


Abstract
MicroRNAs (miRNAs) modulate diverse biological and pathological processes via post-transcriptional gene silencing. High-throughput small RNA sequencing (sRNA-seq) has been widely adopted to investigate the functions and regulatory mechanisms of miRNAs. However, accurate quantification of miRNAs has been limited owing to the severe ligation bias in conventional sRNA-seq methods. Here, we quantify miRNAs and their variants (known as isomiRs) by an improved sRNA-seq protocol, termed AQ-seq (accurate quantification by sequencing), that utilizes adapters with terminal degenerate sequences and a high concentration of polyethylene glycol (PEG), which minimize the ligation bias during library preparation. Measurement using AQ-seq allows us to correct the previously misannotated 5′ end usage and strand preference in public databases. Importantly, the analysis of 5′ terminal heterogeneity reveals widespread alternative processing events which have been underestimated. We also identify highly uridylated miRNAs originating from the 3p strands, indicating regulations mediated by terminal uridylyl transferases at the pre-miRNA stage. Taken together, our study reveals the complexity of the miRNA isoform landscape, allowing us to refine miRNA annotation and to advance our understanding of miRNA regulation. Furthermore, AQ-seq can be adopted to improve other ligation-based sequencing methods including crosslinking-immunoprecipitation-sequencing (CLIP-seq) and ribosome profiling (Ribo-seq).
INTRODUCTION
MicroRNAs (miRNAs) are ∼22 nt-long small non-coding RNAs that regulate gene expression by inducing deadenylation and translational repression of target mRNAs (1). Biogenesis of miRNA involves multiple steps (2). Primary miRNAs (pri-miRNAs) are synthesized by RNA polymerase II and subsequently cleaved by a nuclear ribonuclease (RNase) III enzyme Drosha, releasing small hairpin-shaped precursor miRNAs (pre-miRNAs) (3–6). Pre-miRNAs are exported to the cytoplasm (7,8), where they are further processed by a cytoplasmic RNase III enzyme Dicer into ∼22 nt-long duplex (9–12). The miRNA duplex is loaded onto an Argonaute (Ago) protein, out of which one strand (‘passenger’) gets expelled while the other (‘guide’) remains as the mature miRNA to form a complex called RNA-induced silencing complex (RISC). The strand with uridine or adenosine at its 5′ end binds readily to the 5′ pocket in the MID domain of Ago and is subsequently selected as the guide strand. The strand whose 5′ end is placed in the thermodynamically unstable side of the duplex is also preferentially bound to Ago (13–16). The guide strand base-pairs with the target mRNA mainly through the nucleotides 2–7 relative to the 5′ end of miRNA (so called ‘seed’ sequence), and induces gene silencing in a sequence-specific manner (1).
During maturation, multiple miRNA isoforms (isomiRs) can be generated from a single pri-miRNA hairpin. Alternative cleavage by Drosha or Dicer produces isomiRs with different 5′ and/or 3′ ends (17–20). The 5′ end variation is of particular importance because it changes the seed sequence and, hence, target specificity. Altered cleavage can also affect strand selection, by changing the 5′ end nucleotide and stability of miRNA duplex. Therefore, the 5′ end variation can substantially influence target repertoires.
Another major source of isomiRs is the 3′ end modifications by terminal nucleotidyl transferases. The non-templated nucleotidyl addition (or ‘RNA tailing’) can occur at both the pre- and mature miRNA stages. RNA tailing modulates downstream processing and stability of miRNAs (21–27). For example, when mono-uridylation occurs on pre-let-7 with a 1-nt 3′ overhang (classified into ‘group II’), the U-tail extends the 3′ overhang to make an optimal substrate for Dicer, upregulating pre-let-7 processing (21). In contrast, oligo-uridylation of pre-let-7, which is induced by Lin28, blocks pre-let-7 processing and induces its degradation (22,23,27).
High-throughput small RNA sequencing (sRNA-seq) has been widely adopted to discover and quantify functionally important miRNAs and their variants. sRNA-seq can profile miRNAs at a single nucleotide resolution and detect isomiRs without prior knowledge. However, accurate miRNA profiling has been difficult because certain miRNAs are favored over others in an enzyme-dependent ligation reaction due to the preference of RNA ligases for some sequences and structures, leading to a skewed representation of miRNAs. This can severely compromise quantitative analysis of strand preference and end heterogeneity of a given miRNA (28–39). Recent studies have sought to minimize the ligation bias by adopting randomized adapters, presuming that the increased diversity of adapter sequences raises chances of capturing miRNAs with various sequences (29,31–37,40). Another approach was to apply polyethylene glycol (PEG), which facilitates ligation reaction via molecular crowding effect (30,33,35,36,38,40–42). It has been demonstrated that higher concentrations of PEG lead to better ligation efficiency (30,38,41). Combining both approaches has been recently adopted and has shown to ameliorate the ligation bias (33,35,36,40). However, the optimal condition for their combinatorial use has not been extensively investigated. Furthermore, their performance in detecting isomiRs remains to be examined.
In this study, we systematically evaluate the sequencing bias in sRNA-seq and present a bias-minimized protocol to perform a comprehensive study on miRNA heterogeneity. The results identify misannotated miRNAs and major strands, and reveal previously underappreciated maturation events, notably prevalent alternative processing by RNase III enzymes and uridylation at the pre-miRNA stage.
MATERIALS AND METHODS
Small RNA spike-in control design
Synthetic small RNA spike-in sequences were designed by using a first order Markov chain. The states were separated by the nucleotide position of an RNA. Parameters for the chain were derived from all mature miRNA sequences of Homo sapiens, Mus musculus, Xenopus laevis, and Danio rerio, registered in miRBase release 21. 100,000 randomly generated candidate sequences that are at least 21-nt long were then aligned to the mature miRNA sequences used for modeling by using NCBI BLAST 2.6.0+ with the word size of 4. Candidates with E-values lower than 7.0 were removed. Secondary structures of the survived candidates were predicted using RNAfold in the ViennaRNA suite version 2.1.9 with the ‘--noLP’ option. Candidates with the predicted ΔG value lower than −1.0 kcal/mol were removed from further consideration. The remaining candidates were aligned to genome sequences of H. sapiens (GRCh38), M. musculus (GRCm38), X. laevis (JGI v9.1), and D. rerio (GRCz10), including their mitochondrial genomes and scaffolds included in the top-level DNA sequence bundles provided by ENSEMBL. Sequence alignments were performed using NCBI BLAST 2.6.0+ with the word size of 8. Thirty candidates with the highest maximum E-value to any genome were chosen for final RNA sequences of synthetic spike-ins.
Small RNA sequencing library preparation
Total RNA was isolated from HEK293T and HeLa cells using TRIzol (Invitrogen) and mixed with 1 μl of 10 nM spike-in control oligos, thirty non-human RNA sequences of 21–23 nt in length (Supplementary Table S1). The oligos were obtained from Bioneer Inc., resuspended in distilled water and pooled at equimolar concentrations. The RNA mixture was size-fractionated by 15% urea–polyacrylamide gel electrophoresis and eluted in 0.3 M NaCl to enrich miRNA species using two FAM-labeled markers (17 nt and 29 nt). Small RNA libraries were constructed using either the TruSeq small RNA library preparation kit (Illumina) according to manufacturer's instructions or AQ-seq as follows: miRNA-enriched RNA was ligated to 0.25 μM 3′ randomized adapter using 20 units/μl of T4 RNA ligase 2 truncated KQ (NEB) in 1X T4 RNA ligase reaction buffer (NEB) supplemented with 20% PEG 8000 (NEB) at 25°C for at least 3 h. The ligated RNA was gel-purified on a 15% urea–polyacrylamide gel using two markers (40 nt and 55 nt) to remove the free 3′ adapter and eluted in 0.3 M NaCl. The purified RNA was ligated to 0.18 μM 5′ randomized adapter using 1 unit/μl of T4 RNA ligase 1 (NEB) in 1X T4 RNA ligase reaction buffer supplemented with 1 mM ATP and 20% PEG 8000 (NEB) at 37°C for 1 h. The products were reverse-transcribed using 10 units/μl of SuperScript III reverse transcriptase (Invitrogen) in 1X first-strand buffer (Invitrogen) with 0.2 μM RT primer (RTP, TruSeq kit; Illumina), 0.5 mM dNTP (TruSeq kit; Illumina), and 5 mM DTT (Invitrogen) at 50°C for 1 h. The cDNA was amplified using 0.02 unit/μl of Phusion High-Fidelity DNA Polymerase (Thermo Scientific) in 1X Phusion HF buffer (Thermo Scientific) with 0.5 μM primers (RP1 forward primer and RPIX reverse primer, TruSeq kit; Illumina) and 0.2 mM dNTP (TAKARA). The PCR-amplified cDNA was gel-purified using a 6% polyacrylamide gel to remove adapter dimers and sequenced using MiSeq or HiSeq platforms. Markers for size-fractionation and randomized adapters were obtained from IDT and are listed in Supplementary Table S2.
Analysis of small RNA sequencing
The TruSeq 3′ adapter sequence was removed from FASTQ files using cutadapt (43). For AQ-seq data, 4 nt-long degenerate sequences at the 3′ and 5′ end were trimmed using FASTX-Toolkit (http://hannonlab.cshl.edu/fastx_toolkit/). Next, reads shorter than 18 nt were filtered out, and then low-quality reads (phred quality <20 or <30 in >95% or >50% of nucleotides, respectively) or artifact reads were discarded with FASTX-Toolkit.
Preprocessed reads were first aligned to the spike-in sequences by BWA with the ‘-n 3’ option (44). Reads mapped to spike-in sequences perfectly or with a single mismatch were considered as reliable spike-in reads. The proportion of reads for each spike-in was used for the representation of sRNA-seq bias.
Reads unmapped to spike-in sequences by BWA with the ‘-n 3’ option were subsequently mapped to the human genome (hg38) with the same option. For multi-mapped reads, we selected the alignment results which have the best alignment score, allowing mismatches only at the 3′ end of reads using custom scripts. Reads were classified into annotations from miRBase release 21 (from www.mirbase.org), RefSeq, RepeatMasker (from UCSC genome browser), GtRNAdb (from gtrnadb.ucsc.edu), and Rfam (from rfam.sanger.ac.uk) by intersectBed in BEDTools and used for further analysis (45,46).
Primer extension
The primer was labeled at the 5′ end with T4 polynucleotide kinase (Takara) and [γ-32P] ATP. RNA was extracted using TRIzol reagent (Invitrogen) and then the small RNA fraction was enriched using the mirVana kit (Ambion). The RNA samples were reverse-transcribed with a 5′ end-radiolabeled primer using SuperScript III reverse transcriptase (Invitrogen). The products were separated on a 15% urea-polyacrylamide gel and the radioactive signals were analyzed using a BAS-2500 (FujiFilm). The sequence of RT primer is listed in Supplementary Table S2.
Northern blot analysis
RNA was isolated using either TRIzol (Invitrogen) or the mirVana kit (Ambion), resolved on a 15% urea–polyacrylamide gel, transferred to a Hybond-NX membrane (Amersham) and then crosslinked to the membrane chemically with 1-ethyl-3-(3-dimethylaminopropyl) carbodiimide (47). The probes were labeled at the 5′ end with T4 polynucleotide (Takara) and [γ-32P] ATP. 5′ end-radiolabeled oligonucleotide complementary to the indicated miRNA was hybridized to the membrane. The radioactive signals were analyzed using a BAS-2500. Band intensities were quantified by Multi Gauge software. To strip off probes, the blot was incubated with a pre-boiled solution of 0.5% SDS for 15 min. Synthetic miRNA duplexes (AccuTarget) were obtained from Bioneer. The sequences of probes are listed in Supplementary Table S2.
Quantitative real-time PCR
10 ng of total RNA was reverse-transcribed using the TaqMan miRNA Reverse Transcription kit (Applied Biosystems), and subjected to quantitative real-time PCR with the TaqMan gene expression assay kit (Applied Biosystems) according to manufacturer's instructions. U6 snRNA was used for internal control.
Prediction of miRNA arm ratio
We analyzed miRNA arm ratio as previously described (16) with the following model (Figure 3C):
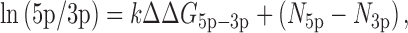 |
where k and N5p(3p) represent the constant for the relative thermodynamic stability and the constant corresponding to the 5′ end identity, respectively.
Figure 3.

Re-assessment of strand preference. (A) Log2-transformed 5p/3p ratios obtained from TruSeq and AQ-seq in HEK293T cells. Dominant arms annotated in miRBase are denoted at the bottom. Bars indicate mean ± standard deviations (s.d.) (n = 2). (B) Northern blot of miR-423-5p and miR-423-3p. Left and middle: Total RNAs from HeLa and HEK293T cells were used for miRNA detection. Synthetic miR-423 duplex was loaded for normalization. Right: A bar plot representing log2-transformed 5p/3p ratio calculated from band intensities of Northern blot shows that the 3p is the major strand, consistent with AQ-seq data. Bars indicate mean ± standard deviations (s.d.) (n = 2). (C) Log2-transformed strand ratio of miR-423 obtained from RT-qPCR-based absolute quantification in HEK293T cells. (D) Two main end properties that determine which strand will be selected: 5′ end identity (N5p(3p)) and thermodynamic stability (ΔG5p(3p)). (E) Comparison of strand ratios between those predicted by the model and those obtained from TruSeq (left panel) or AQ-seq (right panel) in HEK293T cells. See Materials and Methods for details. RPM, reads per million. (F) Linear regression analysis with the indicated equation for strand selection of miRNAs. Bars represent values of the indicated parameters obtained from the model fitted to either TruSeq or AQ-seq data from HEK293T cells.
For prediction, we constructed miRNA duplexes using sequences defined by AQ-seq. If two replicates generated by AQ-seq method reported the identical ends, and the end identities were inconsistent with miRBase, we replaced the miRBase sequences with the those defined by AQ-seq. RNA secondary structure was folded by mfold version 3.6 and thermodynamic stability (ΔG5p(3p)) with dinucleotide subsequences was estimated based on the mfold result (48). Next, we selected top 200 abundant miRNAs (i) which are not duplicated in the genome, (ii) whose 5p and 3p are annotated in miRBase and (iii) whose mature sequences had homogenous 5′ ends (miRNA duplexes whose predominant 5′ ends from 5p and 3p strands accounted for >90% of total reads (5p +3p)). We measured the log ratios of 5p/3p strands using either TruSeq or AQ-seq and performed regression analysis with the previously described equation in R.
RESULTS
Small RNA sequencing optimization using spike-ins
We initially developed spike-in controls which consist of 30 artificial synthetic RNAs of 21–23 nt in order to use them for between-sample normalization (for design of the spike-ins, see Materials and Methods) (Supplementary Table S1). We added the thirty exogenous RNAs to total RNA at equimolar concentrations, performed sRNA-seq library preparation using a widely-used method, called TruSeq, and calculated relative amounts of spike-ins from the sequencing result. We were surprised to observe a strikingly skewed representation of spike-ins in the sequencing result, which reflects severe bias (Figure 1B, top left panel). In light of this observation, we set out to optimize the sRNA-seq protocol by introducing either randomized adapters containing four degenerate nucleotides or PEG (20%). Each modification reduced the bias as expected from the previous studies (29–38,40–42), but not to a satisfying degree (Figure 1B, top right and bottom right panels). Some spike-ins were still grossly overestimated; ∼6 out of 30 spike-ins accounted for >50% of total spike-in reads when only randomized adapters or only PEG was applied. Thus, we tested various combinations of randomized adapters and PEG (Supplementary Figure S1A, lanes 2, 4–15). We found that the addition of PEG only to the 3′ adaptor ligation step does not further significantly mitigate the ligation bias. Less than 20% of PEG—as used in currently available protocols (33,35,36,40)—was not sufficient either. The best result was achieved when we included 20% PEG at both 3′ and 5′ adapter ligation reactions, in combination with randomized adapters (Figure 1A). Importantly, our method produces a minimal amount of adapter dimer contaminants (<0.4% of total reads, Supplementary Figure S1B). Hereafter we refer to this protocol as AQ-seq (accurate quantification by sequencing).
Figure 1.

Small RNA sequencing optimization using spike-ins. (A) Schematic outline of AQ-seq library preparation method. (B) Proportion of 30 spike-ins detected by the indicated protocols.
miRNA and isomiR profiles uncovered by AQ-seq
To evaluate the results from AQ-seq, we examined miRNA abundance in two human cell lines, HEK293T and HeLa, in comparison with TruSeq. While miRNA and isomiR profiles from replicates within each method are highly reproducible (Supplementary Figure S2A), the profiles produced by the two methods correlated poorly with each other (Figure 2). AQ-seq detected ∼1.5-fold more miRNAs than TruSeq did, indicating that AQ-seq is more sensitive than TruSeq is (Figures 2A and B). The expression levels of miRNAs differed substantially between AQ-seq and TruSeq (Figure 2B). For validation, we determined absolute quantities of a subset of miRNAs by quantitative real-time PCR (RT-qPCR) along with synthetic miRNAs of known quantity. The absolute quantity correlated strongly with AQ-seq read counts but not with TruSeq results (Figure 2C and Supplementary Figure S2B).
Figure 2.

miRNA and isomiR profiles uncovered by AQ-seq. (A–G) Comparison of miRNA and isomiR profiles between AQ-seq and TruSeq in HEK293T cells. Abundant miRNAs (>100 RPM in AQ-seq or TruSeq) were included in each analysis, unless otherwise indicated. RPM, reads per million. (A) The number of detected miRNAs in the indicated methods. No abundance filter was applied. Bars indicate mean ± standard deviations (s.d.) (n = 2). (B) Expression profiles. No abundance filter was applied. (C) Comparison between sequencing results and absolute quantities for five miRNAs. The absolute expression levels of the miRNAs were calculated based on the standard curves in Supplementary Figure S2B. (D) The proportion of the 5p strand for a given miRNA calculated by [5p]/([5p]+[3p]), where brackets mean read counts. (E) The proportion of reads whose 5′ end starts at the position as annotated in miRBase for a given miRNA (45). (F and G) Terminal modification frequencies by uridylation (F) or adenylation (G). All terminally modified reads were counted regardless of the tail length.
Accordingly, the strand ratios (5p versus 3p) (Figure 2D) and the isomiR profiles (Figures 2E–G) of many miRNAs were in strong disagreement between the two methods. The 5′-isomiR profile from AQ-seq was markedly different from that of TruSeq (Figure 2E). As for the 3′ terminal modification, highly uridylated miRNAs were identified by both methods, but they were inconsistent (Figure 2F). Adenylation frequencies in the AQ-seq data were mostly lower than those determined by TruSeq (Figure 2G). Collectively, AQ-seq provides the miRNA profiles drastically different from those obtained by TruSeq.
Re-assessment of strand preference
The most striking discrepancy between AQ-seq and TruSeq was found in strand preference (Figure 2D). According to the AQ-seq data, miR-17, miR-106b and miR-151a produce more 5p miRNAs than 3p miRNAs, but TruSeq gave the opposite results (Figure 3A and Supplementary Figure S3A). As for miR-423, 3p is more abundant than 5p according to AQ-seq whereas 5p is supposed to be dominant based on TruSeq and miRBase. Estimation of strand ratio by RT-qPCR was consistent with AQ-seq data rather than TruSeq data (Supplementary Figure S3B). To further validate the strand ratio, we performed Northern blotting using synthetic miR-423 duplex of known quantity as a control. The strand ratio measured by Northern blot analysis matched well to those measured by AQ-seq (Figure 3B). Absolute quantification of each strand also confirmed the AQ-seq result (Figure 3C and Supplementary Figure S2B).
It is known that strand selection is mainly governed by the following rules: (i) the strand whose 5′ end is relatively unstable is favored by Ago and (ii) the strand with uridine or adenosine at its 5′ end is selected as a guide strand (13–16). A recent study using systematic biochemical assays reported that strand ratio is predicted well by these rules and can be expressed by the following equation (Figure 3D):
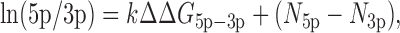 |
where k represents the constant for the relative thermodynamic stability, and N5p and N3p represent the constants corresponding to the 5′ end nucleotide identity of 5p and 3p strands, respectively (16).
To assess the performance of AQ-seq in measuring the strand ratio, we fitted the strand selection model to strand ratio values measured by either AQ-seq or TruSeq (Figure 3E). We then compared the strand ratios predicted by each fitted model with those experimentally obtained. The predicted strand ratios by the linear model indeed fit better with the AQ-seq data than the TruSeq data (Figure 3E and Supplementary Figure S3C). Notably, the fitted model by TruSeq exhibited more dependency on 5′ cytosine than thermodynamic stability (Figure 3F, left and Supplementary Figure S3D, left) whereas the model by AQ-seq relies mainly on relative thermodynamic stability and 5′ uridine (Figure 3F, right and Supplementary Figure S3D, right), which conforms with the previously established strand selection rules. Taken together, our analyses indicated that AQ-seq improves the accuracy in measuring arm ratios.
Correction of the 5′ end annotation
The 5′ end of the guide strand is particularly important for miRNA functionality because the ‘seed’ sequence located at 2–7 nt position relative to the 5′ end of miRNA dictates the specificity of target recognition (1). Since AQ-seq and TruSeq reported inconsistent 5′ ends for many miRNAs (Figures 2D and 4A; 17 in HEK293T and 11 in HeLa cells among miRNAs with reads per million (RPM) over 100), we investigated which method is more reliable in detecting the true start site of miRNAs. For validation, we selected miR-222-5p due to the noticeable discrepancy between the two methods (Figure 4B, upper panel). TruSeq detected the 5′ end of miR-222-5p as annotated in the miRBase, while AQ-seq revealed a different 5′ end that is shifted by 2 nt. To identify the 5′ terminus of miR-222-5p, we performed primer extension experiment and the result was consistent with the AQ-seq data (Figure 4B, lower panel). Of note, the end detected by AQ-seq exactly matches the Drosha cleavage site recently identified by formaldehyde crosslinking, immunoprecipitation, and sequencing (fCLIP-seq) (18) (Figure 4C). These results indicate that the 5′ end of miR-222-5p was indeed correctly identified by AQ-seq and that the miRBase needs to be corrected.
Figure 4.

Correction of the 5′ end annotation. (A) Comparison of miRNA 5′ ends between AQ-seq and TruSeq. The predominant end for a given miRNA among abundant miRNAs (>100 RPM in AQ-seq or TruSeq) was analyzed. RPM, reads per million. (B) Primer extension of miR-222-5p. Identified 5′ ends of miR-222-5p from two sRNA-seq data (AQ-seq and TruSeq) and miRBase are indicated with arrows. The sequence of miR-222-5p deposited in miRBase is colored in red with uppercase. Synthesized oligonucleotides complementary to miR-222-5p with or without 2 nt-extended 5′ end were used as size references. Asterisks mark radiolabeled terminal phosphates. (C) Identification of Drosha cleavage sites of pri-mir-222 using Drosha fCLIP-seq. Top: ∼5.6% of reads mapped on the MIR222 locus were randomly sampled and are denoted as gray bars. The 5′ end of miR-222-5p and the 3′ end of miR-222-3p detected by AQ-seq are indicated with red and blue arrowheads, respectively. Pre-mir-222 is marked as a black bar according to the miRBase-annotated 5′ end of miR-222-5p and the 3′ end of miR-222-3p. Bottom: The stem-loop structure of pri-mir-222. The 5′ and 3′ cleavage sites are indicated with red and blue arrowheads, respectively. The genomic locus and the sequence of miR-222-5p registered in miRBase are colored in red. The uppercase letters represent the sequences of both miR-222-5p and miR-222-3p in miRBase. (D) Comparison of miRNA 5′ ends between AQ-seq and miRBase. Abundant miRNAs (>100 RPM in AQ-seq) with 5′ ends consistently identified in replicates were included in this analysis.
This led us to examine the 5′ end discrepancy between miRBase and AQ-seq data. We found that about 4–6% of abundant miRNAs (>100 RPM) detected in AQ-seq have 5′ ends different from those annotated in miRBase (Figure 4D). Comparison with the Drosha fCLIP-seq data indicates that AQ-seq detected the same 5′ termini of all 5p miRNAs determined by Drosha fCLIP-seq (Supplementary Figure S4). Note that the 5′ end of 3p miRNAs is determined by Dicer, and fCLIP-seq data for Dicer is not currently available. Taken together, these results demonstrate that AQ-seq reliably captures the 5′ ends of miRNAs and offers an opportunity to correct previously misannotated 5′ ends.
Widespread alternative processing
The improved accuracy of AQ-seq in detecting the 5′ ends allowed us to identify alternatively processed miRNAs. Given that even minor 5′-isomiRs are functional and control targets distinct from the targets of the major isoform (17), we analyzed miRNAs with multiple 5′ ends and counted the second most abundant 5′-isomiRs. Notably, a large number of miRNAs show substantial variations at the 5′ end (Figure 5A and Supplementary Figure S5A). Approximately ∼13% of 5p miRNAs and ∼33% of 3p miRNAs (>100 RPM in AQ-seq) produce the 5′-isomiR at >10% frequency (Figure 5B), indicating that alternative processing is more widespread than previously appreciated (20). It is also noteworthy that 3p strands are more variable than 5p strands (Figure 5B), presumably because the 5′ end variation of 5p strands is driven by Drosha only while the 5′ end of 3p strands is determined by both Drosha and Dicer.
Figure 5.

Widespread alternative processing. (A) The fraction of the second most abundant 5′-isomiR for a given miRNA calculated by AQ-seq in HEK293T cells. Since the 5′ ends of 5p and 3p strands are processed by Drosha and Dicer, respectively, they were separately analyzed. RPM, reads per million. (B) The number of miRNAs with unique or alternative 5′ ends. Abundant miRNAs (>100 RPM in AQ-seq) were included in this analysis. If more than two 5′ ends of a given miRNA were detected and each of them accounted for >10% of total reads, the miRNA was considered as an alternatively processed miRNA. Otherwise, the miRNA was considered as a uniquely processed miRNA. Bars indicate mean ± standard deviations (s.d.) (n = 2). (C) Top 10 alternatively processed major strands (>100 RPM in AQ-seq) in HEK293T cells. Bars indicate mean ± standard deviations (s.d.) (n = 2). (D) Illustration of 5′ end usage of four alternatively processed miRNAs. The proportion of sequencing reads with the indicated 5′ end from HEK293T cells is denoted. The sequences of each miRNA deposited in miRBase are colored in red with uppercase.
There are many interesting cases where almost equal amounts of two 5′-isomiRs are produced from the same strand (Figure 5C and Supplementary Figure S5B). For instance, pri-mir-942-5p, pri-mir-192-5p, pri-mir-296-3p, and pri-mir-505-3p undergo alternative processing, resulting in a 1-nt or 2-nt difference in their seed sequences (Figure 5D). Note that the alternative 5′ ends for these miRNAs were underestimated in TruSeq data and missed in miRBase annotations, whereas the Drosha fCLIP-seq showed consistent results to AQ-seq (Supplementary Figure S5C). Taken together, our analyses demonstrate that many miRNA loci produce multiple isoforms with distinct 5′ ends, which increases the diversity of mature miRNAs, consequently expanding their target repertoire.
Highly uridylated miRNAs
Intriguingly, a subset of miRNAs carry non-templated mono-uridine at high frequencies (Figure 6A). For instance, miR-551b-3p, miR-652-3p, miR-760-3p, miR-30e-3p, and miR-324-3p are uridylated at more than ∼50% frequency in HEK293T cells. Notably, uridylation frequencies measured by AQ-seq differ considerably from those by TruSeq (Figure 2E and Supplementary Figure S6). For experimental validation, we chose miR-652-3p which showed a large difference in its uridylation frequency and is abundant enough for Northern blot-based quantification (Figure 6B). The major isoform of miR-652-3p was 1 nt longer than the reference sequence, suggesting that endogenous miR-652-3p is indeed highly mono-uridylated (Figure 6B, left panel). Quantification of the band intensity confirmed that the longer isoform is more abundant than the shorter isoform (Figure 6B, right panel).
Figure 6.

Highly uridylated miRNAs. (A) Top 15 highly uridylated miRNAs among abundant miRNAs (>100 RPM in AQ-seq) in HEK293T and HeLa cells. The type of U-tail is indicated with different color. Light blue, blue, and navy refer to mono-uridylation (U), di-uridylation (UU), and tri-uridylation (UUU), respectively. Bars indicate mean ± standard deviations (s.d.) (n = 2). RPM, reads per million. (B) Northern blot of miR-652-3p detected in the indicated cell lines (left panel). Synthetic miR-652 duplex was loaded as a control. The proportion of the elongated isoform was quantified and compared to uridylation frequencies from TruSeq and AQ-seq (right panel). Bars indicate mean ± standard deviations (s.d.) (n = 2). (C) Northern blot of miR-551b-3p. The miRNA was detected 96 h after transfection of indicated siRNAs in HEK293T cells. Synthetic miR-551b duplex was loaded as a control. (D) Uridylation frequencies calculated by AQ-seq after knockdown of TUT4 and TUT7 in HEK293T cells. Abundant miRNAs (>100 RPM) in the siNC-transfected sample were included in the analysis.
Next, we examined the enzyme(s) responsible for modification of the uridylated miRNAs. It has been reported that two terminal uridylyl transferases (TUTases), TUT4 (ZCCHC11 or TENT3A), and TUT7 (ZCCHC6 or TENT3B), uridylate pre-let-7 and some mature miRNAs (21,23,24,26,27). To test their involvement, we ectopically expressed pri-mir-551b after knockdown of TUT4 and TUT7. Northern blotting shows that, consistent with its high uridylation frequency (∼80%) (Figure 6A), the majority of miR-551b-3p was 1 nt longer than the synthetic miRNA mimic (21 nt). Depletion of TUT4/7 increased the short isoform, indicating that the 1-nt extension was due to mono-uridylation by TUT4/7 (Figure 6C). We also performed AQ-seq after depletion of TUT4/7 and observed a global decrease in uridylation (Figure 6D). These results indicate that the substrates of TUT4 and TUT7 are not limited to group II pre-miRNAs (pre-let-7, pre-mir-98, pre-mir-105-1 and pre-mir-449b) (21), recessed pre-miRNAs (24), and GUAG/UUGU-containing mature miRNAs (let-7, miR-10, miR-99/100 and miR-196 family) (26). Of note, all of the highly uridylated miRNAs detected in our study are 3p miRNAs, originating from the 3′ strand of pre-miRNA (Figure 6A). This confirms the previous notion that TUT4/7 act on pre-miRNAs prior to Dicer-mediated processing.
DISCUSSION
We here present an improved sRNA-seq protocol, termed AQ-seq, which minimizes the ligation bias of sRNA-seq by adopting randomized adaptors in combination with 20% PEG at both 3′ and 5′ ligation steps (Figure 1A). Among commercially available sRNA-seq library preparation kits, NEXTflex (Bioo Scientific) also utilizes randomized adaptors and PEG in both ligation steps. However, the PEG concentrations in 5′ and 3′ adaptor ligation steps are 12.5% and 8.3% in the v2 kit respectively, which we demonstrated are not sufficient to significantly minimize ligation bias (Supplementary Figure S1A). SMARTer® (Clontech) and CATS (Diagenode) were shown to have less bias than NEXTflex since they do not require ligation steps (35). However, the methods employ a tailing reaction which prevents detection of isomiRs derived from 3′ end modification. They also produced a large amount of side products, which we resolved in the AQ-seq protocol (Supplementary Figure S1B) (35).
AQ-seq detects miRNAs of low abundance and reliably defines the terminal sequences of miRNAs undetected when using the conventional sRNA-seq method. Given that AQ-seq data identifies previously undetected termini, it will help us to refine the miRNA annotation in the publicly available databases. It is noteworthy that miRNA target prediction algorithms rely on the complementarity between seed sequences and the targets (1). As the seed sequences are determined by the position relative to the 5′ end of miRNA, incorrect annotation of the 5′ end misleads target identification and functional studies. For instance, the 2-nt shift in the 5′ end of miR-222-5p would result in a distinct set of targets (Figure 4B). The 1-nt or 2-nt offsets found in miR-942-5p, miR-192-5p, miR-296-3p and miR-505-3p are also expected to alter the targets substantially (Figure 5D). Furthermore, accurate quantification of small RNA will help clean up miRBase because a large fraction of its registry is thought to be false (18,49,50). Reliable sRNA-seq data will be imperative to re-evaluate the entries in databases widely used in miRNA research. It is worth noting that AQ-seq also incorporates RNA spike-in controls (51–53). The spike-ins consist of thirty exogenous miRNA-like oligos, which allows us to normalize variations between biological samples as well as to monitor ligation bias and detection sensitivity.
The enhanced performance of AQ-seq data gives us an opportunity to build a reliable catalog of isomiRs and thereby improve our understanding of miRNA biogenesis. In this study, we uncover several interesting maturation events. Firstly, we reliably measure the strand ratio (Figure 3), which will be critical for the studies of alternative strand selection or ‘arm switching’ whose mechanism remains unknown. Secondly, we detect alternative processing in 53 miRNAs in HEK293T or HeLa cells, including miR-101-3p, miR-296-3p, miR-942-5p, miR-505-3p and miR-192-5p (Figure 5). By generating multiple isomiRs from a single hairpin, miRNAs can expand their regulatory capacity. It will be interesting in future studies to investigate whether or not alternative processing of miRNA is regulated in a condition-specific manner, as in the cases of pre-mRNA splicing and alternative polyadenylation. Lastly, we find that more than 15 miRNAs are uridylated at a frequency of over 30% in HEK293T or HeLa cells, and that TUT4/7 mediate the uridylation at the pre-miRNA stage (Figure 6). This indicates that uridylation influences the miRNA pathway well beyond the let-7 family. It will be of interest to study the functional consequences of uridylation of these miRNAs.
Growing evidence suggests that isomiRs are generated in a context-dependent manner and play physiologically relevant roles during developmental and pathological processes (20,54–56). Notably, it was shown that isomiR profiles successfully classify diverse cancer types and, when combined with miRNA profiles, improve discrimination between cancer and normal tissues (57–59), highlighting isomiRs as potential diagnostic biomarkers. Therefore, applying AQ-seq to biosamples may enhance the diagnostic power of sRNA-seq.
It is also important to note that AQ-seq can be adopted to other library preparation methods which involve adapter ligation. In particular, AQ-seq would be beneficial to studies which require ‘within-sample’ comparisons. For example, crosslinking and immunoprecipitation followed by sequencing (CLIP-seq) identifies the protein-RNA interaction sites by quantitatively detecting crosslink sites (60). Since the analysis takes into account relative enrichment of crosslink sites, it is crucial to quantify and compare the abundance of the individual sites within a given sample, which can be severely compromised by ligation bias. Therefore, the inclusion of randomized adapters and PEG in CLIP-seq experiments significantly improves the identification of true binding sites. Another potentially useful application is with ribosome profiling (Ribo-seq) which detects ribosome-protected mRNA fragments (RPFs) by sequencing (61). Ribo-seq is used widely to systematically investigate the mechanism and regulation of translation and the identification of open reading frames. This technique requires unbiased capture of RPFs, which may be achieved by applying randomized adapters and PEG as shown in this study. Taken together, we anticipate AQ-seq will serve as a powerful tool that contributes not only to small RNA studies but also to any experiments that depend on ligation-based library generation.
DATA AVAILABILITY
The small RNA sequencing data in this study are available at the NCBI Gene Expression Omnibus (accession number: GSE123627).
Supplementary Material
ACKNOWLEDGEMENTS
We are grateful to Eunji Kim for technical help. We thank Jaechul Lim, Boseon Kim, Young-Yoon Lee, Young-suk Lee, Hyunjoon Kim, Yongwoo Na and other members of our laboratory for discussion.
Authors contributions: H.K., J.K., K.K., K.Y. and V.N.K. designed experiments. H.C. designed small RNA spike-ins. J.K. and K.Y. generated sRNA-seq libraries. H.K. and J.K. performed biochemical experiments. H.K. carried out computational analyses. H.K., J.K. and V.N.K. wrote the manuscript.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Institute for Basic Science from the Ministry of Science and ICT of Korea [IBS-R008-D1 to H.K., J.K., K.K., H.C., K.Y., V.N.K.]; BK21 Research Fellowships from the Ministry of Education of Korea [to H.K. and K.K.]; NRF (National Research Foundation of Korea) Grant funded by the Korean government [NRF-2015-Global Ph.D. Fellowship Program to H.K.]. Funding for open access charge: Institute for Basic Science from the Ministry of Science and ICT of Korea [IBS-R008-D1].
Conflict of interest statement. None declared.
REFERENCES
- 1. Bartel D.P. Metazoan MicroRNAs. Cell. 2018; 173:20–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Ha M., Kim V.N.. Regulation of microRNA biogenesis. Nat. Rev. Mol. Cell Biol. 2014; 15:509–524. [DOI] [PubMed] [Google Scholar]
- 3. Lee Y., Ahn C., Han J., Choi H., Kim J., Yim J., Lee J., Provost P., Radmark O., Kim S. et al.. The nuclear RNase III Drosha initiates microRNA processing. Nature. 2003; 425:415–419. [DOI] [PubMed] [Google Scholar]
- 4. Denli A.M., Tops B.B., Plasterk R.H., Ketting R.F., Hannon G.J.. Processing of primary microRNAs by the microprocessor complex. Nature. 2004; 432:231–235. [DOI] [PubMed] [Google Scholar]
- 5. Gregory R.I., Yan K.P., Amuthan G., Chendrimada T., Doratotaj B., Cooch N., Shiekhattar R.. The Microprocessor complex mediates the genesis of microRNAs. Nature. 2004; 432:235–240. [DOI] [PubMed] [Google Scholar]
- 6. Han J., Lee Y., Yeom K.H., Kim Y.K., Jin H., Kim V.N.. The Drosha-DGCR8 complex in primary microRNA processing. Genes Dev. 2004; 18:3016–3027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Yi R., Qin Y., Macara I.G., Cullen B.R.. Exportin-5 mediates the nuclear export of pre-microRNAs and short hairpin RNAs. Genes Dev. 2003; 17:3011–3016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Lund E., Guttinger S., Calado A., Dahlberg J.E., Kutay U.. Nuclear export of microRNA precursors. Science. 2004; 303:95–98. [DOI] [PubMed] [Google Scholar]
- 9. Bernstein E., Caudy A.A., Hammond S.M., Hannon G.J.. Role for a bidentate ribonuclease in the initiation step of RNA interference. Nature. 2001; 409:363–366. [DOI] [PubMed] [Google Scholar]
- 10. Grishok A., Pasquinelli A.E., Conte D., Li N., Parrish S., Ha I., Baillie D.L., Fire A., Ruvkun G., Mello C.C.. Genes and mechanisms related to RNA interference regulate expression of the small temporal RNAs that control C. elegans developmental timing. Cell. 2001; 106:23–34. [DOI] [PubMed] [Google Scholar]
- 11. Hutvagner G., McLachlan J., Pasquinelli A.E., Balint E., Tuschl T., Zamore P.D.. A cellular function for the RNA-interference enzyme Dicer in the maturation of the let-7 small temporal RNA. Science. 2001; 293:834–838. [DOI] [PubMed] [Google Scholar]
- 12. Knight S.W., Bass B.L.. A role for the RNase III enzyme DCR-1 in RNA interference and germ line development in Caenorhabditis elegans. Science. 2001; 293:2269–2271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Khvorova A., Reynolds A., Jayasena S.D.. Functional siRNAs and miRNAs exhibit strand bias. Cell. 2003; 115:209–216. [DOI] [PubMed] [Google Scholar]
- 14. Schwarz D.S., Hutvagner G., Du T., Xu Z., Aronin N., Zamore P.D.. Asymmetry in the assembly of the RNAi enzyme complex. Cell. 2003; 115:199–208. [DOI] [PubMed] [Google Scholar]
- 15. Frank F., Sonenberg N., Nagar B.. Structural basis for 5′-nucleotide base-specific recognition of guide RNA by human AGO2. Nature. 2010; 465:818–822. [DOI] [PubMed] [Google Scholar]
- 16. Suzuki H.I., Katsura A., Yasuda T., Ueno T., Mano H., Sugimoto K., Miyazono K.. Small-RNA asymmetry is directly driven by mammalian Argonautes. Nat. Struct. Mol. Biol. 2015; 22:512–521. [DOI] [PubMed] [Google Scholar]
- 17. Chiang H.R., Schoenfeld L.W., Ruby J.G., Auyeung V.C., Spies N., Baek D., Johnston W.K., Russ C., Luo S., Babiarz J.E. et al.. Mammalian microRNAs: experimental evaluation of novel and previously annotated genes. Genes Dev. 2010; 24:992–1009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Kim B., Jeong K., Kim V.N.. Genome-wide mapping of DROSHA cleavage sites on primary MicroRNAs and noncanonical substrates. Mol. Cell. 2017; 66:258–269. [DOI] [PubMed] [Google Scholar]
- 19. Wu H., Ye C., Ramirez D., Manjunath N.. Alternative processing of primary microRNA transcripts by Drosha generates 5′ end variation of mature microRNA. PLoS One. 2009; 4:e7566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Tan G.C., Chan E., Molnar A., Sarkar R., Alexieva D., Isa I.M., Robinson S., Zhang S., Ellis P., Langford C.F. et al.. 5′ isomiR variation is of functional and evolutionary importance. Nucleic Acids Res. 2014; 42:9424–9435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Heo I., Ha M., Lim J., Yoon M.J., Park J.E., Kwon S.C., Chang H., Kim V.N.. Mono-uridylation of pre-microRNA as a key step in the biogenesis of group II let-7 microRNAs. Cell. 2012; 151:521–532. [DOI] [PubMed] [Google Scholar]
- 22. Heo I., Joo C., Cho J., Ha M., Han J., Kim V.N.. Lin28 mediates the terminal uridylation of let-7 precursor MicroRNA. Mol. Cell. 2008; 32:276–284. [DOI] [PubMed] [Google Scholar]
- 23. Heo I., Joo C., Kim Y.K., Ha M., Yoon M.J., Cho J., Yeom K.H., Han J., Kim V.N.. TUT4 in concert with Lin28 suppresses microRNA biogenesis through pre-microRNA uridylation. Cell. 2009; 138:696–708. [DOI] [PubMed] [Google Scholar]
- 24. Kim B., Ha M., Loeff L., Chang H., Simanshu D.K., Li S., Fareh M., Patel D.J., Joo C., Kim V.N.. TUT7 controls the fate of precursor microRNAs by using three different uridylation mechanisms. EMBO J. 2015; 34:1801–1815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Lee M., Choi Y., Kim K., Jin H., Lim J., Nguyen T.A., Yang J., Jeong M., Giraldez A.J., Yang H. et al.. Adenylation of maternally inherited microRNAs by Wispy. Mol. Cell. 2014; 56:696–707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Thornton J.E., Du P., Jing L., Sjekloca L., Lin S., Grossi E., Sliz P., Zon L.I., Gregory R.I.. Selective microRNA uridylation by Zcchc6 (TUT7) and Zcchc11 (TUT4). Nucleic Acids Res. 2014; 42:11777–11791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Hagan J.P., Piskounova E., Gregory R.I.. Lin28 recruits the TUTase Zcchc11 to inhibit let-7 maturation in mouse embryonic stem cells. Nat. Struct. Mol. Biol. 2009; 16:1021–1025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Hafner M., Renwick N., Brown M., Mihailovic A., Holoch D., Lin C., Pena J.T., Nusbaum J.D., Morozov P., Ludwig J. et al.. RNA-ligase-dependent biases in miRNA representation in deep-sequenced small RNA cDNA libraries. RNA. 2011; 17:1697–1712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Jayaprakash A.D., Jabado O., Brown B.D., Sachidanandam R.. Identification and remediation of biases in the activity of RNA ligases in small-RNA deep sequencing. Nucleic Acids Res. 2011; 39:e141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Song Y., Liu K.J., Wang T.H.. Elimination of ligation dependent artifacts in T4 RNA ligase to achieve high efficiency and low bias microRNA capture. PLoS One. 2014; 9:e94619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Sorefan K., Pais H., Hall A.E., Kozomara A., Griffiths-Jones S., Moulton V., Dalmay T.. Reducing ligation bias of small RNAs in libraries for next generation sequencing. Silence. 2012; 3:4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Sun G., Wu X., Wang J., Li H., Li X., Gao H., Rossi J., Yen Y.. A bias-reducing strategy in profiling small RNAs using Solexa. RNA. 2011; 17:2256–2262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Zhang Z., Lee J.E., Riemondy K., Anderson E.M., Yi R.. High-efficiency RNA cloning enables accurate quantification of miRNA expression by deep sequencing. Genome Biol. 2013; 14:R109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Zhuang F., Fuchs R.T., Sun Z., Zheng Y., Robb G.B.. Structural bias in T4 RNA ligase-mediated 3′-adapter ligation. Nucleic Acids Res. 2012; 40:e54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Dard-Dascot C., Naquin D., d’Aubenton-Carafa Y., Alix K., Thermes C., van Dijk E.. Systematic comparison of small RNA library preparation protocols for next-generation sequencing. BMC Genomics. 2018; 19:118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Giraldez M.D., Spengler R.M., Etheridge A., Godoy P.M., Barczak A.J., Srinivasan S., De Hoff P.L., Tanriverdi K., Courtright A., Lu S. et al.. Comprehensive multi-center assessment of small RNA-seq methods for quantitative miRNA profiling. Nat. Biotechnol. 2018; 36:746–757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Fuchs R.T., Sun Z., Zhuang F., Robb G.B.. Bias in ligation-based small RNA sequencing library construction is determined by adaptor and RNA structure. PLoS One. 2015; 10:e0126049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Munafo D.B., Robb G.B.. Optimization of enzymatic reaction conditions for generating representative pools of cDNA from small RNA. RNA. 2010; 16:2537–2552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Linsen S.E., de Wit E., Janssens G., Heater S., Chapman L., Parkin R.K., Fritz B., Wyman S.K., de Bruijn E., Voest E.E. et al.. Limitations and possibilities of small RNA digital gene expression profiling. Nat. Methods. 2009; 6:474–476. [DOI] [PubMed] [Google Scholar]
- 40. Xu P., Bilmeier M., Mohorianu I., Green D., Fraser W.D., Dalmay T.. An improved protocol for small RNA library construction using high definition adapters. Methods Next-Generation Seq. 2015; 2:1–10. [Google Scholar]
- 41. Harrison B., Zimmerman S.B.. Polymer-stimulated ligation: enhanced ligation of oligo- and polynucleotides by T4 RNA ligase in polymer solutions. Nucleic Acids Res. 1984; 12:8235–8251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Shore S., Henderson J.M., Lebedev A., Salcedo M.P., Zon G., McCaffrey A.P., Paul N., Hogrefe R.I.. Small RNA library preparation method for Next-Generation sequencing using chemical modifications to prevent adapter dimer formation. PLoS One. 2016; 11:e0167009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Martin M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet. journal. 2011; 17:10–12. [Google Scholar]
- 44. Li H., Durbin R.. Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics. 2010; 26:589–595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Kozomara A., Griffiths-Jones S.. miRBase: annotating high confidence microRNAs using deep sequencing data. Nucleic Acids Res. 2014; 42:D68–D73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Quinlan A.R., Hall I.M.. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics. 2010; 26:841–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Pall G.S., Hamilton A.J.. Improved northern blot method for enhanced detection of small RNA. Nat. Protoc. 2008; 3:1077–1084. [DOI] [PubMed] [Google Scholar]
- 48. Zuker M. Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res. 2003; 31:3406–3415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Wang X., Liu X.S.. Systematic curation of miRBase annotation using integrated small RNA High-Throughput sequencing data for C. elegans and drosophila. Front. Genet. 2011; 2:25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Fromm B., Billipp T., Peck L.E., Johansen M., Tarver J.E., King B.L., Newcomb J.M., Sempere L.F., Flatmark K., Hovig E. et al.. A uniform system for the annotation of vertebrate microRNA genes and the evolution of the human microRNAome. Annu. Rev. Genet. 2015; 49:213–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Lutzmayer S., Enugutti B., Nodine M.D.. Novel small RNA spike-in oligonucleotides enable absolute normalization of small RNA-Seq data. Sci. Rep. 2017; 7:5913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Locati M.D., Terpstra I., de Leeuw W.C., Kuzak M., Rauwerda H., Ensink W.A., van Leeuwen S., Nehrdich U., Spaink H.P., Jonker M.J. et al.. Improving small RNA-seq by using a synthetic spike-in set for size-range quality control together with a set for data normalization. Nucleic Acids Res. 2015; 43:e89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Fahlgren N., Sullivan C.M., Kasschau K.D., Chapman E.J., Cumbie J.S., Montgomery T.A., Gilbert S.D., Dasenko M., Backman T.W., Givan S.A. et al.. Computational and analytical framework for small RNA profiling by high-throughput sequencing. RNA. 2009; 15:992–1002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Hinton A., Hunter S.E., Afrikanova I., Jones G.A., Lopez A.D., Fogel G.B., Hayek A., King C.C.. sRNA-seq analysis of human embryonic stem cells and definitive endoderm reveals differentially expressed microRNAs and novel IsomiRs with distinct targets. Stem Cells. 2014; 32:2360–2372. [DOI] [PubMed] [Google Scholar]
- 55. Guo L., Liang T., Yu J., Zou Q.. A comprehensive analysis of miRNA/isomiR expression with gender difference. PLoS One. 2016; 11:e0154955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Wang S., Xu Y., Li M., Tu J., Lu Z.. Dysregulation of miRNA isoform level at 5′ end in Alzheimer's disease. Gene. 2016; 584:167–172. [DOI] [PubMed] [Google Scholar]
- 57. Telonis A.G., Loher P., Jing Y., Londin E., Rigoutsos I.. Beyond the one-locus-one-miRNA paradigm: microRNA isoforms enable deeper insights into breast cancer heterogeneity. Nucleic Acids Res. 2015; 43:9158–9175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Telonis A.G., Magee R., Loher P., Chervoneva I., Londin E., Rigoutsos I.. Knowledge about the presence or absence of miRNA isoforms (isomiRs) can successfully discriminate amongst 32 TCGA cancer types. Nucleic Acids Res. 2017; 45:2973–2985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Koppers-Lalic D., Hackenberg M., de Menezes R., Misovic B., Wachalska M., Geldof A., Zini N., de Reijke T., Wurdinger T., Vis A. et al.. Noninvasive prostate cancer detection by measuring miRNA variants (isomiRs) in urine extracellular vesicles. Oncotarget. 2016; 7:22566–22578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Konig J., Zarnack K., Luscombe N.M., Ule J.. Protein-RNA interactions: new genomic technologies and perspectives. Nat. Revi. Genet. 2012; 13:77–83. [DOI] [PubMed] [Google Scholar]
- 61. Ingolia N.T., Ghaemmaghami S., Newman J.R., Weissman J.S.. Genome-wide analysis in vivo of translation with nucleotide resolution using ribosome profiling. Science. 2009; 324:218–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The small RNA sequencing data in this study are available at the NCBI Gene Expression Omnibus (accession number: GSE123627).