

Abstract
Heart failure (HF) is the single largest cause for increased hospitalization after fine particulate matter (PM2.5) exposure. Patients with left HF often progress to right ventricular (RV) failure even with optimal medical care. An increase of PM2.5 of 10 μg per cubic meter was associated with a 76% increase in the risk of death from cardiovascular disease in 4 years' period. However, the role and mechanism of PM2.5 in HF progression are not known. Here we investigated the role of PM2.5 exposure in mice with existing HF mice produced by transverse aortic constriction (TAC). TAC-induced HF caused lung inflammation, vascular remodeling and RV hypertrophy. We found increased PM2.5 profoundly exacerbated lung oxidative stress in mice with existing left HF. To our surprise, PM2.5 exposure had no effect on LV hypertrophy and function, but profoundly exacerbated lung inflammation, vascular remodeling, and RV hypertrophy in mice with existing left HF. These striking findings demonstrate that PM2.5 and/or air pollution is a critical factor for overall HF progression by regulating lung oxidative stress, inflammation and remodeling as well as RV hypertrophy. Improving air quality may save HF patients from a dismal fate.
Keywords: Heart failure, Air pollution, PM2.5, Inflammation, Pulmonary hypertension, Oxidative stress
Abbreviations: HF, Heart failure; TAC, transverse aortic constriction; LV, left ventricular; PM2.5, particulate matter with a median aerodynamic diameter less than 2.5 μm; FA, Filtered air
Graphical abstract
Highlights
-
•
A short PM2.5 exposure profoundly exacerbates heart failure progression
-
•
A short PM2.5 exposure causes multiplicative lung inflammation in heart failure mice
-
•
PM2.5 exposure causes severe lung vessel remodeling in heart failure mice
-
•
PM2.5 exposure causes right ventricular hypertrophy in heart failure mice
-
•
A short PM2.5 exposure does not exacerbate left ventricular function in mice
1. Introduction
Heart failure (HF), also commonly referred to as chronic left ventricular (LV) failure, is a major cause of morbidity and mortality worldwide. HF patients often progress to WHO type-2 pulmonary hypertension (PH) and right ventricular (RV) hypertrophy and/or failure, even with optimal medical care. Recently, we demonstrated that severe LV failure causes profound lung inflammation, vascular remodeling and RV hypertrophy and/or RV failure in experimental animals [[1], [2], [3]]. Severe lung vascular remodeling has been recently reported in human HF patients [4,5]. We showed that, in mice with existing LV failure produced by chronic transverse aortic constriction (TAC), inhibition of inflammation, achieved by the induction of T regulatory cells (Tregs), is effective in halting the transition from LV failure to lung remodeling and RV hypertrophy [6]. Additional studies demonstrates that HF development is attenuated by inhibition of conventional T cell activation in CD28 or B7 knockout mice, and after depletion of CD11c+ antigen-presenting cells [3,7]. Most importantly, two recent retrospective clinical studies showed that inhibition of inflammation by an IL1β inhibitor is highly effective in attenuating major cardiovascular events in patients [8,9], These findings demonstrate that inflammation plays an important role in cardiovascular disease, HF development, and HF progression.
Ambient air pollution and particulate matter (PM), particularly those with a median aerodynamic diameter less than 2.5 μm (PM2.5), have been recognized as a major risk factor for public health including respiratory disease, cancers and heart failure [[10], [11], [12], [13]]. PM2.5 exposure resulted in an increased incidence of myocardial infarction, stroke, arrhythmia and heart failure [14]. An increase of 10 μg per cubic meter PM2.5 was associated with a 76% increase in the risk of death from cardiovascular disease in 4 years' period [14]. HF is the single largest cause for increased hospitalization after acute PM2.5 exposure [15]. We recently showed that prolonged PM2.5 exposure also causes lung inflammation and mild cardiac dysfunction in normal mice [16].
Intrigued by the increased mortality rate in HF patients after short-term air pollution [14], we postulated that air pollution might exert an impact on HF by exacerbating cardiac and lung inflammation. Consequently, we have investigated the role of PM2.5 exposure on LV function, lung inflammation, and RV hypertrophy in a group of mice with existing LV failure.
2. Methods
Detailed methods are available in the online-only Data Supplement.
3. Animals and experimental design
Male Balb/c mice at the age of ∼5 weeks were purchased from the Shanghai Sippr-BK Laboratory Animal Co. Ltd, Shanghai. Mice were subjected to a TAC procedure or sham surgery after at least 7 days adaptation at the research laboratory. Two weeks after TAC, LV ejection fraction (EF) of these mice was determined and the mice were divided into different experimental groups to assure similar initial LV dysfunction.
After the division of the groups, mice were either treated with the local polluted PM2.5 air for 10 h each day for 3 weeks in the research facility at the Haidian district of Beijing or with filtered clean air in the same laboratory. Specifically, the exposure systems include two separate chambers. In the filtered air chamber, a high efficiency particulate air filter (Shanghai Liancheng Purification Equipment CO., LTD, Shanghai) was placed in the inlet valve to remove all the microparticles. In the PM2.5 chamber, a swirler was installed to remove particulate matter with an aerodynamic diameter greater than 2.5 μ. as described previously [17,18]. Echocardiography was performed 5 weeks after TAC using a Visualsonics Vevo 2100 system as previously described [1]. Samples were collected for further biological analysis. The experimental studies were approved by the Institutional Animal Care and Use Committee at Tongji University. All experiments are carried out in accordance with the National Institutes of Health guide for the care and use of Laboratory animals (NIH Publications No. 8023, revised 1978).
4. Statistics
Two-way ANOVA followed by a Bonferroni correction post-hoc test was used to test for differences among more than 2 groups. Student's t-test was used to test for differences between 2 groups. Non-parametric test (Mann-Whitney or Kruskal-Wallis) followed by the Bonferroni post hoc correction was performed. All pairwise p-values are two-sided. The null hypothesis was rejected at P < 0.05.
5. Results
5.1. The general findings related to PM2.5 exposure in normal control and heart failure mice
To best mimic human exposure, mice were exposed to ambient airborne PM2.5 from the Beijing Zhongguancun District (the local Campus of the University of Chinese Academy of Sciences; N39°57′39.83″E116°20′10.97″). As illustrated in Fig. 1A, the PM2.5 exposure was initiated two weeks after the surgery and the exposure duration was 10 h per day for 3 weeks (from April 30th to May 21, 2017) Based on the data obtained from http://www.pm25.com and http://www.zhb.gov.cn, the daily average PM2.5 concentrations of Beijing and the local monitoring station during the study period are presented in Fig. 1B. The linear distance between the Wanliu Station and the study site is ∼3.2 km. The mean airborne PM2.5 concentration during the study period in Beijing city and at the local Wanliu monitoring station was ∼74 μg/m3 (Fig. S1). Since the mice were only exposed to the polluted air for 10 h per day, the average PM2.5 concentration exposed to these mice during the 3 week period was ∼30.8 μg/m3/day (Fig. 1B). The average PM2.5 concentration on day-5 was ∼180 μg/m3/day (Fig. 1B).
Fig. 1.

Moderate PM2.5 exposure did not exacerbate LV dysfunction in mice with existing LV dysfunction. The diagram shows the time of relevant interventions (A). The average of PM2.5 concentrations at the monitoring station (B). Echocardiographic measurements of left ventricle (LV) ejection fraction (C), LV fractional shortening (D), LV end-systolic diameter (E), LV end-diastolic diameter (F) of experimental groups. *P<0.05 between corresponding groups.**P<0.01 between corresponding groups.
5.2. Short term PM2.5 exposure did not exacerbate LV dysfunction in mice with existing LV failure
While TAC caused significant reductions of LV ejection fraction and LV fractional shortening in HF mice, PM2.5 exposure had no detectable effects on LV ejection fraction, LV fractional shortening, LV end-diastolic diameter and LV end-systolic diameter in either sham mice or mice with existing HF (Fig. 1C–F, Fig. S2), indicating that short term PM2.5 did not exacerbate LV failure in these mice.
PM2.5 exposure did not exacerbate TAC-induced LV hypertrophy as evidenced by the similar LV weight (Table S2), and its ratio tibial length in mice with or without PM2.5 exposure (Fig. 2A). PM2.5 exposure had no effect on LV hypertrophy in sham-operated mice as evidenced by the similar LV weight (Table S2), and its ratio to tibial length (Fig. 2A). However, PM2.5 exposure significantly exacerbated TAC-induced increases of LA weight, lung weight, RV weight, and their ratios to tibial length (Fig. 2B and C). PM2.5 exposure did cause a small but significant increase of the ratio of LV weight to bodyweight as compared with HF mice without PM2.5 exposure (Table S2, Fig. S3), an outcome likely due to the reduced bodyweight in mice after PM2.5 exposure. PM2.5 exposure also significantly exacerbated TAC-induced increases of the ratios of LV weight, lung weight and RV weight to bodyweight (Fig. S3).
Fig. 2.

Moderate PM2.5 exposure did not affect LV hypertrophy in mice with existing LV dysfunction but enhanced the increase of the ratio of lung weight to tibial length. Data were collected from sham and TAC mice exposed to either filtered air (FA) or PM2.5. The ratios of LV weight, LA weight, lung weight and RV weight to tibial length of the mice are shown (A–D). Representative images and quantitative data show the RV cardiomyocyte size of mice, as determined by FITC-conjugated wheat germ agglutinin (WGA) staining (E,F). *P<0.05 between corresponding groups.**P<0.01 between corresponding groups. n = 5–6 per group for E-F.
5.3. Short term PM2.5 exposure exacerbated RV hypertrophy in mice with existing LV failure
PM2.5 exposure caused significantly more RV hypertrophy as compared with HF mice treated with the filtered air (Fig. 2D). Furthermore, PM2.5 exposure caused significant RV cardiomyocyte hypertrophy as compared with HF mice treated with the filtered air (Fig. 2E and F).
5.4. PM2.5 exposure profoundly exacerbated lung vascular remodeling and lung fibrosis in mice with existing LV failure
Since RV hypertrophy occurs after lung vascular muscularization and fibrosis [1], to understand the exacerbated RV hypertrophy in HF mice after PM2.5, we determined lung fibrosis and lung vessel muscularization in HF mice with or without PM2.5 exposure. Masson's trichrome stain showed that lung fibrosis increased 9.68-fold in HF mice as compared to that in control mice, and PM2.5 exposure significantly exacerbated lung fibrosis in mice with existing LV failure (Fig. 3A, C). Lung fully muscularized vessels (vessels in which > 75% of the vessel ring is circled by smooth muscle cells), and partially muscularized vessels (vessels with 25%–75% of the vessel ring circled by smooth muscle cells), and total muscularized vessels (including both fully and partially muscularized vessels) were all significantly increased in HF mice as compared to control sham mice (Fig. 3B, D, E). PM2.5 exposure significantly exacerbated the lung vessel muscularization in mice as evidenced by the increased number of muscularized vessels (including both fully and partially muscularized vessels) in the HF mice (Fig. 3D and E), and decreasing the number of nonmuscularized small vessels (vessels with less than 25% of the vessel ring circled by smooth muscle cells) in these HF mice. Moreover, PM2.5 exposure resulted in 1.5 fold increase of lung mRNA level of transforming growth factor-β (TGF-β) in HF mice (Fig. 3F).
Fig. 3.

PM2.5 exposure significantly exacerbated lung fibrosis and lung vessel remodeling in mice with existing LV dysfunction. Data were collected from sham and TAC mice exposed to filtered air (FA) or PM2.5. Representative images and quantitative data of Masson's Trichrome staining (A, C), and smooth muscle a-actin (B, D, E). Quantitative RT-PCR results of TGFβ in lung lysates (F). *P<0.05 between corresponding groups.**P<0.01 between corresponding groups. n = 5 per group for C-E, n = 3 per group for F.
5.5. PM2.5 exposure profoundly exacerbated lung inflammation in mice with existing LV failure
Since our previous study demonstrated that inflammation regulates lung vascular remodeling, fibrosis and RV hypertrophy in mice with existing LV failure, we examined the relative lung leukocyte infiltration in sham-operated and HF mice with or without PM2.5 exposure. We found that PM2.5 exposure caused a small but significant increase of lung CD45+ leukocytes in sham mice, while lung macrophage infiltration was not significantly increased (Fig. 4A and B). Lung CD45+ leukocytes and macrophages were significantly increased in HF mice with or without PM2.5 exposure, but the increases in both CD45+ leukocytes and macrophages were significantly greater in HF mice after PM2.5 exposure (Fig. 4A and B). In addition, we found that lung mRNA contents of pro-inflammatory cytokines, such as monocyte chemoattractant protein-1 (MCP-1), interleukin-1β (IL-1β) and tumor necrosis factor α (TNFα), were all significantly increased in control HF mice, while PM2.5 exposure significantly exacerbated lung pro-inflammatory cytokines in mice with existing LV failure (Fig. 4C–E). HF caused significant increases of lung IL-10 mRNA in HF mice. PM2.5 exposure resulted in a significantly greater increase of lung IL-10 mRNA in HF mice (Fig. 4F). Together, these data demonstrated that PM2.5 exposure promotes lung inflammation in HF mice.
Fig. 4.

PM2.5 exposure exacerbated lung inflammation in mice with existing LV dysfunction. Data were collected from sham-operated and TAC mice exposed to filtered air (FA) or PM2.5. Representative images and quantitative data of CD45 and Mac 2 immunostaining (red) of the lung (A–B). Quantitative reverse transcriptase polymerase chain reaction (RT-PCR) results of interleukin (IL)-1β, tumor necrosis factor (TNF)-α, monocyte chemoattractant protein-1 (MCP)-1, and IL-10 mRNA levels in lung lysates(C-F). *P<0.05 between corresponding groups.**P<0.01 between corresponding groups. n = 4–6 per group for A-B, n = 3 per group for C-F.
5.6. PM2.5 exposure increased VCAM1 and ICAM1 expressions in lung tissues of HF mice
Adhesion molecules, vascular cell adhesion molecule 1 (VCAM1) and intercellular cell adhesion molecule-1 (ICAM1), contribute to leukocyte accumulation in various inflammatory conditions. To understand whether VCAM1 and ICAM1 contribute to the increased lung leukocyte infiltration in mice after PM2.5, we further determined the lung VCAM1 and ICAM1 distribution and expression in each experimental group. Immune staining showed that both VCAM1 and ICAM1 were broadly distributed in lung tissues, and the expression of VCAM1 and ICAM1 proteins were both increased in lung tissue in HF mice exposed to PM2.5. PM2.5 exposure caused significant increases of lung VCAM1 protein expression in both sham mice and in mice with existing LV failure (Figs. S4A and B). PM2.5 exposure did not affect ICAM1 expression in sham mice, but significantly exacerbated its expression in mice with existing LV failure (Figs. S4A and C). Consistent with the increased lung VCAM1 expression in mice after PM2.5 exposure, real-time PCR showed VCAM1 mRNA contents were increased in both sham and HF mice as compared with corresponding control mice exposed to the filtered air (Fig. S4D). PM2.5 exposure did not affect ICAM1 mRNA content in sham mice, but significantly increased ICAM1 mRNA content in HF mice (Fig. S4E).
5.7. PM2.5 exposure caused increases of lung oxidative stress in mice
Since air pollution often causes lung injury through increased oxidative stress, we further determined the relative lung oxidative stress in each experimental group. We first determined lung reactive oxidative species (ROS) by dihydroethidium (DHE) staining. The result demonstrated that PM2.5 exposure caused a significant increase of lung ROS production in both sham and HF mice, while the increase was greater in the HF mice (Fig. 5A; Fig. S5). HF mice showed a significantly greater more increase of lung 3’-NT and 4-HNE as compared with the corresponding sham group (Fig. 5B and C), but PM2.5 exposure caused significantly greater increases of these lung oxidative stress markers 3’-nitrotyrosine (3’-NT) and 4-hydroxynonenal (4-HNE) in both sham and HF mice when compared with corresponding mice treated with filtered air (Fig. 5B and C). Furthermore, PM2.5 exposure caused a significant reduction of the ratio of reduced glutathione (GSH)-to-oxidized glutathione (GSSG) in lung tissues in both sham and HF mice as compared with corresponding sham and HF mice exposed to filtered air(Fig. 5D). Together, these data indicate that PM2.5 exposure causes a significant increase in lung oxidative stress, and the increased oxidative stress might contribute the lung inflammation and remodeling in HF mice.
Fig. 5.

Moderate PM2.5 exposure exacerbates lung oxidative stress in mice with existing LV dysfunction. Data were collected from sham and TAC mice exposed to filtered air (FA) or PM2.5. The level of O2− production in the lung was evaluated with dihydroethidium (DHE) staining (A). The levels of 3′-nitrotyrosine (3’-NT) (B), 4-hydroxynonenal (4-HNE) (C), and the ratio of reduced glutathione to oxidized glutathione (GSH/GSSG) (D). A diagram shows the synergistic effect of heart failure and PM2.5 exposure in mice with existing LV dysfunction (E). *P<0.05 between corresponding groups.**P<0.01 between corresponding groups. n=3 to 5 per group.
6. Discussion
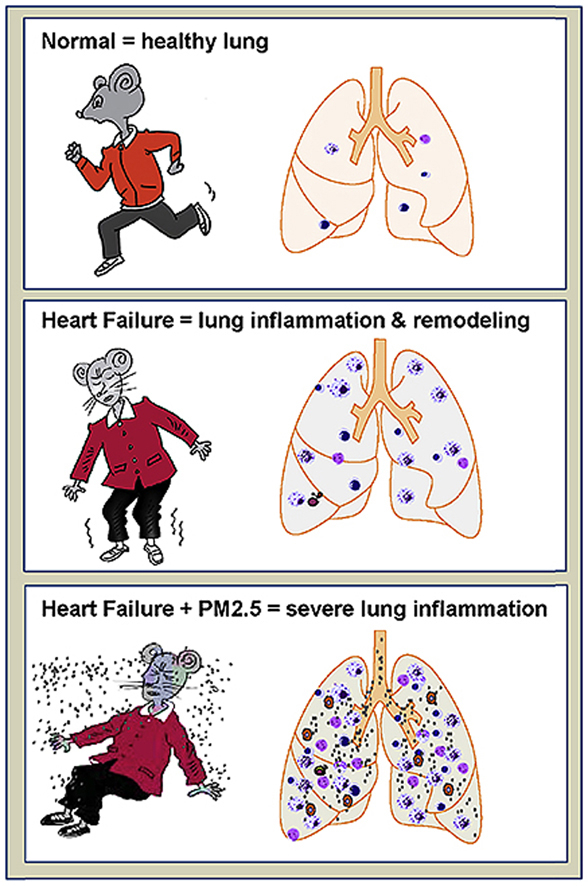
The major finding of our study is that a short PM2.5 exposure did not affect LV hypertrophy and function, but caused a robust lung inflammation, vascular remodeling and fibrosis, as well as significant RV hypertrophy in mice with existing LV failure. These data indicate that PM2.5 synergizes to enhance the effect of LV failure on lung and RV as summarized in Graphical abstract. These results may explain why HF is exacerbated by air pollution. Given the fact that the air quality in many cities during the winter or early spring months is much more toxic than the tested air condition, if HF patients show a similar response to PM2.5 exposure as the HF mice, these findings suggest that PM2.5 could be causing the death of many HF patients. Thus, pollution control instituted by informed policy makers could be an important intervention for HF patients.
Air pollution or PM2.5 has long been recognized as a major public health risk, particular to the vulnerable populations such as patients with preexisting cardiovascular or respiratory diseases. The World Health Organization (WHO) reported that air pollution was responsible for 4.2 million deaths in 2016. It was estimated that 58% of outdoor air pollution-related premature deaths were because of ischemic heart disease and strokes, and only 18% of deaths were because of chronic obstructive pulmonary disease and respiratory infections (http://www.who.int/news-room/fact-sheets/detail/ambient-(outdoor)-air-quality-and-health). Epidemiological studies have consistently demonstrated that air pollution is associated with increased morbidity and mortality related to cardiovascular diseases and respiratory diseases (chronic obstructive pulmonary disease and asthma) [14,19,20]. Similarly, a 16-year follow up study demonstrated that major cardiovascular risk increased ∼18% for every 10 μg/m3 of PM2.5 in the air in USA [21]. PM2.5 air pollution was associated with an increased mortality and incidence of myocardial infarction in the USA [[22], [23], [24]], and an increased hospitalization and heart failure-related mortality related to acute decompensated heart failure in USA [13]. It is estimated that a mean reduction in PM2.5 of 3.9 μg/m3 would prevent 7978 heart failure hospitalizations and dramatically reduce the economic burden to the society [13]. The current study further demonstrates that air quality is a critical factor for overall HF progression through the exacerbation of lung inflammation and RV remodeling.
Since PM2.5 exposure caused a profound lung inflammation, vascular remodeling and the subsequent RV hypertrophy without directly exacerbating LV dysfunction in HF mice, we postulate that the lung inflammation/remodeling and subsequent RV dysfunction might be the major culprit for the increased mortality rate in HF patients after air pollution exposure [14]. While the mechanism by which PM2.5 air pollution exacerbates lung remodeling in HF mice is not totally clear, previous studies from others have demonstrated that PM2.5 exposure resulted in increased lung and cardiac oxidative stress, increased lung and systemic inflammation [17,25], reduced oxygen saturation [26], and reduced vascular endothelial repair capacity [27]. A previous study suggest that lung inflammation after PM2.5 exposure was via a TLR2/TLR4/MyD88-signaling pathway [25]. Our data demonstrate that PM2.5 exposure exacerbated HF progression through increased lung oxidative stress, inflammation, and lung vascular remodeling. In the context that previous studies demonstrated that PM2.5 exposure caused mild LV dysfunction in adult mice [16,28,29], the unchanged LV functions in both sham and HF mice after PM2.5 exposure were not fully anticipated. The unchanged LV function in the current study might relate to the fact that the PM2.5 exposure was only a short period at low concentration.
In this study, since both CHF and PM2.5 exposure caused significantly more reduction of bodyweight as compared with the corresponding group (Table S2), and since the tibial lengths were comparable among 4 experimental groups, to avoid the unwanted influence of different bodyweights in these groups, the ratios to tibial length were used to determine the relative degree of LV hypertrophy in this study. Since the LV weight and its ratio to tibial length was unchanged in HF mice with PM2.5 exposure as compared with HF without PM2.5 exposure, the small but significant increase of the ratio of LV weight to bodyweight in HF mice with PM2.5 exposure is likely an outcome of the reduced bodyweight in this group.
One mechanism to explain the increase in lung inflammation is through the up-regulation of adhesion molecules, such as VCAM-1 and ICAM1. We found that PM2.5 causes up-regulation of lung VCAM-1 and ICAM1 in mice, both under basal conditions and in mice with existing LV failure, but the increase was multiplicative in the presence of LV failure. The dramatic increased lung inflammation after PM2.5 in HF mice indicates that these mice are more vulnerable to PM2.5-induced lung inflammatory response. The significant synergistic increases in lung oxidative stress, which we observed in the HF mice after PM2.5, suggest that oxidative stress is likely to contribute to the lung inflammation, injury, fibrosis and vascular remodeling.
The current study has several limitations. First, the composition and size of polluted air particles vary in different cities and seasons. Consequently, the robust detrimental effects of air pollution observed in the HF mice are likely be different when they are exposed to the polluted air of different cities or of the same city in different seasons. Nevertheless, the findings from the study still provide important insights for the vulnerability of HF patients to air pollution. Second, RV pressure was not determined in the current study. As the lung vessel muscularization and RV hypertrophy are strongly correlated with RV pressure, the severity of the pulmonary hypertension induced by the overall lung remodeling and HF can be surmised from the degree of lung vessel remodeling, lung inflammation, and RV hypertrophy in each experimental group. Third, we only studied male mice in the present study. However, increased lung weight, lung inflammation and vascular remodeling and RV hypertrophy are commonly observed in both male and female mice after HF, we anticipate that PM2.5 would have similar negative impacts in HF progression in both male and female mice.
7. Conclusions
Our findings indicate that a short and moderate PM2.5 exposure is sufficient to cause multiplicative lung inflammation, vascular remodeling and fibrosis, as well as significant RV hypertrophy in the mice with existing LV failure. Since the air qualities in many cities during the winter or early spring times are far worse than our tested conditions, if heart failure patients show similar responses to PM2.5 exposure as the heart failure mice, these findings suggest that air quality is a critical factor for overall HF progression by regulating lung inflammation and remodeling. Thus, improving air quality may save HF patients from a dismal fate.
Submission declaration
The authors declare that the work described has not been published previously.
Declaration of interest
All authors declare that they have no competing interests.
Acknowledgements
We would like to thank Linlin Shi, Linlin Shang, Zezhong Zhang, Chunyan Wu, Hongyun Wang and Yongguang Wu for animal care and technical supports during the study.
Footnotes
Supplementary data to this article can be found online at https://doi.org/10.1016/j.redox.2019.101161.
Contributor Information
Zhongbing Lu, Email: luzhongbing@ucas.ac.cn.
Yawei Xu, Email: xuyawei@tongji.edu.cn.
Yingjie Chen, Email: chenx106@umn.edu.
Sources of funding
This study was supported by Grants 81370197, 81470512, 81570355, 81600308 and 91743104 from National Natural Science Foundation Grants, and a Grant from the American Heart Association.
Appendix A. Supplementary data
The following are the Supplementary data to this article:
References
- 1.Chen Y., Guo H., Xu D., Xu X., Wang H., Hu X., Lu Z., Kwak D., Xu Y., Gunther R., Huo Y., Weir E.K. Left ventricular failure produces profound lung remodeling and pulmonary hypertension in mice: heart failure causes severe lung disease. Hypertension. 2012;59:1170–1178. doi: 10.1161/HYPERTENSIONAHA.111.186072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Liu X., Kwak D., Lu Z., Xu X., Fassett J., Wang H., Wei Y., Cavener D.R., Hu X., Hall J., Bache R.J., Chen Y. Endoplasmic reticulum stress sensor protein kinase r-like endoplasmic reticulum kinase (perk) protects against pressure overload-induced heart failure and lung remodeling. Hypertension. 2014;64:738–744. doi: 10.1161/HYPERTENSIONAHA.114.03811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wang H., Kwak D., Fassett J., Hou L., Xu X., Burbach B.J., Thenappan T., Xu Y., Ge J.B., Shimizu Y., Bache R.J., Chen Y. Cd28/b7 deficiency attenuates systolic overload-induced congestive heart failure, myocardial and pulmonary inflammation, and activated t cell accumulation in the heart and lungs. Hypertension. 2016;68:688–696. doi: 10.1161/HYPERTENSIONAHA.116.07579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Fayyaz A.U., Edwards W.D., Maleszewski J.J., Konik E.A., DuBrock H.M., Borlaug B.A., Frantz R.P., Jenkins S.M. Redfield MM Global pulmonary vascular remodeling in pulmonary hypertension associated with heart failure and preserved or reduced ejection fraction. Circulation. 2018;137:1796–1810. doi: 10.1161/CIRCULATIONAHA.117.031608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Guazzi M., Naeije R. Pulmonary hypertension in heart failure: pathophysiology, pathobiology, and emerging clinical perspectives. J. Am. Coll. Cardiol. 2017;69:1718–1734. doi: 10.1016/j.jacc.2017.01.051. [DOI] [PubMed] [Google Scholar]
- 6.Wang H., Hou L., Kwak D., Fassett J., Xu X., Chen A., Chen W., Blazar B.R., Xu Y., Hall J.L., Ge J.B., Bache R.J., Chen Y. Increasing regulatory t cells with interleukin-2 and interleukin-2 antibody complexes attenuates lung inflammation and heart failure progression. Hypertension. 2016;68:114–122. doi: 10.1161/HYPERTENSIONAHA.116.07084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wang H., Kwak D., Fassett J., Liu X., Yao W., Weng X., Xu X., Xu Y., Bache R.J., Mueller D.L., Chen Y. Role of bone marrow-derived cd11c(+) dendritic cells in systolic overload-induced left ventricular inflammation, fibrosis and hypertrophy. Basic Res. Cardiol. 2017;112:25. doi: 10.1007/s00395-017-0615-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ridker P.M., Everett B.M., Thuren T., MacFadyen J.G., Chang W.H., Ballantyne C., Fonseca F., Nicolau J., Koenig W., Anker S.D., Kastelein J.J.P., Cornel J.H., Pais P., Pella D., Genest J., Cifkova R., Lorenzatti A., Forster T., Kobalava Z., Vida-Simiti L., Flather M., Shimokawa H., Ogawa H., Dellborg M., Rossi P.R.F., Troquay R.P.T., Libby P., Glynn R.J. Group CT Antiinflammatory therapy with canakinumab for atherosclerotic disease. N. Engl. J. Med. 2017;377:1119–1131. doi: 10.1056/NEJMoa1707914. [DOI] [PubMed] [Google Scholar]
- 9.Ridker P.M., MacFadyen J.G., Thuren T., Everett B.M., Libby P., Glynn R.J. Group CT Effect of interleukin-1 beta inhibition with canakinumab on incident lung cancer in patients with atherosclerosis: exploratory results from a randomised, double-blind, placebo-controlled trial. Lancet. 2017;390:1833–1842. doi: 10.1016/S0140-6736(17)32247-X. [DOI] [PubMed] [Google Scholar]
- 10.Brook R.D., Rajagopalan S., Pope C.A., 3rd, Brook J.R., Bhatnagar A., Diez-Roux A.V., Holguin F., Hong Y., Luepker R.V., Mittleman M.A., Peters A., Siscovick D., Smith S.C., Jr., Whitsel L., Kaufman J.D. American Heart Association Council on E, Prevention CotKiCD, Council on Nutrition PA, Metabolism Particulate matter air pollution and cardiovascular disease: an update to the scientific statement from the american heart association. Circulation. 2010;121:2331–2378. doi: 10.1161/CIR.0b013e3181dbece1. [DOI] [PubMed] [Google Scholar]
- 11.Huang C., Moran A.E., Coxson P.G., Yang X., Liu F., Cao J., Chen K., Wang M., He J., Goldman L., Zhao D., Kinney P.L. Gu D Potential cardiovascular and total mortality benefits of air pollution control in urban China. Circulation. 2017;136:1575–1584. doi: 10.1161/CIRCULATIONAHA.116.026487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pope C.A., 3rd Dockery DW Health effects of fine particulate air pollution: lines that connect. J. Air Waste Manag. Assoc. 2006;56:709–742. doi: 10.1080/10473289.2006.10464485. [DOI] [PubMed] [Google Scholar]
- 13.Shah A.S., Langrish J.P., Nair H., McAllister D.A., Hunter A.L., Donaldson K., Newby D.E., Mills N.L. Global association of air pollution and heart failure: a systematic review and meta-analysis. Lancet. 2013;382:1039–1048. doi: 10.1016/S0140-6736(13)60898-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hoek G., Brunekreef B., Fischer P., van Wijnen J. The association between air pollution and heart failure, arrhythmia, embolism, thrombosis, and other cardiovascular causes of death in a time series study. Epidemiology. 2001;12:355–357. doi: 10.1097/00001648-200105000-00017. [DOI] [PubMed] [Google Scholar]
- 15.Calderon-Garciduenas L., Vincent R., Mora-Tiscareno A., Franco-Lira M., Henriquez-Roldan C., Barragan-Mejia G., Garrido-Garcia L., Camacho-Reyes L., Valencia-Salazar G., Paredes R., Romero L., Osnaya H., Villarreal-Calderon R., Torres-Jardon R., Hazucha M.J., Reed W. Elevated plasma endothelin-1 and pulmonary arterial pressure in children exposed to air pollution. Environ. Health Perspect. 2007;115:1248–1253. doi: 10.1289/ehp.9641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wang H., Shen X., Tian G., Shi X., Huang W., Wu Y., Sun L., Peng C., Liu S., Huang Y., Chen X., Zhang F., Chen Y., Ding W., Lu Z. Ampkalpha2 deficiency exacerbates long-term pm2.5 exposure-induced lung injury and cardiac dysfunction. Free Radic. Biol. Med. 2018;121:202–214. doi: 10.1016/j.freeradbiomed.2018.05.008. [DOI] [PubMed] [Google Scholar]
- 17.Xu X., Deng F., Guo X., Lv P., Zhong M., Liu C., Wang A., Tzan K., Jiang S.Y., Lippmann M., Rajagopalan S., Qu Q., Chen L.C. Sun Q Association of systemic inflammation with marked changes in particulate air pollution in beijing in 2008. Toxicol. Lett. 2012;212:147–156. doi: 10.1016/j.toxlet.2012.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chen H., Chen X., Hong X., Liu C., Huang H., Wang Q., Chen S., Chen H., Yang K. Sun Q Maternal exposure to ambient pm2.5 exaggerates fetal cardiovascular maldevelopment induced by homocysteine in rats. Environ. Toxicol. 2017;32:877–889. doi: 10.1002/tox.22287. [DOI] [PubMed] [Google Scholar]
- 19.Brook R.D., Franklin B., Cascio W., Hong Y., Howard G., Lipsett M., Luepker R., Mittleman M., Samet J., Smith S.C., Jr., Tager I. Expert Panel on P, Prevention Science of the American Heart A Air pollution and cardiovascular disease: a statement for healthcare professionals from the expert panel on population and prevention science of the american heart association. Circulation. 2004;109:2655–2671. doi: 10.1161/01.CIR.0000128587.30041.C8. [DOI] [PubMed] [Google Scholar]
- 20.Brunekreef B., Holgate S.T. Air pollution and health. Lancet. 2002;360:1233–1242. doi: 10.1016/S0140-6736(02)11274-8. [DOI] [PubMed] [Google Scholar]
- 21.Pope C.A., 3rd, Muhlestein J.B., May H.T., Renlund D.G., Anderson J.L. Horne BD Ischemic heart disease events triggered by short-term exposure to fine particulate air pollution. Circulation. 2006;114:2443–2448. doi: 10.1161/CIRCULATIONAHA.106.636977. [DOI] [PubMed] [Google Scholar]
- 22.Nawrot T.S., Perez L., Kunzli N., Munters E., Nemery B. Public health importance of triggers of myocardial infarction: a comparative risk assessment. Lancet. 2011;377:732–740. doi: 10.1016/S0140-6736(10)62296-9. [DOI] [PubMed] [Google Scholar]
- 23.Peters A., Dockery D.W., Muller J.E., Mittleman M.A. Increased particulate air pollution and the triggering of myocardial infarction. Circulation. 2001;103:2810–2815. doi: 10.1161/01.cir.103.23.2810. [DOI] [PubMed] [Google Scholar]
- 24.Samet J.M., Dominici F., Curriero F.C., Coursac I., Zeger S.L. Fine particulate air pollution and mortality in 20 u.S. Cities, 1987-1994. N. Engl. J. Med. 2000;343:1742–1749. doi: 10.1056/NEJM200012143432401. [DOI] [PubMed] [Google Scholar]
- 25.He M., Ichinose T., Yoshida Y., Arashidani K., Yoshida S., Takano H., Sun G., Shibamoto T. Urban pm2.5 exacerbates allergic inflammation in the murine lung via a tlr2/tlr4/myd88-signaling pathway. Sci. Rep. 2017;7:11027. doi: 10.1038/s41598-017-11471-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pope CAr, Dockery D.W., Kanner R.E., Villegas G.M. Schwartz J Oxygen saturation, pulse rate, and particulate air pollution: a daily time-series panel study. Am. J. Respir. Crit. Care Med. 1999;159:365–372. doi: 10.1164/ajrccm.159.2.9702103. [DOI] [PubMed] [Google Scholar]
- 27.Haberzettl P., Conklin D.J., Abplanalp W.T., Bhatnagar A., O'Toole T.E. Inhalation of fine particulate matter impairs endothelial progenitor cell function via pulmonary oxidative stress. Arterioscler. Thromb. Vasc. Biol. 2018;38:131–142. doi: 10.1161/ATVBAHA.117.309971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wold L.E., Ying Z., Hutchinson K.R., Velten M., Gorr M.W., Velten C., Youtz D.J., Wang A., Lucchesi P.A., Sun Q., Rajagopalan S. Cardiovascular remodeling in response to long-term exposure to fine particulate matter air pollution. Circ. Heart Fail. 2012;5:452–461. doi: 10.1161/CIRCHEARTFAILURE.112.966580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tanwar V., Gorr M.W., Velten M., Eichenseer C.M., Long V.P., 3rd, Bonilla I.M., Shettigar V., Ziolo M.T., Davis J.P., Baine S.H., Carnes C.A., Wold L.E. Utero particulate matter exposure produces heart failure, electrical remodeling, and epigenetic changes at adulthood. J. Am. Heart Assoc. 2017;6 doi: 10.1161/JAHA.117.005796. pii: e005796. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.