

Abstract
DOT1-like protein (Dot1L) is the sole methyltransferase for methylation of lysine 79 in histone H3. Dot1L-dependent H3K79 methylation is involved in many biological processes, including telomeric silencing, cell cycle regulation, transcriptional activation and DNA repair. Genome-wide sequencing studies have revealed recurrent deletion and mutations of Dot1L gene in many types of human malignancies including ovarian cancer, however the role of Dot1L in ovarian cancer are largely unknown. To demonstrate the role of Dot1L in ovarian cancer, the expression of Dot1L was knocked out in ovarian cancer cells using CRISPR/Cas9 technology in the present study. Dot1L loss showed minimal effect on cell growth, but significantly promoted cell invasion and induced cancer stem-like cell property in ovarian cancer cells. Mechanistically, loss of Dot1L downregulated the expression of tight junction makers E-Cadherin and TJP1 and upregulated the expression of ALDH1A1 through Wnt signaling activation. Our data indicate potential tumor suppressor function of Dot1L in ovarian cancer, which is correlated with observed deletion of Dot1L gene in ovarian cancer patients, further study is granted to elucidate the function of Dot1L in tumorigenesis and progression in ovarian cancer.
Keywords: DOT1-like protein, ovarian cancer, cell invasion, cancer stem cell, Wnt signaling
Introduction
Post-translational modifications of histone are emerging as essential mechanisms to regulate gene expression. Distinct modifications of histone have been identified and well demonstrated, including acetylation, methylation, phosphorylation, ubiquitination and SUMOylation [1,2]. Those modifications interact and crosstalk with each other to concert gene transcription. Methylation was the firstly identified post-translational modification of histone, by adding a methyl group to lysine (K) or arginine (R) residue. Histone methylation is a dynamic and reversible process, which is mediated by Histone methyltransferases and histone demethylases [3,4]. Many histone methyltransferases have been shown to be involved in the initiation and development of human cancers, including ovarian cancers [5,6].
DOT1-like (Dot1L) protein is the human homology of yeast Dot1 (Disruptor of telomeric silencing 1), whose overexpression causes impaired telomeric silencing in yeast [7]. Further study has demonstrated Dot1L as a histone methyltransferase in human. Dot1L is the only known methyltransferase responsible for mono-, di-, and tri-methylation of lysine 79 of histone H3, as knockout of Dot1L led to complete loss of H3K79 methylation in yeast, flies, mice and humans [8,9]. Dot1L-mediated H3K79 methylation has demonstrated a wide range of regulatory functions in many biological processes, including telomeric silencing, cell cycle regulation, transcriptional activation and DNA repair [10,11].
Genome-wide next-generation sequencing studies have revealed recurrent somatic mutations of epigenetic regulators in human cancers. Deletion and somatic mutations of DOT1L gene are found in several types of solid cancers including melanoma, colorectal cancer and ovarian cancer (Figure 1A) [12,13]. Inactivating mutations of Dot1L has been identified in 4.4-15% of melanomas. Loss of Dot1L was shown to reduce H3K79 methylation and promote melanoma development in mice under UVR exposure, indicating a tumor suppressor function of Dot1L [14].
Figure 1.
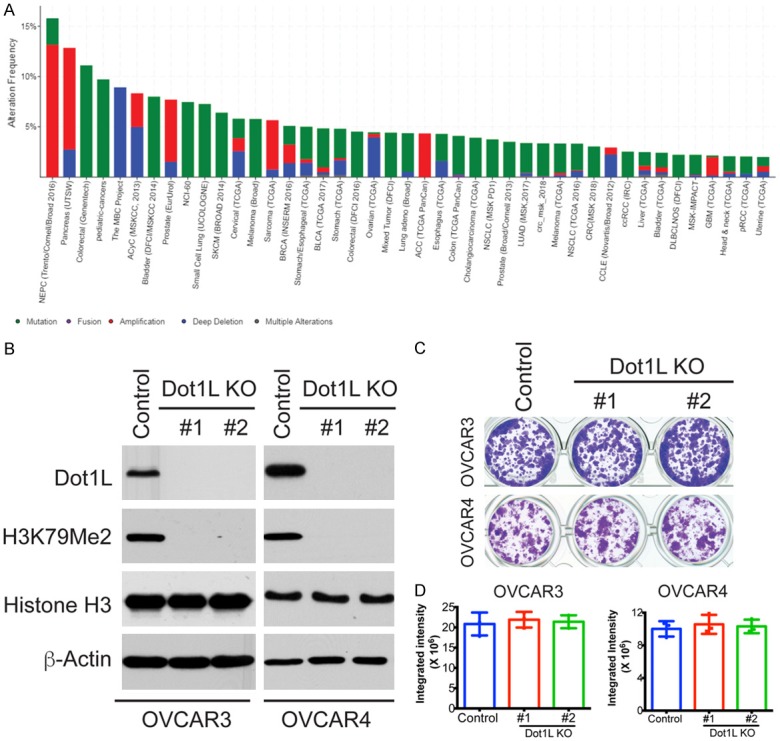
Dot1L knockout in ovarian cancer cells. (A) Dot1L mutations in multiple type of cancers. Dot1L mutations and CNV alterations were analyzed in multiple cancers from TCGA database. Sample number ≥ 50, mutation frequency > 2% were shown here. (B) Dot1L expression was knockout in ovarian cancer cells OVCAR3 and OVCAR4 with two different sgRNAs using the CRISPR methodology. Cells were selected in puromycin for 3 days, the expression of Dot1L and it mediated H3K79 Methylation was examined in Dot1L knockout cells by western blotting, total histone H3 and β-actin are used as loading controls. (C) Colony formation assay of OVCAR3 and OVCAR4 cells with Dot1L knockout by CRISPR. 1000 of indicated cells were plated into 24-well plate, and 7 days later cells were fixed and stained with 0.1% crystal violet. (D) Quantification of (C). Integrated intensity was quantified by Image J software. Mean of three independent experiments with SD were shown.
Here, we demonstrated the role of Dot1L in ovarian cancer by using CRISPR/Cas9 technology. Dot1L loss has minimal effect on cell growth, but significantly induced cancer-stem cells properties and promoted cell invasion ability. Mechanistically, loss of Dot1L enhances Wnt signaling and downregulates tight junction makers E-Cadherin and TJP1. Our results indicate potential tumor suppressor function in ovarian cancer, which is correlated with observed deletion of Dot1L gene in ovarian cancer patients.
Materials and methods
Cell lines, culture conditions and transfection
The ovarian cancer cell lines OVCAR3, OVCAR4 and CAOV4 cells were cultured in RPMI 1640 (Corning Life Sciences) supplemented with 10% fetal bovine serum (FBS; Sigma-Aldrich) and 1% penicillin/streptomycin at 37°C supplied with 5% CO2. Viral packing cell 293FT was cultured in Dulbecco’s modified Eagle’s medium (DMEM) with 10% FBS and 1% penicillin/streptomycin at 37°C supplied with 5% CO2. Mycoplasma testing was performed using LookOut Mycoplasma PCR detection (Sigma-Aldrich) every month. Transfection was performed using Lipofectamine 2000 (Life Technologies) following the manufacturer’s specifications. Each of the experiments was performed in triplicate in three independent experimental repeats unless otherwise stated.
Reagents and antibodies
lentiCRISPRv2 puro was a gift from Brett Stringer (Addgene plasmid # 98290). sgRNAs for Dot1L (#1: GAGACTGAAGTCGCCCGTGG; #2: GACCGGTGAGCGCGGCTTGG) were synthesized in Integrated DNA Technologies. The following antibodies were obtained from the indicated suppliers: anti-β-actin (Sigma-Aldrich, Cat. No. A5441, 1:10,000), Di-Methyl-Histone H3 (Lys79) (D15E8) XP® Rabbit mAb (Cell signaling, #5427 1:2000), anti-Dot1L (D1W4Z) Rabbit mAb (Cell signaling, #77087, 1:1000), mouse monoclonal anti-ALDH1A1 (BD Transduction Laboratories), E-cadherin Antibody (67A4) (santa cruz, sc-21791z), anti-ZO-1 (TJP1) Antibody (H-300) (santa cruz, sc-10804), anti-β-Catenin (D10A8) mAb (Cell signaling, #8480), anti- GAPDH (14C10) Rabbit mAb (Cell signaling, #2118).
Immunoblotting
Protein was isolated with RIPA buffer (50 mM Tris pH 8.0, 150 mM NaCl, 1% Triton X-100, 0.5% sodium deoxycholate and 1 mM PMSF). Protein concentration was measured using Bradford assay. Protein was separated on a SDS-PAGE and transferred to polyvinylidene fluoride membrane (Millipore). Membranes were blocked with 5% non-fat milk (Bio-Rad) in TBS/0.1% Tween 20 (TBST), and then incubated sequentially with primary and secondary antibodies.
Lentivirus production packaging and infection
Lentivirus was packaged using the Virapower Kit from Invitrogen according to the manufacturer’s instructions as described previously [15]. HEK293FT cells were transfected by Lipofectamine 2000. Lentivirus was harvested 48 hours post-transfection. Cells infected with viruses encoding the puromycin resistance gene were selected using 1 µg/ml puromycin for 48 h.
Quantitative RT-PCR (qRT-PCR)
RNA was extracted using Trizol (Invitrogen) according to manufacturer’s instruction, and then DNase treatment (RNeasy columns by Qiagen) was performed. Extracted RNAs expression was determined using the iTaq Universal SYBR Green One-step kit (Bio-Rad Laboratories) on the QuantStudio 3 Real-Time PCR System (Thermo Fisher). The primers sequences used for qRT-PCR are as follow: β-2-microglobulin (B2M) (forward, 5’-GGCATTCCTGAAGCTGACA-3’; reverse, 5’-CTTCAATGTCGGATGGATGAAAC-3’), E-cadherin (CDH1) (forward, 5’-CCCAATACATCTCCCTTCACAG-3’; reverse, 5’-CCACCTCTAAGGCCATCTTTG-3’), TJP1 (forward, 5’-TGCTGAGTCCTTT GGTGATG3’; reverse, 5’-AATTTGGATCTCCGGGAAGAC-3’), B2M was used as an internal control. Each sample was run in triplicate.
Colony formation assay
Colony formation was performed as previously described. Briefly, Cells were cultured in 12-well, 24-well or 96-well plates with different number according to the growth rate. Medium was changed every three days with appropriate drug doses for 12 days or until control wells became confluent. Colonies were washed twice with PBS and fixed with 10% methanol and 10% acetic acid in distilled water. Fixed colonies were stained with 0.05% crystal violet. Analysis was performed using NIH ImageJ software.
Cell invasion assay
Cell invasion capacity was assessed using a 24-well trans-well plate (8-μm pore size coated with matrigel (BD Biosciences). 1 × 105 of Dot1L knockout or control cells were resuspended in serum free medium and added to the upper chamber, the lower chamber was filled with medium supplemented with 10% FBS. Cells were incubated at 37°C for 48 hours. The cells on the upper surface were scraped with cotton swabs and washed away, whereas the invaded cells on the lower surface were fixed, stained with 0.1% crystal violet for 1 h, and quantified by determining the cell number in three randomly chosen visual fields. Assays was performed in duplicate and repeated three independent times.
Aldefluor assay and flow cytometry
Aldefluor assay kit (Stem Cell) was used to measure the aldehyde dehydrogenase enzymatic activity (ALDH) following the manufacturer’s instructions. Briefly, 200,000 cells were re-suspended in aldefluor assay buffer and ALDH substrate and incubated at 37°C for 30 min. Cells were washed in PBS, and the fluorescence intensity was analyzed using a BD FACSCalibur flow cytometer. DEAB treated cells were used as a negative control [15].
Wnt reporter transfection assays
The TOPFLASH reporter plasmid and RLSV40 Renilla plasmid were co-transfected into control or Dot1L knockout cells using Lipofectamine 3000. At 48 h after transfection, luciferase activity was measured using Dual-Luciferase Reporter Assay System (Promega) and Victor X3 2030 Multilabel Reader (Perkin Elmer) according to the manufacturer’s instructions. Data was normalized based upon control Renilla luciferase activity. Each group was repeated in triplicates.
Results
Dot1L knockout in ovarian cancer cells has no effect on cell growth
Multiple Sequence studies from different groups have observed Dot1L mutations and CNV alterations in many types of cancer, including ovarian cancer [12,13]. However, the exact role of Dot1L and its alteration in ovarian cancer in largely unknown. To investigate the role of Dot1L in ovarian cancer, we depleted Dot1L expression using CRISPR-Cas9 technology in ovarian cancer cells OVCAR3 and OVCAR4 to mimic deletion of Dot1L observed in patient samples. The deletion of Dot1L was confirmed by western blotting, both sgRNAs could successfully knockout Dot1L expression in both cell lines (Figure 1B). Dot1L is a unique histone methyltransferase solely responsible for mono-, di- and tri- methylation of histone H3 lysine 79 (H3K79) residue, knockout of Dot1L completely removed H3K79 methylation of histone H3 without affecting total H3 expression level (Figure 1B), further demonstrate that Dot1L is exclusive for H3K79 methylation in mammalian cells. Colony formation assay showed that Dot1L knockout has no significant effect on cell growth in both OVCAR3 and OVCAR4 cells (Figure 1C and 1D). Taken together, those data suggesting that Dot1L knockout didn’t alter cell growth of ovarian cancer cells in vitro.
Dot1L knockout promote cell invasion in ovarian cancer cells
Above results prompt us to further explore the potential biological function of Dot1L in ovarian cancer progression. Cell invasion ability allows cancer cells to enter lymphatic and/or blood vessels for dissemination into the circulation, followed by invasion to distant organs for metastatic growth [16,17], therefore next we sought to determine if Dot1L knockout promote cell invasion. To demonstrate the potential effect of Dot1L on cell invasion in ovarian cancer cells, Matrigel Invasion assay was performed with control or Dot1L knockout cells. Indeed, Dot1L knockout significantly promote cell invasion resulted in about 10-fold and 20-fold increase in the number of invading cells respectively in both OVCAR3 and OVCAR4 cells (Figure 2). Two different sgRNAs targeting Dot1L showed similar results in both cell lines, strongly indicating that Dot1L play essential role in cancer cell invasion.
Figure 2.
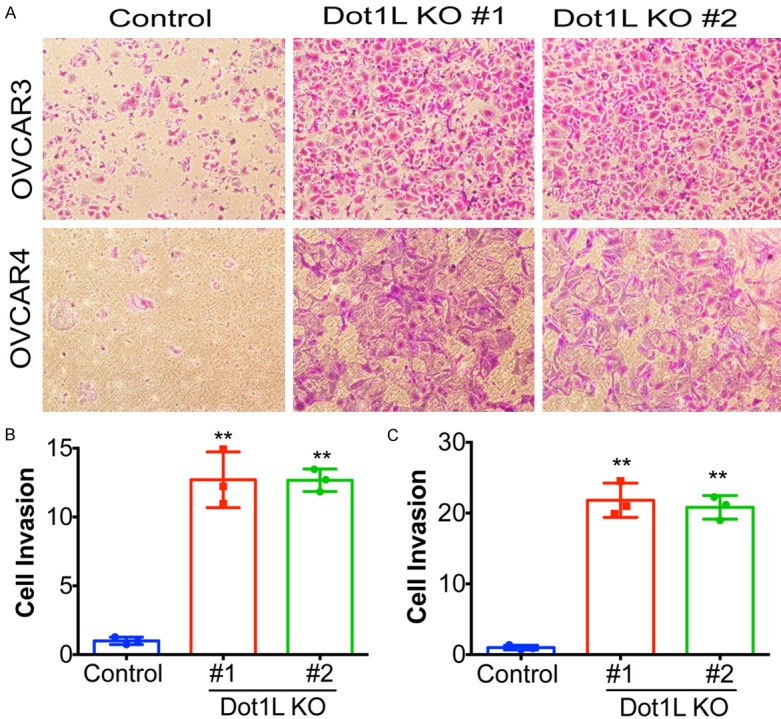
Dot1L knockout promotes cell migration of ovarian cancer cells. (A) OVCAR3 and OVCAR4 cells were infected with the Cas9 and sgRNA-expressing or control (Cas9 only) lentivirus, after selected in puromycin, the cells were subjected to trans-well migration assays. Representative images of cell migration were shown. (B and C) Same as (A) but quantified for crystal violet staining of OVCAR3 cells (B) and OVCAR4 cells (C). A total of 200 cells were examined for each of the indicated groups, error bars represent mean of three independent experiments with SD. **P < 0.01.
Depletion of Dot1L decreases expression of E-cadherin and TJP1
The diminution of cell-cell adhesion is instrumental in the loss of epithelial features occurring in epithelial-mesenchymal transition (EMT) processes associated with the metastatic conversion of epithelial tumor cells [18,19]. At the molecular level, this involves the reorganization of cell-cell adhesion complexes including adherent junctions (AJ) and tight junctions (TJ). Dysregulation of proteins involved in AJs and TJs such as E-cadherin and TJP1 has been the major critical molecule contributing to EMT and cell invasion [16]. Thus, we tested the expression of E-cadherin and TJP1 in Dot1L depleted OVCAR3 and OVCAR4 cells. Our results show that the protein and mRNA level of both E-cadherin and TJP1 are significantly downregulated by Dot1L knockout in ovarian cancer cells (Figure 3A-D). Conversely, restoration of Dot1L expression in Dot1L-deficient CAOV4 cells increased the mRNA level of both E-cadherin and TJP1 (Figure 3E and 3F). Those results together suggest that Dot1L depletion disrupt cell junctions and promote EMT by decreasing E-cadherin and TJP1 expression.
Figure 3.
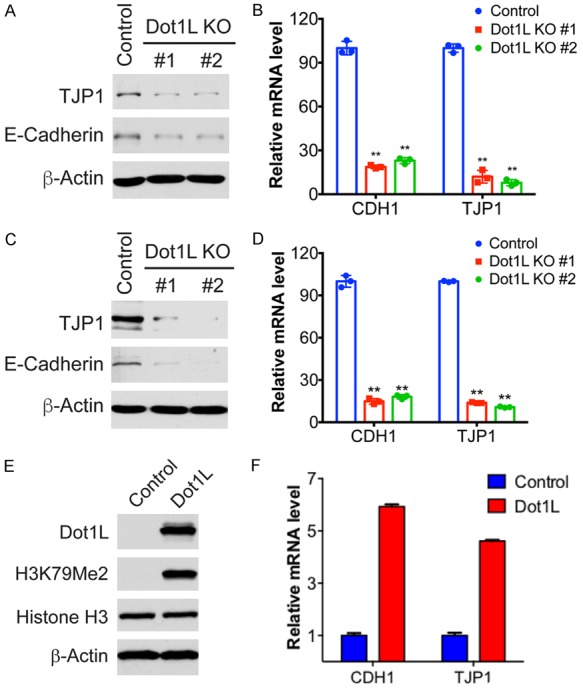
Dot1L knockout and restoration in ovarian cancer cells. Dot1L was knockout in Dot1L-wildtype OVCAR3 and OVCAR4 cells with two different sgRNAs using CRISPR technology, the mRNA and protein level of E-Cadherin and TJP1 were examined by qPCR and western blotting. CDH1 and TJP1 are genes encoding E-Cadherin and TJP1 protein respectively. A and B. Protein and mRNA level of E-Cadherin and TJP1 in OVCAR3 cells with Dot1L knockout. C and D. Protein and mRNA level of E-Cadherin and TJP1 in OVCAR4 cells with Dot1L knockout. E and F. Dot1L expression was restored in Dot1L-deficient CAOV4 ovarian cancer cells, the expression of E-cadherin and TJP1 were determined by qPCR. E. The express ion of Dot1L and H3K79 Methylation was examined in Dot1L-overexpressed CAOV4 cells by western blotting, total histone H3 and β-actin are used as loading controls. F. The mRNA level of CDH1 and TJP1 in Dot1L-overexpressed CAOV4 cells.
Depletion of Dot1L induces cancer stem-like cell property and enhances Wnt pathway activation
Cancer stem cells (CSCs) represent a fraction of undifferentiated cancer cells that exhibit stem cell-like features, they are responsible for tumor initiation and metastasis. Recent studies have highlighted a link between EMT and CSC formation. EMT is relevant to the acquisition and maintenance of stem cell-like characteristics and is sufficient to endow differentiated normal and cancer cells with stem cell properties [20,21]. Thus, we further tested cancer stem cell properties by measuring ALDH activity in Dot1L knockout cells. Indeed, loss of Dot1L significantly increased percentage of ALDH positive cells in both OVCAR3 and OVCAR4 cells (Figure 4A and 4B), this is due to the upregulation of ALDH1A1 protein (Figure 4C).
Figure 4.
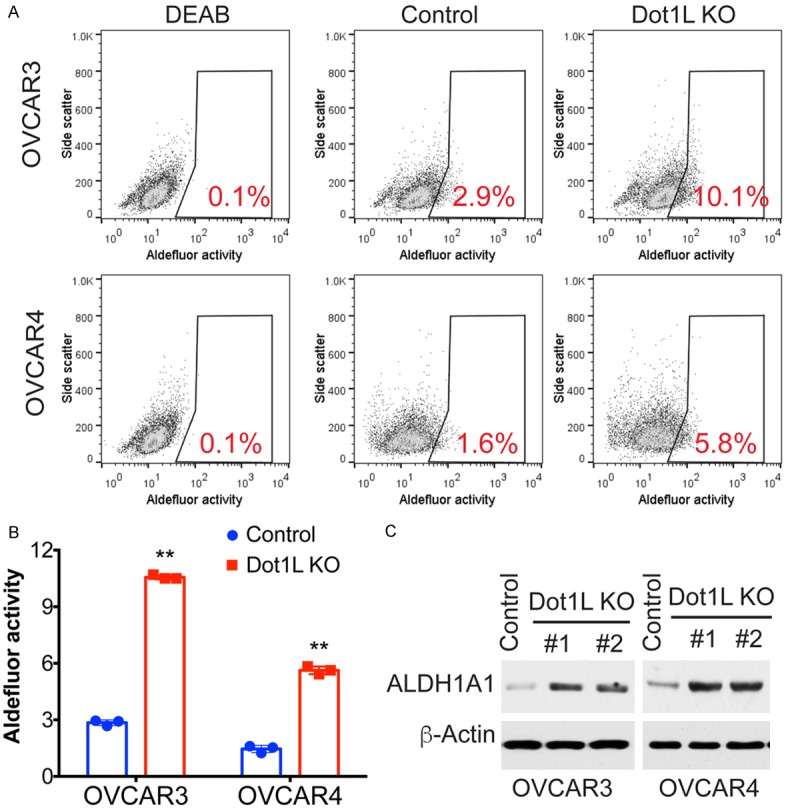
Dot1L knockout promotes cancer stem cell like properties in ovarian cancer cells. (A) ALDH activity was measured by FACS using ALDEFLUORTM Kit in control or Dot1L knockout OVCAR3 and OVCAR4 cells. DEAB was used as negative control for ALDH activity, the percentages of positive cells are indicated. (B) Quantification of (A). Error bars represent mean of three independent experiments with SD. **P < 0.01. (C) The expression level of ALDH1A1 was detected by western blotting in control or Dot1L knockout OVCAR3 and OVCAR4 cells. β-actin was used as loading control here.
Wnt signaling is one of the key cascades regulating development and stemness, which has been tightly associated with cancer progression. The activation of Wnt pathway has been shown be involved in EMT. The expression of E-cadherin is a downstream target gene of Wnt pathway, the cytoplasmic/nuclear re-localization of β-catenin and TJP1 from the adherent and tight junctions are common processes of the epithelial-mesenchymal transition (EMT) associated with tumor invasion [22,23]. Therefore, we sought to test the effect of Dot1L depletion on Wnt signaling. Wnt reporter assay shows that Wnt activity is enhanced in Dot1L depleted ovarian cancer cells, with about 2-3-fold increase compared to control cells (Figure 5A). Dot1L depletion didn’t increase the expression level of β-catenin, but increased the active version of β-catenin, shown by non-phosphorylated β-catenin by western blotting (Figure 5B). These results together suggest Dot1L depletion enhances the activity of Wnt signaling and pro motes cancer stem cell properties in ovarian cancer.
Figure 5.
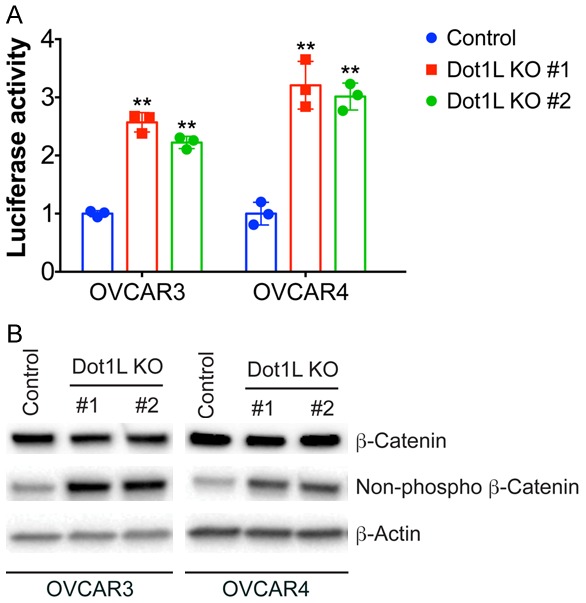
Dot1L knockout promote Wnt signaling pathway. A. The activity of Wnt/β-catenin signaling pathway was assessed by dual-luciferase reporter assays in control and Dot1L knockout cells (n=3). B. The expression of total and activated β-catenin (non-phosphorylation status) was detected by western blotting in control or Dot1L knockout OVCAR3 and OVCAR4 cells.
Discussion
Accumulating studies have indicated the function of Dot1L in cancer development, however the role of Dot1L in cancer are not fully understood. In the present study, we demonstrated a tumor suppressor function of Dot1L in ovarian cancer by knockout Dot1L expression using CRISPR/Cas9 technology. Loss of Dot1L expression and H3K79 methylation in ovarian cancer cells promote cell invasion by downregulating the expression of tight junction factors E-cadherin and TJP1. Restoration of Dot1L expression in Dot1L-defecient cells increased the expression of E-Cadherin and TJP1. Loss of Dot1L also induce cancer-stem cell properties in ovarian cancer cells by upregulation of ALDH1 expression. Our data indicated the potential roles of Dot1L in regulating cancer stem cell and cell invasion, providing potential directions to further elucidate the function of Dot1L in tumorigenesis and progression in ovarian cancer.
Wnt signaling is key pathway in regulation of development and cell stemness, mutations and aberrant activation of Wnt pathway occur frequently in many types of human cancers. Wnt signaling pathway plays important roles in different stage of cancer development, including maintenance of cancer stemness, promoting cancer cell invasion and metastasis, drug resistance and immune surveillance [3,20,24,25]. Recent studies have linked Dot1L dependent H3K79 methylation to Wnt signaling. Dot1L-containing complex is required for the proper regulation of Wnt target genes [26,27]. Consistently, out data also show the regulation of Dot1L on tumor cell invasion and stemness, Dot1L knockout cells show to increase non-phosphorylated β-catenin and Wnt signaling activity, indicating that loss of Dot1L may promote tumorigenesis and progression through Wnt pathway. The future study in needed to elucidate the mechanism whereby Dot1L dependent H3K79 methylation regulate the activation of Wnt pathway and the transcription of its target genes.
The oncogenic function of Dot1L were implicated in many type of human cancers, including prostate cancer, breast cancer, neuroblastoma and colorectal cancers [10]. Requirement of Dot1L during the initiation and maintenance of mixed lineage leukemia (MLL)-rearranged leukemia has been well established. During leukemogenesis, Dot1L is recruited to target genes by MLL fusion proteins to enhance H3K79 methylation level and promote gene transcription [28,29]. Dot1L inhibition is considered as a promising therapeutic strategy for the treatment of MLL-rearranged leukemia, a specific inhibitor of Dot1L, EPZ-5676, is current in clinical trials for MLL-rearranged leukemia [30].
However, the role of Dot1L in human cancers are context dependent. Genome sequencing has identified somatic mutations and deep deletions of Dot1L gene in multiple human cancers, including melanoma, colorectal cancer and ovarian cancer [12,13]. About 4-15% of melanomas are found having loss of function mutations of Dot1L. Further study showed that loss of Dot1L promote ultraviolet radiation (UVR)-induced melanoma in mice, indicating a tumor suppressor function of Dot1L in melanoma [14]. Consistent with the findings in melanoma, our data also indicated a tumor suppressor function of Dot1L in ovarian cancer, further study is granted to elucidate the mechanism whereby loss of Dot1L promote tumorigenesis and progression.
Acknowledgements
This research project was supported by the National Natural Science Foundation of China (Grant No. 81601244).
Disclosure of conflict of interest
None.
References
- 1.Bannister AJ, Kouzarides T. Regulation of chromatin by histone modifications. Cell Res. 2011;21:381–395. doi: 10.1038/cr.2011.22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Stillman B. Histone modifications: insights into their influence on gene expression. Cell. 2018;175:6–9. doi: 10.1016/j.cell.2018.08.032. [DOI] [PubMed] [Google Scholar]
- 3.Audia JE, Campbell RM. Histone modifications and cancer. Cold Spring Harb Perspect Biol. 2016;8:a019521. doi: 10.1101/cshperspect.a019521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.McGrath J, Trojer P. Targeting histone lysine methylation in cancer. Pharmacol Ther. 2015;150:1–22. doi: 10.1016/j.pharmthera.2015.01.002. [DOI] [PubMed] [Google Scholar]
- 5.Karakashev S, Zhu H, Wu S, Yokoyama Y, Bitler BG, Park PH, Lee JH, Kossenkov AV, Gaonkar KS, Yan H, Drapkin R, Conejo-Garcia JR, Speicher DW, Ordog T, Zhang R. CARM1-expressing ovarian cancer depends on the histone methyltransferase EZH2 activity. Nat Commun. 2018;9:631. doi: 10.1038/s41467-018-03031-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Li H, Cai Q, Godwin AK, Zhang R. Enhancer of zeste homolog 2 promotes the proliferation and invasion of epithelial ovarian cancer cells. Mol Cancer Res. 2010;8:1610–1618. doi: 10.1158/1541-7786.MCR-10-0398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Park G, Gong Z, Chen J, Kim JE. Characterization of the DOT1L network: implications of diverse roles for DOT1L. Protein J. 2010;29:213–223. doi: 10.1007/s10930-010-9242-8. [DOI] [PubMed] [Google Scholar]
- 8.Nguyen AT, Zhang Y. The diverse functions of Dot1 and H3K79 methylation. Genes Dev. 2011;25:1345–1358. doi: 10.1101/gad.2057811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wong M, Polly P, Liu T. The histone methyltransferase DOT1L: regulatory functions and a cancer therapy target. Am J Cancer Res. 2015;5:2823–2837. [PMC free article] [PubMed] [Google Scholar]
- 10.Cho MH, Park JH, Choi HJ, Park MK, Won HY, Park YJ, Lee CH, Oh SH, Song YS, Kim HS, Oh YH, Lee JY, Kong G. DOT1L cooperates with the c-Myc-p300 complex to epigenetically derepress CDH1 transcription factors in breast cancer progression. Nat Commun. 2015;6:7821. doi: 10.1038/ncomms8821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wood K, Tellier M, Murphy S. DOT1L and H3K79 methylation in transcription and genomic stability. Biomolecules. 2018;8 doi: 10.3390/biom8010011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA, Jacobsen A, Byrne CJ, Heuer ML, Larsson E, Antipin Y, Reva B, Goldberg AP, Sander C, Schultz N. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012;2:401–404. doi: 10.1158/2159-8290.CD-12-0095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gao J, Aksoy BA, Dogrusoz U, Dresdner G, Gross B, Sumer SO, Sun Y, Jacobsen A, Sinha R, Larsson E, Cerami E, Sander C, Schultz N. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal. 2013;6:pl1. doi: 10.1126/scisignal.2004088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhu B, Chen S, Wang H, Yin C, Han C, Peng C, Liu Z, Wan L, Zhang X, Zhang J, Lian CG, Ma P, Xu ZX, Prince S, Wang T, Gao X, Shi Y, Liu D, Liu M, Wei W, Wei Z, Pan J, Wang Y, Xuan Z, Hess J, Hayward NK, Goding CR, Chen X, Zhou J, Cui R. The protective role of DOT1L in UV-induced melanomagenesis. Nat Commun. 2018;9:259. doi: 10.1038/s41467-017-02687-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yokoyama Y, Zhu H, Lee JH, Kossenkov AV, Wu SY, Wickramasinghe JM, Yin X, Palozola KC, Gardini A, Showe LC, Zaret KS, Liu Q, Speicher D, Conejo-Garcia JR, Bradner JE, Zhang Z, Sood AK, Ordog T, Bitler BG, Zhang R. BET inhibitors suppress ALDH activity by targeting ALDH1A1 super-enhancer in ovarian cancer. Cancer Res. 2016;76:6320–6330. doi: 10.1158/0008-5472.CAN-16-0854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Valastyan S, Weinberg RA. Tumor metastasis: molecular insights and evolving paradigms. Cell. 2011;147:275–292. doi: 10.1016/j.cell.2011.09.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhao H, Hou W, Tao J, Zhao Y, Wan G, Ma C, Xu H. Upregulation of lncRNA HNF1A-AS1 promotes cell proliferation and metastasis in osteosarcoma through activation of the Wnt/beta-catenin signaling pathway. Am J Transl Res. 2016;8:3503–3512. [PMC free article] [PubMed] [Google Scholar]
- 18.Davidson B, Trope CG, Reich R. Epithelial-mesenchymal transition in ovarian carcinoma. Front Oncol. 2012;2:33. doi: 10.3389/fonc.2012.00033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Diepenbruck M, Christofori G. Epithelial-mesenchymal transition (EMT) and metastasis: yes, no, maybe? Curr Opin Cell Biol. 2016;43:7–13. doi: 10.1016/j.ceb.2016.06.002. [DOI] [PubMed] [Google Scholar]
- 20.Singh A, Settleman J. EMT, cancer stem cells and drug resistance: an emerging axis of evil in the war on cancer. Oncogene. 2010;29:4741–4751. doi: 10.1038/onc.2010.215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.De Francesco EM, Sotgia F, Lisanti MP. Cancer stem cells (CSCs): metabolic strategies for their identification and eradication. Biochem J. 2018;475:1611–1634. doi: 10.1042/BCJ20170164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhan T, Rindtorff N, Boutros M. Wnt signaling in cancer. Oncogene. 2017;36:1461–1473. doi: 10.1038/onc.2016.304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Howard S, Deroo T, Fujita Y, Itasaki N. A positive role of cadherin in Wnt/beta-catenin signalling during epithelial-mesenchymal transition. PLoS One. 2011;6:e23899. doi: 10.1371/journal.pone.0023899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Galluzzi L, Spranger S, Fuchs E, Lopez-Soto A. WNT signaling in cancer immunosurveillance. Trends Cell Biol. 2019;29:44–65. doi: 10.1016/j.tcb.2018.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Taciak B, Pruszynska I, Kiraga L, Bialasek M, Krol M. Wnt signaling pathway in development and cancer. J Physiol Pharmacol. 2018;69 doi: 10.26402/jpp.2018.2.07. [DOI] [PubMed] [Google Scholar]
- 26.Mohan M, Herz HM, Takahashi YH, Lin C, Lai KC, Zhang Y, Washburn MP, Florens L, Shilatifard A. Linking H3K79 trimethylation to Wnt signaling through a novel Dot1-containing complex (DotCom) Genes Dev. 2010;24:574–589. doi: 10.1101/gad.1898410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gibbons GS, Owens SR, Fearon ER, Nikolovska-Coleska Z. Regulation of Wnt signaling target gene expression by the histone methyltransferase DOT1L. ACS Chem Biol. 2015;10:109–114. doi: 10.1021/cb500668u. [DOI] [PubMed] [Google Scholar]
- 28.Bernt KM, Zhu N, Sinha AU, Vempati S, Faber J, Krivtsov AV, Feng Z, Punt N, Daigle A, Bullinger L, Pollock RM, Richon VM, Kung AL, Armstrong SA. MLL-rearranged leukemia is dependent on aberrant H3K79 methylation by DOT1L. Cancer Cell. 2011;20:66–78. doi: 10.1016/j.ccr.2011.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nguyen AT, Taranova O, He J, Zhang Y. DOT1L, the H3K79 methyltransferase, is required for MLL-AF9-mediated leukemogenesis. Blood. 2011;117:6912–6922. doi: 10.1182/blood-2011-02-334359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.McLean CM, Karemaker ID, van Leeuwen F. The emerging roles of DOT1L in leukemia and normal development. Leukemia. 2014;28:2131–2138. doi: 10.1038/leu.2014.169. [DOI] [PubMed] [Google Scholar]