virology. 2003;70(1):177–82. [DOI] [PubMed] [Google Scholar]
- 41.Blatter JA, Sweet SC, Conrad C, Danziger-Isakov LA, Faro A, Goldfarb SB, et al. Anellovirus loads are associated with outcomes in pediatric lung transplantation. Pediatric transplantation. 2018;22(1). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bhattacharya T, Ghosh TS, Mande SS. Global Profiling of Carbohydrate Active Enzymes in Human Gut Microbiome. PloS one. 2015;10(11). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kobayashi T, Andoh A. Numerical analyses of intestinal microbiota by data mining. Journal of clinical biochemistry and nutrition. 2018;62(2):124–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Madsen CD, Eugen-Olsen J, Kirk O, Parner J, Kaae Christensen J, Brasholt MS, et al. TTV viral load as a marker for immune reconstitution after initiation of HAART in HIV-infected patients. HIV clinical trials. 2002;3(4):287–95. [DOI] [PubMed] [Google Scholar]
- 45.Okamoto H, Nishizawa T, Takahashi M, Asabe S, Tsuda F, Yoshikawa A. Heterogeneous distribution of TT virus of distinct genotypes in multiple tissues from infected humans. Virology. 2001;288(2):358–68. [DOI] [PubMed] [Google Scholar]
- 46.Devalle S, Rua F, Morgado MG, Niel C. Variations in the frequencies of torque teno virus subpopulations during HAART treatment in HIV-1-coinfected patients. Archives of Virology. 2009;154(8):1285–91. [DOI] [PubMed] [Google Scholar]
- 47.Bzhalava D, Ekström J, Lysholm F, Hultin E, Faust H, Persson B, et al. Phylogenetically diverse TT virus viremia among pregnant women. Virology. 2012;432(2):427–34. [DOI] [PubMed] [Google Scholar]
- 48.Jelcic I, Hotz-Wagenblatt A, Hunziker A, Zur Hausen H, de Villiers E- MM. Isolation of multiple TT virus genotypes from spleen biopsy tissue from a Hodgkin’s disease patient: genome reorganization and diversity in the hypervariable region. Journal of virology. 2004;78(14):7498–507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ninomiya M, Takahashi M, Nishizawa T, Shimosegawa T, Okamoto H. Development of PCR assays with nested primers specific for differential detection of three human anelloviruses and early acquisition of dual or triple infection during infancy. Journal of clinical microbiology. 2008;46(2):507–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Green M, Covington S, Taranto S, Wolfe C, Bell W, Biggins SW, et al. Donor-derived transmission events in 2013: a report of the Organ Procurement Transplant Network Ad Hoc Disease Transmission Advisory Committee. Transplantation. 2015;99(2):282–7. [DOI] [PubMed] [Google Scholar]
- 51.Zheng H, Ye L, Fang X, Li B, Wang Y, Xiang X, et al. Torque teno virus (SANBAN isolate) ORF2 protein suppresses NF-kappaB pathways via interaction with IkappaB kinases. Journal of virology. 2007;81(21):11917–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Bergallo M, Costa C, Terlizzi ME, Astegiano S, Curtoni A, Solidoro P, et al. Quantitative detection of the new polyomaviruses KI, WU and Merkel cell virus in transbronchial biopsies from lung transplant recipients. Journal of clinical pathology. 2010;63(8):722–5. [DOI] [PubMed] [Google Scholar]
- 53.Willner D, Haynes MR, Furlan M, Hanson N, Kirby B, Lim YW, et al. Case studies of the spatial heterogeneity of DNA viruses in the cystic fibrosis lung. American journal of respiratory cell and molecular biology. 2012;46(2):127–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Krishnamurthy SR, Wang D. Origins and challenges of viral dark matter. Virus research. 2017. [DOI] [PubMed] [Google Scholar]
- 55.Yusen RD, Edwards LB, Kucheryavaya AY, Benden C, Dipchand AI, Goldfarb SB, et al. The Registry of the International Society for Heart and Lung Transplantation: Thirty-second Official Adult Lung and Heart-Lung Transplantation Report—2015; Focus Theme: Early Graft Failure. The Journal of Heart and Lung Transplantation. 2015;34(10):1264–77. [DOI] [PubMed] [Google Scholar]
- 56.Stewart S, Fishbein MC, Snell GI, Berry GJ, Boehler A, Burke MM, et al. Revision of the 1996 working formulation for the standardization of nomenclature in the diagnosis of lung rejection. The Journal of heart and lung transplantation. 2007;26(12):1229–42. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Sequence data is collected in BioProject record PRJNA419524.