


Abstract
Experimental studies of Escherichia coli K-12 MG1655 often implicate poorly annotated genes in cellular phenotypes. However, we lack a systematic understanding of these genes. How many are there? What information is available for them? And what features do they share that could explain the gap in our understanding? Efforts to build predictive, whole-cell models of E. coli inevitably face this knowledge gap. We approached these questions systematically by assembling annotations from the knowledge bases EcoCyc, EcoGene, UniProt and RegulonDB. We identified the genes that lack experimental evidence of function (the ‘y-ome’) which include 1600 of 4623 unique genes (34.6%), of which 111 have absolutely no evidence of function. An additional 220 genes (4.7%) are pseudogenes or phantom genes. y-ome genes tend to have lower expression levels and are enriched in the termination region of the E. coli chromosome. Where evidence is available for y-ome genes, it most often points to them being membrane proteins and transporters. We resolve the misconception that a gene in E. coli whose primary name starts with ‘y’ is unannotated, and we discuss the value of the y-ome for systematic improvement of E. coli knowledge bases and its extension to other organisms.
INTRODUCTION
Unannotated genes in model organisms still play important roles in determining cell phenotype. This point was driven home by recent efforts to synthesize a minimal bacterial genome. The resulting syn3.0 organism includes just 473 genes, all of which are essential for growth, and a full 30% of which lack functional annotation (1,2). Even in Escherichia coli K-12 MG1655, perhaps the best-studied model organism, unannotated genes often appear in experimental studies of strain engineering (3), laboratory evolution (4) and pathogenicity (5). Efforts to build predictive models of the genotype-phenotype relationship for whole cells will be hindered by unannotated genes that still affect cell phenotype (6,7).
Historically, unannotated genes in E. coli are known as ‘y-genes’ because they have primary names starting with ‘y’ (8)—not to be confused with ‘Y genes’ which can indicate genes on the human Y chromosome (9). However, genes with primary names that begin with ‘y’ are often functionally annotated. For example, in a recent study where E. coli was engineered to produce fatty acids via reversal of the fatty-acid beta-oxidation pathway, the authors knocked out the genes yqeF and yqhD to increase production of target molecules (3) and included the genes ydiQRST, ydiO and ydbK in a predictive model of the cell (10). Searching for these genes in public knowledge bases such as EcoCyc (11) reveals that they vary greatly in annotation quality. Some (e.g. yqhD) are well-annotated with direct experimental evidence, while others (e.g. ydiO) have limited functional information. The variation of annotation quality among y-genes suggests that a systematic approach to understand the unannotated genes in E. coli is needed which goes beyond the primary gene name.
There are several knowledge bases that represent the collected knowledge of the E. coli K-12 MG1655 genome: EcoCyc (11), EcoGene (12), UniProt (13) and RefSeq (14). Other useful knowledge bases cater to specific classes of gene products, such as the RegulonDB, which contains manually curated functional information about transcription factors in E. coli (15). Our initial review of these knowledge bases yielded conflicting information on gene function and level of annotation for many E. coli genes. Any attempt to systematically assess the function of unannotated genes must therefore draw from multiple knowledge bases and resolve these conflicts.
Many research groups have categorized E. coli genes and proteins by annotation quality as a part of their studies. In 2009, Hu et al. constructed a global functional atlas of E. coli proteins (16). First, they identified all unannotated proteins in the K-12 W3110 and MG1655 genomes. In order for a protein-encoding gene to be considered functionally uncharacterized in their analysis, it had to meet the following criteria: (i) The gene name begins with ‘y’, (ii) the gene does not have a known pathway within EcoCyc and (iii) the gene does not have a functional description in GenProtEC (17) (any gene with a description containing the words ‘predicted’, ‘hypothetical’, or ‘conserved’). Based on these criteria, it was determined that 1431 of 4225 protein coding sequences were not functionally annotated. In 2015, Kim et al. published a database called EcoliNet that curated and predicted cofunctional gene networks for every protein coding gene in the E. coli genome (18). This study also quantified the number of uncharacterized protein coding genes in E. coli. To assess functional annotation, they used the presence of experimentally supported ‘biological process’ annotations in the Gene Ontology database (19). They concluded that ∼2000 protein coding genes in E. coli were not functionally annotated. The most comprehensive effort to assess the level of annotation in bacterial genomes has been Computational Bridges to Experiments (COMBREX) (20,21). The COMBREX knowledge base currently contains information about 4182 protein coding genes in E. coli K-12 MG1655, of which 2378 (57%) have experimentally verified function, 1741 (42%) have predicted but not experimentally verified function and 63 (2%) have no predicted function. These studies of unannotated genes in E. coli K-12 MG1655 provided inspiration for this work. However, our effort covers both protein-coding and nonprotein-coding genes, disregards nomenclature (i.e. whether a gene name begins with ‘y’) as an indicator of annotation quality, and is presented as a reproducible workflow to keep the analysis up-to-date as knowledge bases improve.
In seeking the set of E. coli genes that lack functional annotation, one must determine what level of annotation is sufficient to call a gene ‘well-annotated.’ Experimental evidence is essential for assigning gene functions with confidence. While computational inference of gene function is improving, it is still prone to error (22,23), it cannot distinguish the various roles played generalist proteins in different environments (24,25), and it cannot be used to determine the detailed effects of genes like transcription factors with complex modes of action (26). Taking the example of transcription factors, computational inference can be used to predict that a gene is a transcription factor (26,27). Should this be sufficient to determine that the gene is annotated?
To answer this question, we propose that functional annotation of a gene should establish a mechanistic link to the phenotypic effects of the gene. Thus, a transcription factor is annotated if the regulated genes are known and the mode of regulation (activation, repression) for each gene is established. To take another example, knowing that a gene encodes an oxidoreductase enzyme does not indicate which phenotypes might be enabled by the gene (e.g. which catabolic pathway it might be a part of). But if a specific biochemical activity can be established (e.g. by enzymatic assay), that could clearly establish the contribution of the gene to cell phenotype. Furthermore, an association between a gene and a phenotype is not sufficient if it lacks the mechanistic information that is generally desired when annotating genes (e.g. if a gene is essential for cell division but the mechanism of the effect is unknown, then the gene fails this test). This approach at least provides guidelines on how to assess the level of functional annotation, even if it is difficult to put into practice. This article describes a first approximation of the approach, but to precisely define and enforce this definition of functional annotation will require continued effort, and we describe the next steps in the Discussion.
We propose the term ‘y-ome’ for the set of genes in a genome that lack experimental evidence of function with a mechanism for affecting cell phenotypic. We sought to identify the y-ome of E. coli K-12 MG1655 based on existing knowledge bases. Because we are limited by the annotations that are already available, this y-ome is an approximation based on a heuristic workflow described below. Therefore, we present the E. coli y-ome as a reproducible workflow that can improve over time.
The resulting y-ome includes 35% of E. coli genes. We describe some trends for these y-ome genes, including their enrichment in the termination region of the E. coli chromosome, lower average expression levels than well-annotated genes and evidence that certain types of genes (e.g. transporters) are enriched in the y-ome. Finally, we resolve the misconception that a gene in E. coli whose primary name starts with ‘y’ is necessarily unannotated.
MATERIALS AND METHODS
A heuristic approach to identifying the y-ome
A y-ome workflow was developed to assign genes to the y-ome based on annotations in E. coli knowledge bases. Existing knowledge bases do not explicitly annotate whether a gene has both experimental evidence of function and sufficient evidence to determine the mechanistic effect on cell phenotype. Therefore, we took a heuristic approach to analyze the databases, following the process that one might take if they were manually curating the entire list.
First, we looked for common indications in annotations that genes were very poorly annotated. Often particular keywords or structured annotations were identified—e.g. the keyword ‘hypothetical’ in a gene description—that always appeared along with genes that had minimal annotation. Next, we looked for clear indications that genes were very well annotated, such as entries in a functional evidence ontology. Defining these keywords and annotation rules was a subjective process, so we took a conservative approach whenever possible. Genes that could not be automatically annotated were labeled for manual curation. The full process is detailed in the following sections.
A workflow to determine the E. coli y-ome
We collected data from the following knowledge bases: EcoCyc release 22.5 (11), EcoGene version 3.0 (12), UniProt release 2018_10 for proteome UP000000625 (13) and RegulonDB version 9.4 (15). While data in RegulonDB is also available in EcoCyc, the RegulonDB data downloads were more convenient for extracting annotations of transcription factors. The E. coli K-12 MG1655 NCBI RefSeq genome annotation (accession NC_000913.3) was included for comparison as it is a commonly-used resource in the field (14). Features were extracted from the downloaded data and used to populate a relational (SQLite) database.
Pseudogenes are genes that have lost their function through mutation, and phantom genes are regions of the genome that were once considered genes but are no longer based on better evidence or analysis. Pseudogenes and phantom genes are not included in the y-ome, so they are assigned to the ‘Excluded’ category in the workflow. Annotations of pseudogenes and phantom genes were taken from both EcoGene and EcoCyc. In EcoGene, pseudogenes are indicated by a primary name ending in an apostrophe. EcoCyc explicitly defines lists of pseudogenes and phantom genes, currently available here:
https://ecocyc.org/ECOLI/class-instances?object=Pseudo-Genes
https://ecocyc.org/ECOLI/class-instances?object=Phantom-Genes
Cryptic genes—defined as genes that are phenotypically silent—were not marked ‘Excluded’. These genes might be activated under novel conditions, and therefore they are an interesting component of the y-ome (28).
Keywords were used to automatically categorize genes for each knowledge base feature. To identify the keywords, we read gene entries in the knowledge bases to look for commonly used phrases in the parlance of the particular knowledge base that signified the level of annotation. For example, in EcoCyc, the keywords ‘possibly’, ‘predicted’ and ‘hypothetical’ in the ‘description’ field were used to identify genes with low annotation level, and the keywords ‘assay’, ‘traceable author statement to experimental support’ and ‘reaction blocked in mutant’ in the ‘evidence’ field were used to identify genes with high annotation (well-annotated). The full list of keywords used in the workflow listed in Dataset S5.
Defining these keywords was a subjective process, so we relied on structured data whenever possible, with the following rules. To determine annotation level for UniProt genes, we used the ‘annotation score’ for each associated protein. Annotation scores of two or below were used to indicate ‘y-ome’ and four or above to indicate ‘well-annotated’. Genes with annotation score three were categorized as ‘Not enough information for automated assignment’. Additionally, EcoCyc genes with a reaction_equation annotation, gene complex annotations or explicitly marked as insertion elements were considered ‘well-annotated’.
Consensus rules
After assigning genes in each knowledge base to categories, consensus rules were applied to combine the results from the separate knowledge bases. In general, we checked first for agreement among knowledge bases. For instance, if two knowledge bases indicated a gene was ‘well-annotated’ and the others did not have enough information to assign a category, then the consensus was ‘well-annotated’. When databases disagreed, then no consensus was possible. In these cases, manual annotations were made (for 334 genes) based on reading the knowledge base entries and consulting the literature. There were four exceptions that were identified as heuristics to improve the quality of the final list:
The Evidence section for EcoCyc genes is a high-quality, manually-curated ontology of functional evidence derived from the primary literature (29). Therefore, we gave this section priority over other data in the workflow. Particularly, we looked for Evidence features with keywords ‘assay’, ‘reaction blocked in mutant’ and ‘traceable author statement to experimental support’ which were marked as Well-annotated in the final categorization.
RegulonDB contains curated and experimentally-validated annotations of transcription factors. Thus, genes with ‘Strong’ evidence in RegulonDB were marked as well-annotated in the final categorization.
When EcoCyc and UniProt were both categorized as well-annotated for a given gene, then this gene was automatically marked as well-annotated in the final categorization. This heuristic was helpful to identify cases where EcoGene was missing key evidence that the other knowledge bases had picked up (e.g. dhaM).
Insertion elements, identified in EcoCyc by gene names beginning with ‘ins’, were considered to be well-annotated in the final categorization.
Genes with no information
To identify genes for which no information at all is available, we filtered the database for genes with features drawn from knowledge-base-specific phrase lists that corresponded to genes with no other functional information. For example, ‘Putative uncharacterized’ often appeared in such UniProt entries. As another example, EcoCyc genes with no information have summaries that begin with this phrase ‘No information about this’. (e.g. ‘No information about this protein was found by a literature search conducted on 23 February 2017’ for ybiU). The full list of phrases that were used can be found in Dataset S5.
When genes were annotated only with a protein domain or family, we still included them in the list because such domains (e.g. DUF1479 for ybiU) often themselves have no functional information associated (DUF1479 has the description ‘Protein of unknown function’ on Pfam: https://pfam.xfam.org/family/PF07350).
Gene expression compendium
A compendium of RNA-seq data for E. coli K-12 MG1655 and BW25113 (wild-type, single gene mutants and laboratory evolution endpoints) was used to analyze expression of y-ome genes. All RNA-seq experiments were conducted using the protocol described by Seo et al. (30). Raw sequencing reads were collected from GEO (31) (see Dataset S4 for accession numbers) and mapped to the reference genome (NC_000913.3 for strain MG1655 and CP009273 for BW25113) using bowtie 1.1.2 (32) with the following options: ‘-X 1000 -n 2 -3 3’. Transcript abundance was quantified using summarizeOverlaps from the R GenomicAlignments package, with the following options: ‘mode = ‘IntersectionStrict’, singleEnd = FALSE, ignore.strand = FALSE, preprocess.reads = invertStrand’ (33). Transcripts per Million (TPM) were calculated by DESeq2 (34). The final expression compendium was log-transformed with log2(TPM+1) before analysis, referred to as log-TPM. Biological replicates with R2 < 0.9 between log-TPM were removed to reduce technical noise.
Co-expression analysis
Gene co-expression analysis was performed on the gene expression compendium using IterativeWGCNA (35), an extension of the popular WGCNA algorithm (36). IterativeWGCNA uses iterative pruning steps to improve the robustness of detected gene modules. The method was run with a minimum module size of 10 and otherwise with default parameters.
Chromosome location & density
Gene locations were extracted from gene start sites in the E. coli RefSeq genome annotation NC_000913.3. Gene density plots were created with a circular kernel density estimation method using the von Mises distribution.
RESULTS
A workflow for identifying the E. coli y-ome
To systematically determine an initial y-ome for E. coli K-12 MG1655, we developed a semi-automated approach (Figure 1) to identify unique genes across four E. coli knowledge bases and integrate their annotations. The automated part of this process proceeded in three steps: (i) downloading data from each knowledge base, (ii) extracting text-based features (Dataset S2) and (iii) using keywords to automatically assign each gene annotation in a knowledge base to the categories ‘y-ome,’ ‘Well-annotated’ or ‘Not enough information for automated assignment’ (Figure 2A). Pseudogenes and phantom genes were kept separate and marked ‘excluded’. The rules and keywords used to make these assignments are described in the ‘Materials and Methods’ section.
Figure 1.

A workflow for defining the y-ome of E. coli K-12 MG1655. Data were collected from four E. coli knowledge bases, and automated categorization was applied to determine their annotation level. Next, consensus rules were applied to combine categorizations from multiple databases. When the consensus rules could not be applied, genes were manually curated and placed in one of the categories. Thus, genes were categorized as ‘Well-Annotated’ or ‘y-ome’ according to the definition of the y-ome (see Section ‘Definition of the y-ome’). Pseudogenes and phantom genes were treated separately in the ‘Excluded’ category.
Figure 2.

Gene annotation across knowledge bases. The y-axes represent all unique genes in the database. Gene order is maintained within each subplot so one can track the annotation of a set of genes across knowledge bases. (A) An automated approach was used to categorize genes from each database as ‘Well-annotated’ or ‘y-ome’ based on the definition of the y-ome. Pseudogenes and phantom genes were excluded. The resulting y-ome includes 1600 genes. (B) y-ome categories were compared to the content of the latest E. coli genome-scale ME-model. (C) A total of 173 genes have primary names that start with ‘y’ but are well-annotated, and 462 genes in the y-ome have non-‘y’ primary names.
Based on this analysis, we identified 4623 unique genes across E. coli K-12 MG1655 knowledge bases, and each was assigned to the ‘y-ome’, ‘well-annotated’ or ‘excluded’ categories (Dataset S1). Of these 4623 genes, 2803 have information that indicate a sufficient level of functional evidence to exclude them from the y-ome (Figure 2A), and 1600 genes (34.6%) are in the y-ome of E. coli K-12 MG1655. No individual knowledge base provides information to fully define the y-ome, but EcoCyc comes the closest (Figure 2A). Of the 1600 y-ome genes, there were 111 for which we found no information in the knowledge bases (see ‘Materials and Methods’ section) and 220 that were marked as pseudogenes or phantom genes.
Of the similar studies reviewed in the Introduction, only the article by Hu et al. (16) provided a complete list of unannotated genes. Comparing that set to the y-ome reveals differences in the annotation levels of hundreds of genes, particularly in the cases where Hu et al. relied on the primary name (names ending in ‘y’) to determine the annotation level of the gene (Supplementary Figure S1). By this measure, at least, the y-ome workflow offers a more complete view of the annotation level of E. coli genes.
Gene expression and chromosome location
It was previously observed by Hu et al. that poorly annotated genes tend to be expressed at a lower level than well-annotated genes (16). We confirmed this with the y-ome by comparing gene expression of y-ome genes and well-annotated genes in a compendium of RNA-seq data. The RNA-seq compendium includes expression values for 4385 E. coli genes across 78 conditions, including a variety of carbon sources, nitrogen sources, gene knockouts, stress conditions and laboratory evolution endpoints (conditions are described in Dataset S4). Genes in the y-ome tend to have lower expression across the surveyed conditions (Figure 3), where the t-test P-value < 1 × 10−6. A large-scale quantitative proteomics dataset is also available for E. coli (37), and comparing y-ome protein abundance to well-annotated protein abundance in that dataset reveals the same trend (Supplementary Figure S2). Attempts to annotate y-ome genes may be more successful if priority is given to the highly expressed y-ome genes that have a greater potential to affect observable phenotypes. Alternatively, experiments that identify growth conditions with greater expression of y-ome genes could help elucidate their functions because genes are more likely to have a phenotypic effect under conditions where they are expressed (28).
Figure 3.
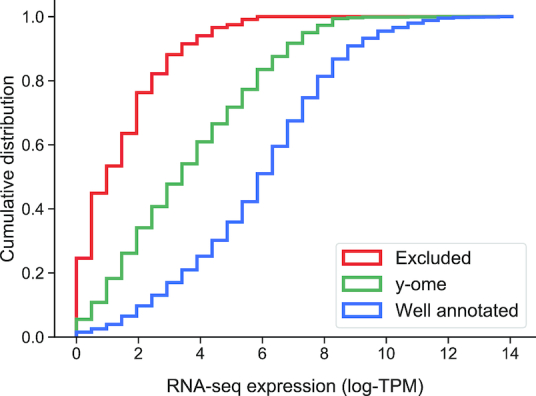
Average gene expression for all genes in a compendium of E. coli RNA-seq data. Cumulative distributions of normalized mean expression levels (mean log-TPM) for ‘y-ome’ (green), ‘Well annotated’ (blue) and ‘Excluded’ (red) genes across the 78 conditions surveyed in a compendium of RNA-Seq data.
We observed a low density of y-ome genes near the origin of replication (ORI) of the E. coli chromosome and a high density of y-ome genes in the termination region (opposite ORI, Figure 4A). Highly expressed genes are known to be enriched near ORI (38–40), which was observed in our gene expression compendium (Figure 4B). It has also been shown that genes whose deletion affect growth phenotypes under stress conditions, so-called ‘responsive genes,’ are enriched near ORI (41). These observations tell a simple story of highly expressed genes that have obvious effects on phenotypes under laboratory conditions and are therefore well-annotated, and lowly expressed genes that do not affect phenotypes enough to be easily characterized. However, the y-ome genes with highest mean expression (top 20th percentile) are split between the origin and termination regions (Figure 4A), which suggests that some other factor might be keeping genes near the termination region from being characterized. High-throughput gene annotation might shed further light on this phenomenon.
Figure 4.

Gene expression by location on the chromosome. (A) The y-ome genes are enriched in the termination region of the E. coli chromosome, opposite the ORI. However, the y-ome genes in the top 20th percentile of expression (mean log-TPM > 7.57) are enriched both near the ORI and the termination region. (B) Highly expressed genes are known to be enriched around the ORI (16), which we confirmed by plotting density of genes in the chromosome with increasing thresholds of mean gene expression (mean log-TPM) across the compendium of RNA-seq data for 78 conditions.
Co-expression analysis
Genes that are co-expressed often have functional relationships, so weighted-gene correlation network analysis (WGCNA) is a popular approach for generating hypotheses of gene functions (18,36). Using an extension of the method called IterativeWGCNA (35), we identified co-expressed gene modules in the RNA-seq compendium.
Well-annotated and y-ome genes tend to appear in modules together, indicating that highly-expressed y-ome genes could be functioning to affect cell phenotypes (Figure 5, Dataset S6). In total, 840 well-annotated genes and 347 y-ome genes appeared in the 56 modules (along with 4 genes marked as pseudogenes or phantom genes; these might actually have functional roles).
Figure 5.

Co-expressed gene modules identified with IterativeWGCNA. The bar plot summarizes the number of genes in each module by category (‘Well-annotated’, ‘y-ome’ and ‘Psuedogene or phantom gene’). The table lists all genes in Module M21—of which only ygiQ is in the y-ome—and the primary names and descriptions from EcoCyc.
As an example, module M21 includes the y-ome gene ygiQ whose only annotation is membership in the radical SAM protein superfamily (Figure 5). All other genes in the module are well-annotated, and they are generally associated with protein translation (e.g. ribosomal subunits, protein elongation factors and transfer RNA-associated proteins). Some radical SAM enzymes are known to be involved in protein translation, e.g. rlmN (42). These observations provide some insight into the potential function of ygiQ. This study is not intended to determine new y-ome functions. Rather, the co-expression analysis demonstrates that y-ome genes are often expressed with well-annotated genes, and these 347 co-expressed y-ome genes are the highest priority targets for experimental characterization (Dataset S6).
Functions of y-ome genes
The most common terms associated with y-ome genes can easily be extracted from E. coli knowledge bases (Table 1). These terms indicate that many membrane-associated proteins (502 genes) and particularly transporters (295 genes) remain to be annotated. Membrane-bound proteins and transporters are particularly hard to characterize with certainty (43), but high-throughput methods might change that, as they already have for enzymatic assays (44,45), gene-environment networks (46) and protein–protein interactions (16). Thus, the y-ome offers a set of candidate transport-associated genes for high-throughput analysis. High-throughput analysis could also be relevant for gene sets related to enzymes (271 genes), signaling (267 genes), lipoproteins (98 genes) and biofilms (74 genes). As evidence accumulates in E. coli knowledge bases, this workflow can be run again to improve the candidate gene sets.
Table 1.
The most common words found in knowledge bases features for y-ome genes.
| Word or word set | y-ome (n = 1600) | Well-annotated (n = 2803) | Excluded (n = 220) |
|---|---|---|---|
| peptide/polypeptide/protein(s) | 1564 | 2571 | 153 |
| inner/outer/membrane/transmembrane | 502 | 751 | 30 |
| binds/binding | 418 | 1480 | 21 |
| regulate(s)/regulated/regulator(y)/regulation/regulon | 367 | 897 | 21 |
| transport/transporter/export/import | 295 | 688 | 35 |
| enzyme | 271 | 1588 | 12 |
| signal | 267 | 264 | 18 |
| initiation | 253 | 409 | 24 |
| phage/prophage | 186 | 185 | 64 |
| oxidoreductase/reductase/reduce | 139 | 361 | 4 |
| transcription/transcriptional | 123 | 382 | 13 |
| promoter | 119 | 204 | 4 |
| periplasm/periplasmic | 113 | 356 | 3 |
| lipoprotein | 98 | 88 | 5 |
| transferase | 92 | 376 | 15 |
| resistance | 92 | 242 | 1 |
| structures/structural | 91 | 206 | 0 |
| cryptic | 76 | 45 | 17 |
| lysis | 75 | 360 | 6 |
| synthesis | 74 | 743 | 3 |
| biofilm | 74 | 139 | 2 |
| phosphate | 70 | 819 | 5 |
| metabolism | 61 | 415 | 6 |
| codon | 58 | 208 | 93 |
| sugar | 58 | 98 | 8 |
| stress | 57 | 186 | 0 |
| production | 57 | 118 | 5 |
| aerobic | 55 | 215 | 0 |
The counts indicate the number of unique genes for which each phrase appears. Similar words are grouped into sets.
DISCUSSION
In 1998, a year after the first E. coli genome sequence was released, Kenneth Rudd proposed a systematic naming scheme for unannotated open reading frames where each was given a unique name starting with the letter ‘y’ (8). This is a convenient system, but the community did not settle on an official mechanism for assigning new names for these y-genes when functions were established. The tradition has been that new primary names are proposed in the first published report of a newly-identified gene function. This leaves it to peer reviewers to call out duplicate names and other issues. Without a central mechanism for standardized naming, many y-genes have been annotated without receiving new names (173 genes, Figure 2C). And poorly annotated genes have received new names not starting with ‘y’ because their function was partially established, determined based on computational predictions or based on presence in an operon (462 genes, Figure 2C). With the y-ome, we can decouple gene names from assessments of functional annotation and provide a more consistent resource for anyone interested in systematic analysis of unannotated genes.
Future y-ome analysis will be far more precise if the y-ome definition can be formalized in biological knowledge bases. Experimental evidence is already being recorded by evidence ontologies in BioCyc (EcoCyc) (29) and ECO (available in UniProt) (47). However, the implementation details of these ontologies are important for their use in the y-ome. This is best explained with the example of yihA/engB, a poorly annotated gene in the y-ome. The ECO evidence ontology describes experimental evidence of GTP binding for yihA/engB, and EcoCyc describes the function of the gene as being inferred from a mutant phenotype. The gene is described as having ‘extremely low GTPase activity’ (EcoCyc) and being ‘necessary for normal cell division and for the maintenance of normal septation’ (UniProt). But, the mechanism by which the gene affects cell phenotype is not described in these knowledge bases, so it is included in the y-ome even though evidence ontology information is available. Evidence ontologies are extremely useful, but, to incorporate them into the y-ome, additional mechanistic information is required. A potential solution is to use systems biology models to encode that mechanistic link.
In systems biology, predictive models can be used to link genotype to phenotype. In these models, the definition of functional annotation is that a gene can be mechanistically linked through a network to a measurable phenotypic effect. The contribution of any gene function to cellular phenotype can now be codified computationally for various cellular systems, including metabolism (48), cell signaling (49), gene expression (50) and replication (7). Comparing the 2803 well-annotated genes in E. coli to the 1678 genes in the latest genome-scale ME-model (one of the most comprehensive predictive models of E. coli to-date) (51), it is clear that the models can grow by over a thousand genes before running up against our lack of knowledge (Figure 2B). To integrate additional biological knowledge, great progress is being made on mechanistic modeling of whole cells (7,52). If the knowledge in whole-cell models can be integrated with evidence ontologies, then it should be possible to greatly improve the precision of the y-ome.
It is worth noting that 75 genes appear in both the ME-model and the y-ome (Figure 2B). Often, genes lacking experimental evidence are included in genome-scale models as part of a ‘gap-filling’ process (53). Usually these genes have some evidence of function (e.g. a known protein family), and the systems biology context indicates that a specific activity is necessary for the cell to function, so the most-likely (low confidence) gene annotation is included in the model. These cases can then be used to drive experimental characterization of low-confidence gene function (25). Thus, integrating predictive models with knowledge bases will provide both clarity on the content of y-ome and insight into the functions of y-ome genes.
The concept of a y-ome can be applied to any genome, and we hope that the y-ome workflow will inspire development of the y-ome of other organisms. For well-characterized model organisms, the workflow presented here can be an initial guide to develop a y-ome. For non-model organisms with few direct experimental studies of gene function, the y-ome will encompass much of the genome. To decrease the size of the y-ome in these organisms, new workflows will be necessary, combining computational genome annotation with systems biology modeling and new high-throughput experimental approaches (26,44,45,54) to establish high-confidence functional annotations across the genome.
DATA AVAILABILITY
All code and data necessary to reproduce this analysis can be found on GitHub and with a permanent DOI on Zenodo:
Supplementary Material
ACKNOWLEDGEMENTS
Special thanks to Marc Abrams for his help with editing, clarification and discussion, to Ye Gao and Donghyuk Kim for providing feedback on the approach and to Devleena Kole for her support and suggestions.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
National Science Foundation Graduate Research Fellowship [DGE-1144086 to S.G.]; Novo Nordisk Foundation through the Center for Biosustainability at the Technical University of Denmark [NNF10CC1016517]. Funding for open access charge: Novo Nordisk Foundation through the Center for Biosustainability at the Technical University of Denmark [NNF10CC1016517].
Conflict of interest statement. None declared.
REFERENCES
- 1. Hutchison C.A. 3rd, Chuang R.-Y., Noskov V.N., Assad-Garcia N., Deerinck T.J., Ellisman M.H., Gill J., Kannan K., Karas B.J., Ma L. et al.. Design and synthesis of a minimal bacterial genome. Science. 2016; 351:aad6253. [DOI] [PubMed] [Google Scholar]
- 2. Danchin A., Fang G.. Unknown unknowns: essential genes in quest for function. Microb. Biotechnol. 2016; 9:530–540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Dellomonaco C., Clomburg J.M., Miller E.N., Gonzalez R.. Engineered reversal of the β-oxidation cycle for the synthesis of fuels and chemicals. Nature. 2011; 476:355–359. [DOI] [PubMed] [Google Scholar]
- 4. Sandberg T.E., Pedersen M., LaCroix R.A., Ebrahim A., Bonde M., Herrgard M.J., Palsson B.O., Sommer M., Feist A.M.. Evolution of Escherichia coli to 42°C and subsequent genetic engineering reveals adaptive mechanisms and novel mutations. Mol. Biol. Evol. 2014; 31:2647–2662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Hufnagel D.A., DePas W.H., Chapman M.R.. The disulfide bonding system suppresses CsgD-independent cellulose production in Escherichia coli. J. Bacteriol. 2014; 196:3690–3699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Bordbar A., Monk J.M., King Z.A., Palsson B.O.. Constraint-based models predict metabolic and associated cellular functions. Nat. Rev. Genet. 2014; 15:107–120. [DOI] [PubMed] [Google Scholar]
- 7. Karr J.R., Sanghvi J.C., Macklin D.N., Gutschow M.V., Jacobs J.M., Bolival B., Assad-Garcia N., Glass J.I., Covert M.W.. A whole-cell computational model predicts phenotype from genotype. Cell. 2012; 150:389–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Rudd K.E. Linkage map of Escherichia coli K-12, edition 10: the physical map. Microbiol. Mol. Biol. Rev. 1998; 62:985–1019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Ballouz S., Dobin A., Gingeras T.R., Gillis J.. The fractured landscape of RNA-seq alignment: the default in our STARs. Nucleic Acids Res. 2018; 46:5125–5138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Cintolesi A., Clomburg J.M., Gonzalez R.. In silico assessment of the metabolic capabilities of an engineered functional reversal of the β-oxidation cycle for the synthesis of longer-chain (C≥4) products. Metab. Eng. 2014; 23:100–115. [DOI] [PubMed] [Google Scholar]
- 11. Keseler I.M., Mackie A., Peralta-Gil M., Santos-Zavaleta A., Gama-Castro S., Bonavides-Martínez C., Fulcher C., Huerta A.M., Kothari A., Krummenacker M. et al.. EcoCyc: fusing model organism databases with systems biology. Nucleic Acids Res. 2013; 41:605–612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Zhou J., Rudd K.E.. EcoGene 3.0. Nucleic Acids Res. 2013; 41:D613–D624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Consortium UniProt. UniProt: a hub for protein information. Nucleic Acids Res. 2015; 43:D204–D212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. O’Leary N.A., Wright M.W., Brister J.R., Ciufo S., Haddad D., McVeigh R., Rajput B., Robbertse B., Smith-White B., Ako-Adjei D. et al.. Reference sequence (RefSeq) database at NCBI: current status, taxonomic expansion, and functional annotation. Nucleic Acids Res. 2016; 44:D733–D745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Gama-Castro S., Salgado H., Santos-Zavaleta A., Ledezma-Tejeida D., Muñiz-Rascado L., García-Sotelo J.S., Alquicira-Hernández K., Martínez-Flores I., Pannier L., Castro-Mondragón J.A. et al.. RegulonDB version 9.0: high-level integration of gene regulation, coexpression, motif clustering and beyond. Nucleic Acids Res. 2016; 44:D133–D143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Hu P., Janga S.C., Babu M., Díaz-Mejía J.J., Butland G., Yang W., Pogoutse O., Guo X., Phanse S., Wong P. et al.. Global functional atlas of Escherichia coli encompassing previously uncharacterized proteins. PLoS Biol. 2009; 7:e96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Serres M.H., Goswami S., Riley M.. GenProtEC: an updated and improved analysis of functions of Escherichia coli K-12 proteins. Nucleic Acids Res. 2004; 32:D300–D302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Kim H., Shim J.E., Shin J., Lee I.. EcoliNet: a database of cofunctional gene network for Escherichia coli. Database. 2015; 2015:bav001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Gene Ontology Consortium Gene Ontology Consortium: going forward. Nucleic Acids Res. 2015; 43:D1049–D1056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Anton B.P., Chang Y.-C., Brown P., Choi H.-P., Faller L.L., Guleria J., Hu Z., Klitgord N., Levy-Moonshine A., Maksad A. et al.. The COMBREX project: design, methodology, and initial results. PLoS Biol. 2013; 11:e1001638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Chang Y.-C., Hu Z., Rachlin J., Anton B.P., Kasif S., Roberts R.J., Steffen M.. COMBREX-DB: an experiment centered database of protein function: knowledge, predictions and knowledge gaps. Nucleic Acids Res. 2015; 44:D330–D335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Roberts R.J., Chang Y.-C., Hu Z., Rachlin J.N., Anton B.P., Pokrzywa R.M., Choi H.-P., Faller L.L., Guleria J., Housman G. et al.. COMBREX: a project to accelerate the functional annotation of prokaryotic genomes. Nucleic Acids Res. 2011; 39:D11–D14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Galperin M.Y., Koonin E.V.. From complete genome sequence to ‘complete’ understanding. Trends Biotechnol. 2010; 28:398–406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Nam H., Lewis N.E., Lerman J. a., Lee D.-H., Chang R.L., Kim D., Palsson B.O.. Network context and selection in the evolution to enzyme specificity. Science. 2012; 337:1101–1104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Guzmán G.I., Utrilla J., Nurk S., Brunk E., Monk J.M., Ebrahim A., Palsson B.O., Feist A.M.. Model-driven discovery of underground metabolic functions in Escherichia coli. Proc. Natl. Acad. Sci. U.S.A. 2015; 112:929–934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Gao Y., Yurkovich J.T., Seo S.W., Kabimoldayev I., Dräger A., Chen K., Sastry A.V., Fang X., Mih N., Yang L. et al.. Systematic discovery of uncharacterized transcription factors in Escherichia coli K-12 MG1655. Nucleic Acids Res. 2018; 46:10682–10696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Eichner J., Topf F., Dräger A., Wrzodek C., Wanke D., Zell A.. TFpredict and SABINE: sequence-based prediction of structural and functional characteristics of transcription factors. PLoS One. 2013; 8:e82238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Tamburini E., Mastromei G.. Do bacterial cryptic genes really exist. Res. Microbiol. 2000; 151:179–182. [DOI] [PubMed] [Google Scholar]
- 29. Karp P.D., Paley S., Krieger C.J., Zhang P.. An evidence ontology for use in pathway/genome databases. Pac. Symp. Biocomput. 2004; 190–201. [DOI] [PubMed] [Google Scholar]
- 30. Seo S.W., Kim D., Latif H., O’Brien E.J., Szubin R., Palsson B.O.. Deciphering Fur transcriptional regulatory network highlights its complex role beyond iron metabolism in Escherichia coli. Nat. Commun. 2014; 5:4910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Barrett T., Wilhite S.E., Ledoux P., Evangelista C., Kim I.F., Tomashevsky M., Marshall K.A., Phillippy K.H., Sherman P.M., Holko M. et al.. NCBI GEO: archive for functional genomics data sets–update. Nucleic Acids Res. 2013; 41:D991–D995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Langmead B., Trapnell C., Pop M., Salzberg S.L.. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009; 10:R25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Lawrence M., Huber W., Pagès H., Aboyoun P., Carlson M., Gentleman R., Morgan M.T., Carey V.J.. Software for computing and annotating genomic ranges. PLoS Comput. Biol. 2013; 9:e1003118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Love M.I., Huber W., Anders S.. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014; 15:550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Greenfest-Allen E., Cartailler J.-P., Magnuson M.A., Stoeckert C.J.. iterativeWGCNA: iterative refinement to improve module detection from WGCNA co-expression networks. bioRxiv. 2017; doi:10.1101/234062. [Google Scholar]
- 36. Langfelder P., Horvath S.. WGCNA: an R package for weighted correlation network analysis. BMC Bioinformatics. 2008; 9:559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Schmidt A., Kochanowski K., Vedelaar S., Ahrné E., Volkmer B., Callipo L., Knoops K., Bauer M., Aebersold R., Heinemann M.. The quantitative and condition-dependent Escherichia coli proteome. Nat. Biotechnol. 2016; 34:104–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Allen T.E., Price N.D., Joyce A.R., Palsson B.Ø.. Long-range periodic patterns in microbial genomes indicate significant multi-scale chromosomal organization. PLoS Comput. Biol. 2006; 2:e2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Bryant J.A., Sellars L.E., Busby S.J.W., Lee D.J.. Chromosome position effects on gene expression in Escherichia coli K-12. Nucleic Acids Res. 2014; 42:11383–11392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Duigou S., Boccard F.. Long range chromosome organization in Escherichia coli: the position of the replication origin defines the non-structured regions and the Right and Left macrodomains. PLoS Genet. 2017; 13:e1006758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Nichols R.J., Sen S., Choo Y.J., Beltrao P., Zietek M., Chaba R., Lee S., Kazmierczak K.M., Lee K.J., Wong A. et al.. Phenotypic landscape of a bacterial cell. Cell. 2011; 144:143–156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Fitzsimmons C.M., Fujimori D.G.. Determinants of tRNA recognition by the radical SAM enzyme RlmN. PLoS One. 2016; 11:e0167298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Herzberg M., Kaye I.K., Peti W., Wood T.K.. YdgG (TqsA) controls biofilm formation in Escherichia coli K-12 through autoinducer 2 transport. J. Bacteriol. 2006; 188:587–598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Fuhrer T., Zampieri M., Sévin D.C., Sauer U., Zamboni N.. Genomewide landscape of gene-metabolome associations in Escherichia coli. Mol. Syst. Biol. 2017; 13:907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Sévin D.C., Fuhrer T., Zamboni N., Sauer U.. Nontargeted in vitro metabolomics for high-throughput identification of novel enzymes in Escherichia coli. Nat. Methods. 2017; 14:187–194. [DOI] [PubMed] [Google Scholar]
- 46. Price M.N., Wetmore K.M., Waters R.J., Callaghan M., Ray J., Liu H., Kuehl J.V., Melnyk R.A., Lamson J.S., Suh Y. et al.. Mutant phenotypes for thousands of bacterial genes of unknown function. Nature. 2018; 557:503–509. [DOI] [PubMed] [Google Scholar]
- 47. Chibucos M.C., Siegele D.A., Hu J.C., Giglio M.. The Evidence and Conclusion Ontology (ECO): supporting GO annotations. Methods Mol. Biol. 2017; 1446:245–259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Reed J.L., Famili I., Thiele I., Palsson B.O.. Towards multidimensional genome annotation. Nat. Rev. Genet. 2006; 7:130–141. [DOI] [PubMed] [Google Scholar]
- 49. Papin J.A., Palsson B.O.. The JAK-STAT signaling network in the human B-cell: an extreme signaling pathway analysis. Biophys. J. 2004; 87:37–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. O’Brien E.J., Lerman J.A., Chang R.L., Hyduke D.R., Palsson B.Ø.. Genome-scale models of metabolism and gene expression extend and refine growth phenotype prediction. Mol. Syst. Biol. 2013; 9:693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Liu J.K., O’Brien E.J., Lerman J.A., Zengler K., Palsson B.Ø, Feist A.M.. Reconstruction and modeling protein translocation and compartmentalization in Escherichia coli at the genome-scale. BMC Syst. Biol. 2014; 8:110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Carrera J., Covert M.W.. Why build Whole-Cell models. Trends Cell Biol. 2015; 25:719–722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Orth J.D., Palsson B.. Gap-filling analysis of the iJO1366 Escherichia coli metabolic network reconstruction for discovery of metabolic functions. BMC Syst. Biol. 2012; 6:30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Rhee S.Y., Mutwil M.. Towards revealing the functions of all genes in plants. Trends Plant Sci. 2014; 19:212–221. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All code and data necessary to reproduce this analysis can be found on GitHub and with a permanent DOI on Zenodo: