

Abstract
Angiogenesis is essential for tumor growth. Vascular endothelial growth factor (VEGF), a crucial factor in tumor angiogenesis, has been reported to be transcriptionally regulated by hypoxia-inducible factor-1 (HIF-1). An 8-oxo-G or apurinic/apyrimidinic (AP) site, which is frequently associated with DNA damage, has been identified in the promoter region of VEGF. However, the detailed molecular mechanisms by which AP sites regulate VEGF gene transcription are largely unknown. The dual functional protein apurinic/apyrimidinic endonuclease 1 (APE1) is both the key enzyme in DNA base excision repair and the redox factor shown to regulate HIF-1 DNA-binding activity. In the present study, we tested the involvement of both the AP endonuclease and redox activity of APE1 in regulating HIF-1 DNA binding and VEGF transcription in HUVECs. By employing two APE1 activity-specific inhibitors and AP-site-containing reporter constructs, we confirmed that both activities of APE1 were involved in regulating VEGF expression under hypoxic conditions. Furthermore, we found that the interaction between APE1 and its downstream repair enzyme, DNA polymerase β, was compromised when the N-terminal structure of APE1 was distorted under oxidative conditions. Our data suggest that the DNA repair and redox activity of APE1 can play a collaborative role in regulating the transcriptional initiation of the AP-site-containing promoter.
Keywords: Hypoxia, HIF-1, APE1, Redox, AP site
Abbreviations: APE1, Apurinic/apyrimidinic endonuclease; AP sites, Apurinic/apyrimidinic sites; BER, Base excision repair; Co-IP, Coimmunoprecipitation; Egr-1, Early growth response protein-1; EMSA, Electrophoretic mobility-shift assay; Fapy, Formamidopyrimidine; HIF-1, Hypoxia-induced factor-1; HRE, Hypoxic response element; HUVEC, Human umbilical vein endothelial cells; NF-κB, Nuclear factor-kappa B; OGG1, DNA Oxoguanine glycosylase 1; Pol β, DNA Polymerase β; qPCR, Quantitative PCR; VEGF, Vascular endothelial growth factor
1. Introduction
Efficient inhibition of solid tumors at the molecular level requires the characterization and a greater understanding of the mechanistic pathways that lead to the development, sustenance, and growth of these tumors. To develop novel and efficient anticancer strategies, it is crucial not only to characterize the primary factors essential for tumorigenesis but also to identify the secondary factors involved in tumor growth and regulation. The status of genomic stability and the regulation of transcription by physiological stress factors, such as a hypoxic environment, are crucial signals that contribute to the development of solid tumors [1]. During the course of growth, a solid tumor requires oxygen and nutrition, and therefore, angiogenesis is a key step in a developing tumor [2]. Vascular endothelial growth factor (VEGF) is a critical factor in tumor angiogenesis [3].
The expression of VEGF is mainly under the control of a key hypoxia-related transcription factor, namely, hypoxia-induced factor 1 (HIF-1) [4]. HIF-1 is a heterodimer that binds to the promoter of hypoxia-responsive genes and associates with other regulators of transcription factors, such as APE1 [5]. HIF-1 is involved in cancer promotion and progression; as its name suggests, HIF-1 is upregulated in hypoxic tissue and highly expressed in certain tumors while being minimally expressed in normoxic tissues or more indolent tumors. Several studies have reported a role for HIF-1 in specific types of cancers (reviewed in [6]). For example, the estrogen and androgen receptor signaling pathways have been postulated to upregulate HIF-1 in breast and prostate cancer, respectively [7], and inhibition of HIF-1 activity has been shown to impair gastric tumor growth, angiogenesis, and vessel maturation [8].
Elucidating the significance of HIF-1-responsive genes and the specific regulation of HIF-1 transcriptional activity is crucial for cancer research because of the central importance of HIF-1 in tumor activity. Under hypoxic conditions, which frequently occur in solid tumors, the transcriptional activity of HIF-1 is regulated by Redox effector factor 1/apurinic/apyrimidinic endonuclease (Ref-1/APE1) [9]. Recently, the role of APE1 has come under scrutiny with respect to its involvement in the regulation of transcriptional complexes associated with hypoxia-induced genes [[10], [11]]. It has been suggested that oxidative damage induced by hypoxia can damage specific bases in the hypoxic response element (HRE) of the VEGF gene [12]. The subsequent strand-modifying/cleaving activity of OGG1/APE1 results in a DNA single-strand break that alters the topology of the HRE. Altering the topology of promoter regions may have a profound effect on nucleosome positioning and potentially allow structural changes in the promoter region that alter the recruitment/activity of relevant transcription factors [13].
Elevations in APE1 levels are associated with chemotherapeutic resistance, a negative prognosis and decreased survival (reviewed in [14]). Another study found that APE1 is critical for the formation of the hypoxia-inducible transcriptional complex on the HRE on the VEGF gene and that the presence of APE1 in the complex is required for the apparent high-affinity association between HIF-1 and its DNA recognition sequence [15]. The specific role of APE1 during hypoxia with respect to the HIF-1-regulated VEGF gene makes it a candidate for further study, specifically regarding how its multiple functions contribute to VEGF gene transcription in cancer promotion. The balance between genomic stability and instability and the potential role(s) of APE1 in affecting the topology of the HRE of the VEGF gene leads us to explore the functional determinants of APE1 with respect to this pathway and their effects on the subsequent expression of hypoxia-inducible genes, such as VEGF. The transcriptional activity of HIF-1, specifically at the VEGF promoter, is thought to be regulated by APE1 in the following manner: oxidative base modifications are introduced into the VEGF gene and HRE by 1) physiological signals (i.e., hypoxia) via an uncharacterized pathway and 2) by redox signaling via a reduction in oxidized cysteine residues in HIF-1. It has been suggested that the 5′ guanine in the HRE is oxidized and produces an 8-oxoguanine that potentially recruits DNA oxoguanine glycosylase 1 (OGG1). The formation of a basic site by OGG1 is then followed by the recruitment of APE-1, the initial enzyme of the base excision repair (BER) pathway that removes the apurinic sugar, affects a single-strand break in the phosphodiester backbone, and generates a 3′-OH as a primer for DNA polymerase β (pol β) (review in [16]). The reduction in topological constraints in the HRE of the VEGF promoter can then affect gene expression by modulating sequence flexibility.
APE-1 is a bifunctional protein, and we sought to determine how altering its two primary activities would impact VEGF expression in the context of hypoxic conditions. We previously showed that APE1 played an important role in angiogenesis, which we then confirmed in subsequent studies [10]. APE1 fosters redox-dependent interactions between HIF-1 and transcriptional coactivators, which form a multiprotein complex that binds to contiguous and sometimes noncontiguous bases in the HRE [17]. Our specific interest was focused on the effect of APE1 on the HRE of VEGF, which is known to be crucial for angiogenesis [[10], [11], [12], [13], [14], [15], [16], [17], [18]]. APE1 is a 318-amino-acid monomeric protein that has been organized into three functionally independent domains. These domains include the redox domain, the DNA-repair domain, and the RNA-binding domain. The DNA-repair function is mediated by residue His309 in the catalytic site near the C terminus. The function of APE1 as a redox protein depends on the Cys65 residue, which is involved in stimulating the DNA-binding activity of several transcription factors that are important for cancer progression, including the HIF-1, nuclear factor-kappa B (NF-κB), early growth response protein-1 (Egr-1), p53, CREB, AP-1, and Pax proteins [19]. APE1 gained redox function over the course of evolution, meaning that both AP endonuclease and redox activity can be coupled during some essential biological processes. A recent study showed that an AP site in the VEGF promoter region increased HIF-1 binding and upregulated VEGF expression [11]. This study highlighted that APE1, as both a regulator of transcription and a critical enzyme in the BER pathway, can play a crucial role in mediating enhanced VEGF transcription via an AP-site-containing promoter. However, how the distinct activities of APE1 are involved in VEGF regulation is unclear and is a subject of our current study.
Therefore, we aimed to determine in greater detail how VEGF expression is regulated at the transcriptional level by focusing on the redox and repair activities of APE1 at the VEGF promoter. In the present study, we first characterized how the redox status of APE1 affected its DNA-repair capacity in the promoter region. We found that oxidized APE1 had a significant loss of affinity to DNA polymerase β, which increased the exposure of single-strand DNA and led to increased flexibility of the promoter region and the transcriptional activity of HIF-1.
2. Materials and Methods
2.1. Materials
Dulbecco's Modified Eagle's Medium (DMEM), Opti-MEM® I Reduced-Serum Medium, fetal bovine serum (FBS), TRIzol RNA isolation reagent, ElectroMAX DH12S competent cells with M13KO7 helper phage, and primers were obtained from Invitrogen (Grand Island, NY or Shanghai, China). E3330, myrecitin, synthetic siRNA against Pol β and other general chemicals were obtained from Sigma-Aldrich (St, Louis, MO, USA). Biotin-conjugated or unconjugated tetrahydrofuran sites containing oligonucleotides were obtained from Takara (Dalian, China). The VEGF ELISA kit was obtained from R&D (Minneapolis, MN, USA). Dual luciferase vector pmirGLO, the Dual-Glo® luciferase assay kit, the FuGENE 6 transfection reagent, T4 polynucleotide kinase (T4 PNK), T4 ligase, T4 polymerase, restriction endonucleases, and high-fidelity Pfu DNA polymerase were purchased from Promega (Madison, WI, USA). The QuickChange mutagenesis kit was purchased from Stratagene (Santa Clara, CA, USA). The Halt protease inhibitor cocktail, Protein A/G agarose beads, a GST protein interaction pulldown kit, LightShift chemiluminescent EMSA kit, SuperSignal West Pico chemiluminescent reagents, horseradish peroxidase-conjugated anti-mouse or anti-rabbit IgG antibodies and other Western blot-related reagents were purchased from Pierce (Rockford, IL, USA).
3. Cell Culture and Hypoxic Incubation
Human Umbilical Vein Endothelial Cells (HUVECs) and HeLa cells were obtained from ATCC and maintained in DMEM supplemented with 10% FBS and 1% penicillin-streptomycin. APE1 knockin HeLa cell lines were kindly provided by Dr. Gianluca Tell. The knockin of APE1 in the APE1WT and APE1C65S cell lines was performed as previously described. All knockin expression assays were performed at day 10 post-doxycycline treatment (1 μg/ml). The hypoxic challenge was performed in a hypoxic incubation chamber (Stem Cell Technologies, Vancouver, BC, Canada) following the manufacturer's instructions. Briefly, a 1% O2 hypoxic condition was obtained by purging the chamber with a N2/CO2 gas mixture at a flow rate of 20 l/min for 5 min.
4. DNA Polβ Eukaryotic Expression Vector
The DNA Polβ coding sequence was amplified from HeLa cDNA using two primers, namely, forward 5′-GCTCTAGAATGAGCAAACGGAAGGCG-3′ and reverse 5′-CCCAAGCTTTCATTTGTCGTCATCATCCTTATAGTCTTCGCTCCGGTCCTTGG-3′, which contains a FLAG tag. The PCR product was purified by gel separation and inserted into the pcDNA3.1 eukaryotic expression vector between the XbaI and HindIII sites. Successful insertion of the target gene into the vector was confirmed by DNA sequencing.
5. Western Blotting
Equal amounts of whole cell lysate were electrophoresed on a 10% SDS-PAGE gel. Proteins were then transferred to PVDF membranes (Bio-Rad, Hercules, CA, USA). After blocking in TBST [50 mM Tris-HCl, pH 7.5, 150 mM NaCl and 0.1% (v/v) Tween 20] containing 5% (w/v) nonfat dry milk for 1 h at room temperature (RT), membranes were incubated with specific primary antibodies. After three washes with TBST, membranes were incubated with appropriate peroxidase-conjugated secondary antibodies for 1 h at RT with shaking. After five washes in TBST, membranes were developed with chemiluminescent reagents and exposed to Biomax-Light films (Kodak, Rochester, NY, USA). Band intensities were analyzed using Quantity One software (Bio-Rad, CA, USA).
The specific antibodies used for Western blotting were as follows: anti-APE1 monoclonal (Novus, CO, USA), 1 h at 37 °C, dilution 1:5000; anti-Flag (M2, Sigma, MO, USA) monoclonal, 1 h at 37 °C, dilution 1:2000; anti-HIF-1α monoclonal (BD Bioscience, USA), overnight at 4 °C, dilution 1:1000; anti-β-actin monoclonal (Sigma, USA), 1 h at 37 °C, dilution 1:2000; anti-Polymerase β polyclonal (Abcam, UK) overnight at 4 °C, dilution 1:500; and anti-Sp1 polyclonal (Santa Cruz, USA) overnight at 4 °C, dilution 1:500.
6. Coimmunoprecipitation Assay
For cell harvests, cells were scraped off dishes and washed once with ice-cold phosphate-buffered saline (PBS). The cell pellets were resuspended and incubated in IP lysis buffer (Beyotime Institute of Biotechnology, Jiangsu, China) supplemented with protease inhibitor cocktail (Pierce, USA) at a cell density of 107 cells/ml on ice for 30 min. After centrifugation at 12,000 ×g for 10 min at 4 °C, the supernatant was collected as the total cell lysate. The protein concentration was determined using the Bradford assay (Bio-Rad, Hercules, CA, USA). Samples were precleared via incubation with protein A/G agarose resin for 30 min on ice and then coimmunoprecipitated (Co-IPed) for 3 h using anti-Flag M2 antibody, APE1 antibody or Polβ antibody according to the manufacturer's instructions. Protein A/G agarose resin was then added and incubated for 1 h at 4 °C. After 3 washes with PBS containing protease inhibitor, agarose beads and the binding proteins in pellets were mixed with sample buffer and boiled at 100 °C for 5 min. The samples were then stored at −80 °C or subjected to Western blot analysis immediately.
7. DNA Affinity Precipitation
DNA affinity precipitation analyses were performed as described with minor modifications [13]. A total of 10 pmol of gel-purified, biotin-labeled 57-mer oligonucleotides, corresponding to a minimal functionally active HIF-1 segment representing the binding site in the promoter region of the VEGF gene, was incubated with 100 μl of streptavidin agarose resin (50 μl of settled resin, Pierce) for 30 min at RT. The oligo sequence was as follows: 5′- TGCATAC-Ap-TGGGTTCACACGGTCGTCTCCCTCCGGCCACTGACTAACTG CTCGGG -3′ (the underlined sequence represents the HIF-1 binding site, and “Ap” represents the AP sites in the position of the original guanine). The biotinylated oligonucleotides bound to the streptavidin agarose were collected by centrifugation at 1000 ×g for 1 min. The bead-associated oligonucleotides were washed two times with Tris-EDTA and equilibrated with PBS. Then, 50 μg of nuclear protein or 10 μM E3330-treated HUVECs were added to the oligonucleotide-streptavidin-agarose pellet and incubated on ice for 2 h. After incubation, the pellet was washed twice with PBS to remove unbound proteins. The protein-DNA complexes were then resuspended in sample buffer and incubated at 100 °C for 5 min to elute the bound proteins. The samples were then subjected to Western blot analysis.
8. Quantitative PCR
Total RNA was isolated using the TRIzol reagent and chloroform/isopropanol precipitation according to the manufacturer's instructions. Genomic DNA (gDNA) was extracted using the DNeasy blood and tissue kit from Qiagen (Hilden, Germany). The RNA and gDNA concentrations were determined by spectrophotometry (Eppendorf AG, Hamburg, Germany), and the qualities were assessed by agarose gel electrophoresis. For quantitative reverse transcription, cDNA was synthesized from 1 μg of total RNA using the ReverTra Ace reversal transcription kit (Toyobo, Osaka, Japan). For genomic DNA PCR, gDNA was first treated with fpg protein (New England BioLabs, Beverley, MA, USA) before performing the PCR for the VEGF promoter region. Because fpg incises DNA at the damaged purine base, leaving a single-strand break, the amplification rate is reduced for the lesion-containing DNA template after fpg treatment. Quantitative PCR (qPCR) was performed using SYBR Premix Ex TaqTM (Takara, Dalian, China) with the LightCycler® 480 Real-Time PCR System (Roche, Indianapolis, IN, USA).
9. Electrophoretic Mobility-Shift Assay (EMSA)
EMSA was performed according to the manufacturer's instructions in the LightShift chemiluminescent EMSA kit with minor modifications. Briefly, 15 μg of nuclear extract was incubated with 3′-biotin-labeled and purified double-stranded oligonucleotide probes containing the HIF-1 consensus: 5′-GACTCCACAGTGCATACGTGGGCTCCAACAGGT-3′ (Sangon, Shanghai, China). After incubation, samples were separated on a 5% polyacrylamide gel at 100 V for 90 min and then transferred to a Zeta-Probe GT nylon membrane (Bio-Rad). The probes were detected by HRP-conjugated streptavidin (1:300), and the bands were visualized by ECL reagents. The resultant bands were quantified using Quantity One imaging software (Bio-Rad).
10. Lesion-Containing Luciferase Reporter Gene Assay
The lesion containing the luciferase reporter vector was constructed according to a previously published protocol [20]. First, the VEGF promoter with 1396 bp (−1233~ + 162) was inserted upstream of the firefly luciferase coding region of the dual luciferase vector (pmirGLO vector) by replacing the PGK promoter. The single-strand DNA (ssDNA) replication origin f1 from the pBluescript-SK vector was inserted into the vector in the correct orientation. The ssDNA was produced using phage-based purification followed by annealing with the AP-site-containing oligo. The synthesis of the second strand was performed by adding T4 DNA polymerase and T4 DNA ligase. The double-strand DNA products were separated on a low-melting agarose gel and recovered by β-agarase I digestion. Targeted cells were plated on 96-well plates at a density of 2000–5000 cells/well. Twenty-four hours later, cells were transfected with 5–20 ng of AP-site-containing luciferase reporter vector using FuGENE 6. Cells were assayed for luciferase expression 24 h post-transfection. The transcriptional activity of the VEGF promoter was assessed using the Promega luciferase assay system following the manufacturer's instructions. Firefly luciferase activity was normalized to Renilla luciferase activity.
11. Tissue Immunohistochemistry
Briefly, sections from paraffin-embedded tissues were incubated with the indicated primary antibodies overnight at 4 °C and then rinsed with PBS and incubated with its associated HRP-conjugated secondary antibody for 30 min at RT. Sections were rinsed with PBS and developed with diaminobenzidine substrate and then counterstained with hematoxylin to visualize the nucleus. Positive staining was recognized as brown. Ten random high-power fields or at least 1000 tumor cells were counted, and the expression of the targeted protein was evaluated based on the following criteria. The immunostained tissue sections were evaluated and scored under a light microscope independently by two pathologists in a blinded fashion. Protein was scored according to four categories: a score of 0, no expression in tumor cells; a score of 1+, faint/barely perceptible partial nuclear expression in 10% of tumor cells; and a score of 3+, strong expression of the entire nucleus in >10% of tumor cells. A score of 2+/3+ was defined as positive, while a score of 0/1+ was defined as negative.
The primary antibodies used in this assay were APE1 (ab137708, Abcam, Cambridge, MA, USA), POLB (ab26343, Abcam, Cambridge, MA, USA), and VEGF (sc-7269, Santa Cruz Biotechnology, Dallas, TX, USA).
11.1. Statistical Analysis
Statistical analysis was performed with SPSS using Student's t-test. P < 0.05 was considered statistically significant.
12. Results
12.1. Cellular APE1 Expression Level Impacts VEGF Transcription under Hypoxic Conditions
The gain and loss of APE1 function in HUVECs was achieved by the utilization of adenovirus the Ad5/F35-shAPE1 and pCMV-APE1 vectors fused with 3 × FLAG. An adenovirus against the scramble sequence and a pCMV-FLAG empty vector were employed as negative controls. APE1 expression in these manipulated HUVECs was confirmed by Western blot (Fig. 1B). Exogenously expressed APE1 was indicated as a band shift on the blot because the expression vector contained a triple FLAG tag fused at the C terminus of the recombinant protein. β-actin was used as a loading control to confirm that equal amounts of whole cell extracts were loaded in each well. VEGF expression in these cells under hypoxic conditions was measured at the mRNA (Fig. 1A) and protein levels (Fig. 1B & 1C). Our results indicated that the VEGF mRNA levels were increased significantly in APE1-overexpressing HUVECs and decreased significantly in APE1 knockdown cells (p = 0.001). Consistently, ELISA analysis further revealed that VEGF secretion was significantly increased in cells overexpressing APE1 but significantly decreased in APE1 knockdown cells (Fig. 1C). These results indicated that APE1 may directly impact VEGF expression under hypoxic conditions.
Fig. 1.

APE1 regulates VEGF gene expression in HUVECs. HUVECs were transfected with adenovirus Ad5 or F35-shAPE1 for 48 h to overexpress or knock down APE1 expression, respectively. Cells were then cultured in hypoxic conditions (1% O2). VEGF mRNA levels were measured by qPCR after 8 h of hypoxic exposure (A). VEGF and APE1 protein levels in whole cell extracts were assessed by Western blot after 24 h of hypoxic exposure (B). VEGF secretion in the culture medium was assessed by ELISA after 48 h of hypoxic exposure (C). * P < 0.01, treated group vs control group by Student's t-test.
12.2. Inhibition of APE1 Activities Regulates the DNA-Binding Activity of HIF-1α at the VEGF Promoter Region
Myrecitin was utilized to inhibit APE1 endonuclease function, and the redox inhibitor E3330 was utilized to repress APE1 redox activation of transcriptional factors [[21], [22]]. HUVECs were treated with Myrecitin or E3330 at 10 μM for 2 h, and the VEGF mRNA levels were measured by qPCR. As indicated in Fig. 2A, inhibition of the redox activity of APE1 appeared to have a greater inhibitory effect on VEGF mRNA expression than inhibition of its endonuclease activity (p = 0.002) (Fig. 2A). The addition of both inhibitors showed an additive effect on inhibiting VEGF mRNA expression (p = 0.05). The intracellular protein levels of APE1 and VEGF were also measured in HUVECs treated with myrecitin and E3330. Compared with control HUVECs (DMSO treated), HUVECs treated with either inhibitor separately or in combination demonstrated a significant decrease in VEGF protein expression (Fig. 2B). Moreover, VEGF protein secretion by HUVECs treated with myrecitin, E3330 or their combination was also significantly reduced compared with that by the controls (Fig. 2C). These results suggest that both repair and redox activity play important roles in regulating the DNA-binding activity of HIF-1α at the VEGF promoter region.
Fig. 2.

APE1 activities affect the gene expression of VEGF. HUVECs were pretreated with myrecitin, E3330 or DMSO at 10 μM for 2 h and were then cultured in hypoxic conditions (1% O2). VEGF mRNA levels were measured by qPCR after 8 h of hypoxic culture (A). VEGF and APE1 protein levels in HUVECs were assessed by Western blot after 24 h of hypoxic culture. β-actin was used as an internal loading control (B). The secretion of VEGF was assessed by ELISA after 48 h of hypoxic culture (C). *P < 0.01, treated group vs control group by Student's t-test.
12.2.1. AP Sites in the VEGF Promoter Region Promote HIF-1α Binding
EMSA demonstrated that HIF-1 has minimal DNA-binding activity at the HRE of the VEGF promoter under normoxic conditions regardless of treatment with APE1 inhibitors (myrecitin or E3330). In contrast, a protein-DNA complex was detected using a probe against the HRE of the VEGF promoter when HUVECs were exposed to hypoxic conditions (4 h). Moreover, the alteration of the endonuclease or redox function of APE1 by myrecitin or E3330 demonstrated a remarkable decrease in DNA binding by HIF-1. Notably, the redox inhibitor E3330 appeared to induce a slightly stronger inhibition of HIF-1 binding activity than myrecitin, whereas the combination of both drugs appeared to produce an additive effect (Fig. 3A).
Fig. 3.

APE1 redox and repair activity are involved in regulating HIF-1 DNA binding to an AP-site containing HRE probe. EMSA of HIF-1 binding with a regular HRE oligo (A) and AP-site-containing HRE oligo (B). Following 2 h of pretreatment with DMSO, myrecitin, E3330 or myrecitin/E3330 at 10 μM for 2 h, HUVECs were cultured in either normal (20%) or hypoxic (1%) conditions for 3 h. The nuclear extracts were prepared for EMSA. HIF-1 antibody was added to the supershift EMSA reaction, whereas ten-fold unlabeled HRE oligos were added to the cold-probe EMSA reaction as an indicator of specificity of the binding between HIF-1 and the HRE oligo. Representative EMSA images are shown, and similar results were obtained in three separate experiments.
A second experiment was designed to elucidate whether HIF-1 binding to the HRE of the VEGF promoter was increased with the utilization of a probe containing an AP site. Interestingly, the introduction of a probe containing an AP site into the HRE in HUVECs resulted in an increase in DNA-protein complexes under both normoxic and hypoxic conditions. Additionally, a supershift band representing a complex containing the HIF-1 protein, AP-containing probe, and antibody to HIF-1 was also observed under hypoxic conditions. EMSA demonstrated the decreased DNA-binding activity of HIF-1 at the HRE of the VEGF promoter when either the endonuclease or the redox function of APE1 was altered by inhibitors under either normoxic or hypoxic conditions. When cells were treated with the combination of myrecitin and E3330, a remarkable decrease in DNA-binding by HIF-1 in hypoxic conditions was observed, whereas an increase in HIF-1 binding activity was also detected in normoxic conditions (Fig. 3B). These results suggest that the combination of both drugs produces an additive effect on inhibition and that AP sites in the VEGF promoter region might promote HIF-1 binding.
12.2.2. AP Sites in the VEGF Promoter Region Affect Transcription Initiation Efficiency by APE1
We first treated genomic DNA under normoxic and hypoxic conditions with fpg (formamidopyrimidine [fapy]-DNA glycosylase, also known as 8-oxoguanine DNA glycosylase), which acts as an N-glycosylase and an AP-lyase. Fpg functions to convert oxidative damage to single-strand breaks that subsequently block DNA replication. We then performed qPCR to measure the oxidative DNA base damage or the “replicable” DNA content. Interestingly, we found that the VEGF promoter region (−1226 ~ −897 containing an HRE) had more AP sites and more DNA oxidative damage in hypoxia-treated HUVECs (Fig. 4A). To further confirm this, we constructed an AP-site-containing HRE-reporter luciferase vector to test the initiation activity. The core HRE binding sequence was constructed to contain a tetrahydrofuran at position −976; a flanking region reporter F1 contained a tetrahydrofuran at position −993; and F2 contained a tetrahydrofuran at position −946 (Fig. 4B). These reporters were then tested under both normoxic (control) and hypoxic conditions. The hypoxic conditions showed the greatest increase in luciferase reporter activity, with an approximately 10-fold increase (p = 0.001). The AP-mimic sites introduced into the HRE as well as at sites upstream (F1) and downstream (F2) of the HRE site all displayed increased luciferase activities, though to a lesser extent than hypoxic conditions alone (Fig. 4C). The results indicated that the AP site in the different regions of the VEGF promoter region could affect transcription initiation efficiency by APE1.
Fig. 4.

AP sites in the VEGF promoter region affect transcription initiation efficiency by APE1. qPCR assay of AP sites and oxidative base lesions in the VEGF promoter region under normoxic or hypoxic conditions (A). AP sites in the different regions of the VEGF promoter gene constructs (F1, HER, F2) were utilized to measure the transcriptional initiation efficiency of these promoters (B and C). The statistical analysis was performed via Student's t-test, * p < 0.01.
12.2.3. Redox Activity of APE1 Affects its Interaction with DNA Polymerase β
An intact N-terminal structure of APE1 is critical for the “hand-off” model in BER [23]. BER is a continuous process that prevents its vulnerable intermediates from being exposed. Because APE1 interacts with DNA Pol β in the BER pathway before transferring its repair intermediates to Pol β, we further tested whether the APE1 transfer to DNA Pol β could be disrupted. The protein-protein interaction between APE1 and Pol β was measured using a GST pulldown assay under both oxidizing and reducing conditions. To mimic the oxidizing or reducing conditions, the pulldown reactions were further incubated with either H2O2 or DTT. We found that APE1 interacted with Pol β under both oxidizing or reducing conditions, especially under reducing conditions (Fig. 5A).
Fig. 5.

The redox modification of APE1 interrupts the interaction between APE1 and DNA Polβ. Co-IP of endogenous APE1 and Polβ. After DTT or hydrogen peroxide treatment, APE1 or Polβ protein was immunoprecipitated (IPed) with APE1 antibody (APE1) or Polβ antibody (Polβ), respectively. Western blotting was performed to detect Polβ or APE1 expression (A). IgG was added as a negative control in the Co-IP system. A Western blot of APE1 and Polβ without IP is shown to indicate the “input”. WT, C65S, and E3330 were added as negative controls, redox-active site mutants or redox inhibitors. The interaction between APE1 and Polβ was measured by Co-IP (B). After E3330 and DMSO treatment overnight, the HUVEC nuclear extract was prepared and subjected to Western blot using antibodies against HIF-1, APE1, and Polβ (C).
Next, we used E3330 to block the C65 or Cys 65 redox-active site mutation (C65S) to determine whether the change at the redox-active site had an impact on its interaction with Polβ. A decreased APE1- Pol β interaction was observed (Fig. 5B). Using these experimental approaches, we could determine whether the E3330 inhibitor blocked the APE1 interaction with Polβ, which would then release repair intermediates containing single-strand breaks. We found that the APE1 and Polβ levels were decreased and that HIF-1 was increased in the nucleus after E3330 treatment (Fig. 5C). These results suggested that the APE1 transfer to DNA Pol β could be disrupted by inhibiting the APE1 redox function.
Using both AP-site-containing luciferase reporters (HRE-AP), we investigated the effect of the loss of the APE1/Polβ interaction on VEGF promoter transcriptional activity and VEGF expression separately. We used an APE1-overexpressing cell line or Polβ knockdown cell line, which either increased the APE1 level or decreased the Polβ level, respectively, rendering an imbalance in APE1/Polβ in the BER pathway. The overexpression and knockdown efficiency were confirmed by Western blot (Fig. 6A). Both the APE1-overexpressing cell line and the Polβ knockdown cell line showed increased luciferase activity. Conversely, overexpression of Pol β resulted in a remarkable decrease in luciferase activity. In addition, the increase in luciferase activity in the model in which both APE1 and Pol β were increased was ranked behind that in which only APE1 was increased. These results suggested that the imbalance in APE1/Polβ at the promoter region affected the transcription regulatory function of APE1. Further experiments were focused on exploring the effects of the loss of the APE1/Polβ interaction on VEGF levels (Fig. 6B). We used the APE1-overexpressing cell line together with the cell line overexpressing both APE1 and Polβ. The VEGF levels were significantly increased in both cell models, especially in the APE1-increased model (Fig. 6C). The above results suggested that the imbalance in APE1/Polβ at the promoter region affected VEGF promoter transcriptional activity and VEGF expression.
Fig. 6.

The balance between APE1 and DNA Polβ affects VEGF expression under hypoxic conditions. After 48 h of transfection of vectors with APE1 overexpression (APE1 WT), Polβ overexpression (Polβ WT) or Polβ knockdown (Polβ KD), HUVECs were transfected with an AP-containing luciferase reporter construct (HRE-AP site) and cultured under hypoxic conditions for 24 h. The normalized luciferase readings were measured (A). The VEGF mRNA levels of the APE1-overexpressing cells and the APE1/Polβ co-overexpressing cells were also measured by qPCR (B and C). The statistical analysis was performed via Student's t-test to compare two groups, * indicates that the difference between the indicated groups is statistically significant (p < 0.01).
12.2.4. Expressional Imbalance between APE1 and Polβ Is Associated with VEGF Expression in Human Cancer Tissue
To further investigate the correlation between imbalanced APE1/Polβ expression and VEGF expression in human tumor tissues, a cohort containing 77 non-small cell lung cancer tissues was subjected to IHC using antibodies against APE1, Polβ and VEGF. All samples were further categorized according to the staining score of APE1 and Polβ as four subgroups: APE1-high/Polβ-high, APE1-high/Polβ-low, APE1-low/Polβ-high and APE1-low/Polβ-low. The VEGF IHC scores were analyzed in the above-mentioned subgroups. As shown in Fig. 7A, APE1 and Polβ staining was basically localized to the nuclei, while VEGF staining was localized to the cytoplasm. The VEGF staining scores were significantly higher (mostly scores of 3) in the APE1-high/Polβ-low group and significantly lower in the APE1-low/Polβ-high group. Evenly distributed VEGF staining was observed in the other two groups with balanced expression of APE1 and Polβ. The results clearly suggest that imbalanced APE1/Polβ coordination plays a key role in controlling VEGF expression, which further confirms our findings in cells.
Fig. 7.
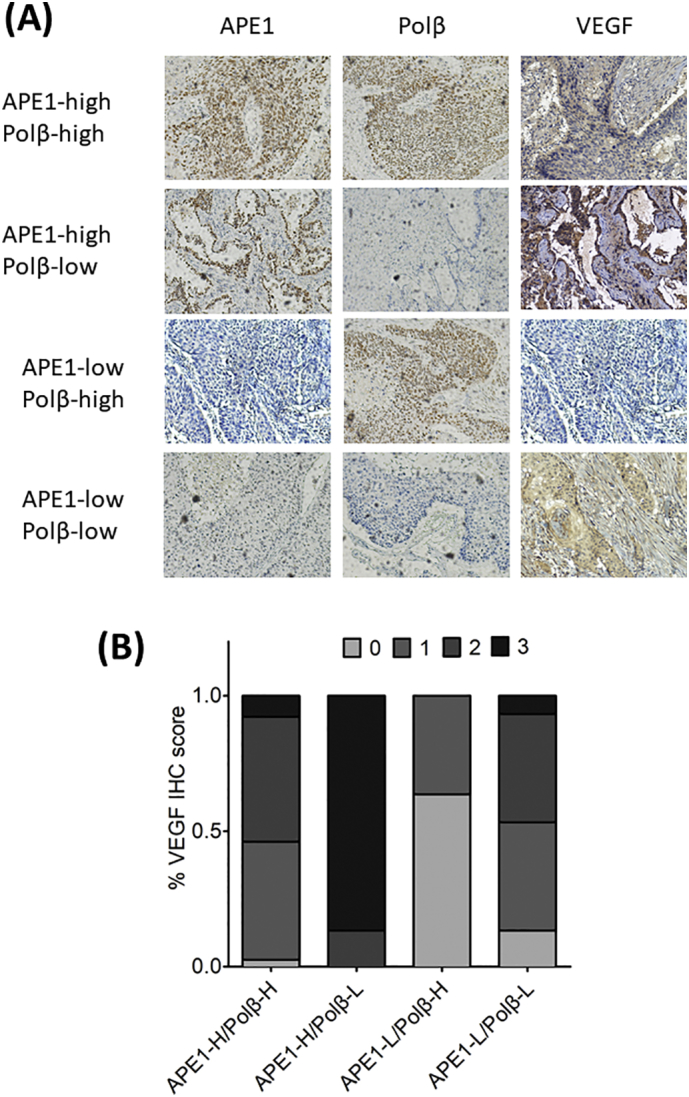
An imbalance between APE1 and Polβ is associated with VEGF expression in human cancer tissue. The samples from patients with non-small cell lung cancer were subjected to IHC using antibodies against APE1, Polβ and VEGF (A). All samples were further categorized according to the staining score of APE1, Polβ and VEGF as the following four subgroups: APE1-high/Polβ-high, APE1-high/Polβ-low, APE1-low/Polβ-high and APE1-low/Polβ-low. Quantitative analysis of the VEGF staining scores of the four subgroups (B).
13. Discussion
The oxidative damage resulting in AP sites at the promoter region serves as a new type of regulatory element for gene expression under oxidative stress. BER is responsible for the repair of these types of DNA lesions and is initiated by 8-oxoguanine DNA glycosylase (OGG1) and AP sites. AP sites enable the melting of the duplex to unmask the potential G-quadruplex-forming sequences (PQS), adopting a G-quadruplex fold in which APE1 binds, but inefficiently cleaves, the AP sites for activation of VEGF [24]. How this repair machinery processes AP sites in the promoter region remains unknown. Our data indicate that the key BER enzyme, APE1, plays a central regulatory role in the repair of AP sites in the promoter region. By employing activity-specific inhibitors and functional mutants of APE1, we validate that the specific redox activity of APE1 is involved in enhancing transcriptional efficiency at the AP-site-containing VEGF promoter, whereas the role of APE1 in DNA repair still needs further exploration. Further mechanistic studies show that oxidative modification of the N terminus, in terms of the redox domain of APE1, disrupts the interaction between APE1 and DNA Polβ, a crucial step in BER following AP endonuclease activity. We also confirmed the early findings that the association and quantitative balance between APE1 and DNA Polβ were important for the transcriptional activity of an AP-site-containing VEGF promoter under hypoxic conditions. The current study reveals the novel finding that both activities of APE1 play distinct roles in regulating the transcription of the stress-responsive gene VEGF.
DNA modifications, such as DNA methylation, are some of the most effective ways to regulate gene expression. Previous studies have shown that AP sites, which are usually considered to be lesions that block replication, are not distributed evenly throughout the genome. Under certain types of genotoxic stress, AP sites accumulate in the promoter regions of specific stress-responsive genes. The regulation of HIF-VEGF was used as a model system to study the effects of AP sites on gene expression. For instance, Gillespie and colleagues found that under hypoxic conditions, increased AP sites in the VEGF promoter caused an increase in VEGF gene expression [12]. In our present study, we confirmed this phenomenon using an AP-site-containing luciferase reporter vector. In this particular methodology, we inserted a 1396-bp VEGF promoter sequence, which contained most of the VEGF promoter region and active HIF binding site in the distal region. The AP sites in the HRE and its flanking regions effectively increased luciferase expression. Interestingly, the AP site in the HRE increased transcription initiation more effectively than the AP site in the flanking regions. In addition, even without further distal regions included in the reporter vector, the AP sites in the HRE could still effectively promote transcription. This observation suggests that the AP site in the promoter region is more likely to increase the flexibility of the DNA-binding element to the transcription factor than to promote the folding of the DNA to approximate the distal region with the transcription start point. Further structural studies are needed to characterize the binding affinity of HIF-1 between regular and AP-site-containing HREs.
BER is the main pathway to address AP sites and oxidative base damage, which requires a step-by-step enzymatic process with scaffold proteins as coordinators [23]. In the context of oxidative base lesions, the BER of damaged bases begins with their removal by a DNA glycosylase to generate an AP site, and the APE1 protein hydrolyzes at the 5′-phosphodiester. The resulting 3′-hydroxyl nucleotide supports DNA-repair synthesis, mostly by DNA Polβ in mammalian cells. Polβ also removes the APE1 product 5′-deoxyribose-5-phosphate (5′-dRp) by means of a separate lyase activity [25]. Given that the repair intermediates, which contain AP sites and single-strand breaks, are usually genotoxic, the entire BER process is usually well coordinated to avoid the release of the repair intermediates. Previous reports indicate that the balance of APE1 and DNA Polβ is critical for BER capacity and that the interaction between these two proteins is an essential feature for the “passing the baton” model and functions to protect the repair intermediates from exposure [[26], [27]]. However, our results indicated the presence of variations in the interaction between APE1 and DNA Polβ under different conditions in the promoter region. The protein interaction assay indicated that the interaction between APE1 and DNA Polβ decreased under oxidative conditions and was restored under reducing conditions. In addition, when the key redox modification residue, C65, was mutated, the interaction with DNA Polβ was diminished. These results suggest that the distorted N-terminal domain plays an important role in the coordination of the AP endonuclease and polymerase activity steps in the BER pathway.
We then focused on the unique redox modification of APE1, which had been shown to promote the DNA-binding activity of HIF-1. APE1 is a unique dual-function protein. The N terminus of APE1 is recognized as the redox domain, and the C65 residue is the critical active site for its redox regulatory function. APE1 regulates the activity of several transcription factors, such as HIF-1α, STAT3 and NF-κB [25]. Recently, more posttranscriptional modifications in the N terminus of APE1 have been identified and characterized, highlighting the regulatory role of the APE1 N terminus in AP endonuclease activity, which resides in the C terminus of APE1 (reviewed in [28]; redox modifications are specifically addressed in [29]). These studies imply that these two evolutionarily combined distinct activities are closely related to each other; however, the path by which the separate activities function as a complex in a coordinated biological process is still unknown. The redox activity of APE1 has been recently characterized in detail. Reduced C65 and C93 can interact with active sites in transcription factors. The formation of the disulfide bond between C65 and C93 changes the configuration of the APE1 N terminus. Our data showed that under oxidative conditions, with increased formation of the intramolecular disulfide bond, the interaction between APE1 and DNA Polβ is disrupted. Interestingly, the C65S mutation also interrupts the interaction between APE1 and DNA Polβ observed by Co-IP. Previous studies showed that the C65A and C65S mutations had reduced transcriptional activity as assessed by EMSA but rather normal AP endonuclease activity either in vivo or in purified mutant proteins. We postulated that C65S might also change the configuration of the N terminus of APE1, which may further affect the coordination of BER. Due to the central role of the APE1 N terminus and C65 residue in APE1 redox activity, our observations suggest that the redox modification of the APE1 N terminus has a significant impact on its repair efficiency by interrupting the coordination of BER. Therefore, we propose that in the promoter region, APE1 is first recruited and functions as a transcription regulator under hypoxic conditions. In the meantime, APE1 recognizes the AP sites and functions as an AP endonuclease. However, at the promoter region, most of the APE1 molecules are oxidized at the N terminus, which results in failure to interact with the downstream repair enzyme DNA Polβ and leads to exposure of the single-strand break, thereby increasing affinity to the transcription factor.
To our knowledge, the present study is the first to demonstrate that the two distinct activities of APE1 are closely related to each other in the context of gene regulation in terms of affecting transcription factor-promoter binding. Our data provide a plausible explanation for the mechanism by which increased gene transcription can be induced by the accumulation of AP sites in the promoter region and shed light on the possible application of utilizing AP sites as epigenetic markers for transcriptional regulation.
Conflicts of Interest
The authors declare no conflicts of interest regarding this study.
Acknowledgments
This work was supported by the National Natural Science Foundation of China to (NFSC.30801368), (NSFC 81372373). The authors are grateful to Dr. Gianluca Tell for sending us the APE1 knockin HeLa cell lines and to Dr. Hualiang Xiao, Dr. Zengpeng Li, Ms. Yuxin Yang, Mr. Yi Cheng, and Mr. Linli Zeng for their technical support.
References
- 1.Pouyssegur J., Dayan F., Mazure N.M. Hypoxia signalling in cancer and approaches to enforce tumour regression. Nature. 2006;441:437–443. doi: 10.1038/nature04871. [DOI] [PubMed] [Google Scholar]
- 2.Harris A.L. Hypoxia--a key regulatory factor in tumour growth. Nat Rev Cancer. 2002;2:38–47. doi: 10.1038/nrc704. [DOI] [PubMed] [Google Scholar]
- 3.Waldner M.J., Neurath M.F. Targeting the vegf signaling pathway in cancer therapy. Expert Opin Ther Targets. 2012;16:5–13. doi: 10.1517/14728222.2011.641951. [DOI] [PubMed] [Google Scholar]
- 4.Forsythe J.A., Jiang B.H., Iyer N.V. Activation of vascular endothelial growth factor gene transcription by hypoxia-inducible factor 1. Mol Cell Biol. 1996;16:4604–4613. doi: 10.1128/mcb.16.9.4604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Semenza G.L. Hif-1: upstream and downstream of cancer metabolism. Curr Opin Genet Dev. 2010;20:51–56. doi: 10.1016/j.gde.2009.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rankin E.B., Giaccia A.J. The role of hypoxia-inducible factors in tumorigenesis. Cell Death Differ. 2008;15:678–685. doi: 10.1038/cdd.2008.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kimbro K.S., Simons J.W. Hypoxia-inducible factor-1 in human breast and prostate cancer. Endocr Relat Cancer. 2006;13:739–749. doi: 10.1677/erc.1.00728. [DOI] [PubMed] [Google Scholar]
- 8.Stoeltzing O., McCarty M.F., Wey J.S. Role of hypoxia-inducible factor 1alpha in gastric cancer cell growth, angiogenesis, and vessel maturation. J Natl Cancer Inst. 2004;96:946–956. doi: 10.1093/jnci/djh168. [DOI] [PubMed] [Google Scholar]
- 9.Lando D., Pongratz I., Poellinger L., Whitelaw M.L. A redox mechanism controls differential DNA binding activities of hypoxia-inducible factor (hif) 1alpha and the hif-like factor. J Biol Chem. 2000;275:4618–4627. doi: 10.1074/jbc.275.7.4618. [DOI] [PubMed] [Google Scholar]
- 10.Wang D., Zhong Z.Y., Li M.X., Xiang D.B., Li Z.P. Vector-based ape1 small interfering rna enhances the sensitivity of human osteosarcoma cells to endostatin in vivo. Cancer Sci. 2007;98:1993–2001. doi: 10.1111/j.1349-7006.2007.00616.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ziel K.A., Grishko V., Campbell C.C., Breit J.F., Wilson G.L., Gillespie M.N. Oxidants in signal transduction: impact on DNA integrity and gene expression. FASEB J. 2005;19:387–394. doi: 10.1096/fj.04-2805com. [DOI] [PubMed] [Google Scholar]
- 12.Pastukh V., Ruchko M., Gorodnya O., Wilson G.L., Gillespie M.N. Sequence-specific oxidative base modifications in hypoxia-inducible genes. Free Radic Biol Med. 2007;43:1616–1626. doi: 10.1016/j.freeradbiomed.2007.08.027. [DOI] [PubMed] [Google Scholar]
- 13.Breit J.F., Ault-Ziel K., Al-Mehdi A.B., Gillespie M.N. Nuclear protein-induced bending and flexing of the hypoxic response element of the rat vascular endothelial growth factor promoter. FASEB J. 2008;22:19–29. doi: 10.1096/fj.07-8102com. [DOI] [PubMed] [Google Scholar]
- 14.Abbotts R., Madhusudan S. Human ap endonuclease 1 (ape1): from mechanistic insights to druggable target in cancer. Cancer Treat Rev. 2010;36:425–435. doi: 10.1016/j.ctrv.2009.12.006. [DOI] [PubMed] [Google Scholar]
- 15.Ziel K.A., Campbell C.C., Wilson G.L., Gillespie M.N. Ref-1/ape is critical for formation of the hypoxia-inducible transcriptional complex on the hypoxic response element of the rat pulmonary artery endothelial cell vegf gene. FASEB J. 2004;18:986–988. doi: 10.1096/fj.03-1160fje. [DOI] [PubMed] [Google Scholar]
- 16.Fromme J.C., Verdine G.L. Base excision repair. Adv Protein Chem. 2004;69:1–41. doi: 10.1016/S0065-3233(04)69001-2. [DOI] [PubMed] [Google Scholar]
- 17.Carrero P., Okamoto K., Coumailleau P., O'Brien S., Tanaka H., Poellinger L. Redox-regulated recruitment of the transcriptional coactivators creb-binding protein and src-1 to hypoxia-inducible factor 1alpha. Mol Cell Biol. 2000;20:402–415. doi: 10.1128/mcb.20.1.402-415.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zou G.M., Karikari C., Kabe Y., Handa H., Anders R.A., Maitra A. The ape-1/ref-1 redox antagonist e3330 inhibits the growth of tumor endothelium and endothelial progenitor cells: therapeutic implications in tumor angiogenesis. J Cell Physiol. 2009;219:209–218. doi: 10.1002/jcp.21666. [DOI] [PubMed] [Google Scholar]
- 19.Evans A.R., Limp-Foster M., Kelley M.R. Going ape over ref-1. Mutat Res. 2000;461:83–108. doi: 10.1016/s0921-8777(00)00046-x. [DOI] [PubMed] [Google Scholar]
- 20.Bregeon D., Doetsch P.W. Assays for transcriptional mutagenesis in active genes. Methods Enzymol. 2006;409:345–357. doi: 10.1016/S0076-6879(05)09020-8. [DOI] [PubMed] [Google Scholar]
- 21.Laev S.S., Salakhutdinov N.F., Lavrik O.I. Inhibitors of nuclease and redox activity of apurinic/apyrimidinic endonuclease 1/redox effector factor 1 (ape1/ref-1) Bioorg Med Chem. 2017;25:2531–2544. doi: 10.1016/j.bmc.2017.01.028. [DOI] [PubMed] [Google Scholar]
- 22.Simeonov A., Kulkarni A., Dorjsuren D. Identification and characterization of inhibitors of human apurinic/apyrimidinic endonuclease ape1. PLoS One. 2009;4:e5740. doi: 10.1371/journal.pone.0005740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Prasad R., Beard W.A., Batra V.K., Liu Y., Shock D.D., Wilson S.H. A review of recent experiments on step-to-step "hand-off" of the DNA intermediates in mammalian base excision repair pathways. Mol Biol. 2011;45:586–600. [PMC free article] [PubMed] [Google Scholar]
- 24.Fleming A.M., Ding Y., Burrows C.J. Oxidative DNA damage is epigenetic by regulating gene transcription via base excision repair. Proc Natl Acad Sci U S A. 2017;114:2604–2609. doi: 10.1073/pnas.1619809114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Quinones J.L., Thapar U., Yu K., Fang Q., Sobol R.W., Demple B. Enzyme mechanism-based, oxidative DNA-protein cross-links formed with DNA polymerase beta in vivo. Proc Natl Acad Sci U S A. 2015;112:8602–8607. doi: 10.1073/pnas.1501101112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Prasad R., Shock D.D., Beard W.A., Wilson S.H. Substrate channeling in mammalian base excision repair pathways: passing the baton. J Biol Chem. 2010;285:40479–40488. doi: 10.1074/jbc.M110.155267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wilson S.H., Kunkel T.A. Passing the baton in base excision repair. Nat Struct Biol. 2000;7:176–178. doi: 10.1038/73260. [DOI] [PubMed] [Google Scholar]
- 28.Busso C.S., Lake M.W., Izumi T. Posttranslational modification of mammalian ap endonuclease (ape1) Cell Mol Life Sci. 2010;67:3609–3620. doi: 10.1007/s00018-010-0487-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Luo M., Zhang J., He H. Characterization of the redox activity and disulfide bond formation in apurinic/apyrimidinic endonuclease. Biochemistry. 2012;51:695–705. doi: 10.1021/bi201034z. [DOI] [PMC free article] [PubMed] [Google Scholar]