

Abstract
Background
Even though liver kinase B1 (LKB1) is usually described as a tumor suppressor in a wide variety of tissues, it has been shown that LKB1 aberrant expression is associated with bad prognosis in Hepatocellular Carcinoma (HCC).
Methods
Herein we have overexpressed LKB1 in human hepatoma cells and by using histidine pull-down assay we have investigated the role of the hypoxia-related post-translational modification of Small Ubiquitin-related Modifier (SUMO)ylation in the regulation of LKB1 oncogenic role. Molecular modelling between LKB1 and its interactors, involved in regulation of LKB1 nucleocytoplasmic shuttling and LKB1 activity, was performed. Finally, high affinity SUMO binding entities-based technology were used to validate our findings in a pre-clinical mouse model and in clinical HCC.
Findings
We found that in human hepatoma cells under hypoxic stress, LKB1 overexpression increases cell viability and aggressiveness in association with changes in LKB1 cellular localization. Moreover, by using site-directed mutagenesis, we have shown that LKB1 is SUMOylated by SUMO-2 at Lys178 hampering LKB1 nucleocytoplasmic shuttling and fueling hepatoma cell growth. Molecular modelling of SUMO modified LKB1 further confirmed steric impedance between SUMOylated LKB1 and the STe20-Related ADaptor cofactor (STRADα), involved in LKB1 export from the nucleus. Finally, we provide evidence that endogenous LKB1 is modified by SUMO in pre-clinical mouse models of HCC and clinical HCC, where LKB1 SUMOylation is higher in fast growing tumors.
Interpretation
Overall, SUMO-2 modification of LKB1 at Lys178 mediates LKB1 cellular localization and its oncogenic role in liver cancer.
Fund
This work was supported by grants from NIH (US Department of Health and Human services)-R01AR001576-11A1 (J.M.M and M.L.M-C.), Gobierno Vasco-Departamento de Salud 2013111114 (to M.L.M.-C), ELKARTEK 2016, Departamento de Industria del Gobierno Vasco (to M.L.M.-C), MINECO: SAF2017–87301-R and SAF2014–52097-R integrado en el Plan Estatal de Investigación Cientifica y Técnica y Innovación 2013–2016 cofinanciado con Fondos FEDER (to M.L.M.-C and J.M.M., respectively), BFU2015–71017/BMC MINECO/FEDER, EU (to A.D.Q. and I.D.M.), BIOEF (Basque Foundation for Innovation and Health Research): EITB Maratoia BIO15/CA/014; Instituto de Salud Carlos III:PIE14/00031, integrado en el Plan Estatal de Investigación Cientifica y Técnica y Innovacion 2013–2016 cofinanciado con Fondos FEDER (to M.L.M.-C and J.M.M), Asociación Española contra el Cáncer (T.C.D, P·F-T and M.L.M-C), Daniel Alagille award from EASL (to T.C.D), Fundación Científica de la Asociación Española Contra el Cancer (AECC Scientific Foundation) Rare Tumor Calls 2017 (to M.L.M and M.A), La Caixa Foundation Program (to M.L.M), Programma di Ricerca Regione-Università 2007–2009 and 2011–2012, Regione Emilia-Romagna (to E.V.), Ramón Areces Foundation and the Andalusian Government (BIO-198) (A.D.Q. and I.D.M.), ayudas para apoyar grupos de investigación del sistema Universitario Vasco IT971–16 (P.A.), MINECO:SAF2015–64352-R (P.A.), Institut National du Cancer, FRANCE, INCa grant PLBIO16–251 (M.S.R.), MINECO - BFU2016–76872-R to (E.B.). Work produced with the support of a 2017 Leonardo Grant for Researchers and Cultural Creators, BBVA Foundation (M.V-R). Finally, Ciberehd_ISCIII_MINECO is funded by the Instituto de Salud Carlos III. We thank MINECO for the Severo Ochoa Excellence Accreditation to CIC bioGUNE (SEV-2016-0644). Funding sources had no involvement in study design; in the collection, analysis, and interpretation of data; in the writing of the report; and in the decision to submit the paper for publication.
Keywords: LKB1, SUMO, HCC, SIRT1, STRADα
Highlights
-
•
Overexpression of LKB1 in human hepatoma cells during hypoxic stress induces deregulated cell growth and survival.
-
•
SUMO-2 modifications of LKB1 at Lys178 occur in human hepatoma cells hampering its nucleocytoplasmic shuttling.
-
•
LKB1 SUMOylation is augmented in pre-clinical mouse models and clinical HCC, being a hallmark of more aggressive HCC tumors.
Research in context.
Evidence before this study
LKB1 is a serine threonine kinase protein with an ambiguous role in cancer progression. LKB1 expression or activity is augmented in bad prognosis and late stage liver cancer. LKB1 cellular localization is relevant for its activity. LKB1 has been shown to be a target of SUMO post-translational modifications. SUMO-mediated modifications are known to play an important role in protein target subcellular localization during cancer progression.
Added value of this study
In this study, we show that LKB1 overexpression in human hepatoma cells during hypoxic stress promotes tumor cell growth and survival. Moreover, we provide evidence that endogenous LKB1 is modified by SUMO in pre-clinical mouse models of HCC as well as in liver biopsies of HCC patients. Finally, our data suggest that LKB1 SUMOylation is aberrant in liver cancer being essential for the regulation of its subcellular localization and oncogenic role.
Implications of all the available evidence
Our results support a potential role for LKB1 SUMOylation as a novel oncogenic mechanism in HCC and thereby the putative therapeutic potential of protein-based, peptidyl and small molecule inhibitors of various SUMO specific proteases isoforms.
Alt-text: Unlabelled Box
1. Introduction
Liver Kinase B1 (LKB1) is a 50 kDa serine/threonine kinase ubiquitously expressed in adult and fetal tissues, particularly in pancreas, liver, testes and skeletal muscle [1]. LKB1 is an upstream activator of 14 kinases from the ARK (AMP-activated protein kinase-related kinase) family including AMP-activated protein kinase (AMPK). In fact, the defective activation of AMPK in LKB1-null cells can be rescued by re-expression of LKB1 [2,3]. AMPK functions as a cellular energy sensor to provide metabolic adaptations under ATP-deprived conditions such as starvation and oxygen deprivation (hypoxia) [4]. Under these circumstances, AMPK activation results in stimulation of bioenergetic pathways and inhibition of ATP- and NADPH-consuming processes such as biosynthesis and proliferation. It is particularly relevant in this respect that AMPK inhibits the mammalian target of rapamycin complex 1 (mTORC1) pathway [5] with concomitant downregulation of the glycolytic pathway [6,7]. Indeed, silencing LKB1 in mouse embryonic fibroblasts promotes a metabolic switch to aerobic glycolysis, also called the Warburg effect, a hallmark of highly proliferative tumor cells, supported by mTOR activity and driven by hypoxia inducible factor (HIF)-1α [8]. Moreover, AMPK can regulate energy expenditure by modulating mitochondrial biogenesis and controlling the expression of oxidative enzymes [[9], [10], [11]]. Alternatively, LKB1-induced activation of AMPK signaling can offer a protective effect by allowing the cell time to attempt to reverse the aberrantly high ratio of AMP/ATP, that otherwise can cause cell death [12].
Germline mutations or deletions in the LKB1 gene are responsible for the Peutz-Jeghers Syndrome (PTS), a cancer-prone autosomal dominant inherited disorder [1,13]. Likewise, somatic mutations of LKB1 gene are involved in the development of sporadic cancers, such as cervical, prostate and lung cancers, among others [[14], [15], [16]]. Although genetic evidence supports the tumor-suppressive role for LKB1, other evidence revealed that LKB1 may also exhibit pro-oncogenic functions. In the context of liver disease, a controlled balance in hepatic LKB1 levels has been described as a gatekeeper of hepatocyte proliferation during regeneration [17,18]. Furthermore, LKB1 expression or activity have been previously shown to be augmented in Hepatocellular Carcinoma (HCC), the most common type of liver cancer, especially related to bad prognosis and late stage HCC [19,20]. The mechanisms underlying the oncogenic role of LKB1 in HCC remain rather unexplored.
LKB1 localization within the cell is critical for the regulation of its activity. LKB1 has a N-terminal regulatory domain in the most N-terminal region after the kinase domain and a C-terminal regulatory domain [21]. LKB1 has also two specific recognition sequences called nuclear localization sequences (NLS) in its N-terminal region [22], that allow the shuttling between cytoplasm and nucleus of LKB1 in mammalian cells. LKB1 nucleocytoplasmic shuttling is mediated by cofactors, such as the STe20-Related ADaptor (STRADα) and mouse protein 25 (MO25) [2]. Briefly, STRADα induces relocalization of LKB1 from the nucleus to the cytoplasm whereas MO25 stabilizes the STRADα-LKB1 interaction [23,24]. On the other hand, STRADα inhibits nuclear import of LKB1 by competing with karyopherin importin-α for binding to LKB1 [25]. To date, several regulating mechanisms have been proposed to explain LKB1 cellular localization: i) aberrant expression of STRADα was associated with nuclear LKB1 during corticogenesis [26]; ii) the orphan nuclear receptor Nur77 can bind and sequester LKB1 in the nucleus [27]; and finally iii) reversible post-translational modifications of LKB1 have been shown to regulate its stability and activity [19,28,29].
Post-translational modification is a critical event in the dynamic regulation of protein stability, location, structure, function, activity and interaction with other proteins. Recently, Ritho and colleagues have described for the first time LKB1 Small Ubiquitin-related MOdifier (SUMO)-mediated modifications and its implication under metabolic stress circumstances where SUMO-mediated modification of LKB1 is essential in promoting its interaction with its downstream target AMPK via a SUMO-interacting motif (SIM) essential for AMPK activation [29]. In addition, the SUMO protein modification has an extensive and critical role in the adaptive cellular response to hypoxia [[30], [31], [32], [33], [34]]. Chronic hypoxia is an important micro-environmental factor for establishing the aggressiveness of HCC by promoting tumor invasion and metastasis [35,36]. Thus, SUMOylation appears to be upregulated in many types of cancer, including HCC [[37], [38], [39]]. Previously, expression of both the SUMO E2 conjugating enzyme (Ubc9) and the E3 SUMO-protein ligase CBX4 were found to be upregulated and related to poor prognosis in HCC [38,39].
In this study, we aimed to explore the functional role of LKB1 in hepatocarcinogenesis and progression with special focus on the role played by SUMOylated LKB1 in the modulation of its cellular localization in hepatoma cells during hypoxic stress and in liver cancer.
2. Materials and methods
2.1. Cell lines
Huh-7, Hep G2, and PLC/PRF/5, human hepatoma cell lines; and MLP-29, mouse liver progenitor cells were used. All cells were grown at 37 °C in a humidified atmosphere of 5% CO2–95% air and cultured in Dulbecco's Modified Eagle Medium (DMEM) supplemented with 10% FBS (Gibco, Thermo Fisher Scientific, Spain), unless otherwise stated.
2.2. Cell transfection
Cell lines were transiently transfected using Lipofectamine 2000 (Invitrogen, USA), according to the manufacturer's instructions. Briefly, Lipofectamine 2000 (2.5 μl/1 μg DNA) was diluted in OPTI-MEM (Gibco) medium for transfections and incubated for 5 min. After incubation, the mixture was added to OPTI-MEM containing plasmid DNA (1 to 6 μg depending on the experiment), and incubated for at least 20 min at room temperature (RT) to allow for the formation of the DNA-Lipofectamine complexes. DNA-lipofectamine complexes previously formed were added to the culture plates containing the corresponding culture medium with 10% FBS but without antibiotics and with the cells in suspension. Plasmid DNA used are listed in Supplemental Table 1. Putative SUMO sequence binding sites on LKB1 were analyzed using the SUMOplot Analysis Program (http://www.abgent.com/SUMOplot, Abgent, USA). The 4 highest ranking sites predicted by SUMOplot were used to create the mutants (lysines 96, 97, 178 and 235). Lysines were mutated to arginines (R). An additional acetylation LKB1 mutant (K48R) was also created. The mutant LKB1 plasmid constructs were created using the QuickChange kit for directed mutagenesis (Stratagene, USA), according to the manufacturer's instructions, with two complementary oligonucleotides and with the pcDNA3-FLAG LKB1 plasmid as a template. The products were sequenced (STABvida, Portugal).
2.3. Cell treatments
The selective inhibitor of sirtuin 1 (SIRT1), Ex-527 (Sigma-Aldrich) in DMSO, was given to cells for 24 h at 30 μM. Cycloheximide, a eukaryote protein synthesis inhibitor was added to cells at 50 μg/ml in H2O.
2.4. Hypoxia in vitro
For hypoxia experiments, cells were incubated in an Invivo2 400 hypoxia Workstation (Baker Ruskinn, USA) at 1% O2 for until 72 h and lysed/fixed in the hypoxia chamber.
2.5. Serum deprivation in vitro
Cells were grown without FBS for a 24-hour period and compared to cells maintained on 10%FBS.
2.6. Mouse models
All animal experiments were performed according to the ARRIVE guidelines and carried out in accordance with the National Institutes of Health guide for the care and use of Laboratory animals (NIH Publications N0.8023, revised 1978) and the guidelines of European Research Council for animal care and use. Adult male C57BL/6 mice and mouse models that develop HCC at 8-months old, such as the glycine N-methyltransferase (Gnmt) deficient (Gnmt−/−) and wild type (Gnmt+/+) mice, were bred and housed in the animal unit of CIC bioGUNE, which is an AAALAC-accredited facility. Animals were housed under controlled temperature (22 °C) and humidity conditions in a 12 h light/dark cycle with ad libitum access to food and water.
2.7. In vivo hypoxia treatment
Three-month-old C57BL/6 mice males were exposed to systemic hypoxia in a sealed workstation (Baker-Ruskinn InvivO2 400, Cultek). The final 10% O2 environment was reached after a 2 h30 period in which oxygen levels were gradually reduced. Feeding and light cycles were kept uniform in the hypoxia workstation. Control mice were also exposed to the same chamber but under a 21% O2 environment. Mice were euthanized by cervical dislocation inside the chamber, and the dissected organs were directly fixed.
2.8. Human HCC samples
The work described has been carried out in accordance with the code of ethics of the World Medical Association (Declaration of Helsinki) for experiments involving humans after obtaining informed consent. Surgically resected liver specimens of 22 patients with HCC (10 Hepatitis C, 10 ASH and 2 NASH) were examined. The Basque Biobank (http://www.biobancovasco.org) provided the data and type of biospecimen. In addition, we have used frozen paired HCC tumor and surrounding tissue liver biopsies obtained during tumor resection (n = 6) as well as HCC liver biopsies from a set of samples previously classified as fast growing (n = 3) or slow growing (n = 3). The latter samples were included in a previously published larger study, which included two cohorts of patients with cirrhosis of any etiology on ultrasound surveillance (training set = 78 and validation set = 54). At first identification of HCC, they underwent two CT scans 6 weeks apart with no treatment in-between. Fast growing patients has a median tumor doubling time of 42 days while for the slow growing patient's median tumor doubling time was 97 days [40]. US-guided liver biopsies were initially obtained and used to generate a microarray (Agilent Whole Human Genome Oligo) according to the MIAME guidelines. Gene expression data is available at the Gene Expression Omnibus website (http://www.ncbi.nlm.nih.gov/geo) under the accession number: GSE54236.
2.9. Protein extraction and western blotting analysis
Protein extraction and Western blotting analysis was performed as previously described [41]. Primary antibodies and their optimal incubation conditions are detailed in Supplemental Table 2.
2.10. Crystal violet viability assay
Cell viability was assessed using crystal violet staining (Sigma-Aldrich).
2.11. Scratching wound-healing assay
Cells were seeded to confluence over 12 mm coverslip after an overnight transfection. A pipette tip (200 μl) was used to scratch a straight line through all the wells. Media was changed twice to remove dead and unattached cells. The wells were then placed in hypoxia during 72 h. Pictures of the scratch were taken using an Eclipse TS100 microscope (Nikon, Japan).
2.12. Subcellular proteome extraction kit
ProteoExtract® Subcellular Proteome Extraction Kit (S-PEK) from Merck (Spain) was used.
2.13. Immunocytofluorescence
Cells seeded on 12 mm coverslips were fixed in PBS 4% paraformaldehyde (Santa Cruz Biotechnology, USA). Coverslips were then blocked and permeabilized with PBS containing 0.1% BSA, 10% goat serum and 0·2% Triton X-100 for 30 min at RT. After blocking, the coverslips were washed in PBS and incubated overnight in a humid chamber with the primary antibody (FLAG 1:100) in PBS. Coverslips were washed in PBS and incubated during 1 h at RT in blocking solution with DAPI but no Triton X-100 with secondary antibody (dilution 1:200, Cy3 conjugated anti-mouse or FITC-conjugated anti-rabbit, Jackson ImmunoResearch laboratories, USA). Coverslips were mounted in Dako fluorescence mounting medium (Dako, Denmark). Five Images from each experimental condition were taken using an Axioimager D1 (Zeiss). Blind quantification of LKB1 positive nuclear cells versus total number of stained cells was performed manually by an immunohistochemistry technician.
2.14. Nickel-Histidine affinity purification using nickel-nitriolotriacetic acid (Ni2+-NTA) beads
Cells were transfected with the respective constructs and His6-SUMO as described. After treatments, His6-SUMOylated proteins were purified as previously described by using low density Ni2+-NTA-agarose beads (ABT, Spain) [19,42].
2.15. Protein immunoprecipitation assays
Total protein extracts were immunoprecipitated with 5 μg of IgG1 (BD Pharmigen), anti-LKB1 or anti-STRADα antibodies by using A/G PLUS-Agarose Beads (Santa Cruz).
2.16. SUMO binding entities (SUBEs)
SUBEs, earlier described and validated [43], were used as a capturing system for endogenous SUMOylated LKB1. Liver tissue (75 mg) from Gnmt−/− and Gnmt+/+ as well as from HCC patients were used. Briefly, livers were lysed in lysis buffer [50 mM Tris pH 8.5; 150 mMNaCl, 5 mM EDTA, 1% Igepal, supplemented with 1× protease inhibitor cocktail (Roche) and 50 μM of PR-619 (ubiquitin and ubiquitin-like isopeptidases inhibitor, LifeSensors)]. Lysates were centrifuged at 14000 ×g and the supernatant was incubated with 50 μl of GST-agarose beads containing 50 μg of SUBEs or GST and 1 mM DTT (Dithiothreitol) for 2 h, at 4 °C. Beads were then pulled down by centrifugation, 1000 ×g for 5 min, and 1/10 of the unbound fraction was saved for western blot analysis (flow through-FT). Washes were carried out using 30 column volumes of wash buffer (50 mM Tris pH 8.5; 50 mM NaCl, 5 mM EDTA and 1% Igepal). Elutions were performed in one column volume of 2 Laemmli Buffer. For Western blot analysis, samples were separated in NUPAGE 4–12% BT Gels, 1.5 mm, 15Well.
2.17. Modelling
Computational analysis of the various LKB1 species used the X-ray diffraction coordinates of the LKB1-STRADα-MO25α complex by Zequiraj et al. (2009; pdb 2WTK). Highly mobile regions of LKB1 were absent and, therefore, needed modelling. For this purpose, 250 structures were generated using Modeller 9v7 [44] and classified according to their Discrete Optimized Protein Energy (zDOPE) scores to select the model with its lowest value. Molecular Dynamics trajectories were computed with the AMBER 16 package [45], using the 14SB force field [46]. For the acetylated lysine residue, Papamokos' parameterizations [47] were used. Simulations run under periodic boundary conditions in orthorhombic boxes. Initially, the minimum distance between protein and cell faces was 10 Å. PME electrostatics were set with the Ewald summation cut-off at 9 Å. Sodium counter-ions neutralized the charges of the system. The structures were solvated with SPC water molecules [48]. Protein side-chains were energy-minimized (100 steepest descent and 1400 conjugate gradient steps) down to a RMS energy gradient of 0·01 kJ mol-1 Å-1. Afterwards, solvent was subjected to 1000 steps of steepest descent minimization followed by 500 ps NPT-MD computations using isotropic molecule position scaling and a pressure relaxation time of 2 ps at 298 K. Temperature was regulated with Berendsen's heat bath algorithm [49], with a coupling time constant equal to 0·5 ps. The density of the system reached a plateau after ca. 150 ps simulation. Then, for each protein, the whole system was energy minimized and submitted to NVT-MD at 298 K, using 2·0 fs integration time steps. Snapshots were saved every 100 ps. SHAKE algorithm (Ryckaert et al., 1977) was used to constrain bonds involving hydrogen atoms. Coordinate files were processed using CPPTRAJ [50]. Further processing was made in Origin 16 (Originlab) and graphic displays were built in UCSF Chimera [51].
2.18. Immunohistochemistry
Paraffin-embedded sections (5 μm thick) of formalin-fixed liver samples were initially deparafinized in xylene or xylene-substitute and rehydrated through graded alcohol solutions. Specific antibodies and experimental conditions used in immunohistochemistry can be found in supplemental Table 3. For the analysis, images were taken with an upright light microscope (Zeiss, Germany). The average sum of intensities and stained area percentage of each sample was calculated using FRIDA software (http://bui3.win.ad.jhu.edu/frida/, John Hopkins University).
2.19. Statistical analysis
Data is expressed as mean ± SEM (standard error of the mean). Statistical significance was estimated using the Mann-Whitney U test. A p value of <0·05 was considered significant.
3. Results
3.1. LKB1 offers survival and invasiveness advantage to human hepatoma cells during hypoxic stress
LKB1 is endogenously expressed in human hepatoma cells (Suppl. Fig. 1a). These results are in agreement with earlier evidence showing augmented expression of LKB1 in rodent hepatoma cells [[52], [53], [54]]. In order to further explore the functional role of LKB1 in HCC progression, we transiently overexpressed LKB1 in Huh-7 human hepatoma cells both after serum deprivation (0% FBS) or under hypoxic (1% O2) conditions in comparison with cells under normoxia (21% O2) and cultured with 10% FBS. LKB1 ectopic transient overexpression in Huh-7 hepatoma cells results in comparable levels of LKB1 after 24 h of hypoxic, serum deprivation or normoxic stimuli (Fig. 1a, Suppl. Fig. 1b).
Fig. 1.

Liver Kinase B1 (LKB1) offers survival and invasiveness advantage to human hepatoma cells during hypoxic stress. LKB1 overexpression was induced in human hepatoma Huh-7 cell line by using the pcDNA3-FLAG-LKB1 Wild Type plasmid (LKB1) and compared to control overnight transfection with the pcDNA™3.3-TOPO® plasmid (Ctrl), followed by 24 h treatment under control conditions of normoxia and complete media (21%oxygen, 10% serum), serum deprivation (SD) and hypoxia (1% oxygen, 10% serum). a. Representative Western blot of LKB1, its downstream target AMP-activated protein (AMPK) and phosphorylated AMPK at Thr172 and hypoxia inducible factor (HIF1α), a hypoxic marker, are shown. [β-actin was used as loading control]. Quantifications are shown in Suppl. Fig. 1b; b. Cell viability as detected by staining of attached cells with crystal violet dye; c. Time-course of cell viability and cell migration using a wound-healing scratch assay after LKB1 overexpression under hypoxia; d. Representative immunofluorescence staining for LKB1 (FLAG) in Huh-7 hepatoma cells and quantification of the percentage of LKB1 nuclear positive staining cells. Scale bar corresponds to 50 μm; and e. Western blot of LKB1 levels in cytoplasmic and nuclear fractions [Glyceraldehyde 3-phosphate (GAPDH) was used as loading control for cytoplasmic fractions and Histone H3 for nuclear fractions]. Quantifications are shown in Suppl. Fig. 1b. At least triplicates were used per experimental condition. Data is shown as mean ± SEM. *p < 0·05 and **p < 0·01 are indicated (Mann-Whitney U test). (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
LKB1 is an upstream activator of AMPK promoting the phosphorylation of AMPK Thr-172 in the activation loop of its α subunit in response to metabolic stress in order to inhibit biosynthesis and proliferation [2,3]. Herein, LKB1 overexpression increased phosphorylation of AMPK at Thr-172 except during hypoxic stress, a condition characterized by high HIF-1α levels (Fig. 1b, Suppl. Fig. 1b). Interestingly, after 24 h of hypoxia, LKB1 upregulation was associated with increased cell viability in comparison with control and serum deprived cells, both in Huh-7 (Fig. 1b) and another cell line of mouse liver progenitor cells, the MLP-29 cells (Suppl. Fig. 2a). A time course indicates that whereas hepatoma cells growth is hampered during hypoxia, LKB1 overexpression is able to induce cell growth under these conditions (Fig. 1c). Furthermore, hypoxia is known to unleash the invasive potential of tumor cells. Scratch wound-healing assay revealed that LKB1 overexpression also provides and invasiveness advantage to tumor cells under hypoxia (Fig. 1c).
Fig. 2.

Liver Kinase B1 (LKB1) is SUMOylated by SUMO-2 in human hepatoma cells. a. Ni2+-NTA agarose bead pulldown in Huh-7 human hepatoma cells after transfection with His-SUMO-1, 2, or 3, with the pcDNA3-FLAG-LKB1 Wild type plasmid (LKB1 WT) in the presence and absence of ubiquitin conjugating enzyme 9 (UBC9). b. Ni2+-NTA agarose bead pulldown in Huh-7 human hepatoma cells after transfection with His-SUMO-2 with the LKB1 WT plasmid in the presence of the different SUMO E3 ligases PIAS 1, 2α, 2β, and 4. c. Ni2+-NTA agarose bead pulldown in Huh-7 human hepatoma cells after transfection with His-SUMO-2 and with the LKB1 WT plasmid in the presence of the different SUMO-specific proteases, SENPs 1–7. d. Ni2+-NTA agarose bead pulldown in Huh-7 human hepatoma cells after transfection with His-SUMO-2 and with the LKB1 WT plasmid or the pcDNA3-FLAG-LKB1 Kinase Dead K78I plasmid. e. Immunoprecipitation assay between LKB1 and the STe20-Related ADaptor (STRADα) cofactor after STRADα overexpression in Huh-7 human hepatoma cells. Normalized quantifications relative to inputs are shown below the panel.
LKB1 cellular localization plays an important role its activity. During serum deprivation and normoxia conditions LKB1 actively shuttles between the nucleus and cytoplasm whereas under hypoxia, the LKB1 nucleocytoplasmic shuttling is hampered and LKB1 is more present in the nucleus (Fig. 1d, e, Suppl. Fig. 1c).
Overall, LKB1 overexpression provides growth survival and invasiveness advantage to hepatoma cells during hypoxic stress which agrees with previous evidence from our laboratory and others showing that LKB1 expression is induced in HCC tumors [19,20], tumors characterized by a highly hypoxic environment.
3.2. Increased LKB1 SUMOylation in human hepatoma cells
As previously mentioned, SUMOylation post-translational modifications are critical during hypoxia and thereby relevant in HCC [38,39]. In mammals, there are five SUMO paralogues, being SUMO-1, -2 and -3 more studied, compared to SUMO-4 and -5 [[55], [56], [57]]. Hepatic SUMO-1 protein levels are low, on the contrary to other tissues such as lung, uterus and prostate, whereas SUMO-2/3 levels in the liver are high [58]. On this regard, we have found that mice exposed to hypoxia show increased liver LKB1 nuclear expression and SUMO-2/3 levels (Suppl. Fig. 3a). Furthermore, we show that endogenous LKB1 SUMOylation by SUMO-2/3 is induced in Huh-7 hepatoma cells after 24 h of hypoxia in comparison with cells grown under normoxic conditions (Suppl. Fig. 3b). To further explore the role of SUMO-mediated modifications of LKB1 in hepatoma cells, Ni2+-NTA agarose bead-pulldowns were performed in Huh-7 human hepatoma cells after co-transfection of pcDNA3-FLAG-LKB1 wild type (WT) and pcDNA3-His6-SUMO1, pcDNA3-His6-SUMO2 or pcDNA3-His6-SUMO3 plasmids. Our results show that LKB1 is mostly modified by SUMO-2, in a SUMO-conjugating enzyme UBC9 dependent process, both in Huh-7 human hepatoma cells (Fig. 2a) and in MLP-29 cells (Suppl. Fig. 4a). Co-transfection with the E3 SUMO-protein ligases PIAS, especially PIAS 1, further increased LKB1 SUMOylation by SUMO-2 in Hu7–7 cells whereas co-transfection with SUMO-specific proteases, particularly SENP2, and SENP-1 and -3, decreased LKB1 SUMOylation (Fig. 2b,c). In agreement, SENP2 has been previously reported to play a critical role in the control of HCC cell growth [59]. The SUMOylation of LKB1 does not depend on LKB1 kinase activity domain as a kinase dead (KD) mutant, pcDNA3-FLAG-LKB1 K78I, was equally modified by SUMO-2 (Fig. 2d e). Finally, SUMO-2-mediated modification of LKB1 reduced the interaction between LKB1 and STRADα, a co-factor involved in LKB1 nuclear export, as shown by immunoprecipitation assay, during STRADα overexpression (Fig. 2e).
Fig. 3.

Liver Kinase B1 (LKB1) is SUMOylated by SUMO-2 at Lys178 in human hepatoma cells. a. Schematic representation of LKB1 showing the Nuclear (NLS) localization and the Kinase domain (KDN). SUMOylation LKB1 mutants used are also shown and described. b. Ni2+-NTA agarose bead pulldown in Huh-7 human hepatoma cells after transfection with His-SUMO-2 and the LKB1 SUMO mutants, LKB1 K96R, LKB1 K97R, LKB1 K178R and LKB1 K235R. Normalized quantifications relative to inputs are shown below each panel.
Fig. 4.

Liver Kinase B1 (LKB1) SUMOylation at Lys178 by SUMO-2 regulates human hepatoma cell survival by hampering LKB1 nucleocytoplasmic shuttling. LKB1 overexpression was induced in Huh-7 cells with pcDNA3-FLAG-LKB1 Wild Type plasmid (LKB1 WT) or the pcDNA3-FLAG-LKB1 K178R plasmid (LKB1 K178R) in the presence of His-SUMO-2. a. Western blot analysis of LKB1, [Glyceraldehyde 3-phosphate (GAPDH) was used as loading control]. Quantifications are shown in Suppl. Fig. 4a; b and c. Cell viability as detected by staining of attached cells with crystal violet dye and number of cells; d. Immunoprecipitation assay between LKB1 WT and LKB1 K178R and the STe20-Related ADaptor (STRADα) cofactor after STRADα overexpression in Huh-7 human hepatoma cells. Normalized quantifications relative to inputs are shown below the panel; e. Representative immunofluorescence staining for LKB1 (FLAG) in Huh-7 cells and quantification of the percentage of LKB1 nuclear positive staining cells. Scale bar corresponds to 50 μm. f. LKB1 levels in cytoplasmic (Cyto) and nuclear fractions (Nuc) [Glyceraldehyde 3-phosphate (GAPDH) was used as loading control for cytoplasmic fractions and Histone H3 for nuclear fractions]. Quantifications are shown in Suppl. Fig. 4b. At least triplicates were used per experimental condition. Data is shown as mean ± SEM. *p < 0·05 is indicated (Mann-Whitney U test). (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
3.3. LKB1 is modified by SUMO-2 at Lys178 in human hepatoma cells
By using the SUMOplot™ (http://www.abgent.com/sumoplot) and JASSA Analysis Programs [60] to predict and score the potential SUMOylation sites in LKB1, we found that LKB1 contains 3 high scoring sites for possible SUMO binding sites in its amino acid sequence (Fig. 3a). By using site-directed mutagenesis we created LKB1 arginine mutants of the 3 highest scoring SUMO binding sites. Transfection of human hepatoma Huh-7 (Fig. 3b) and MLP-29 (Suppl. Fig. 4b) cells with pcDNA3-His6-SUMO2 and the different mutants showed that LKB1 is mostly SUMOylated by SUMO-2 at Lys178. In spite of this, other physiological or pathological relevant LKB1 SUMO-binding lysines in LKB1 cannot be excluded. Even though the LKB1 SUMOylation mutant at lysine 178 has been previously shown to display catalytic activity [29], the potential role of SUMO-2 mediated modification at Lys178 of LKB1 on this protein nucleocytoplasmic shuttling in hepatoma cell has not been addressed.
3.4. LKB1 SUMOylation at Lys178 by SUMO-2 regulates human hepatoma cell survival by hampering LKB1 nucleocytoplasmic shuttling
As previously observed under hypoxic stress, transfection with the LKB1 wild type plasmid in the presence of SUMO-2, increased human hepatoma cell survival. On the other hand, transfection with the LKB1 SUMOylation mutant at Lys178 (pcDNA3-FLAG-LKB1 K178R) under the same conditions did not account for increased tumor cell viability (Fig. 4a–c, Suppl. Fig. 5a). Cycloheximide Chase Assay confirmed that protein stability between pcDNA3-FLAG-LKB1 wild type (WT) and pcDNA3-FLAG-LKB1 K178R proteins is similar (Suppl. Fig. 6a). In addition, LKB1 mediated SUMO-2 modifications promoted the loss of the ability of LKB1 to bind to STRADα, whereas pcDNA3-FLAG-LKB1 K178R shows higher affinity for STRADα (Fig. 4d). Finally, transfection of human hepatoma cells with the pcDNA3-FLAG-LKB1 WT in the presence of pcDNA3-His6-SUMO2 induced LKB1 nuclear retention contrary to what occurs with the SUMOylation mutant (Fig. 4e,f, Suppl. Fig. 5b). Transfection of human hepatoma cells with either the pcDNA3-FLAG-LKB1 WT plasmid and with the SUMOylation mutant LKB1 K178R, that is not able to interact with AMPK and phosphorylate it as previously shown [29], show reduced phosphorylation of AMPK (data not shown).
Fig. 5.

Liver Kinase B1 (LKB1) is modified by SUMO-2 in Lys178 after its acetylation at Lys48 in human hepatoma cells. a. Schematic representation of LKB1 showing the Nuclear (NLS) localization, acetylation domain (AD) and the Kinase domain (KDN). Acetylation LKB1 mutant used is shown. b. Ni2+-NTA agarose bead pulldown in Huh-7 human hepatoma cells after transfection with the pcDNA3-FLAG-LKB1 Wild type plasmid (LKB1 WT), His-SUMO-2 and treatment with sirtuin 1 (SIRT1). c. Ni2+-NTA agarose bead pulldown in Huh-7 human hepatoma cells after transfection the LKB1 WT, LKB1 acetylation mutant, LKB1 K48R or the LKB1 SUMOylation mutant, LKB1 K178R, with His-SUMO-2, in the presence and absence of the Ex-527, the SIRT1 inhibitor. Normalized quantifications relative to inputs are shown below each panel; d. Representative immunofluorescence staining for FLAG and quantifications in Huh-7 hepatoma cells after transfection with the LKB1 WT or the LKB1 SUMOylation mutant LKB1 K178R and SUMO-2 in the presence and absence of Ex-527. Scale bar corresponds to 50 μm. At least triplicates were used per experimental condition. Data is shown as mean ± SEM. *p < 0·05 and **p < 0·01 are indicated (Mann-Whitney U test).
Fig. 6.

Model of structural and dynamic changes on Liver Kinase B1 (LKB1) upon post-translational changes. a. Structure of LKB1 in the context of its ternary complex with STRADα and MO25 [80] (pdb code: 2WTJ). Residues of LKB1 closer within 6 Å of either STRADα or Mo25 are in yellow, those closer than 4 Å are in orange. MO25 interacts with LKB1 through the A-loop, while STRADα interacts with the β2-β3 loop and the CFTL region. b. Comparison of the structures along the molecular dynamics computations. The Root Mean Square Deviation of trajectory snapshots upon alignment to the initial, energy minimized structure. Upper: data corresponding to unmodified LKB1 is in dark cyan, and that of K178-SUMOylated LKB1 is in blue. Lower: the trajectory of K48-acetylated LKB1 is in ochre, and dark red dots represent that computed for the K48-acetylated and K178-sumoylated. c. Per-residue atomic fluctuations computed along the last 250 ns of the trajectories. Colour attributes in b apply also to this panel. d. Map of changes in atomic fluctuations between unmodified LKB1 and fully modified (ALY at position 48, SUMO at position 178) onto the model obtained by simulated annealing of the mobile parts of LKB1 using the 2WTJ coordinates. Residues in red are more mobile in the unmodified protein, those in blue are more mobile in the fully modified one. e. Overlay of the structures of unmodified (cyan) and fully modified (red; SUMO in yellow) LKB1. Ribbons represent the coordinates closest to averages of the analyzed trajectory intervals. f. Detail of the terminal part of the CFTL region of the acetylated and SUMOylated STK domain of LKB1, according to the structure closest to the average of the last 250 ns of MD computations. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
Overall, LKB1 SUMOylation at Lys178 by SUMO-2 regulates cell growth in human hepatoma cells by hampering LKB1 interaction with STRADα and thereby its nucleocytoplasmic shuttling.
3.5. LKB1 SUMO-2 modification at Lys178 is favored by its acetylation at Lys48 in human hepatoma cells
Crosstalk between SUMOylation and acetylation pathways is crucial for the regulation of protein activity. On this basis, LKB1 has been previously shown to be acetylated at Lys48 [61]. Importantly, we observed that SIRT1 overexpression, a NAD dependent sirtuin that deacetylates LKB1 at the key acetylation Lys48, as well as transfection with the LKB1 acetylation mutant, pcDNA3-FLAG-LKB1 K48R, both precluding LKB1 acetylation, reduced LKB1 SUMOylation by SUMO-2. Of notice, pcDNA3-FLAG-LKB1 K48R stability is similar to the WT plasmid (Suppl. Fig. 6b). On the other hand, treatment with Ex-527, a SIRT1 inhibitor [62] increasing LKB1 acetylation, significantly augmented LKB1 SUMOylation by SUMO-2 (Fig. 5a–c).
LKB1 acetylation at Lys48 has been previously shown to prevent LKB1 binding to STRADα hampering LKB1 nucleocytoplasmic export [61]. In spite of this, in our in vitro model of human hepatoma cells, treatment with Ex-527, which accounts for increased LKB1 acetylation and promoting LKB1 SUMOylation, further boosts LKB1 nuclear accumulation, whereas on the other hand, transfection with LKB1 K178R SUMOylation mutant, localizes LKB1 to the cytoplasm independent of the LKB1 acetylation status (Fig. 5d,e). These results suggest that in human hepatoma cells, LKB1 is mostly modified by SUMO-2 in Lys178 after its acetylation at Lys48, and that SUMOylation of acetylated LKB1 plays an important role in hampering LKB1 nucleocytoplasmic shuttling.
3.6. Linking structural dynamics of SUMOylated/acetylated LKB1 to biologic effects
Several regions of the LKB1 STK domain interact with either STRADα or MO25 (Fig. 6a). Assessing the effects of post-translational modifications on the affinity LKB1 towards their partners required to analyze their effect on the structure and in particular, the above regions. However, several loop and mobile regions in LKB1 were missing in the set of coordinates. Modelling them by simulated annealing provided a reasonable model from a total of 250 structures (zDOPE < −1). This model was used in four molecular dynamics (MD) computations. Two simulations aided to assess the behavior of the STK domain in the absence of acetylation. The first one corresponded to unmodified STK domain; the second, to the domain SUMOylated at Lys 178. Another pair of simulations were computed upon acetylation of Lys 48: with and without SUMOylation at Lys 178. Fig. 6b displays the time course of the root mean square deviations (RMSD) for the core of the STK domain along the trajectories, taking the energy-minimized structures as reference. None of the proteins showed substantial changes along the trajectories, being stable along the 300 ns computed. However, SUMOylated proteins displayed lower RMSD values, probably because the SUMO adduct damps the motions of nearby regions of the STK domain. The N- and C- termini, which resulted to be highly dynamic, were excluded from this analysis, for a better assessment of atomic fluctuations (Fig. 6c,d). The RMSF patterns indicate that both C- and N-terminus are highly flexible (Fig. 6c). In addition, several sequence stretches display enhanced fluctuations. Among them stand out the β2-β3 loop (residues 68–73), the A-loop (201–208), the EF-αF loop (226–230), the C-lobe and the adjacent α-helix (255–277), as well as the C-terminal Flanking Tail (CFTL, 314–347) and the C-terminus. Differences in mobility between the unmodified and the double modified (K48-acetylated and K178-SUMOylated) protein are further illustrated (Fig. 6d). Both, acetylation and SUMOylation restrain the mobility of most of these regions (Fig. 6c,d). Notably, acetylation of K48 induces a decrease in the mobility of the A-loop and the C-lobe, which lay far from this residue, similar to the effect of SUMO attachment. This suggests a concertation of the N-terminus motions with those in regions that will neighbor the SUMO moiety. The conformational entropy loss due to acetylation could favor SUMOylation, probably providing a pre-pay for the entropy loss intrinsic to the SUMOylated LKB1 species.
The overall structure of the STK domain of LKB1 barely changes along the four trajectories, according to the low RMSD values (Fig. 6e). However, substantial differences are observable at various loops and flexible regions. The most significant ones map at the A-loop, the αEF-αF loop (residues 226–233), the C-lobe and a region of the CFT spanning from residues 328 to 339, shown in Fig. 6f. Notably, this region comprises two targets for phosphorylation (S334 and T336), besides being involved in the interaction with STRADα. Additionally, this sequence stretch locates within an element (331RWRSMTVVPYL343) known to be a target of 14–3-3ζ [63].
In summary, both K48 acetylation and K178 SUMOylation affect both, the structure and the dynamics of several regions involved in binding STRADα, thereby providing a rationale for the biological effects observed related to LKB1 nucleocytoplasmic shuttling.
3.7. LKB1 is modified by SUMO-2 in mouse models of Hepatocellular Carcinoma
Mice deficient in Glycine-N-methyltransferase (Gnmt) spontaneously develop steatosis at 3 months of age, HCC early foci at 6 months old and multifocal HCC around 8 months of age [64]. Eight-month old Gnmt−/− mice show higher staining area for LKB1 (highly nuclear), SUMO-2/3 and the HIF-1α hypoxic marker that their wild-type littermates (Fig. 7a). In these animals, by using by using high affinity SUMO binding entities (SUBEs)-based technology [43], traps that can only recognize endogenous SUMOylated proteins and do not bind to any other member of the ubiquitin family we show that augmented polySUMOylated species of LKB1 are found in the Gnmt−/− liver tumors (Fig. 7b). Likewise, an immunoprecipitation assay revealed that LKB1 is mostly modified by SUMO-2 in the Gnmt−/− mice bearing liver tumors (Fig. 7c). In summary, these data highlight that LKB1 is modified by SUMO-2 in pre-clinical mouse models of HCC.
Fig. 7.

Liver Kinase B1 (LKB1) is modified by SUMO-2 in mouse models of Hepatocellular Carcinoma (HCC). a. Representative immunohistochemical analysis and quantification of LKB1, SUMO-2/3 and HIF1α staining. Scale bar corresponds to 50 μm; At least 5 animals per group were used; b. Western blot analysis and quantification of LKB1 by using SUMO binding entities (SUBEs) to capture endogenous SUMOylated LKB1; and c. Immunoprecipitation assay and quantification for SUMO-2 and LKB1 in tumor and non-tumor livers of wild type and Glycine-N-methyltransferase (Gnmt−/−) HCC mice. Three animals per group are shown. Data is shown as mean ± SEM. *p < 0·05 is indicated (Mann-Whitney U test).
3.8. LKB1 SUMOylation in clinical HCC
Herein, we have evaluated LKB1 and SUMO-2/3 levels in HCC biopsies. Our results show that LKB1 levels are induced as well as SUMO-2/3 expression in HCC in comparison to healthy controls (Fig. 8a). Also, by using SUBEs we show that LKB1 is more SUMOylated in the tumor region versus the surrounding tissue (Fig. 8b). We have moved far beyond and have analyzed the role of SUMOylation in HCC survival. To address this, we have used a previously published microarray study comparing the expression of genes both in aggressive and less aggressive tumors [40]. The majority of SUMOylation related genes, as well as hypoxia and hypoxia responsiveness genes are significantly augmented in patients showing worst survival, with more aggressive tumors (Fig. 8b). These results are in agreement with earlier evidence showing that the hypoxia-inducible factors HIFs are master regulators of angiogenesis [65] and neoagiogenesis-related genes are hallmarks of fast-growing HCC [40]. In agreement, analysis of the gene array retrieved that SUMO-2, PIAS-1 and SENP-3 are significantly augmented in more aggressive HCC, in agreement with our data in human hepatoma cells showing that LKB1 SUMOylation at Lys 178 is mostly regulated by PIAS-1 and SENP-3 activities. Also, in representative paraffin sections of these two groups of patients, we have observed that LKB1 is highly localized in the nucleus of more aggressive tumors (Fig. 8c). Finally, by using SUBEs we show that the levels of SUMOylated LKB1 are induced in more aggressive HCC tumors (Fig. 8d).
Fig. 8.

LKB1 SUMOylation in clinical HCC is associated with aggressiveness of the tumor. a. Representative immunohistochemical analysis and quantification of LKB1 and SUMO-2/3 staining in clinical HCC patients (n = 22) and healthy controls (n = 5). Scale bar corresponds to 50 μm. b. Western blot analysis and quantification of LKB1 by using SUMO binding entities (SUBEs) to capture endogenous SUMOylated LKB1 in liver biopsies of HCC patients comparing the tumor with the surrounding tissue. 5 paired samples were used. Surrounding tissue (ST) and tumor (T). c. Volcano plot of gene expression of the main genes involved in SUMO pathway, hypoxia and hypoxia responsiveness genes retrieved from a previous published microarray where HCC patients were separated into more aggressive and less aggressive HCC, according to the tumor doubling time and survival [40]; d. Representative LKB1 staining in more aggressive and less aggressive HCC tumors. Scale bar corresponds to 50 μm. e. Western blot analysis and quantification of LKB1 by using SUBEs to capture endogenous SUMOylated LKB1 in liver biopsies of HCC patients classified as more aggressive or less aggressive tumors (3 samples each were used for less aggressive and more aggressive tumors). Data is shown as mean ± SEM. *p < 0·05 is indicated (Mann-Whitney U test).
In summary, these data highlighting that LKB1 is modified by SUMO-2 in clinical HCC tumors further supports a potential role for LKB1 SUMOylation as a novel oncogenic mechanism in a subtype of HCC patients.
4. Discussion
LKB1 is usually considered a tumor suppressor in a wide variety of tissues once the hereditary and somatic loss of function mutations of this gene are associated with an increased risk of cancer development [1,[13], [14], [15], [16]]. However, in HCC, one of the most frequent malignant tumors and an important cause of cancer death, only sporadically genetic alterations of the LKB1 gene together with one LKB1 missense mutation and allelic loss were identified [66]. In fact, previous findings from our group and others have shown that LKB1 levels are high in HCC, especially in association with bad prognosis, and showing increased activation of LKB1 at late stages of the disease [19,20,52]. In agreement, in xenograft orthotopic tumors in severe combined immunodeficiency (SCID) mice, LKB1 suppression decreased tumor growth [52]. The role for LKB1 as an oncogenic driver in HCC hepatocarcinogenesis and progression is therefore controversial on the light of current literature.
LKB1 is an upstream activator of AMPK. Indeed, lack of LKB1 is usually associated with inactivation of AMPK and deregulated cell growth [67,68] whereas activation of LKB1 inhibits the proliferation of many different tumor cell types [69]. Most of the current analysis of LKB1 function has focused on its regulation of AMPK and mTOR signaling. Failure of AMPK phosphorylation by LKB1-deficient cells exhibit hyperactivated mTORC1 and elevated HIF signaling which in turn stimulates aerobic glycolysis and lowers oxidative phosphorylation dependence inducing deregulated cell proliferation [7]. In alternative, suppression of LKB1 leads to apoptosis in cell lines where Akt is constitutively active suggesting that LKB1 is necessary for Akt-mediated phosphorylation of proapoptotic proteins [70]. Regarding HCC, we have previously shown that LKB1 is essential for liver tumor pathogenesis in the methionine adenosyltransferase A1 (Mat1A−/−) HCC mice by regulating Akt-mediated survival independent of PI3K, AMPK and mTORC2 [53]. In here, we have moved far beyond and showed that LKB1 overexpression in human hepatoma cells is associated with increased cell viability and invasiveness during hypoxic stress, an important signature of more aggressive HCC [38,39]. Whether under hypoxia LKB1 offers growth advantage to hepatoma cells or accounts for increased survival needs to be further investigated.
The activity of LKB1 is tightly controlled by its cellular localization, a process that relies on the binding of LKB1 to STRADα and stabilization by MO25 [23,24]. We have observed that LKB1 overexpression in hepatoma cells and concomitant increased cell viability during hypoxia is associated with failure of LKB1 binding to STRADα and its retention in the nuclear region. Post-translational modifications of LKB1 play an important role on the assembly of the LKB1/STRADα complex and therefore may regulate LKB1 nucleocytoplasmic shuttling. For example, the phosphorylation of LKB1 at serine 428 was previously shown to induce the nuclear export of LKB1 and activation of AMPK in endothelial cells [71,72]. On the other hand, in melanoma cells transformed by mutations of oncogenic B-Raf kinase, LKB1 becomes phosphorylated at two sites that compromise the ability of this enzyme to bind an phosphorylate AMPK [73]. LKB1 has also been shown to be a neddylation target in HCC, a post-translational modification that increases LKB1 stability through the covalent binding of the ubiquitin-like molecule Nedd8 [19]. Loss of LKB1 farnesylation in mice results in decreased AMPK activation by promoting the interaction and co-localization of these two enzymes at the plasma membrane [28]. In alternative, both LKB1 acetylation [61] and the K63-linked LKB1 polyubiquitination by Skp2-SCF ubiquitin ligase [20] have been shown to be critical for LKB1 activation by maintaining the LKB1-STRADα-MO25 complex. More recently, Ritho and colleagues have described that energy stress triggers an increase in the modification of LKB1 by SUMO-1 at lysine 178, being this modification essential in promoting LKB1-AMPK interaction whereas absence of SUMOylation is associated with defective AMPK signaling inducing apoptosis in starved cells [29].
In this study, we have shown for the first time that SUMO-2 modifications of LKB1 at Lys178 occur in human hepatoma cells during hypoxia hindering the LKB1 nucleocytoplasmic shuttling by hampering the LKB1/STRADα binding. These set of evidence were further supported by molecular modelling data. In hepatoma cells, SUMO-1 mediated modifications of LKB1 were not detected, being SUMO-2 mediated modifications of LKB1 prevalent. This may be related to the fact that SUMO-1 levels are low in liver whereas SUMO-2/3 levels are high, on the contrary to other tissues such as lung and prostate [58]. Modifications of LKB1 by different paralogues of SUMO may play different roles as for example SUMO-1 acts as a chain terminator on SUMO-2/3 polymers [74] whereas polymeric SUMO chain formation is particularly relevant in the regulation of the subcellular localization of its substrate proteins [75]. Importantly, we show that LKB1 SUMOylation was augmented both in pre-clinical mouse models of HCC as well as in more aggressive HCC clinical tumors.
Herein we also describe that LKB1 acetylation at Lys48 is essential for its posterior SUMOylation by SUMO-2 at Lys178 in hepatoma cells. However, we provide evidence that even under circumstances where LKB1 is acetylated, as after the treatment with Ex-527, the SIRT1 inhibitor, failure of LKB1 SUMOylation at Lys178 promotes LKB1 cytoplasmic localization. These results suggest that LKB1 SUMOylation and not its acetylation is the main player in the regulation of LKB1 cellular shuttling in human hepatoma cells.
In the last years, LKB1 has been shown to function both in the cytoplasm and nucleus. The presence of LKB1 in the nucleus during liver cancer described in here can explain how LKB1 fails to activate cytoplasmic AMPK and induce tumor cell growth and invasiveness potential in a tumor hypoxic environment. Likewise, cancer invasion is impeded when STRADα is depleted and enhanced when STRADα is overexpressed [76]. Thus, the observed increased invasiveness as a result of LKB1 overexpression in hepatoma cells can also be a direct result of lack of association between STRADα and LKB1. Finally, the nuclear role for LKB1 during hypoxia in hepatoma cells and liver tumors is not fully understood and appears to be crucial. Although LKB1 was previously shown to bind and stabilize p53 in the nucleus, arresting cell cycle and promoting apoptosis in some types of cells [77], p53 is not required for induction of apoptosis in mouse hepatoma cells [78]. Alternatively, we hypothesize that when present in the nucleus, LKB1 is able to interact, through its SUMO-2 polymeric chains, with other proteins containing SUMO-interaction motifs and DNA binding domains. On this basis, we previously reported that LKB1 is able to activate the oncogenic pathway of RAS through the expression of RAS guanyl releasing protein-3 (RASGRP3) [52]. Interestingly, RasGRP3 was shown to be localized in the cytoplasm of the cells whereas the active form exhibited mostly nuclear immunoreactivity [79]. Based on our premises, nuclear LKB1 could attach to the promoter region of RasGRP3 as well as to other intermediates of oncogenic pathways increasing its expression and related Ras oncogenic activity, at least under hypoxic conditions. Chronic Ras activation can alternatively promote the Skp-2-mediated joint polyubiquitination of Lys48 and other lysines, and regulate LKB1 activity by maintaining LKB1-STRADα-MO25 complex integrity thereby increasing the activation of LKB1 observed at late HCC [20]. Further studies are necessary in order to identify possible binding nuclear LKB1 interactors present in hypoxic liver tumors.
In summary, we propose that LKB1 SUMOylation by SUMO-2 offers an alternative mechanism for hampering the binding of LKB1 to STRADα, possibly due to some type of steric hindrance. LKB1 SUMOylation promotes the nuclear sequestering of LKB1 and concomitant hepatoma cell growth and invasiveness advantage in hypoxic HCC tumors (Fig. 9). On this basis, we can hypothesize that first of all SUMO-2-mediated LKB1 modifications may account for the rather unique role of LKB1 as an oncogenic driver in liver cancer and secondly speculate about the valuable therapeutic potential for some subtypes of HCC of protein-based, peptidyl and small molecule inhibitors of various SUMO specific proteases isoforms.
Fig. 9.

SUMOylation regulates Liver Kinase B1 (LKB1) nucleocytoplasmic shuttling and its oncogenic potential in liver cancer. LKB1 acetylation at Lys48 and posterior SUMOylation at Lys178 by SUMO-2 account for the nuclear retention of LKB1 in liver cancer by hampering the binding of LKB1 to STRADα.
The following are the supplementary data related to this article.
Supplemental Fig. 1.

Liver Kinase B1 (LKB1) expression in human hepatoma cells a. Western blot analysis of LKB1 in several human hepatoma cells, Huh-7, Hep G2 and PLC/PRF/5. [α-Tubulin was used as loading control]. b. and c. Quantification of the Western blot shown in Fig. 1a and e, respectively. [Glyceraldehyde 3-phosphate (GAPDH), and β-actin were used as loading control for total and cytoplasmic fractions and Histone H3 for nuclear fractions]. At least triplicates were used per experimental condition. Data is shown as mean ± SEM. *p < 0·05 and **p < 0·01 versus Ctrl are indicated (Mann-Whitney U test).
Supplemental Fig. 2.

Liver Kinase B1 (LKB1) SUMOylation of mouse liver progenitor MLP-29 cells offers survival advantage during hypoxic stress. a. LKB1 overexpression was induced in human MLP-29 cell line by using the pcDNA3-FLAG-LKB1 Wild Type plasmid (LKB1) and compared to control overnight transfection with the pcDNA™3.3-TOPO® plasmid (Ctrl), followed by 24 h treatment under control conditions of normoxia and complete media (21%oxygen, 10% serum), serum deprivation and hypoxia (1% oxygen, 10% serum). Quantification of cell viability assessed by Crystal violet assay are shown. At least triplicates were used per experimental condition. Data is shown as mean ± SEM. **p < 0·01 is indicated (Mann-Whitney U test).
Supplemental Fig. 3.

LKB1 and SUMO in hypoxia. a. Representative immunohistochemical analysis and quantification of LKB1 and SUMO-2/3 staining in liver biopsies of mice after 2 h30 hypoxia (10% O2 environment) and control mice (21% O2 environment). Scale bar corresponds to 50 μm. Data is shown as mean ± SEM. *p < 0·05 is indicated (Mann-Whitney U test). b. Immunoprecipitation assay between LKB1 and SUMO-2/3 in Huh-7 human hepatoma cells during normoxia (21%oxygen, 10% serum) and hypoxia (1% oxygen, 10% serum) conditions for 24 h.
Supplemental Fig. 4.

Liver Kinase B1 (LKB1) is SUMOylated by SUMO-2 at Lys178 in mouse liver progenitor MLP-29 cells. a. Ni2+-NTA agarose bead pulldown in MLP-29 cells after transfection with His-SUMO-1, 2, or 3, with the pcDNA3-FLAG-LKB1 Wild type plasmid (LKB1 WT) in the presence and absence of ubiquitin conjugating enzyme 9 (UBC9). b. Ni2+-NTA agarose bead pulldown in Huh-7 human hepatoma cells after transfection with His-SUMO-2 and the LKB1 SUMO mutants, LKB1 K96R, LKB1 K97R, LKB1 K178R and LKB1 K235R. Normalized quantifications relative to inputs are shown below each panel.
Supplemental Fig. 5.
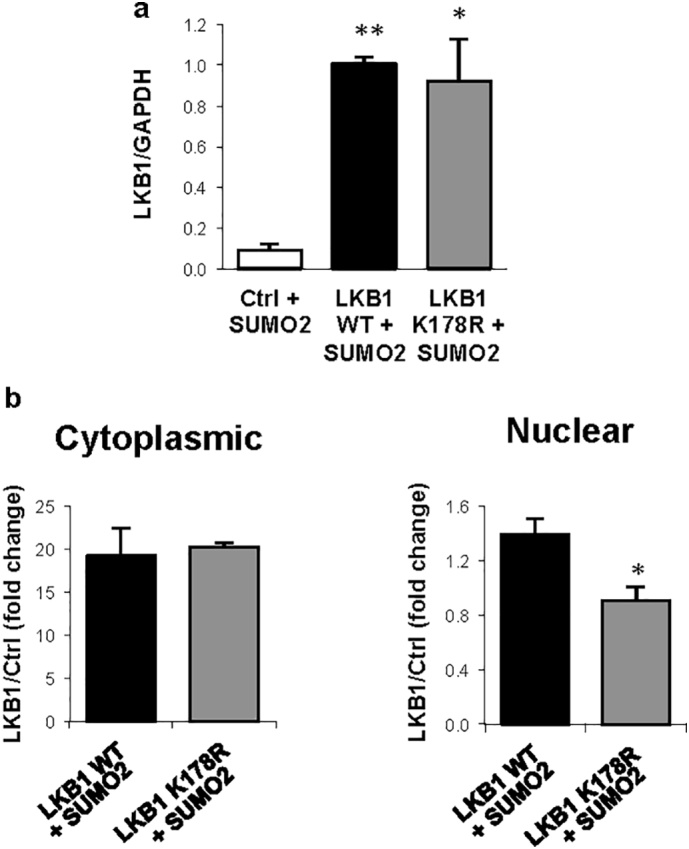
Quantifications of the Western blots shown in a. Fig. 4a and b. Fig. 4e. At least triplicates were used per experimental condition. Data is shown as mean ± SEM. *p < 0·05 and **p < 0·01 are indicated (Mann-Whitney U test).
Supplemental Fig. 6.

Cycloheximide Chase Assay. a. Stability of pcDNA3-FLAG-LKB1 wild type (WT) and b. pcDNA3-FLAG-LKB1 K178R or c. pcDNA3-FLAG-LKB1 K48R proteins as percentage of protein levels remaining after treatment with cycloheximide (CHX). Densitometric analysis and quantifications was obtained from Western blot data. LKB1 WT and mutant proteins at different time points between 0 and 6 h after CHX treatment. Vertical bars are indicative of the calculated half-life for each protein. At least triplicates were used per experimental condition. Data is shown as mean ± SEM.
List of commercial and non-commercial DNA plasmids used.
List of comercial antibodies used for Western blot and Immunoprecipitation and pull-down assays.
List of antibodies used for immunohistochemistry and immunofluorescence.
Gene expression of genes related to SUMO pathway, hypoxia and hypoxia responsiveness genes in HCC tumors classified as aggressive or less aggressive [1]. Whole gene expression data is available at the Gene Expression Omnibus website (http://www.ncbi.nlm.nih.gov/geo) under the accession number: GSE54236.
Acknowledgments
Acknowledgements
The authors would like to acknowledge Drs. Arkaitz Carracedo from CIC bioGUNE and Dr. Rodriguez-Nieto from IDIBELL for providing plasmids and Dr. Rosa Bario and Dr. James Sutherland from CIC bioGUNE for consultation and guidance about SUMOylation experiments. We also thank Laurie Bonnafé for her technical support in the production of SUBEs. Finally, the authors would like to acknowledge all the patients and healthcare personnel and the Basque Biobank.
Declaration of interests
Dr. M. Mato reports other from ABBOTT, other from OWL, other from GALMED, during the conduct of the study; Dr. Martínez-Chantar reports grants from Mitotherapeutix LLC, outside the submitted work.
Author contributions
Conceptualization: TCD MLMC FLO.
Funding acquisition: MVR EV JMM IDM ADQ TCD MLMC.
Investigation: IZF JLGR FLO MS JS PFT LBT DFR VGJ SLD OC AB JA EV DC CM EB PA NB MVR MA MSR IDM ADQ TCD.
Supervision: TCD MLMC.
Writing – original draft: TCD MLMC.
Writing – review & editing: IDM ADQ TCD MLMC.
Contributor Information
Teresa C. Delgado, Email: tcardoso@cicbiogune.es.
María L. Martínez-Chantar, Email: mlmartinez@cicbiogune.es.
References
- 1.Hemminki A., Markie D., Tomlinson I., Avizienyte E., Roth S., Loukola A. A serine/threonine kinase gene defective in Peutz-Jeghers syndrome. Nature. 1998;391(6663):184–187. doi: 10.1038/34432. [DOI] [PubMed] [Google Scholar]
- 2.Hawley S.A., Boudeau J., Reid J.L., Mustard K.J., Udd L., Makela T.P. Complexes between the LKB1 tumor suppressor, STRAD alpha/beta and MO25 alpha/beta are upstream kinases in the AMP-activated protein kinase cascade. J Biol. 2003;2(4):28. doi: 10.1186/1475-4924-2-28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Woods A., Johnstone S.R., Dickerson K., Leiper F.C., Fryer L.G., Neumann D. LKB1 is the upstream kinase in the AMP-activated protein kinase cascade. Curr Biol. 2003;13(22):2004–2008. doi: 10.1016/j.cub.2003.10.031. [DOI] [PubMed] [Google Scholar]
- 4.Lee M., Hwang J.T., Lee H.J., Jung S.N., Kang I., Chi S.G. AMP-activated protein kinase activity is critical for hypoxia-inducible factor-1 transcriptional activity and its target gene expression under hypoxic conditions in DU145 cells. J Biol Chem. 2003;278(41):39653–39661. doi: 10.1074/jbc.M306104200. [DOI] [PubMed] [Google Scholar]
- 5.Bolster D.R., Crozier S.J., Kimball S.R., Jefferson L.S. AMP-activated protein kinase suppresses protein synthesis in rat skeletal muscle through down-regulated mammalian target of rapamycin (mTOR) signaling. J Biol Chem. 2002;277(27):23977–23980. doi: 10.1074/jbc.C200171200. [DOI] [PubMed] [Google Scholar]
- 6.Kishton R.J., Barnes C.E., Nichols A.G., Cohen S., Gerriets V.A., Siska P.J. AMPK is essential to balance glycolysis and mitochondrial metabolism to control T-ALL cell stress and survival. Cell Metab. 2016;23(4):649–662. doi: 10.1016/j.cmet.2016.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Shackelford D.B., Vasquez D.S., Corbeil J., Wu S., Leblanc M., Wu C.L. mTOR and HIF-1alpha-mediated tumor metabolism in an LKB1 mouse model of Peutz-Jeghers syndrome. Proc Natl Acad Sci U S A. 2009;106(27):11137–11142. doi: 10.1073/pnas.0900465106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Faubert B., Vincent E.E., Griss T., Samborska B., Izreig S., Svensson R.U. Loss of the tumor suppressor LKB1 promotes metabolic reprogramming of cancer cells via HIF-1alpha. Proc Natl Acad Sci U S A. 2014;111(7):2554–2559. doi: 10.1073/pnas.1312570111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Canto C., Auwerx J. PGC-1alpha, SIRT1 and AMPK, an energy sensing network that controls energy expenditure. Curr Opin Lipidol. 2009;20(2):98–105. doi: 10.1097/MOL.0b013e328328d0a4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Canto C., Gerhart-Hines Z., Feige J.N., Lagouge M., Noriega L., Milne J.C. AMPK regulates energy expenditure by modulating NAD+ metabolism and SIRT1 activity. Nature. 2009;458(7241):1056–1060. doi: 10.1038/nature07813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Winder W.W., Holmes B.F., Rubink D.S., Jensen E.B., Chen M., Holloszy J.O. Activation of AMP-activated protein kinase increases mitochondrial enzymes in skeletal muscle. J Appl Physiol. 1985;2000;88(6):2219–2226. doi: 10.1152/jappl.2000.88.6.2219. [DOI] [PubMed] [Google Scholar]
- 12.Shaw R.J., Kosmatka M., Bardeesy N., Hurley R.L., Witters L.A., Depinho R.A. The tumor suppressor LKB1 kinase directly activates AMP-activated kinase and regulates apoptosis in response to energy stress. Proc Natl Acad Sci U S A. 2004;101(10):3329–3335. doi: 10.1073/pnas.0308061100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jenne D.E., Reimann H., Nezu J., Friedel W., Loff S., Jeschke R. Peutz-Jeghers syndrome is caused by mutations in a novel serine threonine kinase. Nat Genet. 1998;18(1):38–43. doi: 10.1038/ng0198-38. [DOI] [PubMed] [Google Scholar]
- 14.Sanchez-Cespedes M., Parrella P., Esteller M., Nomoto S., Trink B., Engles J.M. Inactivation of LKB1/STK11 is a common event in adenocarcinomas of the lung. Cancer Res. 2002;62(13):3659–3662. [PubMed] [Google Scholar]
- 15.McCabe M.T., Powell D.R., Zhou W., Vertino P.M. Homozygous deletion of the STK11/LKB1 locus and the generation of novel fusion transcripts in cervical cancer cells. Cancer Genet Cytogenet. 2010;197(2):130–141. doi: 10.1016/j.cancergencyto.2009.11.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pearson H.B., McCarthy A., Collins C.M., Ashworth A., Clarke A.R. Lkb1 deficiency causes prostate neoplasia in the mouse. Cancer Res. 2008;68(7):2223–2232. doi: 10.1158/0008-5472.CAN-07-5169. [DOI] [PubMed] [Google Scholar]
- 17.Vazquez-Chantada M., Ariz U., Varela-Rey M., Embade N., Martinez-Lopez N., Fernandez-Ramos D. Evidence for LKB1/AMP-activated protein kinase/endothelial nitric oxide synthase cascade regulated by hepatocyte growth factor, S-adenosylmethionine, and nitric oxide in hepatocyte proliferation. Hepatology. 2009;49(2):608–617. doi: 10.1002/hep.22660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Maillet V., Boussetta N., Leclerc J., Fauveau V., Foretz M., Viollet B. LKB1 as a gatekeeper of hepatocyte proliferation and genomic integrity during liver regeneration. Cell Rep. 2018;22(8):1994–2005. doi: 10.1016/j.celrep.2018.01.086. [DOI] [PubMed] [Google Scholar]
- 19.Barbier-Torres L., Delgado T.C., Garcia-Rodriguez J.L., Zubiete-Franco I., Fernandez-Ramos D., Buque X. Stabilization of LKB1 and Akt by neddylation regulates energy metabolism in liver cancer. Oncotarget. 2015;6(4):2509–2523. doi: 10.18632/oncotarget.3191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lee S.W., Li C.F., Jin G., Cai Z., Han F., Chan C.H. Skp2-dependent ubiquitination and activation of LKB1 is essential for cancer cell survival under energy stress. Mol Cell. 2015;57(6):1022–1033. doi: 10.1016/j.molcel.2015.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nakano A., Takashima S. LKB1 and AMP-activated protein kinase: regulators of cell polarity. Genes Cells. 2012;17(9):737–747. doi: 10.1111/j.1365-2443.2012.01629.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sapkota G.P., Boudeau J., Deak M., Kieloch A., Morrice N., Alessi D.R. Identification and characterization of four novel phosphorylation sites (Ser31, Ser325, Thr336 and Thr366) on LKB1/STK11, the protein kinase mutated in Peutz-Jeghers cancer syndrome. Biochem J. 2002;362(Pt 2):481–490. doi: 10.1042/0264-6021:3620481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Baas A.F., Boudeau J., Sapkota G.P., Smit L., Medema R., Morrice N.A. Activation of the tumour suppressor kinase LKB1 by the STE20-like pseudokinase STRAD. EMBO J. 2003;22(12):3062–3072. doi: 10.1093/emboj/cdg292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Boudeau J., Baas A.F., Deak M., Morrice N.A., Kieloch A., Schutkowski M. MO25alpha/beta interact with STRADalpha/beta enhancing their ability to bind, activate and localize LKB1 in the cytoplasm. EMBO J. 2003;22(19):5102–5114. doi: 10.1093/emboj/cdg490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dorfman J., Macara I.G. STRADalpha regulates LKB1 localization by blocking access to importin-alpha, and by association with Crm1 and exportin-7. Mol Biol Cell. 2008;19(4):1614–1626. doi: 10.1091/mbc.E07-05-0454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Orlova K.A., Parker W.E., Heuer G.G., Tsai V., Yoon J., Baybis M. STRADalpha deficiency results in aberrant mTORC1 signaling during corticogenesis in humans and mice. J Clin Invest. 2010;120(5):1591–1602. doi: 10.1172/JCI41592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhan Y.Y., Chen Y., Zhang Q., Zhuang J.J., Tian M., Chen H.Z. The orphan nuclear receptor Nur77 regulates LKB1 localization and activates AMPK. Nat Chem Biol. 2012;8(11):897–904. doi: 10.1038/nchembio.1069. [DOI] [PubMed] [Google Scholar]
- 28.Houde V.P., Ritorto M.S., Gourlay R., Varghese J., Davies P., Shpiro N. Investigation of LKB1 Ser431 phosphorylation and Cys433 farnesylation using mouse knockin analysis reveals an unexpected role of prenylation in regulating AMPK activity. Biochem J. 2014;458(1):41–56. doi: 10.1042/BJ20131324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ritho J., Arold S.T., Yeh E.T. A critical SUMO1 modification of LKB1 regulates AMPK activity during energy stress. Cell Rep. 2015;12(5):734–742. doi: 10.1016/j.celrep.2015.07.002. [DOI] [PubMed] [Google Scholar]
- 30.Cai Q., Verma S.C., Kumar P., Ma M., Robertson E.S. Hypoxia inactivates the VHL tumor suppressor through PIASy-mediated SUMO modification. PLoS One. 2010;5(3) doi: 10.1371/journal.pone.0009720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sun L., Li H., Chen J., Iwasaki Y., Kubota T., Matsuoka M. PIASy mediates hypoxia-induced SIRT1 transcriptional repression and epithelial-to-mesenchymal transition in ovarian cancer cells. J Cell Sci. 2013;126(Pt 17):3939–3947. doi: 10.1242/jcs.127381. [DOI] [PubMed] [Google Scholar]
- 32.Carbia-Nagashima A., Gerez J., Perez-Castro C., Paez-Pereda M., Silberstein S., Stalla G.K. RSUME, a small RWD-containing protein, enhances SUMO conjugation and stabilizes HIF-1alpha during hypoxia. Cell. 2007;131(2):309–323. doi: 10.1016/j.cell.2007.07.044. [DOI] [PubMed] [Google Scholar]
- 33.Xu Y., Zuo Y., Zhang H., Kang X., Yue F., Yi Z. Induction of SENP1 in endothelial cells contributes to hypoxia-driven VEGF expression and angiogenesis. J Biol Chem. 2010;285(47):36682–36688. doi: 10.1074/jbc.M110.164236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Nunez-O'Mara A., Berra E. Deciphering the emerging role of SUMO conjugation in the hypoxia-signaling cascade. Biol Chem. 2013;394(4):459–469. doi: 10.1515/hsz-2012-0319. [DOI] [PubMed] [Google Scholar]
- 35.Wu X.Z., Xie G.R., Chen D. Hypoxia and hepatocellular carcinoma: the therapeutic target for hepatocellular carcinoma. J Gastroenterol Hepatol. 2007;22(8):1178–1182. doi: 10.1111/j.1440-1746.2007.04997.x. [DOI] [PubMed] [Google Scholar]
- 36.Lin D., Wu J. Hypoxia inducible factor in hepatocellular carcinoma: a therapeutic target. World J Gastroenterol. 2015;21(42):12171–12178. doi: 10.3748/wjg.v21.i42.12171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Seeler J.S., Dejean A. SUMO and the robustness of cancer. Nat Rev Cancer. 2017;17(3):184–197. doi: 10.1038/nrc.2016.143. [DOI] [PubMed] [Google Scholar]
- 38.Tomasi M.L., Tomasi I., Ramani K., Pascale R.M., Xu J., Giordano P. S-adenosyl methionine regulates ubiquitin-conjugating enzyme 9 protein expression and sumoylation in murine liver and human cancers. Hepatology. 2012;56(3):982–993. doi: 10.1002/hep.25701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Li J., Xu Y., Long X.D., Wang W., Jiao H.K., Mei Z. Cbx4 governs HIF-1alpha to potentiate angiogenesis of hepatocellular carcinoma by its SUMO E3 ligase activity. Cancer Cell. 2014;25(1):118–131. doi: 10.1016/j.ccr.2013.12.008. [DOI] [PubMed] [Google Scholar]
- 40.Villa E., Critelli R., Lei B., Marzocchi G., Camma C., Giannelli G. Neoangiogenesis-related genes are hallmarks of fast-growing hepatocellular carcinomas and worst survival. Results from a prospective study. Gut. 2016;65(5):861–869. doi: 10.1136/gutjnl-2014-308483. [DOI] [PubMed] [Google Scholar]
- 41.Embade N., Fernandez-Ramos D., Varela-Rey M., Beraza N., Sini M., Gutierrez de Juan V. Murine double minute 2 regulates Hu antigen R stability in human liver and colon cancer through NEDDylation. Hepatology. 2012;55(4):1237–1248. doi: 10.1002/hep.24795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Rodriguez M.S., Desterro J.M., Lain S., Midgley C.A., Lane D.P., Hay R.T. SUMO-1 modification activates the transcriptional response of p53. EMBO J. 1999;18(22):6455–6461. doi: 10.1093/emboj/18.22.6455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Da Silva-Ferrada E., Xolalpa W., Lang V., Aillet F., Martin-Ruiz I., de la Cruz-Herrera C.F. Analysis of SUMOylated proteins using SUMO-traps. Sci Rep. 2013;3:1690. doi: 10.1038/srep01690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Eswar N., Eramian D., Webb B., Shen M.Y., Sali A. Protein structure modeling with MODELLER. Methods Mol Biol. 2008;426:145–159. doi: 10.1007/978-1-60327-058-8_8. [DOI] [PubMed] [Google Scholar]
- 45.Case D.A., Cheatham T.E., 3rd, Darden T., Gohlke H., Luo R., Merz K.M., Jr. The Amber biomolecular simulation programs. J Comput Chem. 2005;26(16):1668–1688. doi: 10.1002/jcc.20290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Maier J.A., Martinez C., Kasavajhala K., Wickstrom L., Hauser K.E., Simmerling C. ff14SB: improving the accuracy of protein side chain and backbone parameters from ff99SB. J Chem Theory Comput. 2015;11(8):3696–3713. doi: 10.1021/acs.jctc.5b00255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Papamokos G.V., Tziatzos G., Papageorgiou D.G., Georgatos S.D., Politou A.S., Kaxiras E. Structural role of RKS motifs in chromatin interactions: a molecular dynamics study of HP1 bound to a variably modified histone tail. Biophys J. 2012;102(8):1926–1933. doi: 10.1016/j.bpj.2012.03.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Berendsen H.J.C., Postma J.P.M., van Gunsteren W.F., Hermans J. In: Intermolecular Forces. Pullman e B., editor. Reidel, Dordrecht; 1981. [Google Scholar]
- 49.Berendsen H.J.C., Postma J.P.M., Vangunsteren W.F., Dinola A., Haak J.R. Molecular-dynamics with coupling to an external bath. J Chem Phys. 1984;81(8):3684–3690. [Google Scholar]
- 50.Roe D.R., Cheatham T.E., 3rd. PTRAJ and CPPTRAJ: software for processing and analysis of molecular dynamics trajectory data. J Chem Theory Comput. 2013;9(7):3084–3095. doi: 10.1021/ct400341p. [DOI] [PubMed] [Google Scholar]
- 51.Pettersen E.F., Goddard T.D., Huang C.C., Couch G.S., Greenblatt D.M., Meng E.C. UCSF Chimera—a visualization system for exploratory research and analysis. J Comput Chem. 2004;25(13):1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- 52.Martinez-Lopez N., Garcia-Rodriguez J.L., Varela-Rey M., Gutierrez V., Fernandez-Ramos D., Beraza N. Hepatoma cells from mice deficient in glycine N-methyltransferase have increased RAS signaling and activation of liver kinase B1. Gastroenterology. 2012;143(3) doi: 10.1053/j.gastro.2012.05.050. (787-98 e1-13) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Martinez-Lopez N., Varela-Rey M., Fernandez-Ramos D., Woodhoo A., Vazquez-Chantada M., Embade N. Activation of LKB1-Akt pathway independent of phosphoinositide 3-kinase plays a critical role in the proliferation of hepatocellular carcinoma from nonalcoholic steatohepatitis. Hepatology. 2010;52(5):1621–1631. doi: 10.1002/hep.23860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Vazquez-Chantada M., Fernandez-Ramos D., Embade N., Martinez-Lopez N., Varela-Rey M., Woodhoo A. HuR/methyl-HuR and AUF1 regulate the MAT expressed during liver proliferation, differentiation, and carcinogenesis. Gastroenterology. 2010;138(5):1943–1953. doi: 10.1053/j.gastro.2010.01.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Sarge K.D., Park-Sarge O.K. SUMO and its role in human diseases. Int Rev Cell Mol Biol. 2011;288:167–183. doi: 10.1016/B978-0-12-386041-5.00004-2. [DOI] [PubMed] [Google Scholar]
- 56.Da Silva-Ferrada E., Lopitz-Otsoa F., Lang V., Rodriguez M.S., Matthiesen R. Strategies to identify recognition signals and targets of SUMOylation. Biochem Res Int. 2012;2012:875148. doi: 10.1155/2012/875148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Liang Y.C., Lee C.C., Yao Y.L., Lai C.C., Schmitz M.L., Yang W.M. SUMO5, a novel Poly-SUMO isoform, Regulates PML Nuclear Bodies. Sci Rep. 2016;6:26509. doi: 10.1038/srep26509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Zhang F.P., Mikkonen L., Toppari J., Palvimo J.J., Thesleff I., Janne O.A. Sumo-1 function is dispensable in normal mouse development. Mol Cell Biol. 2008;28(17):5381–5390. doi: 10.1128/MCB.00651-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Jiang Q.F., Tian Y.W., Shen Q., Xue H.Z., Li K. SENP2 regulated the stability of beta-catenin through WWOX in hepatocellular carcinoma cell. Tumour Biol. 2014;35(10):9677–9682. doi: 10.1007/s13277-014-2239-8. [DOI] [PubMed] [Google Scholar]
- 60.Beauclair G., Bridier-Nahmias A., Zagury J.F., Saib A., Zamborlini A. JASSA: a comprehensive tool for prediction of SUMOylation sites and SIMs. Bioinformatics. 2015;31(21):3483–3491. doi: 10.1093/bioinformatics/btv403. [DOI] [PubMed] [Google Scholar]
- 61.Lan F., Cacicedo J.M., Ruderman N., Ido Y. SIRT1 modulation of the acetylation status, cytosolic localization, and activity of LKB1. Possible role in AMP-activated protein kinase activation. J Biol Chem. 2008;283(41):27628–27635. doi: 10.1074/jbc.M805711200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Gertz M., Fischer F., Nguyen G.T., Lakshminarasimhan M., Schutkowski M., Weyand M. Ex-527 inhibits Sirtuins by exploiting their unique NAD±dependent deacetylation mechanism. Proc Natl Acad Sci U S A. 2013;110(30):E2772–E2781. doi: 10.1073/pnas.1303628110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Lu Y., Ding S., Zhou R., Wu J. Structure of the complex of phosphorylated liver kinase B1 and 14-3-3zeta. Acta Crystallogr F Struct Biol Commun. 2017;73(Pt 4):196–201. doi: 10.1107/S2053230X17003521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Martinez-Chantar M.L., Vazquez-Chantada M., Ariz U., Martinez N., Varela M., Luka Z. Loss of the glycine N-methyltransferase gene leads to steatosis and hepatocellular carcinoma in mice. Hepatology. 2008;47(4):1191–1199. doi: 10.1002/hep.22159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Krock B.L., Skuli N., Simon M.C. Hypoxia-induced angiogenesis: good and evil. Genes Cancer. 2011;2(12):1117–1133. doi: 10.1177/1947601911423654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Kim C.J., Cho Y.G., Park J.Y., Kim T.Y., Lee J.H., Kim H.S. Genetic analysis of the LKB1/STK11 gene in hepatocellular carcinomas. Eur J Cancer. 2004;40(1):136–141. doi: 10.1016/s0959-8049(03)00659-2. [DOI] [PubMed] [Google Scholar]
- 67.Rhodes L.V., Tate C.R., Hoang V.T., Burks H.E., Gilliam D., Martin E.C. Regulation of triple-negative breast cancer cell metastasis by the tumor-suppressor liver kinase B1. Oncogene. 2015;4 doi: 10.1038/oncsis.2015.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.George S.H., Milea A., Sowamber R., Chehade R., Tone A., Shaw P.A. Loss of LKB1 and p53 synergizes to alter fallopian tube epithelial phenotype and high-grade serous tumorigenesis. Oncogene. 2016;35(1):59–68. doi: 10.1038/onc.2015.62. [DOI] [PubMed] [Google Scholar]
- 69.Sengupta S., Nagalingam A., Muniraj N., Bonner M.Y., Mistriotis P., Afthinos A. Activation of tumor suppressor LKB1 by honokiol abrogates cancer stem-like phenotype in breast cancer via inhibition of oncogenic Stat3. Oncogene. 2017;36(41):5709–5721. doi: 10.1038/onc.2017.164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Zhong D., Liu X., Khuri F.R., Sun S.Y., Vertino P.M., Zhou W. LKB1 is necessary for Akt-mediated phosphorylation of proapoptotic proteins. Cancer Res. 2008;68(18):7270–7277. doi: 10.1158/0008-5472.CAN-08-1484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Xie Z., Dong Y., Zhang M., Cui M.Z., Cohen R.A., Riek U. Activation of protein kinase C zeta by peroxynitrite regulates LKB1-dependent AMP-activated protein kinase in cultured endothelial cells. J Biol Chem. 2006;281(10):6366–6375. doi: 10.1074/jbc.M511178200. [DOI] [PubMed] [Google Scholar]
- 72.Xie Z., Dong Y., Scholz R., Neumann D., Zou M.H. Phosphorylation of LKB1 at serine 428 by protein kinase C-zeta is required for metformin-enhanced activation of the AMP-activated protein kinase in endothelial cells. Circulation. 2008;117(7):952–962. doi: 10.1161/CIRCULATIONAHA.107.744490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Zheng B., Jeong J.H., Asara J.M., Yuan Y.Y., Granter S.R., Chin L. Oncogenic B-RAF negatively regulates the tumor suppressor LKB1 to promote melanoma cell proliferation. Mol Cell. 2009;33(2):237–247. doi: 10.1016/j.molcel.2008.12.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Matic I., van Hagen M., Schimmel J., Macek B., Ogg S.C., Tatham M.H. In vivo identification of human small ubiquitin-like modifier polymerization sites by high accuracy mass spectrometry and an in vitro to in vivo strategy. Mol Cell Proteomics. 2008;7(1):132–144. doi: 10.1074/mcp.M700173-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Hilgarth R.S., Murphy L.A., Skaggs H.S., Wilkerson D.C., Xing H., Sarge K.D. Regulation and function of SUMO modification. J Biol Chem. 2004;279(52):53899–53902. doi: 10.1074/jbc.R400021200. [DOI] [PubMed] [Google Scholar]
- 76.Eggers C.M., Kline E.R., Zhong D., Zhou W., Marcus A.I. STE20-related kinase adaptor protein alpha (STRADalpha) regulates cell polarity and invasion through PAK1 signaling in LKB1-null cells. J Biol Chem. 2012;287(22):18758–18768. doi: 10.1074/jbc.M111.316422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Zeng P.Y., Berger S.L. LKB1 is recruited to the p21/WAF1 promoter by p53 to mediate transcriptional activation. Cancer Res. 2006;66(22):10701–10708. doi: 10.1158/0008-5472.CAN-06-0999. [DOI] [PubMed] [Google Scholar]
- 78.Unger C., Buchmann A., Bunemann C.L., Kress S., Schwarz M. Wild-type function of the p53 tumor suppressor protein is not required for apoptosis of mouse hepatoma cells. Cell Death Differ. 1998;5(1):87–95. doi: 10.1038/sj.cdd.4400321. [DOI] [PubMed] [Google Scholar]
- 79.Nagy Z., Kovacs I., Torok M., Toth D., Vereb G., Buzas K. Function of RasGRP3 in the formation and progression of human breast cancer. Mol Cancer. 2014;13:96. doi: 10.1186/1476-4598-13-96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Zeqiraj E., Filippi B.M., Goldie S., Navratilova I., Boudeau J., Deak M. ATP and MO25alpha regulate the conformational state of the STRADalpha pseudokinase and activation of the LKB1 tumour suppressor. PLoS Biol. 2009;7(6) doi: 10.1371/journal.pbio.1000126. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
List of commercial and non-commercial DNA plasmids used.
List of comercial antibodies used for Western blot and Immunoprecipitation and pull-down assays.
List of antibodies used for immunohistochemistry and immunofluorescence.
Gene expression of genes related to SUMO pathway, hypoxia and hypoxia responsiveness genes in HCC tumors classified as aggressive or less aggressive [1]. Whole gene expression data is available at the Gene Expression Omnibus website (http://www.ncbi.nlm.nih.gov/geo) under the accession number: GSE54236.