

Abstract
Ectopic mineralization is a global problem and leading cause of morbidity and mortality. The pathomechanisms of ectopic mineralization are poorly understood. Recent studies on heritable ectopic mineralization disorders with defined gene defects have been helpful in elucidation of the mechanisms of ectopic mineralization in general. The prototype of such disorders is pseudoxanthoma elasticum (PXE), a late-onset, slowly progressing disorder with multisystem clinical manifestations. Other conditions include generalized arterial calcification of infancy (GACI), characterized by severe, early-onset mineralization of the cardiovascular system, often with early postnatal demise. In addition, arterial calcification due to CD73 deficiency (ACDC) occurs late in life, mostly affecting arteries in the lower extremities in elderly individuals. These three conditions, PXE, GACI, and ACDC, caused by mutations in ABCC6, ENPP1, and NT5E, respectively, are characterized by reduced levels of inorganic pyrophosphate (PPi) in plasma. Because PPi is a powerful antimineralization factor, it has been postulated that reduced PPi is a major determinant for ectopic mineralization in these conditions. These and related observations on complementary mechanisms of ectopic mineralization have resulted in development of potential treatment modalities for PXE, including administration of bisphosphonates, stable PPi analogs with antimineralization activity. It is conceivable that efficient treatments may soon become available for heritable ectopic mineralization disorders with application to common calcification disorders.
Ectopic Mineralization: A Global Problem
Ectopic mineralization (ie, deposition of calcium/phosphate complexes on connective tissues in aberrant locations) is a subject that has attracted much attention because of it being a leading cause of morbidity and mortality. Of special interest is its wide association with several acquired clinical conditions, such as aging, cancer, diabetes, and autoimmune diseases.1, 2 There are two forms of ectopic mineralization (ie, dystrophic and metastatic calcification). Dystrophic calcification occurs in pathologically altered tissues with normal serum levels of calcium and phosphate, whereas metastatic calcification occurs in normal tissues associated with increased serum levels of calcium and phosphate. The pathomechanistic details of ectopic mineralization are poorly understood. However, recent studies on Mendelian ectopic mineralization disorders caused by mutations in distinct genes, with defined pathomechanistic consequences, have been extremely helpful toward elucidation of the mechanisms of ectopic mineralization processes in general.
Pseudoxanthoma Elasticum: The Prototype of Heritable Ectopic Mineralization Disorders
The prototype of heritable ectopic mineralization diseases with normal calcium and phosphate homeostasis is pseudoxanthoma elasticum (PXE), an autosomal recessive disorder characterized by protean manifestations in the skin, arterial blood vessels, and the eyes (Figure 1).4 A diagnostic histopathologic feature in PXE is accumulation of fragmented pleiomorphic elastotic structures in the skin, which become progressively mineralized. These observations initially suggested that the mutations in PXE may reside in genes encoding elastin or elastin-associated microfibrils. However, early genetic linkage studies largely excluded these genes as the site of causative mutations in this disorder.5, 6
Figure 1.
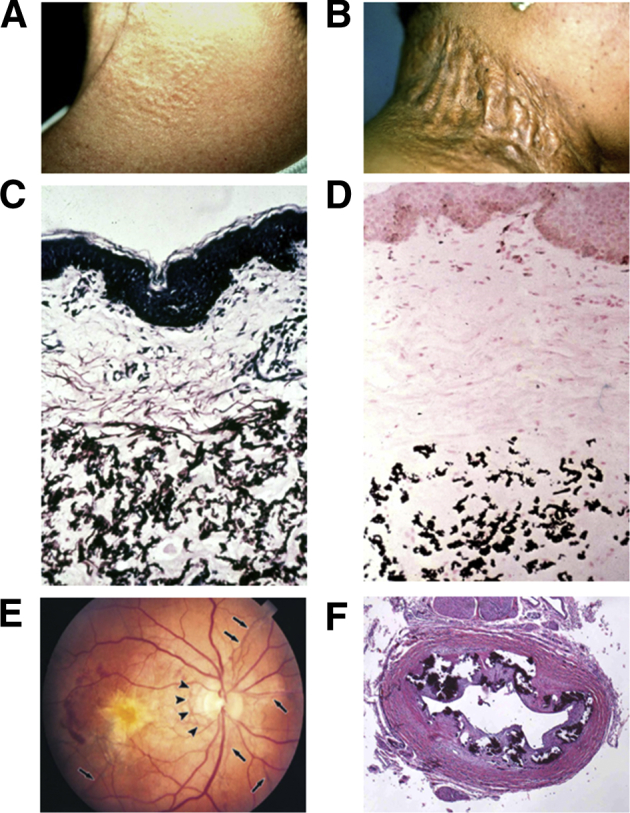
Clinical features and histopathology of ectopic mineralization in pseudoxanthoma elasticum (PXE). A: Characteristic early cutaneous signs of PXE consist of discrete yellowish papules at predilection sites, such as sides of the neck. B: These early lesions coalesce into plaques of inelastic, leathery skin. C: Histopathology of the affected area of skin reveals accumulation of pleiomorphic elastotic material in the mid dermis, as visualized by Verhoeff–van Gieson stain. D: Staining of a parallel section with von Kossa stain reveals mineralization of the elastotic structures. E: Characteristic ocular findings consist of angioid streaks, breaks in the Bruch membrane behind the retina (arrows), which allow neovascularization (arrowheads) of the retina, leading to loss of visual acuity and blindness. F: Histopathology of the left renal artery in a generalized arterial calcification of infancy patient with ABCC6 mutations reveals extensive mineralization (hematoxylin-eosin stain).
Adapted with permission from Uitto et al.3
PXE is caused in most cases by mutations in the ABCC6 gene that encodes ABCC6 (ATP-binding cassette, subfamily C, member 6), a transmembrane efflux transporter. The substrate(s) of ABCC6 remain unknown; however, the tissue-specific expression pattern of ABCC6 has been extensively studied. An initially puzzling observation was that liver is the primary site of ABCC6 expression distal from the tissues directly affected by mineralization in PXE.7 This paradox, together with studies using knockout mouse and rat models as a platform, was resolved by suggestions that PXE is a metabolic disorder with the primary molecular defect residing in the liver.8, 9
The estimated prevalence of PXE is approximately 1:50,000, which implies that there are approximately 7000 affected individuals in the United States. If extrapolated to the global population, the number of patients experiencing PXE increases to approximately 150,000 worldwide. More than 300 distinct loss-of-function mutations have been encountered in the ABCC6 gene, including recurrent p.R1141X and g.del23-29, which account for up to approximately 45% of all mutations.10
The Spectrum of Ectopic Mineralization Disorders
There are several heritable ectopic mineralization disorders with overlapping phenotypic features.2, 11 Compared with PXE, patients with generalized arterial calcification of infancy (GACI) develop severe and early-onset mineralization of the cardiovascular system. Most children die from cardiovascular collapse within the first 6 months of life if left untreated.12 Arterial calcification due to deficiency of CD73 (ACDC) is a rare, adult-onset ectopic mineralization disorder affecting the arteries of the lower extremities and periarticular ligaments.13, 14 Mutations in the ABCC6, ENPP1, and NT5E genes underlie ectopic mineralization for the classic forms of PXE, GACI, and ACDC, respectively (Figure 2). Although these three conditions are distinct diagnostic entities with a different natural history, there are similarities in clinical presentations, particularly the vascular involvement. Some patients with ABCC6 mutations have manifestations similar to GACI, and some patients with ENPP1 mutations present with features of PXE.11, 15 These observations have led to the hypothesis that PXE and GACI represent the two ends of a clinical spectrum but share alterations in the same pathways. In fact, recent studies by us, and confirmed by others, have indicated a unifying pathomechanistic feature in PXE, GACI, and ACDC involving reduction of the circulating antimineralization factor, inorganic pyrophosphate (PPi) (Figure 2).
Figure 2.

Different pathomechanisms controlling ectopic mineralization. Hyperphosphatemic familial tumoral calcinosis (HFTC) is associated with altered Pi homeostasis (marked hyperphosphatemia) because of mutations in one of the three proteins involved in the regulation of inorganic phosphate (Pi) excretion in the kidney (GALNT3, FGF23, and KLOTHO). Normophosphatemic familial tumoral calcinosis (NFTC) is caused by mutations in the SAMD9 gene with unknown function in prevention of ectopic mineralization without altered serum phosphate levels. ATP-binding cassette, subfamily C, member 6 (ABCC6), ectonucleotide pyrophosphotase/phosphodiesterase 1 (ENPP1), and 5’-ecto nucleotidase (CD73) are plasma membrane–associated proteins controlling the synthesis and degradation of inorganic pyrophosphate (PPi). ENPP1 is the principal enzyme that generates PPi from ATP hydrolysis. ABCC6 works upstream of ENPP1 by mediating release of ATP, a substrate for ENPP1. CD73 cleaves AMP to adenosine and Pi, with adenosine being an inhibitor of tissue nonspecific alkaline phosphatase (TNAP), which degrades PPi. Mutations in these proteins result in pseudoxanthoma elasticum (PXE), generalized arterial calcification of infancy (GACI), and arterial calcification due to deficiency of CD73 (ACDC), respectively, all characterized by reduced plasma PPi levels. Loss-of-function mutations in the gene encoding TNAP, which degrades PPi to Pi, result in hypophosphatasia (HOPS) attributable to increased plasma PPi levels. Adapted with permission from Uitto et al.3 EC, extracellular; IC, intracellular.
In addition to the diseases involving reduced PPi in the pathomechanisms of ectopic mineralization, several heritable conditions with distinct gene defects, and presumably different pathomechanisms, have been characterized. One of such conditions is the PXE-like phenotype with multiple coagulation factor deficiency. These patients manifest with cutaneous findings reminiscent of PXE with ultrastructurally demonstrable depositions of calcium complexes in the dermis. However, the skin findings also include severe skin laxity with thick and leathery skin folds, findings reminiscent of cutis laxa. Thus, these patients demonstrate overlapping cutaneous features of PXE and cutis laxa.16 The patients also demonstrate deficiency in vitamin K–dependent coagulation factors (II, VII, IX, and X). No mutations were found in the ABCC6 or VKORC1 genes in these patients.16, 17 Instead, mutations have been disclosed in GGCX, a gene encoding γ-glutamyl carboxylase, together with reduced carboxylation of matrix gla protein. Thus, reduced matrix gla protein carboxylation allows mechanistically tissue mineralization in the skin to occur with phenotypic consequences. These studies established GGCX as the second gene locus causing PXE and provided additional mechanistic details for ectopic mineralization.16, 17 Interestingly, in one family with PXE-like cutaneous features, some individuals were heterozygous for a missense mutation p.V255M in GGCX and heterozygous for a null mutation p.R1141X in ABCC6, suggesting the digenic nature of their skin findings and implying a role for multiple genetic factors in pathologic tissue mineralization in general.18 Another ectopic mineralization disorder, familial tumoral calcinosis, exists in two broad forms (ie, hyperphosphatemic due to altered Pi homeostasis and normophosphatemic with normal serum Pi levels) (Figure 2). Hyperphosphatemic familial tumoral calcinosis is characterized by ectopic mineralization and hyperphosphatemia due to disturbed renal Pi reabsorption. The disease is caused by mutations in any one of three genes, GALNT3, KL, and FGF23, resulting in progressive mineralization in periarticular spaces and soft tissues.19 Normophosphatemic familial tumoral calcinosis manifests with extensive ectopic mineralization of the cutaneous tissues, associated with inflammatory manifestations preceding mineralization. Normophosphatemic familial tumoral calcinosis is caused by loss-of-function mutations in the SAMD9 gene with no currently known function.20, 21
Development of Animal Models to Study Ectopic Mineralization Disorders
The Abcc6−/− mouse was developed as a model for PXE in 2005.22, 23 Subsequently, several naturally occurring Abcc6 mutant mice have been characterized.24, 25, 26 The focus has been on phenotypic characterization of four mouse strains, KK/HlJ, C3H/HeJ, DBA/2J, and 129S1/SvImJ, that harbor the same single-nucleotide polymorphism (rs32756904) in the Abcc6 gene interfering with pre-mRNA splicing and resulting in frameshift of translation, but histopathology demonstrates highly variable mineralization phenotypes affecting several connective tissues. Attesting to the effect of the genetic background on the phenotype are observations that the KK/HlJ strain displays the most severe mineralization phenotype, whereas C3H/HeJ and DBA/2J strains with the same Abcc6 mutation demonstrate extremely mild mineralization. The acceleration of ectopic mineralization in these mice by an experimental diet, enriched in phosphate and low in magnesium, reinforces the role of dietary factors in modulation of the ectopic mineralization phenotype.26 Collectively, these mice have served as novel spontaneous models for PXE, and they have been used as a platform for examining the pathophysiology as well as for identification of genetic factors that modify the PXE phenotype.
Two mutant mouse strains, Enpp1asj and Enpp1asj-2J, have also been characterized as novel models for GACI.27, 28 The asj allele was shown to harbor a homozygous missense mutation, p.V246D, in the Enpp1 gene, resulting in marked reduction in the corresponding protein in the liver, associated with extensive mineralization of the skin and arterial blood vessels. Genetic characterization of the asj-2J allele revealed an insertion/deletion mutation in the Enpp1 gene, leading to complete loss of ENPP1 enzymatic activity and, consequently, the plasma PPi concentration was essentially undetectable, resulting in extensive ectopic mineralization of the arterial blood vessels. Thus, Enpp1asj and Enpp1asj-2J mice serve as novel models recapitulating the mineralization phenotype seen in patients with GACI. In further studies, an Nt5e−/− knockout mouse has been characterized as a model of ACDC.29 These mice demonstrated mineralization in the juxta-articular joint capsules but not in vascular tissues. These mouse models attest to the presence of complex promineralization/antimineralization networks that are required under physiological homeostatic conditions to prevent ectopic tissue mineralization.
The mouse models of PXE have been useful for detailed examination of pathomechanisms of ectopic mineralization in general, and they have provided a platform to develop novel treatment approaches. Mice, however, are too small to examine certain fundamental aspects of pathogenesis by experimental procedures, such as liver and kidney transplantation. Recently, a knockout rat model for PXE was developed by targeted inactivation of the Abcc6 gene by zinc finger nuclease, for the purpose of providing a larger animal model for pathophysiological studies.9 This rat model has been shown to develop ectopic mineralization similar to PXE in mice and humans, and their plasma PPi levels are reduced to <30% of the wild-type controls. Liver and kidney perfusion studies in this rat model suggested a critical role for hepatic ABCC6-dependent formation of PPi under physiological conditions, and ABCC6 expression in the liver has been identified as the main source of PPi in circulation.9
In addition to the rodent models for PXE, zebrafish have provided another system to study ectopic mineralization disorders. There are a total of four zebrafish mutants for the human ABCC6 ortholog abcc6a in zebrafish that have been characterized, including a stable abcc6a knockout zebrafish.30 Although there are similarities in the axial skeleton hypermineralization phenotype in this and three other abcc6a zebrafish models,31, 32, 33 none of them develops ectopic mineralization in the skin, eyes, and vasculature in a similar pattern as seen in patients with PXE and Abcc6−/− murine models of PXE.
Identification of Antimineralization Compounds in Mouse Models of Ectopic Mineralization: Bisphosphonates
The unifying pathogenic feature in mouse models for PXE, GACI, and ACDC is reduced plasma levels of PPi, and this has also been demonstrated in patients with PXE and GACI.2, 3 Because PPi is a strong antimineralization factor, whereas Pi promotes mineralization, a reduced PPi/Pi ratio results in ectopic mineralization. A straightforward approach to counteract ectopic mineralization in these mice would be to normalize the PPi levels in plasma by direct PPi infusion. However, because of instability and the short half-life of PPi, stable PPi analogs, bisphosphonates, have been tested in the murine model systems of ectopic mineralization (Figure 3).34, 35 Bisphosphonates have two major pharmacologic properties.36 On one hand, they prevent the ectopic mineralization, whereas on the other hand, they prevent osteoporosis by inhibiting osteoclast activity. The balance between the two effects is dependent on their chemical structure. The newer, nitrogen-containing bisphosphonates predominantly inhibit osteoclast activity, whereas the older, non–nitrogen-containing bisphosphonates prevent ectopic mineralization and are far less potent inhibitors of osteoclasts. The potential efficacy of bisphosphates to prevent ectopic mineralization was tested in the Abcc6−/− mouse model for PXE.34 The treatment consisted of feeding the mice a diet containing etidronate or injecting the mice with etidronate subcutaneously. The mice were treated at 4 weeks of age with etidronate, before the mineralization process ensues, and the degree of mineralization in muzzle skin was assessed at 12 weeks by either semiquantitative histopathology or direct chemical assay of calcium. Etidronate treatment in 12× quantities of the human doses for treating osteoporosis resulted in a significant reduction in the amount of mineralization with concomitant changes in the bone microarchitecture in the Abcc6−/− mice (Figure 3). Interestingly, the nitrogen-containing bisphosphonate, alendronate, did not show a therapeutic effect in the Abcc6−/− mice, which was attributed to its lower antimineralization potency.34
Figure 3.

Treatment of Abcc6−/− mice, a model of pseudoxanthoma elasticum, with etidronate, a bisphosphonate analog of inorganic pyrophosphate (PPi), prevents ectopic mineralization. A: Bisphosphonates (BPs) are stable analogs of PPi because of the presence of a carbon bond, instead of oxygen, and the presence of side chains (R1 and R2). B: Top left panel:Abcc6−/− mice at 12 weeks of age demonstrate extensive mineralization of the connective sheath of vibrissae in the muzzle skin, as demonstrated by Alizarin Red stain. Administration of etidronate either orally (p.o.; top middle panel) or by s.c. injections (top right panel) during weeks 4 to 12 significantly reduced the mineral deposits. Administration of etidronate also significantly increased the density of trabecular bone both in male (middle row) and in female (bottom row) mice.
Adapted with permission from Li et al.34
This study was accompanied by further characterization of the effects of bisphosphonates on ectopic mineralization combined with analysis of the bone microarchitecture in the Enpp1asj mouse model of GACI.35 Treatment with etidronate demonstrated dual beneficial effects in significantly reduced mineralization in the skin and aorta as well as corrected the hypomineralization of bone in Enpp1asj mice.35 A novel approach for treatment of GACI in Enpp1asj mice has been suggested by administration of recombinant human ENPP1 to compensate for the loss of the mouse endogenous ENPP1. This enzyme replacement therapy resulted in elevated plasma PPi levels, reduced the extent of ectopic mineralization, and prevented mortality.37
The clinical efficiency of etidronate to counteract ectopic mineralization was recently also determined in a cohort of PXE patients in the Treatment of Ectopic Mineralization in Pseudoxanthoma Elasticum Trial (TEMP). In this double-blinded study, etidronate was found to inhibit arterial media calcification and to reduce the appearance of ocular complications.38 These were remarkable results, considering the slowly progressive nature of PXE and the relatively limited time (1 year) the patients were treated. Although the results of the TEMP Trial are encouraging, more work is needed to determine the efficacy and especially safety of etidronate in PXE patients.
Dietary Magnesium as an Inhibitor of Ectopic Mineralization
Another recently completed clinical trial attempted to test the effect of oral magnesium on progression and severity of PXE. This study was predicated on previous demonstrations in the Abcc6−/− mouse model of PXE that increase of the dietary magnesium, fivefold over the standard rodent diet, completely prevented the ectopic mineralization in these animals.39 Conversely, lowering of the magnesium content of the diet to 20% of the standard, together with a twofold increase in phosphorus content, significantly accelerated the ectopic mineralization.40 The patients were treated with diets supplemented with magnesium, 1.2 g/day, and the phenotypic changes were assessed by clinical examination, histopathology, and imaging studies. Although some improvement was noted in patients receiving magnesium supplementation, the results did not reach statistical significance, potentially implying that 1-year follow-up over the natural history of PXE may not be long enough to document relatively small changes (M. Lebwohl, unpublished data). The adverse effects of magnesium supplementation include gastrointestinal disturbances, and it is unclear at this point whether magnesium supplementation plays a role as a potential treatment modality for PXE.
Functionality of ABCC6 as a Putative Efflux Transporter
The unifying pathogenic feature in mouse models for PXE and GACI is reduced plasma levels of PPi. Recent studies demonstrated ABCC6-dependent release of ATP, which is then converted to PPi, an effective inhibitor of calcification.41, 42 These studies used an in vitro model based on the functional reestablishment of polarity in freshly isolated (primary) hepatocyte cultures. In such cultures, the cell-cell contacts are retained in a similar manner as observed in liver tissue. It was shown that hepatocytes from wild-type mouse livers are able to release ATP over their sinusoidal membrane, followed by the appearance of PPi in medium of primary hepatocyte sandwich cultures, but significantly lower levels were detected in medium of hepatocytes lacking ABCC6. These results implied that hepatocytes release ATP in an ABCC6-dependent manner and are able to convert it to PPi. Similar observations were made when liver perfusates were analyzed in Abcc6−/− mice and rats,9, 42 indicating that hepatic ABCC6-mediated ATP release is the main source of circulating PPi, and loss of function of ABCC6 in PXE patients leads to plasma PPi deficiency with subsequent ectopic mineralization of connective tissues.
Although ABC proteins have been shown to transport cyclic nucleotides43, 44, 45 and nucleotide analogs,43, 46 ATP is not among the currently known transported substrates. Cells are known to release ATP via exocytosis or through anion channels, such as pannexins, CalH1, and connexin hemichannels. The published data now support the role of ABCC6 in release of ATP from (liver) cells.9, 41, 42 The ABCC6-dependent conduit allows passage of nucleoside triphosphates in general, among which ATP is quantitatively the most important.41 This is a peculiar function for an ABC transporter: Most ABC proteins use the energy released by intracellular ATP hydrolysis to transport specific substrates across membranes, often against steep concentration gradients.47 What makes ABCC6-mediated cellular ATP release intriguing is that ATP might not need an active transport mechanism to leave cells because there is a huge concentration gradient for ATP across the plasma membrane, and a channel would suffice to get ATP out of the cell.
In patients with PXE and in mutant Abcc6 mouse models, the PPi levels are approximately 30% of the controls, indicating the presence of other pathways independent of ABCC6 for release of ATP and generation of plasma PPi.48 In addition, the work by us and others suggests that PPi deficiency is the major, but not exclusive, cause of ectopic mineralization in PXE, and other as yet unknown mechanisms exist that are independent of PPi by which ABCC6 prevents ectopic mineralization under physiologic conditions.49, 50 To this end, recent observations suggest that ABCC6 is also involved in extracellular nucleotide metabolism, suggesting a central role for ABCC6 in purinergic signaling in the development of ectopic mineralization.51, 52
Potential Mechanisms of ABCC6-Dependent Release of ATP from Hepatocytes
It is presently unclear how ABCC6 mediates ATP release. One of the following mechanisms most likely accounts for the ATP release mediated by ABCC6: i) Similar to ABCC7, which encodes the cystic fibrosis transmembrane regulator, an ATP-gated chloride channel,53 ABCC6 might function as an ATP-dependent ATP channel. ii) ABCC6 could regulate an as yet unidentified ATP channel. Two examples within the C branch of the ABC superfamily that function via a similar mechanism are ABCC8 and ABCC9, both regulating the opening and closing of complex potassium channels.54 iii) ABCC6 might regulate exocytosis of ATP-loaded vesicles.55 iv) Most of the currently available data suggested that ABCC6 is simply an ATP-dependent efflux pump for ATP and other nucleotide triphosphates. Most members of the C subfamily of the ATP-biding cassette superfamily of membrane proteins, including the closest homolog of ABCC6, ABCC1, are bona fide organic anion efflux transporters,46, 56 and ABCC6 has also been shown to transport a few organic anions,57, 58 albeit sluggishly. ATP efflux rates found in HEK293-ABCC6 cells9 are compatible with direct transport because they are similar to the rates by which ABCC1 pumps morphine-3-glucuronide out of HEK293 cells.59 Intriguing differences in the nucleotide-binding properties between ABCC6 and ABCC1 also provide circumstantial evidence that ABCC6 transports ATP: Specifically, under conditions that support ATP hydrolysis (ie, the presence of substrate and temperature at 37°C), the nucleotide-binding domains of ABC transporters, including ABCC1, can be labeled with the photoactive ATP analog 8-azido-ATP. Labeling depends on the presence of phosphate analogs, such as orthovanadate or beryllium fluoride, to trap 8-azido-ADP at the nucleotide-binding domains. However, ABCC6 is also labeled in the absence of phosphate analogs.60 Moreover, although phosphate analogs increase 8-azido-ATP labeling of ABCC6, unlike ABCC1, this does not require the addition of a specific substrate.61 A plausible explanation for these results is that 8-azido-ATP binds not only to the nucleotide-binding domains of ABCC6, but also to its substrate-binding/translocation site.
Providing experimental proof for direct ABCC6-mediated ATP transport has proved to be challenging because of technical difficulties in following ATP transport in vesicular uptake experiments, which are considered the gold standard to prove that a specific molecule is a substrate of an ABC transporter. A major problem with following ATP transport in the vesicular uptake experiments is the fact that high concentrations (mmol/L range) of ATP are needed to drive transport by ABC proteins. Only a fraction of the added ATP will be transported into the vesicles under these conditions, resulting in an unfavorable signal/noise ratio. The vesicles used in vesicular transport experiments are generated from cells overproducing ABCC6.41 These cells also express other channels that allow passage of ATP and contribute to the high background signal in vesicular uptake experiments.55, 62 A solution for these problems might be the use of proteoliposomes with purified ABCC6, which will result in a more favorable signal/noise ratio. Despite the technical challenges, we expect that the mechanistic details of ABCC6-mediated cellular ATP release will soon be uncovered.
Demonstration of Elevated Plasma PPi Levels in PXE Mice and in Human Volunteers after Oral Administration of PPi
Reduced plasma PPi levels in PXE could be corrected by direct i.v. administration of PPi, but considering the possibility that the patients need to be continuously treated for prevention of the mineralization process, oral administration of PPi would be preferred. This approach has not been tested previously, presumably because it was assumed, on the basis of statements in the older literature, that PPi does not get absorbed from the gut.63 This assumption has been recently challenged by preliminary studies obtained in mice. The results of these studies demonstrated attenuation of ectopic mineralization in Abcc6−/− mice as a result of oral PPi administration.64
In subsequent studies, 10 healthy human volunteers ingested a bolus of tetrasodium pyrophosphate solution; six individuals served as controls, drinking water only. Elevated plasma PPi levels in each individual were noted 30 and 60 minutes after ingestion of the PPi solution.64 These studies attest to the fact that PPi can be absorbed from the gut after oral ingestion, associated with increased plasma PPi levels. However, some caution is warranted before advocating oral PPi as a treatment for PXE patients. The human data obtained so far show that a large dose is needed to achieve a relatively modest increase in plasma PPi concentration,64 with the major part of ingested PPi being converted into inorganic Pi, a potent promineralization factor. More important, our studies have shown that a diet with high amounts of Pi accelerates ectopic mineralization in Abcc6−/− rats and mice.9, 65 More work is, therefore, needed to determine at which dose oral PPi might have beneficial effects on soft tissue mineralization.
Collectively, these preliminary studies support the hypothesis that restoration of plasma PPi levels can be elicited by different mechanisms, including enhanced release of ATP from cells in an ABCC6-independent manner and by direct administration of PPi to the animal models and patients with PXE.
Outstanding Issues and Experimental Challenges
Although much progress has been made in the past decade related to the understanding of the pathomechanisms of PXE, developing effective therapies for systematic manifestations of PXE continues to be a challenge. One of the issues relates to the intrafamilial and interfamilial heterogeneity in PXE in families with the same mutations in the same gene. The most striking example of such phenotypic severity is the demonstration that some patients diagnosed as having GACI with extremely severe vascular calcification at the early postnatal period have ABCC6 mutations, whereas precisely the same mutation has been identified in families with slowly progressing late-onset manifestations of classic PXE.15 One plausible explanation is the presence of modifier genes that act on the phenotype predicated on biallelic ABCC6 mutations.3 Furthermore, extensive mouse studies have identified genomic regions that may harbor modifier genes, as noted by the extensive variability in the degree of ectopic mineralization as a result of the same Abcc6 single-nucleotide polymorphism in different strains of mice.3 Collectively, the ectopic mineralization phenotype in PXE is highly variable, reflecting the types and combinations of mutations in different genes, juxtaposed into the environmental factors and lifetime variables at the environment-genome interface.
Although some progress has been made in the development of potential treatment modalities for PXE, to the extent that some of them have entered early clinical trials, these approaches seem to be able to prevent development of mineral deposits; however, none of these therapies has proved efficacious in removing extensive pathologic mineralization. This notion is particularly important because the patients at the time of diagnosis have already extensive ectopic mineralization with accompanying clinical findings. In this regard,3 experimental treatments for patients with GACI consist of feeding the pregnant mother with etidronate, followed by treatment of the newborn affected individual with a combination of bisphosphonate and sodium thiosulfate, a US Food and Drug Administration–approved agent, with the ability to increase the solubility of calcium as used for the treatment of calciphylaxis.
Another approach to potentially reverse the existing tissue mineralization will take advantage of the ability of EDTA to chelate calcium in a targeted microenvironment. Specifically, the mineral deposits in PXE reside in elastic connective tissues in the skin, in the Bruch membrane of the retina, and in media of the arterial blood vessels. As a consequence, there is evidence that mineralization of these structures leads to elastorrhexis (ie, fragmentation of the elastic structures), exposing their elastin core. Thus, targeting EDTA to the sites of aberrant mineralization can be achieved by delivery in albumin nanoparticles, conjugated with antielastin antibodies. This concept has recently been used in in vitro and ex vivo studies and was shown to result in release of the calcium ions from arterial tissue.66, 67, 68 Consequently, in addition to preventing further mineralization by approaches such as etidronate, reversal of the calcium deposits is of utmost importance to alleviate the clinical findings that led to the original diagnosis of PXE.
The latter observations emphasize that there are several issues that are critical for successful development of clinical trials for PXE. First, there is no reliable serum biomarker that would allow accurate monitoring of the disease activity as a result of drug treatment. Furthermore, the natural history of PXE is protean, and even a 1-year follow-up may not be sufficient to capture net changes in the phenotype that may accompany the therapy. Finally, recent rodent studies with mouse and rat models of PXE have suggested that in addition to the pathway involving alterations in the plasma level of PPi, additional mechanisms complementary to this pathway may exist.49, 50, 51
In summary, PXE is a complex ectopic mineralization disorder. With the advent of enhanced understanding of the pathomechanisms of ectopic mineralization as a result of mutations in ABCC6, several potential approaches are currently being tested toward development of treatment modalities for this currently intractable disorder. Some of these treatment approaches have already reached the early clinical trial levels, and it is conceivable that novel efficient treatments may become available soon.
Acknowledgments
This Mini-Review is dedicated to the honor of Professor Andras Váradi on his 70th birthday.
We thank Carol Kelly for assistance in manuscript preparation.
Footnotes
Supported by the NIH/National Institute of Arthritis and Musculoskeletal and Skin Diseases grants K01AR64766 (Q.L.), R01AR28450 (J.U.), R01AR55225 (J.U., Q.L.), and R01AR72695 (Q.L., J.U., K.v.d.W.); and PXE International (K.v.d.W.).
Disclosures: None declared.
Supplemental material for this article can be found at https://doi.org/10.1016/j.ajpath.2018.09.014.
Supplemental Data
References
- 1.Budoff M.J., Shaw L.J., Liu S.T., Weinstein S.R., Mosler T.P., Tseng P.H., Flores F.R., Callister T.Q., Raggi P., Berman D.S. Long-term prognosis associated with coronary calcification: observations from a registry of 25,253 patients. J Am Coll Cardiol. 2007;49:1860–1870. doi: 10.1016/j.jacc.2006.10.079. [DOI] [PubMed] [Google Scholar]
- 2.Li Q., Uitto J. Mineralization/anti-mineralization networks in the skin and vascular connective tissues. Am J Pathol. 2013;183:10–18. doi: 10.1016/j.ajpath.2013.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Uitto J., Li Q., van de Wetering K., Varadi A., Terry S.F. Insights into pathomechanisms and treatment development in heritable ectopic mineralization disorders: summary of the PXE International Biennial Research Symposium-2016. J Invest Dermatol. 2017;137:790–795. doi: 10.1016/j.jid.2016.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Neldner K.H. Pseudoxanthoma elasticum. Clin Dermatol. 1988;6:1–159. doi: 10.1016/0738-081x(88)90003-x. [DOI] [PubMed] [Google Scholar]
- 5.Christiano A.M., Lebwohl M.G., Boyd C.D., Uitto J. Workshop on pseudoxanthoma elasticum: molecular biology and pathology of the elastic fibers: Jefferson Medical College, Philadelphia, Pennsylvania, June 10, 1992. J Invest Dermatol. 1992;99:660–663. doi: 10.1111/1523-1747.ep12668156. [DOI] [PubMed] [Google Scholar]
- 6.Raybould M.C., Birley A.J., Moss C., Hulten M., McKeown C.M. Exclusion of an elastin gene (ELN) mutation as the cause of pseudoxanthoma elasticum (PXE) in one family. Clin Genet. 1994;45:48–51. doi: 10.1111/j.1399-0004.1994.tb03990.x. [DOI] [PubMed] [Google Scholar]
- 7.Belinsky M.G., Kruh G.D. MOAT-E (ARA) is a full-length MRP/cMOAT subfamily transporter expressed in kidney and liver. Br J Cancer. 1999;80:1342–1349. doi: 10.1038/sj.bjc.6690527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jiang Q., Endo M., Dibra F., Wang K., Uitto J. Pseudoxanthoma elasticum is a metabolic disease. J Invest Dermatol. 2009;129:348–354. doi: 10.1038/jid.2008.212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Li Q., Kingman J., van de Wetering K., Tannouri S., Sundberg J.P., Uitto J. Abcc6 knockout rat model highlights the role of liver in PPi homeostasis in pseudoxanthoma elasticum. J Invest Dermatol. 2017;137:1025–1032. doi: 10.1016/j.jid.2016.11.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pfendner E.G., Vanakker O.M., Terry S.F., Vourthis S., McAndrew P.E., McClain M.R., Fratta S., Marais A.S., Hariri S., Coucke P.J., Ramsay M., Viljoen D., Terry P.F., De Paepe A., Uitto J., Bercovitch L.G. Mutation detection in the ABCC6 gene and genotype-phenotype analysis in a large international case series affected by pseudoxanthoma elasticum. J Med Genet. 2007;44:621–628. doi: 10.1136/jmg.2007.051094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Nitschke Y., Baujat G., Botschen U., Wittkampf T., du Moulin M., Stella J., Le Merrer M., Guest G., Lambot K., Tazarourte-Pinturier M.F., Chassaing N., Roche O., Feenstra I., Loechner K., Deshpande C., Garber S.J., Chikarmane R., Steinmann B., Shahinyan T., Martorell L., Davies J., Smith W.E., Kahler S.G., McCulloch M., Wraige E., Loidi L., Hohne W., Martin L., Hadj-Rabia S., Terkeltaub R., Rutsch F. Generalized arterial calcification of infancy and pseudoxanthoma elasticum can be caused by mutations in either ENPP1 or ABCC6. Am J Hum Genet. 2012;90:25–39. doi: 10.1016/j.ajhg.2011.11.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rutsch F., Boyer P., Nitschke Y., Ruf N., Lorenz-Depierieux B., Wittkampf T., Weissen-Plenz G., Fischer R.J., Mughal Z., Gregory J.W., Davies J.H., Loirat C., Strom T.M., Schnabel D., Nurnberg P., Terkeltaub R. Hypophosphatemia, hyperphosphaturia, and bisphosphonate treatment are associated with survival beyond infancy in generalized arterial calcification of infancy. Circ Cardiovasc Genet. 2008;1:133–140. doi: 10.1161/CIRCGENETICS.108.797704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.St Hilaire C., Ziegler S.G., Markello T.C., Brusco A., Groden C., Gill F., Carlson-Donohoe H., Lederman R.J., Chen M.Y., Yang D., Siegenthaler M.P., Arduino C., Mancini C., Freudenthal B., Stanescu H.C., Zdebik A.A., Chaganti R.K., Nussbaum R.L., Kleta R., Gahl W.A., Boehm M. NT5E mutations and arterial calcifications. N Engl J Med. 2011;364:432–442. doi: 10.1056/NEJMoa0912923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Markello T.C., Pak L.K., St Hilaire C., Dorward H., Ziegler S.G., Chen M.Y., Chaganti K., Nussbaum R.L., Boehm M., Gahl W.A. Vascular pathology of medial arterial calcifications in NT5E deficiency: implications for the role of adenosine in pseudoxanthoma elasticum. Mol Genet Metab. 2011;103:44–50. doi: 10.1016/j.ymgme.2011.01.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Li Q., Brodsky J.L., Conlin L., Pawel B., Glatz A., Gafni R.I., Schurgers L.J., Uitto J., Hakonarson H., Deardoff M.A., Levine M. Mutations in the ABCC6 gene as a cause of generalized arterial calcification of infancy: genotypic overlap with pseudoxanthoma elasticum. J Invest Dermatol. 2014;134:658–665. doi: 10.1038/jid.2013.370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Vanakker O.M., Martin L., Gheduzzi D., Leroy B.P., Loeys B.L., Guerci V.I., Matthys D., Terry S.F., Coucke P.J., Pasquali-Ronchetti I., De Paepe A. Pseudoxanthoma elasticum-like phenotype with cutis laxa and multiple coagulation factor deficiency represents a separate genetic entity. J Invest Dermatol. 2007;127:581–587. doi: 10.1038/sj.jid.5700610. [DOI] [PubMed] [Google Scholar]
- 17.Li Q., Schurgers L.J., Smith A.C., Tsokos M., Uitto J., Cowen E.W. Co-existent pseudoxanthoma elasticum and vitamin K-dependent coagulation factor deficiency: compound heterozygosity for mutations in the GGCX gene. Am J Pathol. 2009;174:534–540. doi: 10.2353/ajpath.2009.080865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Li Q., Grange D.K., Armstrong N.L., Whelan A.J., Hurley M.Y., Rishavy M.A., Hallgren K.W., Berkner K.L., Schurgers L.J., Jiang Q., Uitto J. Mutations in the GGCX and ABCC6 genes in a family with pseudoxanthoma elasticum-like phenotypes. J Invest Dermatol. 2009;129:553–563. doi: 10.1038/jid.2008.271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Farrow E.G., Imel E.A., White K.E. Miscellaneous non-inflammatory musculoskeletal conditions: hyperphosphatemic familial tumoral calcinosis (FGF23, GALNT3 and alphaKlotho) Best Pract Res Clin Rheumatol. 2011;25:735–747. doi: 10.1016/j.berh.2011.10.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Topaz O., Indelman M., Chefetz I., Geiger D., Metzker A., Altschuler Y., Choder M., Bercovich D., Uitto J., Bergman R., Richard G., Sprecher E. A deleterious mutation in SAMD9 causes normophosphatemic familial tumoral calcinosis. Am J Hum Genet. 2006;79:759–764. doi: 10.1086/508069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sprecher E. Familial tumoral calcinosis: from characterization of a rare phenotype to the pathogenesis of ectopic calcification. J Invest Dermatol. 2010;130:652–660. doi: 10.1038/jid.2009.337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Klement J.F., Matsuzaki Y., Jiang Q.J., Terlizzi J., Choi H.Y., Fujimoto N., Li K., Pulkkinen L., Birk D.E., Sundberg J.P., Uitto J. Targeted ablation of the Abcc6 gene results in ectopic mineralization of connective tissues. Mol Cell Biol. 2005;25:8299–8310. doi: 10.1128/MCB.25.18.8299-8310.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gorgels T.G., Hu X., Scheffer G.L., van der Wal A.C., Toonstra J., de Jong P.T., van Kuppevelt T.H., Levelt C.N., de Wolf A., Loves W.J., Scheper R.J., Peek R., Bergen A.A. Disruption of Abcc6 in the mouse: novel insight in the pathogenesis of pseudoxanthoma elasticum. Hum Mol Genet. 2005;14:1763–1773. doi: 10.1093/hmg/ddi183. [DOI] [PubMed] [Google Scholar]
- 24.Berndt A., Li Q., Potter C.S., Liang Y., Silva K.A., Kennedy V., Uitto J., Sundberg J.P. A single-nucleotide polymorphism in the Abcc6 gene associates with connective tissue mineralization in mice similar to targeted models for pseudoxanthoma elasticum. J Invest Dermatol. 2013;133:833–836. doi: 10.1038/jid.2012.340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Li Q., Berndt A., Guo H., Sundberg J., Uitto J. A novel animal model for pseudoxanthoma elasticum: the KK/HlJ mouse. Am J Pathol. 2012;181:1190–1196. doi: 10.1016/j.ajpath.2012.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Li Q., Guo H., Chou D.W., Berndt A., Sundberg J.P., Uitto J. Mouse models for pseudoxanthoma elasticum: genetic and dietary modulation of the ectopic mineralization phenotypes. PLoS One. 2013;9:e89268. doi: 10.1371/journal.pone.0089268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Li Q., Guo H., Chou D.W., Berndt A., Sundberg J.P., Uitto J. Mutant Enpp1asj mouse as a model for generalized arterial calcification of infancy. Dis Model Mech. 2013;6:1227–1235. doi: 10.1242/dmm.012765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Li Q., Pratt C.H., Dionne L.A., Fairfield H., Karst S.Y., Sundberg J.P., Uitto J. Spontaneous asj-2J mutant mouse as a model for generalized arterial calcification of infancy: a large deletion/insertion mutation in the Enpp1 gene. PLoS One. 2014;9:e113542. doi: 10.1371/journal.pone.0113542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Li Q., Price T.P., Sundberg J.P., Uitto J. Juxta-articular joint-capsule mineralization in CD73 deficient mice: similarities to patients with NT5E mutations. Cell Cycle. 2014;13:2609–2615. doi: 10.4161/15384101.2014.943567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.van Gils M., Willaert A., De Vilder E., Coucke P., Vanakker O. Generation and validation of the first complete knockout model of abcc6a in zebrafish. J Invest Dermatol. 2018;138:2333–2342. doi: 10.1016/j.jid.2018.06.183. [DOI] [PubMed] [Google Scholar]
- 31.Li Q., Sadowski S., Frank M., Chai C., Varadi A., Ho S.Y., Lou H., Dean M., Thisse C., Thisse B., Uitto J. The abcc6a gene expression is required for normal zebrafish development. J Invest Dermatol. 2010;130:2561–2568. doi: 10.1038/jid.2010.174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Busch-Nentwich E., Kettleborough R., Harvey S., Collins J., Ding M., Dooley C., Fenyes F., Gibbons R., Herd C., Mehroke S., Scahill C., Sealy I., Wali N., White R., Stemple D.L. ZFIN Direct Data; 2012. Sanger Institute Zebrafish Mutation Project Mutant, Phenotype and Image Data Submission. ZDB-PUB-120207-1. [Google Scholar]
- 33.Mackay E.W., Apschner A., Schulte-Merker S. Vitamin K reduces hypermineralisation in zebrafish models of PXE and GACI. Development. 2015;142:1095–1101. doi: 10.1242/dev.113811. [DOI] [PubMed] [Google Scholar]
- 34.Li Q., Sundberg J.P., Levine M.A., Terry S.F., Uitto J. The effects of bisphosphonates on ectopic soft tissue mineralization caused by mutations in the ABCC6 gene. Cell Cycle. 2015;14:1082–1089. doi: 10.1080/15384101.2015.1007809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Li Q., Kingman J., Sundberg J.P., Levine M.A., Uitto J. Dual effects of bisphosphonates on ectopic skin and vascular soft tissue mineralization versus bone microarchitecture in a mouse model of generalized arterial calcification of infancy. J Invest Dermatol. 2016;136:275–283. doi: 10.1038/JID.2015.377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Russell R.G. Bisphosphonates: from bench to bedside. Ann N Y Acad Sci. 2006;1068:367–401. doi: 10.1196/annals.1346.041. [DOI] [PubMed] [Google Scholar]
- 37.Albright R.A., Stabach P., Cao W., Kavanagh D., Mullen I., Braddock A.A., Covo M.S., Tehan M., Yang G., Cheng Z., Bouchard K., Yu Z.X., Thorn S., Wang X., Folta-Stogniew E.J., Negrete A., Sinusas A.J., Shiloach J., Zubal G., Madri J.A., De La Cruz E.M., Braddock D.T. ENPP1-Fc prevents mortality and vascular calcifications in rodent model of generalized arterial calcification of infancy. Nat Commun. 2015;6:10006. doi: 10.1038/ncomms10006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kranenburg G., de Jong P.A., Bartstra J.W., Lagerweij S.J., Lam M.G., Ossewaarde-van Norel J., Risseeuw S., van Leeuwen R., Imhof S.M., Verhaar H.J., de Vries J.J., Slart R.H.J.A., Luurtsema G., den Harder A.M., Visseren F.L.J., Mali W.P., Spiering W. Etidronate for prevention of ectopic mineralization in patients with pseudoxanthoma elasticum. J Am Coll Cardiol. 2018;71:1117–1126. doi: 10.1016/j.jacc.2017.12.062. [DOI] [PubMed] [Google Scholar]
- 39.LaRusso J., Li Q., Jiang Q., Uitto J. Elevated dietary magnesium prevents connective tissue mineralization in a mouse model of pseudoxanthoma elasticum (Abcc6−/−) J Invest Dermatol. 2009;129:1388–1394. doi: 10.1038/jid.2008.391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Jiang Q., Uitto J. Restricting dietary magnesium accelerates ectopic connective tissue mineralization in a mouse model of pseudoxanthoma elasticum (Abcc6−/−) Exp Dermatol. 2012;21:694–699. doi: 10.1111/j.1600-0625.2012.01553.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Jansen R.S., Kucukosmanoglu A., de Haas M., Sapthu S., Otero J.A., Hegman I.E., Bergen A.A., Gorgels T.G., Borst P., van de Wetering K. ABCC6 prevents ectopic mineralization seen in pseudoxanthoma elasticum by inducing cellular nucleotide release. Proc Natl Acad Sci U S A. 2013;110:20206–20211. doi: 10.1073/pnas.1319582110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Jansen R.S., Duijst S., Mahakena S., Sommer D., Szeri F., Varadi A., Plomp A., Bergen A.A., Oude Elferink R.P., Borst P., van de Wetering K. ABCC6-mediated ATP secretion by the liver is the main source of the mineralization inhibitor inorganic pyrophosphate in the systemic circulation-brief report. Arterioscler Thromb Vasc Biol. 2014;34:1985–1989. doi: 10.1161/ATVBAHA.114.304017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Reid G., Wielinga P., Zelcer N., De Haas M., Van Deemter L., Wijnholds J., Balzarini J., Borst P. Characterization of the transport of nucleoside analog drugs by the human multidrug resistance proteins MRP4 and MRP5. Mol Pharmacol. 2003;63:1094–1103. doi: 10.1124/mol.63.5.1094. [DOI] [PubMed] [Google Scholar]
- 44.Wielinga P.R., van der Heijden I., Reid G., Beijnen J.H., Wijnholds J., Borst P. Characterization of the MRP4- and MRP5-mediated transport of cyclic nucleotides from intact cells. J Biol Chem. 2003;278:17664–17671. doi: 10.1074/jbc.M212723200. [DOI] [PubMed] [Google Scholar]
- 45.de Wolf C.J., Yamaguchi H., van der Heijden I., Wielinga P.R., Hundscheid S.L., Ono N., Scheffer G.L., de Haas M., Schuetz J.D., Wijnholds J., Borst P. cGMP transport by vesicles from human and mouse erythrocytes. FEBS J. 2007;274:439–450. doi: 10.1111/j.1742-4658.2006.05591.x. [DOI] [PubMed] [Google Scholar]
- 46.Borst P., de Wolf C., van de Wetering K. Multidrug resistance-associated proteins 3, 4, and 5. Pflugers Arch. 2007;453:661–673. doi: 10.1007/s00424-006-0054-9. [DOI] [PubMed] [Google Scholar]
- 47.Borst P., Elferink R.O. Mammalian ABC transporters in health and disease. Annu Rev Biochem. 2002;71:537–592. doi: 10.1146/annurev.biochem.71.102301.093055. [DOI] [PubMed] [Google Scholar]
- 48.Lohman A.W., Billaud M., Isakson B.E. Mechanisms of ATP release and signalling in the blood vessel wall. Cardiovasc Res. 2012;95:269–280. doi: 10.1093/cvr/cvs187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Pomozi V., Brampton C., van de Wetering K., Zoll J., Calio B., Pham K., Owens J.B., Marh J., Moisyadi S., Varadi A., Martin L., Bauer C., Erdmann J., Aherrahrou Z., Le Saux O. Pyrophosphate supplementation prevents chronic and acute calcification in ABCC6-deficient mice. Am J Pathol. 2017;187:1258–1272. doi: 10.1016/j.ajpath.2017.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zhao J., Kingman J., Sundberg J.P., Uitto J., Li Q. Plasma PPi deficiency is the major, but not the exclusive, cause of ectopic mineralization in an Abcc6(−/−) mouse model of PXE. J Invest Dermatol. 2017;137:2336–2343. doi: 10.1016/j.jid.2017.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kauffenstein G., Yegutkin G.G., Khiati S., Pomozi V., Le Saux O., Leftheriotis G., Lenaers G., Henrion D., Martin L. Alteration of extracellular nucleotide metabolism in pseudoxanthoma elasticum. J Invest Dermatol. 2018;138:1862–1870. doi: 10.1016/j.jid.2018.02.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ziegler S.G., Ferreira C.R., MacFarlane E.G., Riddle R.C., Tomlinson R.E., Chew E.Y., Martin L., Ma C.T., Sergienko E., Pinkerton A.B., Millan J.L., Gahl W.A., Dietz H.C. Ectopic calcification in pseudoxanthoma elasticum responds to inhibition of tissue-nonspecific alkaline phosphatase. Sci Transl Med. 2017;9:eaal1669. doi: 10.1126/scitranslmed.aal1669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Liu F., Zhang Z., Csanady L., Gadsby D.C., Chen J. Molecular structure of the human CFTR ion channel. Cell. 2017;169:85–95.e8. doi: 10.1016/j.cell.2017.02.024. [DOI] [PubMed] [Google Scholar]
- 54.Bryan J., Munoz A., Zhang X., Dufer M., Drews G., Krippeit-Drews P., Aguilar-Bryan L. ABCC8 and ABCC9: ABC transporters that regulate K+ channels. Pflugers Arch. 2007;453:703–718. doi: 10.1007/s00424-006-0116-z. [DOI] [PubMed] [Google Scholar]
- 55.Lazarowski E.R. Vesicular and conductive mechanisms of nucleotide release. Purinergic Signal. 2012;8:359–373. doi: 10.1007/s11302-012-9304-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Borst P., Evers R., Kool M., Wijnholds J. The multidrug resistance protein family. Biochim Biophys Acta. 1999;1461:347–357. doi: 10.1016/s0005-2736(99)00167-4. [DOI] [PubMed] [Google Scholar]
- 57.Belinsky M.G., Chen Z.S., Shchaveleva I., Zeng H., Kruh G.D. Characterization of the drug resistance and transport properties of multidrug resistance protein 6 (MRP6, ABCC6) Cancer Res. 2002;62:6172–6177. [PubMed] [Google Scholar]
- 58.Ilias A., Urban Z., Seidl T.L., Le Saux O., Sinko E., Boyd C.D., Sarkadi B., Varadi A. Loss of ATP-dependent transport activity in pseudoxanthoma elasticum-associated mutants of human ABCC6 (MRP6) J Biol Chem. 2002;277:16860–16867. doi: 10.1074/jbc.M110918200. [DOI] [PubMed] [Google Scholar]
- 59.van de Wetering K., Zelcer N., Kuil A., Feddema W., Hillebrand M., Vlaming M.L., Schinkel A.H., Beijnen J.H., Borst P. Multidrug resistance proteins 2 and 3 provide alternative routes for hepatic excretion of morphine-glucuronides. Mol Pharmacol. 2007;72:387–394. doi: 10.1124/mol.107.035592. [DOI] [PubMed] [Google Scholar]
- 60.Cai J., Daoud R., Alqawi O., Georges E., Pelletier J., Gros P. Nucleotide binding and nucleotide hydrolysis properties of the ABC transporter MRP6 (ABCC6) Biochemistry. 2002;41:8058–8067. doi: 10.1021/bi012082p. [DOI] [PubMed] [Google Scholar]
- 61.Cai J., Daoud R., Georges E., Gros P. Functional expression of multidrug resistance protein 1 in Pichia pastoris. Biochemistry. 2001;40:8307–8316. doi: 10.1021/bi010093c. [DOI] [PubMed] [Google Scholar]
- 62.Clairmont C.A., De Maio A., Hirschberg C.B. Translocation of ATP into the lumen of rough endoplasmic reticulum-derived vesicles and its binding to luminal proteins including BiP (GRP 78) and GRP 94. J Biol Chem. 1992;267:3983–3990. [PubMed] [Google Scholar]
- 63.Orriss I.R., Arnett T.R., Russell R.G. Pyrophosphate: a key inhibitor of mineralisation. Curr Opin Pharmacol. 2016;28:57–68. doi: 10.1016/j.coph.2016.03.003. [DOI] [PubMed] [Google Scholar]
- 64.Dedinszki D., Szeri F., Kozak E., Pomozi V., Tokesi N., Mezei T.R., Merczel K., Letavernier E., Tang E., Le Saux O., Aranyi T., van de Wetering K., Varadi A. Oral administration of pyrophosphate inhibits connective tissue calcification. EMBO Mol Med. 2017;9:1463–1470. doi: 10.15252/emmm.201707532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Li Q., Uitto J. The mineralization phenotype in Abcc6−/− mice is affected by Ggcx gene deficiency and genetic background: a model for pseudoxanthoma elasticum. J Mol Med (Berl) 2010;88:173–181. doi: 10.1007/s00109-009-0522-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Lei Y., Nosoudi N., Vyavahare N. Targeted chelation therapy with EDTA-loaded albumin nanoparticles regresses arterial calcification without causing systemic side effects. J Control Release. 2014;196:79–86. doi: 10.1016/j.jconrel.2014.09.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Lei Y., Grover A., Sinha A., Vyavahare N. Efficacy of reversal of aortic calcification by chelating agents. Calcif Tissue Int. 2013;93:426–435. doi: 10.1007/s00223-013-9780-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Sinha A., Shaporev A., Nosoudi N., Lei Y., Vertegel A., Lessner S., Vyavahare N. Nanoparticle targeting to diseased vasculature for imaging and therapy. Nanomedicine. 2014;10:1003–1012. doi: 10.1016/j.nano.2014.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.