


Abstract
A key step in the Fanconi anemia pathway of DNA interstrand crosslink (ICL) repair is the ICL unhooking by dual endonucleolytic incisions. SLX4/FANCP is a large scaffold protein that plays a central role in ICL unhooking. It contains multiple domains that interact with many proteins including three different endonucleases and also acts in several other DNA repair pathways. While it is known that its interaction with the endonuclease XPF-ERCC1 is required for its function in ICL repair, which other domains act in this process is unclear. Here, we used Xenopus egg extracts to determine ICL repair specific features of SLX4. We show that the SLX4-interacting endonuclease SLX1 is not required for ICL repair and demonstrate that all essential SLX4 domains are located at the N-terminal half of the protein. The MLR domain is crucial for the recruitment of XPF-ERCC1 but also has an unanticipated function in recruiting SLX4 to the site of damage. Although we find the BTB is not essential for ICL repair in our system, dimerization of SLX4 could be important. Our data provide new insights into the mechanism by which SLX4 acts in ICL repair.
INTRODUCTION
DNA interstrand crosslinks (ICLs) are toxic DNA lesions that covalently attach both strands of the duplex, thereby blocking the progression of DNA and RNA polymerases. Due to their toxicity, especially for proliferating cells, DNA crosslinking agents such as cisplatin derivatives and nitrogen mustards, are widely used in cancer chemotherapies (1). However, endogenous metabolites, such as reactive aldehydes, can also induce ICLs (2).
While ICLs can be repaired in the G1 phase of the cell cycle, most of the repair takes place in S-phase and is coupled to DNA replication (3–5). In higher eukaryotes, a complex pathway has evolved to repair ICLs, which is called the Fanconi anemia (FA) pathway. This pathway is linked to the cancer predisposition syndrome Fanconi anemia (FA) that is caused by biallelic mutations in any one of the 22 currently known FA genes. Cells from FA patients are remarkably sensitive to ICL inducing agents, consistent with the FA proteins being involved in the repair of DNA interstrand crosslinks (6,7). Indeed, it has been shown that exogenous ICLs, for example caused by cisplatin, are repaired by the FA pathway (8). Although the source of the endogenous ICL that requires the FA pathway for its repair is currently not known, genetic evidence points towards reactive aldehydes (9–13). However, aldehydes induce several other types of DNA damage (2,14,15) and direct evidence that the aldehyde-induced ICL is repaired by the FA pathway is currently missing.
Based on experiments in Xenopus egg extracts, we and others have previously described a mechanism of FA pathway-dependent ICL repair in S-phase. This requires dual replication fork convergence, in at least a subset of molecules followed by replication fork reversal, ICL unhooking by structure-specific endonucleases, translesion synthesis (TLS), and homologous recombination (Supplemental Figure S1A, (3,16–20)). In some cases, a single fork can bypass an ICL without unhooking, generating a similar X-shaped structure that is the substrate for ICL unhooking (21). Recently, an alternative replication-dependent ICL repair pathway was identified that involves unhooking of the ICL by the glycosylase Neil3, preventing the formation of a double-strand break (22). This process is independent of FA pathway activation and specifically repairs abasic site-, and psoralen/UV-induced ICLs. This suggests that the choice for a specific ICL repair pathway is, at least in part, dependent on the type of ICL. A critical step in ICL repair by the FA pathway is the unhooking of the crosslink from one of the two DNA strands. This step requires activation of the pathway by ubiquitylation of FANCI-FANCD2 that promotes the recruitment of the incision complex consisting of the endonuclease XPF(FANCQ)-ERCC1 and the scaffold protein SLX4(FANCP) (8,19,23). However, mechanistic details of this important step are currently missing.
SLX4 is a large scaffold protein that interacts with many proteins including the three endonucleases XPF-ERCC1, MUS81-EME1 and SLX1 (Figure 1A). In addition to its role in ICL repair, SLX4 acts in several other genome maintenance pathways such as homologous recombination, telomere maintenance, and the resolution of stalled replication forks (24). These different functions are thought to be mediated by specific interactions with its binding partners. The three endonucleases bound to SLX4 affect each other's activity and substrate specificity, and together they can cleave essentially all branched DNA structures (25). The complex of SLX4–SLX1 and MUS81–EME1 is activated in the G2/M cell cycle phase and mediates the resolution of late replication intermediates and Holliday junctions (HJs) (26,27). The interactions of SLX4 with XPF-ERCC1, and possibly SLX1, are required for ICL repair (28–30). In addition to the endonucleases, SLX4 interacts with the mismatch repair proteins MSH2 and MSH3, the uncharacterized open reading frame C20orf94, the telomere-binding factor TRF2, the regulator of DNA damage response TopBP1, and the mitotic kinase PLK1 (31–35). Finally, SLX4 contains several ubiquitin- and SUMO-interaction domains (UBZs and SIMs, respectively), which are implicated in ICL repair, HJ resolution and replication stress response (Figure 1A) (28,29,36–38).
Figure 1.

SLX4 depletion inhibits ICL repair in Xenopus egg extracts. (A) Schematic illustration of the domain organization and interaction partners of SLX4 mapped on the Xenopus laevis SLX4 sequence. Protein interactions are represented by dashed lines. Interaction partners with unknown SLX4 interaction sites are shown in grey. Endonucleases are marked with scissors. Domain boundaries are based on previous reports (28,29,31,33,62). Interaction sites that do not align well with the Xenopus laevis sequence are not numbered and may not be conserved. Especially TRF2 interaction may be human specific (56). (B) Mock- and SLX4-depleted nucleoplasmic egg extract (NPE) were analyzed by western blot using α-SLX4 antibody. A dilution series of undepleted extract was loaded on the same blot to determine the degree of depletion. A relative volume of 100 corresponds to 0.4 μl NPE. (C) As in (B) but using α-XPF (upper panel) or α-ERCC1 antibody (lower panel). (D) Mock-depleted (Mock), SLX4-depleted (ΔSLX4), and SLX4-depleted NPE complemented with wild-type SLX4 (ΔSLX4+SLX4WT) were analyzed by western blot using α-SLX4 antibody (right panel). These extracts, with SLX4-depleted high-speed supernatant (HSS) extract, were used to replicate pICL. Repair efficiency was calculated and plotted (left panel). A higher than endogenous concentration of recombinant SLX4 was required for complete rescue, likely due to partially loss of function of the recombinant protein during purification. (E) SLX4-depleted (ΔSLX4), and SLX4-depleted NPE complemented with wild-type SLX4 (ΔSLX4+SLX4WT) or wild-type SLX4 and XPF-ERCC1 (ΔSLX4+SXE) were analyzed by western blot using α-SLX4 or α-XPF antibodies (right panel). These extracts, with SLX4-depleted HSS, were used to replicate pICL. Repair efficiency was calculated and plotted (left panel). Line within blot indicates position where irrelevant lanes were removed. *, background band. #, SapI fragments from contaminating uncrosslinked plasmid present in varying degrees in different pICL preparations. See also Supplementary Figure S1.
We previously demonstrated that XPF-ERCC1 is required for ICL unhooking and is recruited to the site of damage by SLX4 (19). The interaction of SLX4 with XPF seems to be mostly mediated by the MUS312/MEI9 interaction-like (MLR) domain of SLX4, but the Bric-a-brac, Tramtrack and Broad complex (BTB) domain has also been implicated (Figure 1A) (28,39–41). In addition, the BTB domain plays a role in SLX4 dimerization (28,29,32). So, while the recruitment of XPF-ERCC1 by SLX4 is crucial for ICL repair, the molecular basis for this recruitment is not fully understood.
Another important outstanding question concerns the identity of the second endonuclease that is likely required for ICL unhooking. Although in vitro reconstitution experiments have shown that XPF-ERCC1 can promote dual unhooking incisions on fork-like DNA templates (23,42), most models of ICL repair envision the involvement of a second, 5′ flap, endonuclease (24,43–45). Since SLX1 incises 5′ flap substrates and interacts with SLX4, this is a plausible candidate. Consistent with a role for SLX1 in ICL repair it has been shown that SLX1 deficient cells are sensitive to ICL inducing agents (30,31,40,46).
The Xenopus egg extract system has been used by us and others to dissect biochemical details of replication-coupled DNA interstrand crosslink repair (3,8,16–18,47,48). The use of a plasmid containing a sequence specific cisplatin ICL in this system circumvents the requirement of ICL inducing agents that generate a large fraction of non-ICL DNA lesions. Here, we used this system to examine the function of nuclease-interacting domains of SLX4 in the repair of cisplatin ICLs. We find that while SLX1 strongly associates with SLX4 in our extract, it is not required for ICL repair. Furthermore, the C-terminal half of SLX4, including the BTB domain, the SIMs, and interaction sites for MUS81, PLK1, and TopBP1 are dispensable for repair. In contrast, XPF-binding to the MLR domain of SLX4 is crucial for XPF recruitment and subsequent ICL repair. In addition to XPF-interaction, we demonstrate that the MLR domain also plays a role in efficient recruitment of SLX4 to the site of damage. Together, our findings provide novel insights into the role of SLX4 and its interaction partners in replication-dependent ICL repair.
MATERIALS AND METHODS
Xenopus egg extracts and DNA replication and repair assay
DNA replication assays and preparation of Xenopus egg extracts were performed as described previously (49,50). Preparation of plasmid with a site-specific cisplatin ICL (pICL), and ICL repair assays were performed as described (3,51). Briefly, pICL was first incubated in a high-speed supernatant (HSS) of egg cytoplasm for 20 min, which promotes the assembly of prereplication complexes on the DNA. Addition of two volumes nucleoplasmic egg extract (NPE), which also contained 32P-α-dCTP, triggers a single round of DNA replication. Aliquots of replication reactions (5 μl) were stopped at various times with nine volumes Stop II solution (0.5% SDS, 10 mM EDTA, 50 mM Tris pH 7.5). Samples were incubated with RNase (0.13 μg/μl) for 30 min at 37°C followed by proteinase K (0.5 μg/μl) overnight (O/N) at RT. DNA was extracted using phenol/chloroform, ethanol-precipitated in the presence of glycogen (30 mg/ml) and resuspended in 5 μl 10 mM Tris pH 7.5. ICL repair was analyzed by digesting 1 μl extracted DNA with HincII, or HincII and SapI, separation on a 0.8% native agarose gel, and quantification using autoradiography. Repair efficiency was calculated as described (52). As repair kinetics and absolute efficiency is dependent on the egg extract preparation and depletion conditions, we always use a positive and negative control condition in each experiment using the same extract.
Antibodies and immunodepletions
Antibodies against xlXPF and xlERCC1 were previously described (19). An antibody against xlSLX4 that was previously described (19) was used for detection by western blot. An additional SLX4 antibody was raised against residues 275–509 of xlSLX4. The antigen was overexpressed in bacteria, purified by his-tag affinity purification and denaturing PAGE, and used for immunization of rabbits (PRF&L, Canadensis, USA). This antibody was used for immunodepletions and immunoprecipitations of SLX4. The antibody against SLX1 was raised against residues 93–282 of xlSLX1 and generated similar to the xlSLX4 antibody. Specificity of the antiserum was confirmed using western blot (Supplemental Figure S2F). The anti-FLAG M2 antibody was purchased from Sigma and anti-His antibody from Westburg. To deplete egg extracts of SLX4, one volume protein A sepharose Fast Flow (PAS) (GE Healthcare) was bound to 2.5 volumes α-SLX4 serum or pre-immune serum and washed extensively: twice with PBS, once with ELB (10 mM HEPES–KOH pH 7.7, 50 mM KCl, 2.5 mM MgCl2 and 250 mM Sucrose), twice with ELB + 0.5 M NaCl, and twice with ELB. One volume antibody-bound PAS mixture was then mixed with 5.0 or 6.5 volumes NPE or HSS, respectively, incubated for 20 min at room temperature (RT), after which the extract was harvested. This procedure was repeated twice for NPE and once for HSS. To deplete egg extracts of SLX1, one volume PAS was bound to 3.5 volumes α-SLX1 serum or pre-immune serum and washed as previously described. One volume antibody-bound PAS mixture was mixed with 4.5 volumes NPE or HSS, incubated for 20 min at RT, after which the extract was harvested. This procedure was repeated twice for NPE and once for HSS. After the last depletion round, extracts were collected and immediately used for DNA replication assays. The mock, SLX1 or SLX4 depleted HSS was diluted 2x prior to addition to NPE to reduce protein levels further.
Recombinant protein expression and purification
Xenopus laevis SLX4 containing an N-terminal FLAG- or his-tag, and a C-terminal Strep-tag, was cloned into pDONR201 (Life Technologies). Xenopus laevis SLX1 containing a C-terminal FLAG-tag was also cloned into pDONR201. Point mutations for SLX4SBD*, SLX4MLR* and SLX4BTB* mutants were introduced in pDONR-FLAG-strep-SLX4 using QuikChange site-directed mutagenesis (Agilent Technologies). Similarly, a stop codon was cloned at position 559 in pDONR-FLAG-strep-SLX4 for the generation of the SLX41–558 mutant, which consequently lacks the C-terminal Strep-tag. The MLR domain was deleted by PCR amplification of flanking regions that were ligated by the introduction of a short linker containing a KpnI restriction site. An in-gene reverse primer was used to clone SLX41–840 in pDONR201, this deletion mutant does not contain a C-terminal Strep-tag to maximize resemblance to mini-SLX4 (23). Baculoviruses were produced using the BaculoDirect system following manufacturer's protocol (Life Technologies). Proteins were expressed in suspension cultures of Sf9 insect cells by infection with xlSLX4 viruses for 65 h. Cells from 150 ml culture were collected by centrifugation, resuspended in 6 ml lysis buffer (50 mM Tris pH 8.0, 250 mM NaCl, 0.1% NP-40, 5% glycerol, 0.4 mM PMSF, 1 tablet/10 ml Complete Mini EDTA-free Protease Inhibitor Cocktail (Roche)), and lysed by sonication. The soluble fraction obtained after centrifugation (20 000 × g for 20 min at 4°C) was incubated for 1 h at 4°C with 250 μl anti-FLAG M2 affinity gel (Sigma) that was pre-washed with lysis buffer. After incubation, the beads were washed with 40 ml wash buffer I (50 mM Tris pH 8.0, 250 mM NaCl, 0.1% NP-40, 5% glycerol, 0.4 mM PMSF) and subsequently with 30 ml wash buffer II (20 mM Tris pH 8.0, 200 mM NaCl, 0.1% NP-40, 5% glycerol, 0.1 mM PMSF, 10 μg/ml apropotin/leupeptin). The xlSLX4 protein was eluted in wash buffer II containing 100 μg/ml 3× FLAG peptide (Sigma). The protein was aliquoted, flash frozen and stored at −80°C. Expression and purification of xlSLX4 mutant proteins were identical to the wild-type protein. Proteins analyzed by circular dichroism (CD) were separately expressed and purified to avoid the presence of chloride ions in the buffers. (NH4)2SO4 was used instead of NaCl and Tris buffers were adjusted with phosphoric acid instead of HCl. Expression and purification of xlSLX4–xlSLX1 protein complexes were similar to xlSLX4 proteins, with only minor deviations listed here. Proteins were expressed by co-infection with xlSLX4 and xlSLX1 viruses. Lysis buffer and wash I buffer contained 1 mM PMSF; wash II buffer contained 40 mM Tris pH 8.0, 0.08% NP-40, 4% glycerol and 1 mM PMSF. The xlXPF-hsERCC1 complex was prepared as previously described (19).
Immunoprecipitations
Immunoprecipitations (IPs) were performed in cell lysates from Sf9 cells expressing recombinant proteins, or from Xenopus egg extracts. For IPs from insect cell lysates, proteins were individually expressed in adherent Sf9 insect cell cultures by infection with xlSLX1 or xlSLX4 (or xlSLX4 mutants) viruses for 65 h. Cells were collected, resuspended in 550 μl lysis buffer (50 mM Tris pH 8.0, 300 mM NaCl, 1% Triton, 4 mM EDTA, 10 μg/ml apropotin/leupeptin), and lysed by sonication. After centrifugation (20 000 × g for 20 min at 4°C) the soluble fractions were collected. The xlSLX1 soluble fraction (250 μl) was mixed with the xlSLX4 (or xlSLX4 mutants) soluble fraction (250 μl) and incubated for 1 h at 4°C to allow protein binding. Five volumes PAS were mixed with one volume α-SLX4 or α-SLX1 sera, incubated at 4°C for 1 h, and washed with lysis buffer. Antibody-bound PAS (8 μl) was added to 200 μl of the soluble fraction mixtures followed by a 30-min incubation at 4°C. The beads were washed using 2 ml lysis buffer, taken up in 30 μl 2× SDS sample buffer, and incubated for 4 min at 95°C. Proteins were separated by SDS-PAGE and visualized by western blot using indicated antibodies.
For IPs from Xenopus egg extract, xlSLX4 (or xlSLX4 mutants) was added to HSS at a concentration of 5 ng/μl. To each 10 μl extract, 55 μl IP buffer (1× ELB salts, 0.25 M sucrose, 75 mM NaCl, 2 mM EDTA, 10 μg/ml apropotin/leupeptin, 0.1% NP-40) and 10 μl pre-washed FLAG M2 beads (Sigma-Aldrich) were added. Beads were incubated for 60 min at 4°C and subsequently washed using 2.5 ml IP buffer. Beads were taken up in 30 μl 2× SDS sample buffer and incubated for 4 min at 95°C. Proteins were separated by SDS-PAGE and visualized by western blot using the indicated antibodies.
Proteins were expressed in adherent cultures of Sf9 insect cells in 6-well plates by co-infection with His-xlSLX4 and FLAG-xlSLX4 (or FLAG-xlSLX4 mutants) viruses for 65 h. Cells were resuspended in medium and collected by centrifugation, resuspended in 250 μl lysis buffer (50 mM Tris pH 8.0, 300 mM NaCl, 1% Triton, 4 mM EDTA, 10 μg/ml apropotin/leupeptin), and lysed by sonication. After centrifugation (20 000 × g for 20 min at 4°C), 200 μl soluble fraction was incubated for 30 min at 4°C with 8 μl FLAG M2 beads (Sigma-Aldrich) that were pre-washed with lysis buffer. When benzonase treatment was included in the experiment the fractions were split in two, 0.25 μl benzonase (Sigma-Aldrich) or buffer was added and incubation was continued for another 30 min. After incubation, the beads were washed using 2 ml lysis buffer. Beads were taken up in 30 μl 2× SDS sample buffer and incubated for 5 min at 95°C. Proteins were separated by SDS-PAGE and visualized by western blot using respective antibodies.
Chromatin immunoprecipitation
Chromatin immunoprecipitation (ChIP) was performed as described previously (53). Briefly, reaction samples were crosslinked with formaldehyde, sonicated to yield DNA fragments of ∼100–500 bp, and immunoprecipitated with the indicated antibodies. Protein–DNA crosslinks were reversed and DNA was phenol/chloroform-extracted for analysis by quantitative real-time PCR with the following primers: pICL (5′-AGCCAGATTTTTCCTCCTCTC-3′ and 5′-CATGCATTGGTTCTGCACTT-3′) and pQuant (5′-TACAAATGTACGGCCAGCAA-3′ and 5′-GAGTATGAGGGAAGCGGTGA-3′). pQuant was analyzed to determine non-specific localization to undamaged DNA. The values from pQuant primers were subtracted from the values for pICL primers to establish the specific recruitment to ICL sites.
Circular dichroism
Far-UV circular dichroism (CD) experiments were performed on a J-810 spectropolarimeter (Jasco) using a quartz glass cuvette with an optical pathlength of 1 mm at room temperature. Experimental parameters included a wavelength increment of 2 nm and SLX4 concentrations ranging from 55–137 ng/μl as determined by Coomassie staining. In some cases the protein was recovered after analysis, diluted 2.5 times with 5 M GuHCl or demineralized water and measured again. The resulting spectra are buffer- and concentration-corrected averages of 4 scans in the range of 200–250 nm (native) or 212–250 nm (denatured). The reported mean residue ellipticity (MRE) values were obtained using the molecular mass, total number of amino acids and protein concentration of each SLX4 preparation.
RESULTS
SLX4 depletion does not co-deplete essential ICL repair factors
SLX4 is a multi-domain protein that interacts with many factors and acts in several genome maintenance pathways (Figure 1A) (24,31). Its role in DNA repair is mediated by the interaction with the endonucleases XPF-ERCC1, MUS81-EME1 and SLX1. Although SLX4 and its binding partners have been analyzed biochemically (25), a clear view of which of the SLX4 domains are important for which DNA repair pathway is missing. To study the role of SLX4 in ICL repair we made use of the Xenopus egg extract system that recapitulates replication-coupled ICL repair in vitro (3). We previously demonstrated that immunodepletion of XPF-ERCC1 co-depletes the vast majority of SLX4 from extract, and that ICL repair in XPF-ERCC1 depleted extracts is only rescued when both XPF-ERCC1 and SLX4 are supplemented (19). To biochemically dissect the function of SLX4 we raised an antibody against Xenopus laevis SLX4 (xlSLX4) and immunodepleted it from egg extract. Immunodepletion of SLX4 removed ∼90% of the protein from extract and co-depleted ∼50% of XPF-ERCC1 (Figure 1B and C), confirming that XPF-ERCC1 is present in excess compared to SLX4 (19).
To investigate the effect of SLX4-depletion on ICL repair we replicated a plasmid containing a cisplatin interstrand crosslink (pICL) in a mock- and SLX4-depleted egg extract. Replication intermediates were isolated and repair efficiency was determined by measuring the regeneration of a SapI recognition site that is blocked by the ICL prior to repair (3). In contrast to mock depletion, depletion of SLX4 completely abrogated ICL repair (Figure 1D, Supplemental Figure S1D). This defect was rescued upon addition of recombinant xlSLX4 (Figure 1D, Supplemental Figure S1B and D) indicating that no essential factors were co-depleted. To assess whether the 50% co-depletion of XPF-ERCC1 had a negative effect on the repair efficiency we added additional recombinant XPF-ERCC1 (Supplemental Figure S1C), however this did not affect the repair efficiency (Figure 1E, Supplemental Figure S1E). This indicates that the residual XPF-ERCC1 left after SLX4 depletion interacts with recombinant SLX4 to promote efficient ICL repair. Next, we used this experimental setup to study the role of the various SLX4 domains and interactors in ICL repair.
The endonuclease SLX1 is not required for ICL repair
SLX1 is a 5′ flap endonuclease that is only active when in complex with SLX4 (54). Several reports have demonstrated a cellular sensitivity to ICL-inducing agents in the absence of SLX1 (30,40,46). Accordingly, a model has been suggested in which SLX1 acts together with XPF-ERCC1 to perform the dual unhooking incisions during ICL repair (45). However, SLX1-deficient cells are less sensitive to ICL-inducing agents compared to SLX4-deficient cells, and mouse models display a stronger phenotype when SLX4 is compromised (55). In addition, the stability of SLX1 seems to depend on the presence of SLX4 (30,46,56), which further complicates the conclusions based on sensitivity data. To determine whether SLX1 is specifically required for ICL repair we first generated an SLX1 interaction mutant of SLX4, SLX4SBD* (Figure 2A, Supplemental Figure S2A and B). This xlSLX4 C1753R mutation is equivalent to previously reported mutations situated in the C-terminal SLX1-binding domain (SBD) of the mouse and human proteins (30,56). We immunodepleted SLX4 from egg extract, complemented the extract with SLX4SBD* or wildtype SLX4 (SLX4WT), and examined ICL repair efficiency (Supplemental Figure S2C). Strikingly, the SLX4SBD* mutant complemented the ICL repair defect of the SLX4-depleted extract with the same efficiency as SLX4WT, suggesting that the interaction with SLX1 is not required for ICL repair (Figure 2B, Supplemental Figure S2E).
Figure 2.

SLX1 is not required for ICL repair in Xenopus egg extracts. (A) Schematic illustration of the xlSLX4SBD* mutant protein. The C1753R mutation, indicated by a red line, is predicted to disrupt interaction with SLX1. (B) SLX4-depleted (ΔSLX4), and SLX4-depleted NPE complemented with wild-type SLX4 (ΔSLX4+SLX4WT) or mutant SLX4 (ΔSLX4+SLX4SBD*) were analyzed by western blot using α-SLX4 antibody (right panel). These extracts, with SLX4-depleted HSS, were used to replicate pICL. Repair efficiency was calculated and plotted (left panel). Line within blot indicates position where irrelevant lanes were removed. (C) SLX1, wild-type and mutant SLX4 (SLX4WT and SLX4SBD* respectively) were individually expressed in Sf9 insect cells. Cells were lysed and indicated lysates were mixed and incubated. SLX1 was immunoprecipitated using PAS-conjugated α-SLX1 antibodies. The input (IN), flow-through (FT), and bound fractions (B) were analyzed by western blot using α-FLAG (upper panel) and α-SLX1 antibodies (lower panel). Line within blot indicates position where irrelevant lanes were removed. (D) Mock- and SLX1-depleted NPE were analyzed by western blot using α-SLX1 (upper panel) and α-SLX4 antibodies (lower panel). A dilution series of undepleted extract was loaded on the same blots to determine the degree of depletion. A relative volume of 100 corresponds to 0.4 μl NPE. (E) Mock- and SLX4-depleted NPE were analyzed by western blot using α-SLX1 antibody. A dilution series of undepleted extract was loaded on the same blot to determine the degree of depletion. A relative volume of 100 corresponds to 0.4 μl NPE. (F) Mock-depleted (Mock), SLX1-depleted (ΔSLX1), and SLX1-depleted NPE complemented with wild-type SLX4 (ΔSLX1+SLX4WT) or SLX4-SLX1 complex (ΔSLX4+SLX4/1) were analyzed by western blot using α-SLX1 (upper right panel) and α-SLX4 antibodies (lower right panel). These extracts, with SLX1-depleted HSS, were used to replicate pICL. Repair efficiency was calculated and plotted (left panel). *, background band. #, SapI fragments from contaminating uncrosslinked plasmid present in varying degrees in different pICL preparations. See also Supplementary Figure S2.
To confirm that SLX4SBD* disrupts the interaction with SLX1 efficiently we raised antibodies against Xenopus laevis SLX1 (Supplemental Figure S2F) and performed binding assays using recombinant proteins. To this end, SLX1, SLX4WT and SLX4SBD* were overexpressed in insect cells, and their interaction was examined by immunoprecipitation with the SLX1 antibody. To our surprise, SLX4WT and SLX4SBD* co-precipitated with SLX1 with similar efficiency (Figure 2C), indicating that SLX4SBD* mutant still interacts with SLX1. Although it has been shown that this mutant fails to rescue the reduced stability of SLX1 upon SLX4 depletion in mouse cells, a direct interaction assay was not reported (30,56). Therefore, this mutant could affect the SLX1 affinity enough to destabilize SLX1 in SLX4 deficient cells but could still favour interaction in our overexpression setting. Nonetheless, we reasoned another method to test the function of SLX1 in ICL repair was required. Therefore, we directly immunodepleted SLX1 from Xenopus egg extract. This not only depleted over 95% of SLX1, but also co-depleted ∼90% SLX4 and ∼50% XPF-ERCC1 (Figure 2D, Supplemental Figure S2G). Vice versa, SLX4 depletion co-depleted SLX1 to a large extent, indicating that SLX1 and SLX4 are mostly in complex in Xenopus egg extract (Figure 2E). We then examined ICL repair in the SLX1-depleted extract and detected a moderate repair defect compared to mock-depleted extract (Figure 2F, Supplemental Figure S2H). This defect was fully restored by addition of recombinant SLX4-SLX1 but also by SLX4 alone (Supplemental Figure S2D). This shows that the repair defect upon SLX1 depletion was caused by co-depletion of SLX4. We therefore conclude that SLX1 is not required for ICL repair in our system. Notably, these data also show that SLX1 does not play a role in the resolution of HR intermediates during ICL repair.
The C-terminal domains of SLX4 are dispensable for ICL repair
We previously demonstrated that the endonuclease MUS81-EME1 is not essential for ICL repair in Xenopus egg extracts (19). Both MUS81-EME1 and SLX1 interact with the C-terminus of SLX4, suggesting that this region may not be required for ICL repair. Consistently, a truncated form of SLX4, lacking the C-terminal half, can partially rescue the sensitivity of SLX4 deficient cells to the ICL inducing agent mitomycin C (23). To assess the importance of the C-terminal domain in ICL repair directly, we generated the equivalent truncation mutant xlSLX41–840 (Figure 3A, Supplemental Figure S3A). In addition to the MUS81 and SLX1 interaction sites, this mutant also lacks the SUMO interaction motifs (SIMs), and the PLK1 and TopBP1 interaction sites. To verify that this mutant does not interact with SLX1 we overexpressed SLX1, SLX4WT, SLX4SBD* and SLX41–840 in insect cells and tested interaction by SLX4 immunoprecipitations. While wildtype SLX4 and the SBD mutant co-precipitated SLX1 efficiently, the C-terminal truncation mutant did not (Figure 3B). We then purified SLX41–840 protein and added this mutant, or SLX4WT, to an SLX4-depleted extract (Supplemental Figure S3B). The truncation mutant rescued the ICL repair defect of the SLX4-depleted extract with the same efficiency as SLX4WT (Figure 3C, Supplemental Figure S3C), indicating that the C-terminal half of SLX4 is dispensable for its function in ICL repair. These data show that the N-terminal half of SLX4 contains all essential domains for ICL repair, and that the SIMs, and the interactions with SLX1, MUS81, PLK1 and TopBP1 are not required.
Figure 3.
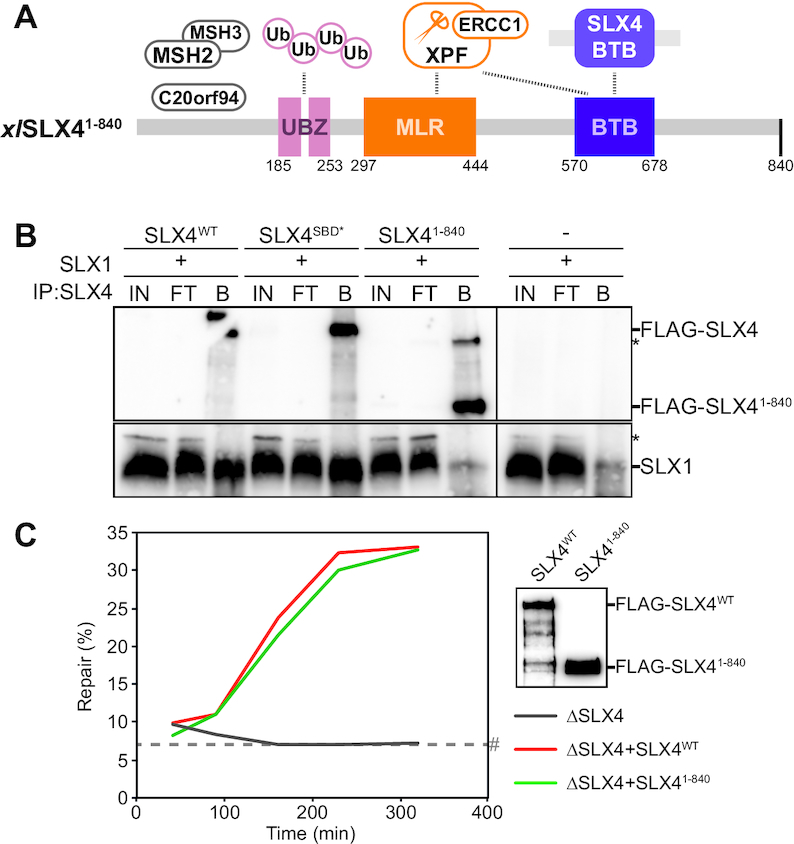
The N-terminal half of SLX4 is sufficient to support ICL repair. (A) Schematic illustration of the xlSLX41–840 mutant protein. This protein is truncated at residue 840. (B) SLX1, wild-type SLX4 (SLX4WT) and SLX4 mutants (SLX4MUT) were individually expressed in Sf9 insect cells. Cells were lysed and indicated lysates were mixed and incubated. SLX4 was immunoprecipitated using PAS-conjugated α-SLX4 antibodies. The input (IN), flow-through (FT), and bound fractions (B) were analyzed by western blot using α-FLAG (upper panel) and α-SLX1 antibodies (lower panel). Line within blot indicates position where irrelevant lanes were removed. (C) SLX4-depleted (ΔSLX4), and SLX4-depleted NPE complemented with wild-type SLX4 (ΔSLX4+SLX4WT) or mutant SLX4 (ΔSLX4+SLX41–840), were used with SLX4-depleted HSS to replicate pICL. Repair efficiency was calculated and plotted (left panel). Protein dilutions of wild-type SLX4 (SLX4WT) and mutant SLX4 (SLX41–840) were analyzed by western blot using α-FLAG antibody to ensure equivalent protein concentrations (right panel). *, background band. #, SapI fragments from contaminating uncrosslinked plasmid present in varying degrees in different pICL preparations. See also Supplementary Figure S3.
The MLR domain is crucial for XPF-ERCC1 recruitment and important for localization of SLX4 to ICLs
During ICL repair, SLX4 recruits the endonuclease XPF-ERCC1 to the site of damage, which promotes ICL unhooking (19). The major XPF interaction site on SLX4 has been mapped to the MUS312/MEI9 interaction-like (MLR) domain, while the BTB domain likely contains a secondary interaction site (28,29,39,40,57,58). Mutations in the MLR domain of SLX4 sensitize cells to ICL-inducing agents, suggesting this domain is important for ICL repair (28,29,59). To test this directly we purified xlSLX4ΔMLR (Figure 4A, Supplemental Figure S4A and D) and examined XPF interaction in Xenopus egg extract. We found that while SLX4WT and SLX41–840 interacted with XPF, SLX4ΔMLR did not (Figure 4E, Supplemental Figure S3D, and (39)). Importantly, we analyzed the mutant protein by circular dichroism and showed that the spectra for the mutant and wildtype protein are highly similar, indicating that the deletion does not change the overall structure (Supplemental Figure S4F). We then tested whether the xlSLX4ΔMLR protein was able to rescue the ICL repair defect after SLX4 depletion. In contrast to SLX4WT, SLX4ΔMLR failed to rescue repair efficiency (Figure 4C, Supplemental Figure S4G). This confirms that the MLR domain of SLX4 is crucial for ICL repair, most likely by recruiting XPF-ERCC1 to the site of damage.
Figure 4.

The MLR domain ensures XPF-binding and contributes to SLX4-recruitment to ICLs. (A) Schematic illustration of the xlSLX4ΔMLR mutant protein. The residues 286–444, including the MLR domain, are replaced by a short linker, indicated by a black line. This deletion is predicted to disrupt interaction with XPF-ERCC1. (B) Schematic illustration of the xlSLX4MLR* mutant protein. The four point mutations in the MLR domain, T418A, W420A, Y434A, and Y435A, are indicated by two red lines and predicted to disrupt interaction with XPF-ERCC1. (C) SLX4-depleted (ΔSLX4), and SLX4-depleted NPE complemented with wild-type SLX4 (ΔSLX4+SLX4WT) or mutant SLX4 (ΔSLX4+SLX4ΔMLR) were analyzed by western blot using α-SLX4 antibody (right panel). These extracts, with SLX4-depleted HSS, were used to replicate pICL. Repair efficiency was calculated and plotted (left panel). Line within blot indicates position where irrelevant lanes were removed. (D) SLX4-depleted (ΔSLX4), and SLX4-depleted NPE complemented with wild-type SLX4 (ΔSLX4+SLX4WT) or mutant SLX4 (ΔSLX4+SLX4ΔMLR) were analyzed by western blot using α-SLX4 (upper right panel) and α-XPF antibodies (lower right panel). These extracts, with SLX4-depleted HSS, were used to replicate pICL. Samples were taken at various times and immunoprecipitated with α-XPF (left panel) or α-SLX4 antibodies (middle panel). Co-precipitated DNA was isolated and analyzed by quantitative PCR using the pICL or pQuant primers. Recovery values of pQuant were subtracted from pICL recovery values. The resulting data were plotted as the percentage of peak value with the highest value within one experiment set to 100%. (E) Purified wild-type SLX4 (SLX4WT) or mutant SLX4 (SLX4MUT) were added to HSS. After incubation, recombinant SLX4 was immunoprecipitated using anti-FLAG M2 affinity gel (Sigma). The input (IN), flow-through (FT), and bound fractions (B) were analyzed by western blot using α-FLAG (upper panel) and α-XPF antibodies (lower panel). Line within blot indicates position where irrelevant lanes were removed. (F) As in (D) but using the xlSLX4MLR* mutant protein. (G) As in (C) but using the xlSLX4MLR* mutant protein. Line within blot indicates position where irrelevant lanes were removed. *, background band. #, SapI fragments from contaminating uncrosslinked plasmid present in varying degrees in different pICL preparations. See also Supplementary Figure S4.
While XPF-ERCC1 depends on SLX4 for its localization to the ICL, the localization of SLX4 to the site of damage is independent of XPF-ERCC1 (19). This leads to the prediction that SLX4ΔMLR localizes to ICLs but is unable to recruit XPF-ERCC1. To test this, we employed chromatin immunoprecipitations (ChIP) of XPF and SLX4. Because our crosslinked plasmid contains a site-specific ICL we can monitor SLX4 and XPF recruitment specifically to the damage site during repair (16,19,39,47,60). We replicated pICL in an SLX4-depleted extract supplemented with SLX4ΔMLR or SLX4WT and performed immunoprecipitations with XPF and SLX4 antibodies followed by quantitative PCR. As predicted, XPF was recruited to the ICL site during repair in the presence of SLX4WT but not in the presence of SLX4ΔMLR (Figure 4D left panel, Supplemental Figure S4H). However, when we performed ChIP on the same samples with an SLX4 antibody we found that the SLX4ΔMLR protein itself was also poorly recruited to the site of damage compared to SLX4WT (Figure 4D middle panel, Supplemental Figure S4H). This suggests that the MLR domain is involved in SLX4 recruitment to the ICL. Consequently, we cannot distinguish whether the lack of recruitment of XPF-ERCC1 to the ICL, and the failure to support ICL repair, is caused by a defect in SLX4 or XPF localization to the ICL.
To clarify this, we generated an additional SLX4 mutant with point mutations in residues that were shown to be involved in XPF-binding (28,59) (Figure 4B, Supplemental Figure S4B and C) and purified this xlSLX4MLR* mutant from insect cells (Supplemental Figure S4E). Similar to SLX4ΔMLR, SLX4MLR* failed to co-precipitate XPF from Xenopus egg extract (Figure 4E), confirming that this mutant is defective in XPF interaction. We then tested the recruitment of the SLX4MLR* mutant to the ICL by ChIP and found that, in contrast to SLX4ΔMLR, this mutant was recruited to the ICL efficiently (Figure 4F middle panel, Supplemental Figure S4I). However, recruitment of XPF in presence of the SLX4MLR* mutant was severely compromised (Figure 4F left panel, Supplemental Figure S4I). Consistent with this, SLX4MLR* did not rescue the ICL repair defect after SLX4-depletion (Figure 4G, Supplemental Figure S4J). These data show that efficient interaction of SLX4 with XPF-ERCC1 via its MLR domain is crucial for ICL repair. Moreover, it indicates that, in addition to XPF recruitment, the MLR domain harbours an additional function in localizing SLX4 to the ICL.
The BTB domain contributes to SLX4-dimerization but is not crucial for ICL repair
In addition to the MLR domain, the BTB domain of SLX4 has also been associated with XPF interaction (28,39,40), as well as with dimerization (28,32,61). BTB mutants are mildly sensitive to ICL inducing agents (28,29,32) suggesting a function for this domain in ICL repair. However, whether this is mediated by dimerization or XPF interaction is not clear. To address the role of the BTB domain in ICL repair, we purified xlSLX4BTB*, a mutant that was reported to affect SLX4 homodimerization and XPF interaction (28,32) (Figure 5A, Supplemental Figures S4F and S5A, C, D). First, we added this mutant to egg extract and examined XPF interaction by immunoprecipitation. SLX4BTB* precipitated similar levels of XPF compared to SLX4WT (Figure 5C), indicating that this mutant does not affect XPF-SLX4 interaction in our extract. To assess the effect of BTB-domain mutation on ICL repair, we added purified SLX4BTB* or SLX4WT to SLX4-depleted extract and monitored repair efficiency. We found that mutating the BTB domain only had a minor effect on ICL repair (Figure 5D, Supplemental Figure S5F). To test whether SLX4BTB* is compromised in dimerization we co-expressed His-tagged SLX4WT and FLAG-tagged SLX4WT or SLX4BTB* in Sf9 cells and examined their co-precipitation. Surprisingly, we found no reduction in dimerization for the SLX4BTB* mutant in the presence or absence of benzonase, indicating that this interaction is not mediated through DNA (Figure 5E, compare lanes 9, 12, 15 and 18).
Figure 5.

The BTB domain is not absolutely required for ICL repair but contributes to SLX4 dimerization. (A) Schematic illustration of the xlSLX4BTB* mutant protein. The two point mutations in the BTB domain, F644A and Y646A, are indicated by a red line and predicted to affect SLX4 dimerization and XPF positioning. (B) Schematic illustration of the xlSLX41–558 mutant protein. This protein is truncated at residue 558. (C) Purified wild-type SLX4 (SLX4WT), mutant SLX4 (SLX4BTB*) or buffer (-) were added to HSS. After incubation, recombinant SLX4 was immunoprecipitated using anti-FLAG M2 affinity gel (Sigma). The input (IN), flow-through (FT), and bound fractions (B) were analyzed by western blot using α-FLAG (upper panel) and α-XPF antibodies (lower panel). Line within blot indicates position where irrelevant lanes were removed. (D) SLX4-depleted (ΔSLX4), and SLX4-depleted NPE complemented with wild-type SLX4 (ΔSLX4+SLX4WT) or mutant SLX4 (ΔSLX4+SLX4BTB*) were analyzed by western blot using α-SLX4 antibody (right panel). These extracts, with SLX4-depleted HSS, were used to replicate pICL. Repair efficiency was calculated and plotted (left panel). Line within blot indicates position were irrelevant lanes were removed. (E) Wild-type (SLX4WT) or mutant SLX4 (SLX4BTB*) containing an N-terminal FLAG-tag and a C-terminal Strep-tag, were co-expressed with wild-type SLX4 containing an N-terminal His-tag and a C-terminal Strep-tag (His-SLX4) in Sf9 insect cells. Cells were lysed and FLAG-tagged SLX4 was immunoprecipitated using anti-FLAG M2 affinity gel (Sigma). To examine the contribution of DNA to SLX4-dimerization the cell lysates were split during the immunoprecipitation step and treated with benzonase (Sigma) (+) or buffer (–). The input (IN), flow-through (FT), and bound fractions (B) were analyzed by western blot using α-FLAG and α-His antibodies. Line within blot indicates position where irrelevant lanes were removed. (F) SLX4-depleted (ΔSLX4), and SLX4-depleted NPE complemented with wild-type SLX4 (ΔSLX4+SLX4WT) or mutant SLX4 (ΔSLX4+SLX41–558), were used with SLX4-depleted HSS to replicate pICL. Repair efficiency was calculated and plotted (left panel). Protein dilutions of wild-type SLX4 (SLX4WT) and mutant SLX4 (SLX41–558) were analyzed by western blot using α-FLAG antibody to ensure equivalent protein concentrations (right panel). (G) As in (E) but using the xlSLX41–558 mutant protein and without benzonase treatment. *, background band. #, SapI fragments from contaminating uncrosslinked plasmid present in varying degrees in different pICL preparations. See also Supplementary Figure S5.
Since the SLX4BTB* mutant had no dimerization or XPF interaction defect, and no major defect in ICL repair we decided to generate another SLX4 truncation mutant, containing residues 1–558, lacking the C-terminus including the entire BTB domain (Figure 5B, Supplemental Figure S5B and E). This truncation was based on a mutation found in a Fanconi anemia patient (62). SLX41–558 interacted normally with XPF (Supplemental Figure S5G) and surprisingly, it fully rescued ICL repair efficiency after SLX4 depletion (Figure 5F, Supplemental Figure S5H). This indicates that ICL repair can occur in the absence of the BTB domain. When testing dimerization we observed that SLX41–558 was still able to dimerize, although this was reduced compared to SLX4WT (Figure 5G, Supplemental Figure S5I, compare lanes 6 and 9). This suggests that the BTB domain is involved in dimerization but other sites on xlSLX4 also play a role in this. Collectively, this suggests that although the BTB domain of SLX4 is not crucial for ICL repair, dimerization of SLX4 could still play a role.
DISCUSSION
A key step in the Fanconi anemia pathway for ICL repair is the ICL unhooking by backbone incisions on either side of the crosslink. SLX4(FANCP) exerts a central role in this step; upon its localization to the ICL, which is facilitated by ubiquitylation of FANCI-FANCD2, it directly recruits XPF-ERCC1 to promote unhooking incisions. It has been suggested that SLX4 has additional roles during ICL repair but this has not been shown directly. Here, we use Xenopus egg extracts to define ICL repair specific functions of SLX4. Our data indicate that the SLX4 interacting endonuclease SLX1 is not required for ICL repair, and therefore that SLX4 does not position two different endonucleases around the crosslink for unhooking. In addition, we find that the BTB domain of SLX4 does not play a major role in ICL repair. Finally, an essential domain for ICL repair is the MLR domain located on the N-terminal half of SLX4. We show that the MLR domain provides the major interaction site for XPF but it is also involved in the localization of SLX4 to the site of damage.
While it has been reported that SLX1 knockdown causes cellular sensitivity to crosslinking agents we demonstrated that efficient ICL repair can take place in the absence of SLX1 in Xenopus egg extracts (Figure 2) (30,31,40,46). This can be explained by the fact that SLX1 is required to repair other types of DNA damage that are induced by crosslinking agents. In support of this view, it has been shown that SLX1–SLX4 interaction is required for resistance to camptothecin and PARP inhibitors, and SLX1, together with SLX4 and MUS81, can process Holliday junction intermediates (25,29,30). It is also consistent with the reports showing that cells deficient in SLX4, a known player in ICL repair, are much more sensitive to, and show more chromosomal instability upon treatment with ICL-inducing agents, compared to SLX1 deficient cells (30,31,40,57,63). Although we show that interstrand crosslinks can be repaired without SLX1, we do not exclude that SLX1 plays a role in the repair of a subset of ICLs, or in specific cell types or conditions. In addition, another nuclease could act redundantly with SLX1 in Xenopus egg extract while this backup mechanism is less active in cells.
An important outstanding question is which nucleases play a role in ICL unhooking in the FA pathway. To date, the only nuclease found crucial for this step is XPF-ERCC1, further supported by the identification of XPF/FANCQ as a FA complementation group (19,64,65). A model in which two different nucleases act in ICL unhooking has been mostly envisioned (43,45,66). This second nuclease is not likely to be SLX1, MUS81 or FAN1 (Figure 2 and (19)), although it is still possible that some of these could act redundantly. In addition, models have been proposed in which the first incision by XPF-ERCC1 is followed by exonuclease activity of SNM1A beyond the ICL leading to unhooking (20,67). Finally, XPF-ERCC1 could perform both unhooking incisions, as suggested based on experiments with fork-like DNA templates and purified proteins (23,42,68). In fact, SLX4 dimerization could promote a similar model in which two XPF-ERCC1 complexes are recruited to each ICL to perform dual incisions. Although this might not be the most likely model due to the strict substrate specificity of XPF-ERCC1, a recent study has suggested a similar model where SLX4 dimerization acts to bridge two MUS81 molecules for controlled breakage of stalled replication intermediates (35).
The role of SLX4 in ICL repair includes, but is not necessarily limited to, unhooking incisions. Downstream events in the pathway include double strand break (DSB) repair via homologous recombination (HR). In cells, SLX4-SLX1 promotes homologous recombination by Holliday junction resolution at G2/M phase, when CDK1 and PLK1 activate MUS81-EME1 by phosphorylation. This induces binding to SLX4–SLX1–XPF–ERCC1, thereby forming an active HJ resolving enzyme (25,34). Our finding that the C-terminal half of SLX4 is not required for ICL repair, supported by data that MUS81 and SLX1 are dispensable for ICL repair, suggests that SLX4 plays no preferred role during HR in the FA pathway (Figures 2 and 3, (19)).
Our observation that SLX41–840 is fully functional in ICL repair further suggests that SLX4 interaction with PLK1, TopBP1 and SUMOylated factors is dispensable for this process. Instead, SLX4 likely employs these factors for its roles in other genome maintenance pathways (26,28,32,33,38). Mutation of the three SUMO interaction motifs in SLX4 has led to variable effects on ICL sensitivity: in one study it had no effect (38), while in two other studies it caused a very mild sensitivity to MMC (28,37). We show that the SIMs are not required for the repair of cisplatin interstrand crosslinks (Figure 3), however, they could play a role in other types of ICLs such as those induced by MMC.
The MLR domain of SLX4 has been shown to mediate the recruitment of XPF-ERCC1 to damage foci in cells and we previously demonstrated that it directly promotes the interaction with XPF-ERCC1 (29,39,58). Using MLR domain mutants we now show that the interaction between SLX4 and XPF-ERCC1 is crucial for ICL repair (Figure 4). In addition to preventing XPF-interaction, deletion of the MLR domain prevents efficient SLX4-recruitment to ICL sites. Since SLX4 is recruited to ICLs independently of XPF (19), this indicates a novel function for the MLR domain in SLX4 localization. It is currently believed that recruitment of SLX4 to ICL damage sites is mediated by the UBZ domains (36,62), however, whether this is through a direct interaction with ubiquitylated FANCI-FANCD2, or mediated by another factor is currently unclear (36,69). Our finding that an additional SLX4 domain is involved in ICL recruitment is important for further studies on how SLX4 is recruited to ICLs.
To examine the importance of the BTB domain of SLX4 in ICL repair we generated a C-terminal truncation lacking this domain based on an FA patient mutation. Notably, in the cells of this patient the mutant protein was not detected indicating that the absence of SLX4 protein is likely the cause of the FA phenotype (29,62). Overexpression of this patient mutant, consisting of the N-terminal 671 residues of SLX4 followed by 119 non-SLX4 residues, partially rescued MMC sensitivity. In addition, disruption of the SLX4 BTB domain in human cells has been shown to cause a very minor (32) to mild sensitivity to ICL-inducing agents (28,29), suggesting that the BTB domain is not absolutely required for ICL repair. Consistent with this, we find that deletion of the BTB domain does not affect ICL repair in our system. The mild effect of BTB mutations in cells could be caused by reduced XPF interaction, and/or defective SLX4 dimerization as was demonstrated previously (28). However, we and others have not observed a defect in XPF interaction in BTB mutants (Figure 5C, Supplementary Figure S5I and (29)). Although our previous work suggested that there is a transient interaction between XPF and the SLX4 BTB domain, this could also be mediated by another site on SLX4 (39). Mutations in the BTB domain seem to have a greater effect on dimerization of the human SLX4 compared to the Xenopus laevis SLX4 (28,32). However, we find that xlSLX4 lacking the BTB domain is still able to dimerize, albeit less efficient compared to wildtype. It is possible that an alternative dimerization site plays a more important role in Xenopus laevis SLX4 which could explain why the BTB mutants still support efficient ICL repair in our system.
Supplementary Material
ACKNOWLEDGEMENTS
We thank Koichi Sato and Alice Bolner for critical reading of the manuscript and Eefjan Breukink (Utrecht University) for help with the circular dichroism experiments. We thank Julie Schouten for technical assistance, Daisy Klein Douwel for helpful discussions, the Hubrecht animal caretakers for animal support, and the other members of the Knipscheer laboratory for feedback.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
This work was supported by the Netherlands organization for Scientific Research (VIDI 700.10.421 to P.K. and the gravitation program CancerGenomiCs.nl). This work is part of the Oncode Institute which is partly financed by the Dutch Cancer Society and was funded by a grant from the Dutch Cancer Society (KWF HUBR 2015-7736 to P.K.).
Conflict of interest statement. None declared.
REFERENCES
- 1. Deans A.J., West S.C.. DNA interstrand crosslink repair and cancer. Nat. Rev. Cancer. 2011; 11:467–480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Voulgaridou G.-P., Anestopoulos I., Franco R., Panayiotidis M.I., Pappa A.. DNA damage induced by endogenous aldehydes: Current state of knowledge. Mutat. Res. 2011; 711:13–27. [DOI] [PubMed] [Google Scholar]
- 3. Raschle M., Knipscheer P., Enoiu M., Angelov T., Sun J., Griffith J.D., Ellenberger T.E., Scharer O.D., Walter J.C.. Mechanism of replication-coupled DNA interstrand crosslink repair. Cell. 2008; 134:969–980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Akkari Y.M., Bateman R.L., Reifsteck C.A., Olson S.B., Grompe M.. DNA replication is required To elicit cellular responses to psoralen-induced DNA interstrand cross-links. Mol. Cell. Biol. 2000; 20:8283–8289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Williams H.L., Gottesman M.E., Gautier J.. The differences between ICL repair during and outside of S phase. Trends Biochem. Sci. 2013; 38:386–393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Kottemann M.C., Smogorzewska A.. Fanconi anaemia and the repair of Watson and Crick DNA crosslinks. Nature. 2013; 493:356–363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Walden H., Deans A.J.. The Fanconi anemia DNA repair pathway: structural and functional insights into a complex disorder. Annu. Rev. Biophys. 2014; 43:257–278. [DOI] [PubMed] [Google Scholar]
- 8. Knipscheer P., Raschle M., Smogorzewska A., Enoiu M., Ho T.V., Scharer O.D., Elledge S.J., Walter J.C.. The Fanconi anemia pathway promotes replication-dependent DNA interstrand cross-link repair. Science. 2009; 326:1698–1701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Ridpath J.R., Nakamura A., Tano K., Luke A.M., Sonoda E., Arakawa H., Buerstedde J.M., Gillespie D.A., Sale J.E., Yamazoe M. et al.. Cells deficient in the FANC/BRCA pathway are hypersensitive to plasma levels of formaldehyde. Cancer Res. 2007; 67:11117–11122. [DOI] [PubMed] [Google Scholar]
- 10. Hira A., Yabe H., Yoshida K., Okuno Y., Shiraishi Y., Chiba K., Tanaka H., Miyano S., Nakamura J., Kojima S. et al.. Variant ALDH2 is associated with accelerated progression of bone marrow failure in Japanese Fanconi anemia patients. Blood. 2013; 122:3206–3209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Garaycoechea J.I., Crossan G.P., Langevin F., Daly M., Arends M.J., Patel K.J.. Genotoxic consequences of endogenous aldehydes on mouse haematopoietic stem cell function. Nature. 2012; 489:571–575. [DOI] [PubMed] [Google Scholar]
- 12. Langevin F., Crossan G.P., Rosado I.V., Arends M.J., Patel K.J.. Fancd2 counteracts the toxic effects of naturally produced aldehydes in mice. Nature. 2011; 475:53–58. [DOI] [PubMed] [Google Scholar]
- 13. Rosado I.V., Langevin F., Crossan G.P., Takata M., Patel K.J.. Formaldehyde catabolism is essential in cells deficient for the Fanconi anemia DNA-repair pathway. Nat. Struct. Mol. Biol. 2011; 18:1432–1434. [DOI] [PubMed] [Google Scholar]
- 14. Wang M., McIntee E.J., Cheng G., Shi Y., Villalta P.W., Hecht S.S.. Identification of DNA adducts of acetaldehyde. Chem. Res. Toxicol. 2000; 13:1149–1157. [DOI] [PubMed] [Google Scholar]
- 15. Cheng G., Shi Y., Sturla S.J., Jalas J.R., McIntee E.J., Villalta P.W., Wang M., Hecht S.S.. Reactions of formaldehyde plus acetaldehyde with deoxyguanosine and DNA: formation of cyclic deoxyguanosine adducts and formaldehyde cross-links. Chem. Res. Toxicol. 2003; 16:145–152. [DOI] [PubMed] [Google Scholar]
- 16. Long D.T., Raschle M., Joukov V., Walter J.C.. Mechanism of RAD51-dependent DNA interstrand cross-link repair. Science. 2011; 333:84–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Budzowska M., Graham T.G., Sobeck A., Waga S., Walter J.C.. Regulation of the Rev1-pol zeta complex during bypass of a DNA interstrand cross-link. EMBO J. 2015; 34:1971–1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Zhang J., Dewar J.M., Budzowska M., Motnenko A., Cohn M.A., Walter J.C.. DNA interstrand cross-link repair requires replication-fork convergence. Nat. Struct. Mol. Biol. 2015; 22:242–247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Klein Douwel D., Boonen R.A., Long D.T., Szypowska A.A., Raschle M., Walter J.C., Knipscheer P.. XPF-ERCC1 acts in Unhooking DNA interstrand crosslinks in cooperation with FANCD2 and FANCP/SLX4. Mol. Cell. 2014; 54:460–471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Amunugama R., Willcox S., Wu R.A., Abdullah U.B., El-Sagheer A.H., Brown T., McHugh P.J., Griffith J.D., Walter J.C.. Replication fork reversal during DNA interstrand crosslink repair requires CMG unloading. Cell Rep. 2018; 23:3419–3428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Huang J., Liu S., Bellani M.A., Thazhathveetil A.K., Ling C., de Winter J.P., Wang Y., Wang W., Seidman M.M.. The DNA translocase FANCM/MHF promotes replication traverse of DNA interstrand crosslinks. Mol. Cell. 2013; 52:434–446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Semlow D.R., Zhang J., Budzowska M., Drohat A.C., Walter J.C.. Replication-Dependent unhooking of DNA interstrand Cross-Links by the NEIL3 glycosylase. Cell. 2016; 167:498–511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Hodskinson M.R., Silhan J., Crossan G.P., Garaycoechea J.I., Mukherjee S., Johnson C.M., Scharer O.D., Patel K.J.. Mouse SLX4 is a tumor suppressor that stimulates the activity of the nuclease XPF-ERCC1 in DNA crosslink repair. Mol. Cell. 2014; 54:472–484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Kim Y. Nuclease delivery: versatile functions of SLX4/FANCP in genome maintenance. Mol. Cells. 2014; 37:569–574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Wyatt H.D., Laister R.C., Martin S.R., Arrowsmith C.H., West S.C.. The SMX DNA repair Tri-nuclease. Mol. Cell. 2017; 65:848–860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. West S.C., Blanco M.G., Chan Y.W., Matos J., Sarbajna S., Wyatt H.D.. Resolution of recombination Intermediates: Mechanisms and regulation. Cold Spring Harb. Symp. Quant. Biol. 2015; 80:103–109. [DOI] [PubMed] [Google Scholar]
- 27. West S.C., Chan Y.W.. Genome instability as a consequence of defects in the resolution of recombination intermediates. Cold Spring Harb. Symp. Quant. Biol. 2017; 82:207–212. [DOI] [PubMed] [Google Scholar]
- 28. Guervilly J.H., Takedachi A., Naim V., Scaglione S., Chawhan C., Lovera Y., Despras E., Kuraoka I., Kannouche P., Rosselli F. et al.. The SLX4 complex is a SUMO E3 ligase that impacts on replication stress outcome and genome stability. Mol. Cell. 2015; 57:123–137. [DOI] [PubMed] [Google Scholar]
- 29. Kim Y., Spitz G.S., Veturi U., Lach F.P., Auerbach A.D., Smogorzewska A.. Regulation of multiple DNA repair pathways by the Fanconi anemia protein SLX4. Blood. 2013; 121:54–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Castor D., Nair N., Declais A.C., Lachaud C., Toth R., Macartney T.J., Lilley D.M., Arthur J.S., Rouse J.. Cooperative control of holliday junction resolution and DNA repair by the SLX1 and MUS81-EME1 nucleases. Mol. Cell. 2013; 52:221–233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Svendsen J.M., Smogorzewska A., Sowa M.E., O’Connell B.C., Gygi S.P., Elledge S.J., Harper J.W.. Mammalian BTBD12/SLX4 assembles a Holliday junction resolvase and is required for DNA repair. Cell. 2009; 138:63–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Yin J., Wan B., Sarkar J., Horvath K., Wu J., Chen Y., Cheng G., Wan K., Chin P., Lei M. et al.. Dimerization of SLX4 contributes to functioning of the SLX4-nuclease complex. Nucleic Acids Res. 2016; 44:4871–4880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Gritenaite D., Princz L.N., Szakal B., Bantele S.C., Wendeler L., Schilbach S., Habermann B.H., Matos J., Lisby M., Branzei D. et al.. A cell cycle-regulated Slx4-Dpb11 complex promotes the resolution of DNA repair intermediates linked to stalled replication. Genes Dev. 2014; 28:1604–1619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Wyatt H.D., Sarbajna S., Matos J., West S.C.. Coordinated actions of SLX1-SLX4 and MUS81-EME1 for Holliday junction resolution in human cells. Mol. Cell. 2013; 52:234–247. [DOI] [PubMed] [Google Scholar]
- 35. Duda H., Arter M., Gloggnitzer J., Teloni F., Wild P., Blanco M.G., Altmeyer M., Matos J.. A mechanism for controlled breakage of Under-replicated chromosomes during mitosis. Dev. Cell. 2016; 39:740–755. [DOI] [PubMed] [Google Scholar]
- 36. Lachaud C., Castor D., Hain K., Munoz I., Wilson J., MacArtney T.J., Schindler D., Rouse J.. Distinct functional roles for the two SLX4 ubiquitin-binding UBZ domains mutated in Fanconi anemia. J. Cell Sci. 2014; 127:2811–2817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Gonzalez-Prieto R., Cuijpers S.A., Luijsterburg M.S., van Attikum H., Vertegaal A.C.. SUMOylation and PARylation cooperate to recruit and stabilize SLX4 at DNA damage sites. EMBO Rep. 2015; 16:512–519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Ouyang J., Garner E., Hallet A., Nguyen H.D., Rickman K.A., Gill G., Smogorzewska A., Zou L.. Noncovalent interactions with SUMO and ubiquitin orchestrate distinct functions of the SLX4 complex in genome maintenance. Mol. Cell. 2015; 57:108–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Klein Douwel D., Hoogenboom W.S., Boonen R.A., Knipscheer P.. Recruitment and positioning determine the specific role of the XPF-ERCC1 endonuclease in interstrand crosslink repair. EMBO J. 2017; 36:2034–2046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Andersen S.L., Bergstralh D.T., Kohl K.P., LaRocque J.R., Moore C.B., Sekelsky J.. Drosophila MUS312 and the vertebrate ortholog BTBD12 interact with DNA structure-specific endonucleases in DNA repair and recombination. Mol. Cell. 2009; 35:128–135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Yildiz O., Majumder S., Kramer B., Sekelsky J.J.. Drosophila MUS312 interacts with the nucleotide excision repair endonuclease MEI-9 to generate meiotic crossovers. Mol. Cell. 2002; 10:1503–1509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Kuraoka I., Kobertz W.R., Ariza R.R., Biggerstaff M., Essigmann J.M., Wood R.D.. Repair of an interstrand DNA cross-link initiated by ERCC1-XPF repair/recombination nuclease. J. Biol. Chem. 2000; 275:26632–26636. [DOI] [PubMed] [Google Scholar]
- 43. Crossan G.P., Patel K.J.. The Fanconi anaemia pathway orchestrates incisions at sites of crosslinked DNA. J. Pathol. 2012; 226:326–337. [DOI] [PubMed] [Google Scholar]
- 44. Cybulski K.E., Howlett N.G.. FANCP/SLX4: a Swiss army knife of DNA interstrand crosslink repair. Cell Cycle. 2011; 10:1757–1763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Zhang J., Walter J.C.. Mechanism and regulation of incisions during DNA interstrand cross-link repair. DNA Repair (Amst.). 2014; 19:135–142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Munoz I.M., Hain K., Declais A.C., Gardiner M., Toh G.W., Sanchez-Pulido L., Heuckmann J.M., Toth R., Macartney T., Eppink B. et al.. Coordination of structure-specific nucleases by human SLX4/BTBD12 is required for DNA repair. Mol. Cell. 2009; 35:116–127. [DOI] [PubMed] [Google Scholar]
- 47. Fu Y.V., Yardimci H., Long D.T., Ho T.V., Guainazzi A., Bermudez V.P., Hurwitz J., van Oijen A., Scharer O.D., Walter J.C.. Selective bypass of a lagging strand roadblock by the eukaryotic replicative DNA helicase. Cell. 2011; 146:931–941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Hoogenboom W.S., Klein Douwel D., Knipscheer P.. Xenopus egg extract: A powerful tool to study genome maintenance mechanisms. Dev. Biol. 2017; 428:300–309. [DOI] [PubMed] [Google Scholar]
- 49. Walter J., Sun L., Newport J.. Regulated chromosomal DNA replication in the absence of a nucleus. Mol. Cell. 1998; 1:519–529. [DOI] [PubMed] [Google Scholar]
- 50. Tutter A.V., Walter J.C.. Chromosomal DNA replication in a soluble cell-free system derived from Xenopus eggs. Methods Mol. Biol. 2006; 322:121–137. [DOI] [PubMed] [Google Scholar]
- 51. Enoiu M., Jiricny J., Scharer O.D.. Repair of cisplatin-induced DNA interstrand crosslinks by a replication-independent pathway involving transcription-coupled repair and translesion synthesis. Nucleic Acids Res. 2012; 40:8953–8964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Knipscheer P., Raschle M., Scharer O.D., Walter J.C.. Replication-coupled DNA interstrand cross-link repair in Xenopus egg extracts. Methods Mol. Biol. 2012; 920:221–243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Pacek M., Tutter A.V., Kubota Y., Takisawa H., Walter J.C.. Localization of MCM2-7, Cdc45, and GINS to the site of DNA unwinding during eukaryotic DNA replication. Mol. Cell. 2006; 21:581–587. [DOI] [PubMed] [Google Scholar]
- 54. Gaur V., Wyatt H.D., Komorowska W., Szczepanowski R.H., de Sanctis D., Gorecka K.M., West S.C., Nowotny M.. Structural and mechanistic analysis of the Slx1-Slx4 endonuclease. Cell Rep. 2015; 10:1467–1476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Wyatt H.D., West S.C.. Holliday junction resolvases. Cold Spring Harb. Perspect. Biol. 2014; 6:a023192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Wilson J.S., Tejera A.M., Castor D., Toth R., Blasco M.A., Rouse J.. Localization-dependent and -independent roles of SLX4 in regulating telomeres. Cell Rep. 2013; 4:853–860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Fekairi S., Scaglione S., Chahwan C., Taylor E.R., Tissier A., Coulon S., Dong M.Q., Ruse C., Yates J.R. 3rd, Russell P. et al.. Human SLX4 is a Holliday junction resolvase subunit that binds multiple DNA repair/recombination endonucleases. Cell. 2009; 138:78–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Crossan G.P., van der Weyden L., Rosado I.V., Langevin F., Gaillard P.H., McIntyre R.E., Sanger Mouse Genetics P., Gallagher F., Kettunen M.I., Lewis D.Y. et al.. Disruption of mouse Slx4, a regulator of structure-specific nucleases, phenocopies Fanconi anemia. Nat. Genet. 2011; 43:147–152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Hashimoto K., Wada K., Matsumoto K., Moriya M.. Physical interaction between SLX4 (FANCP) and XPF (FANCQ) proteins and biological consequences of interaction-defective missense mutations. DNA Repair (Amst.). 2015; 35:48–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Long D.T., Joukov V., Budzowska M., Walter J.C.. BRCA1 promotes unloading of the CMG helicase from a stalled DNA replication fork. Mol. Cell. 2014; 56:174–185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Perez-Torrado R., Yamada D., Defossez P.A.. Born to bind: the BTB protein-protein interaction domain. Bioessays. 2006; 28:1194–1202. [DOI] [PubMed] [Google Scholar]
- 62. Kim Y., Lach F.P., Desetty R., Hanenberg H., Auerbach A.D., Smogorzewska A.. Mutations of the SLX4 gene in Fanconi anemia. Nat. Genet. 2011; 43:142–146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Sarbajna S., Davies D., West S.C.. Roles of SLX1-SLX4, MUS81-EME1, and GEN1 in avoiding genome instability and mitotic catastrophe. Genes Dev. 2014; 28:1124–1136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Bogliolo M., Schuster B., Stoepker C., Derkunt B., Su Y., Raams A., Trujillo J.P., Minguillon J., Ramirez M.J., Pujol R. et al.. Mutations in ERCC4, encoding the DNA-repair endonuclease XPF, cause Fanconi anemia. Am. J. Hum. Genet. 2013; 92:800–806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Kashiyama K., Nakazawa Y., Pilz D.T., Guo C., Shimada M., Sasaki K., Fawcett H., Wing J.F., Lewin S.O., Carr L. et al.. Malfunction of nuclease ERCC1-XPF results in diverse clinical manifestations and causes Cockayne syndrome, xeroderma pigmentosum, and Fanconi anemia. Am. J. Hum. Genet. 2013; 92:807–819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Sengerova B., Wang A.T., McHugh P.J.. Orchestrating the nucleases involved in DNA interstrand cross-link (ICL) repair. Cell Cycle. 2011; 10:3999–4008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Wang A.T., Sengerova B., Cattell E., Inagawa T., Hartley J.M., Kiakos K., Burgess-Brown N.A., Swift L.P., Enzlin J.H., Schofield C.J. et al.. Human SNM1A and XPF-ERCC1 collaborate to initiate DNA interstrand cross-link repair. Genes Dev. 2011; 25:1859–1870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Fisher L.A., Bessho M., Bessho T.. Processing of a psoralen DNA interstrand cross-link by XPF-ERCC1 complex in vitro. J. Biol. Chem. 2008; 283:1275–1281. [DOI] [PubMed] [Google Scholar]
- 69. Yamamoto K.N., Kobayashi S., Tsuda M., Kurumizaka H., Takata M., Kono K., Jiricny J., Takeda S., Hirota K.. Involvement of SLX4 in interstrand cross-link repair is regulated by the Fanconi anemia pathway. PNAS. 2011; 108:6492–6496. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.