

Abstract
Chronic rejection acts as the most formidable obstacle for organ transplantation in clinical settings. Herein we demonstrated in a cardiac transplantation model that blockade of Janus kinase 2 (Jak2) provides protection for cardiac allografts against chronic rejection. Specifically, loss of Jak2 almost completely abolished the production of IFN-γ+ Th1 cells, while the percentage of Foxp3+ regulatory T cells (Tregs) was significantly increased. As a result, loss of Jak2 significantly prolonged allograft survival (58 ± 30.6 days vs. 7 ± 0.3 days). Particularly, 4 out of 13 Jak2 deficient recipients (30%) showed long-term acceptance of allografts as manifested by the graft survival time > 100 days. Cellular studies revealed that Jak2 deficiency did not impact the intrinsic proliferative capability for CD4+ T cells in response to nonspecific polyclonal and allogenic stimulation. Mechanistic studies documented that the impaired Th1 development was caused by the attenuated IFN-γ/STAT1 and IL-12/STAT4 signaling along with repressed expression of Th1 transcription factors T-bet, Hlx and Runx3. However, the IL-2/STAT5 signaling remained intact, which ensured normal Treg development in Jak2-/- naïve CD4 T cells. Together, our data support that blockade of Jak2 may have therapeutic potential for prevention and treatment of allograft rejection in clinical settings.
Keywords: Jak2, cardiac transplantation, chronic rejection, regulatory T cells, allograft
Introduction
Cardiac transplantation is a common clinical strategy for treatment of patients with end-stage heart failure. Although the survival rate for cardiac allografts has recently been greatly improved, long-term survival, however, remains disappointed due to the toxicity of anti-rejection drugs and shortage of effective therapeutic strategies to manage chronic rejection. More recently, regulatory T cell (Treg) based therapies appeared to be a promising therapeutic alternative to attenuate chronic rejection while free of side effect [1]. However, problems with the expansion, instability and antigen specificity of Tregs hindered its clinical applications [2,3]. Therefore, a better understanding of the underlying molecular mechanisms would be instrumental for the development of Treg based strategies to achieve clinical transplant tolerance.
As non-receptor tyrosine kinases, the mammalian Janus kinase (Jak) family comprises four evolutionarily conserved members, Jak1, Jak2, Jak3 and tyrosine kinase 2 (Tyk2). Upon the engagement of cytokines with cell surface receptors, Jaks undergo autophosphorylation on tyrosine residues, which generates docking sites to phosphorylate signal transducers and activators of transcription (STAT). Jak phosphorylated STAT members next form homo- or heterodimers along with nuclear translocation to regulate the expression of immune responsive genes [4]. It is noteworthy that selective Jak3 inhibitors have recently been shown to be noninferior to the currently used immunosuppressants for treatment of transplant rejection and autoimmune disorders [5-7]. Nevertheless, graft protective Tregs were also significantly suppressed due to blockage of the IL-2-Jak3-STAT5 axis [8], which significantly limited their application in the setting of chronic rejection.
As an important member in Jak family, Janus kinase 2 (Jak2) is involved in the regulation of various processes relevant to cell survival, proliferation, activation and differentiation. Unlike Jak3, the research interest for Jak2 was mainly focused on its role in hematologic malignancies relevant to the V617F gain-of-function mutation [9]. Preceding studies on cell lines revealed that Jak2 plays an indispensable role in interleukin-3 (IL-3), interleukin-5 (IL-5), interleukin-12 (IL-12), interferon-γ (IFN-γ) and granulocyte-macrophage colony-stimulating factor (GM-CSF) signaling [10-12]. Given that mice deficient in Jak2 are embryonic lethal, the above observations might not fully resemble the enzymatic coupling that happens in vivo. Previously, we demonstrated that loss of Jak2 in adult mice impairs dendritic cell (DC) development and maturation [13], while its role in adaptive immune response, particularly in T helper 1 (Th1) response, is yet to be fully addressed. We thus in the current report induced Jak2 deficiency in adult mice and then assessed its role in adaptive immune response in the setting of cardiac allograft rejection. Loss of Jak2 significantly suppressed Th1 development, which led to a preferential increase of Tregs and, as a result, cardiac allografts were protected from chronic rejection.
Materials and methods
Mice
Jak2fl/fl (H-2b) mice were generated as described previously [14], which were backcrossed with Cre-ERT2 transgenic mice under the control of human ubiquitin C promoter to generate Cre+-Jak2fl/fl mice. Jak2 deficiency in Cre+-Jak2fl/fl mice was induced by i.p. injection of tamoxifen (25 mg/kg body weight) for five consecutive days. Littermates administered with equal volume of carrier solution (corn oil) were used as controls. BALB/c (H-2d) mice were purchased from the Animal Experimental Center of Hubei Province (Wuhan, China). Male mice at the age of 8-week (8 wk) old were used for experimental purpose. The mice were housed in a SPF facility at the Tongji Medical College, and all studies were conducted in accordance with the NIH guidelines and approved by the Animal Care and Use Committee (ACUC) in Tongji Hospital.
Abdominal cardiac transplantation
Cardiac allografts harvested from BALB/c mice were heterotopically transplanted into Jak2-/- and control recipients as previously reported [15]. Briefly, the ascending aorta on the graft side was anastomosed with the abdominal artery on the recipient side, while the pulmonary artery from the graft was then sutured with inferior vena cava of the recipient juxtaposed with the abdominal artery. Upon closure of abdominal wall, the recipient was placed on the heated cushion of the temperature controller to maintain its anal temperature at 37°C until its full resuscitation. Graft survival was blindly monitored by palpation two times a day. Cessation of transplanted heart beat was further validated by direct visualization.
Flow cytometry analysis
Single cell suspensions were freshly prepared from spleens, lymph nodes and peripheral blood or recovered from cell cultures. Staining of surface markers (e.g., CD4) and intracellular molecules (e.g., IFN-γ or Foxp3) was conducted using the established techniques [16]. Flow cytometry was performed using a FACSCalibur cytometer (BD Biosciences, San Jose, CA, USA), and the data were analyzed with the FlowJo version 7.6 software as instructed. FITC anti-CD3e, APC anti-CD25 and PE anti-CD8a were purchased from the Miltenyi Biotec (Auburn, CA, USA). PE anti-CD4, Alexa Fluor® 647 anti-CD4, APC anti-CD62L, FITC anti-CD44, APC anti-IFN-γ and APC anti-CD11c antibodies were purchased from the BD Biosciences (San Jose, CA, USA), while Alexa Fluor® 647 anti-Foxp3 was obtained from the eBioscience (San Diego, CA, USA).
Real-time PCR analysis
The apical part of cardiac grafts or cell suspensions were collected and subjected to RNA isolation using the TRIzol (Invitrogen, Carlsbad, CA, USA) reagent as instructed. Complementary DNA was synthesized from 1 μg RNA using a first-strand DNA synthesis kit (Fermentas Life Sciences, St Leon-Rot, Germany). Real-time PCR analysis of each target gene was then carried out using the SYBR Premix Ex TaqTM II (TaKaRa, Liaoning, China) on a LightCycler 480 Real-time PCR system (Roche, PA, USA). The analyses included IFN-γ (5’-GGC ACA GTC ATT GAA AGC CTA-3’ and 5’-CTG CAG GAT TTT CAT GTC ACC-3’), Tumor Necrosis Factor-α (TNF-α, 5’-GCC TCC CTC TCA TCA GTT CT-3’ and 5’-CAC TTG GTG GTT TGC TAC GA-3’), CC chemokine ligand 2 (CCL-2, 5’-ACC TGC TGC TAC TCA TTC ACC-3’ and 5’-CCC ATT CCT TCT TGG GGT CA-3’), IL-2 (5’-CCT GAG CAG GAT GGA GAA TTA CA-3’ and 5’-TCC AGA ACA TGC CGC AGA G-3’), IL-6 (5’-AGT TGC CTT CTT GGG ACT GA-3’ and 5’-TCC ACG ATT TCC CAG AGA AC-3’), and IL-12p40 (5’-GGA AGC ACG GCA GCA GAA TA-3’ and 5’-AAC TTG AGG GAG AAG TAG GAA TGG-3’). Glyceraldehyde-3-phosphate dehydrogenase (GAPDH, 5’-TGG CAT TGT GGA AGG GCT CA-3’, 5’-GCA CCA GTG GAT GCA GGG AT-3’) was used for normalization. Relative expression levels for each of the above target genes were calculated by using the 2-ΔΔCt method as previously reported [17].
CD4+ T cell proliferation assay
CD4+ T cells were purified from spleens and lymph nodes of Jak2-/- or control mice using a mouse CD4+ T cell isolation kit (StemCell, Seattle, WA) by negative selection as reported [17], and the purity for the isolated cells was > 90%. CD4+ T cell proliferation was determined by labeling the cells with 5, 6-carboxyfluorescein diacetate succinimidyl ester (CFSE) as instructed. Briefly, the cell pellets were resuspended in PBS working solution (1×106/ml to 2×107/ml) containing 0.5 mM CFSE (Molecular Probes, Eugene, OR, USA). After incubation at 37°C for 8 min, the cells were washed 3 times with RPMI 1640 medium supplemented with 10% heat-inactivated fetal bovine serum, 2-mercaptoethanol, 2-hydroxyethyl, non-essential amino acids, penicillin, streptomycin, and L-glutamine. T cell depleted splenocytes from control mice were treated with mitomycin C (final concentration, 50 ug/ml, Sigma-Aldrich, St. Louis, MO, USA) to serve as accessory cells following washes. The CFSE labeled cells were next stimulated with 10 ng/ml phorbol-12-myristate-13-acetate (PMA) (Sigma-Aldrich, St. Louis, MO, USA) and 250 ng/ml ionomycin (Sigma-Aldrich, St. Louis, MO, USA), or anti-CD3 (0.5 ug/ml, eBioscience, San Diego, USA) and anti-CD28 (0.5 ug/ml, BD Biosciences, San Jose, CA, USA) under 5% CO2 for 72 h in the presence of accessory cells, followed by staining with an Alexa Fluor 647 labeled anti-CD4 antibody (eBioscience, San Diego, CA, USA). Cell proliferation was analyzed by flow cytometry by assessing the halving of CFSE fluorescence intensity as instructed.
Mixed lymphocyte reaction (MLR)
Bone marrow dendritic cells (BMDCs) were generated from BALB/c mice as described in detail previously [13]. The purity for CD11c+ BMDCs in the current study was > 90% as determined by flow cytometry. For alloantigen specific MLR, 4×104 mitomycin C treated BALB/c BMDCs were co-cultured with 4×105 CFSE labeled splenic cells originated from Jak2-/- or control mice for 72 h in 96-well round bottom plates, followed by flow cytometry analysis of CD4+ T cell proliferation as described above.
Treg suppression assay
CD4+CD25+ Tregs and CD4+CD25- effector T cells (Teff) were isolated using a mouse CD4+CD25+ regulatory T cell isolation kit (Miltenyi, San Diego, CA, USA) as instructed. CFSE labeled CD4+CD25- T cells were served as T responder cells (Tresp) and seeded in 96-well round bottom plates with a fixed number of 5×104 cells/well. T cell depleted splenic cells (1×105 cells/well) originated from control mice were treated with mitomycin C as above to serve as accessory cells. Tregs derived from Jak2-/- and control mice were mixed with Tresp at the ratio of 0:1, 1:8, 1:4, 1:2, 1:1 and 2:1, respectively. The mixed cells were next stimulated with anti-CD3 antibodies (0.5 ug/ml), followed by flow cytometry analysis of cell proliferation as above.
Naïve T cell polarization
CD4+CD44lowCD62Lhigh naïve T cell were isolated from Jak2-/- and control mice with a mouse naïve CD4+ T cell isolation kit (StemCell, Seattle, WA, USA), and the purity of isolated cells was > 85%. For induction of Th1 cells, naïve T cells were activated with 1 μg/ml anti-CD3, 1 μg/ml anti-CD28, 10 ng/ml IL-2 (Gemini, West Sacramento, CA, USA), 20 ng/ml IL-12 (eBioscience, San Diego, CA, USA), 2 μg/ml anti-IL-4 (eBioscience, San Diego, CA, USA), with or without 50 ng/ml IFN-γ (Gemini, West Sacramento, CA, USA). For induction of Tregs, naïve T cells were incubated with 1 μg/ml anti-CD3, 20 ng/ml IL-2, 10 ng/ml TGF-β (PeproTech, Rocky Hill, NJ, USA), with or without 1 μg/ml anti-CD28. The cells were undergone half volume change of culture medium along with the addition of fresh antibodies and cytokines on day 3, followed by two additional days of culture as previously described [17]. The cells were next subjected to flow cytometry analysis of CD4+IFN-γ+ and CD4+Foxp3+ cells as above.
Western blot analysis
CD4+ T cells were purified as described above. The isolated cells were first rested under normal medium for 3 h, followed by IFN-γ (80 ng/ml, Gemini, West Sacramento, CA, USA), IL-2 (80 ng/ml, Gemini, West Sacramento, CA, USA), and IL-12 (80 ng/ml, eBioscience, San Diego, CA, USA) stimulation for 25 min. Naïve T cells were isolated and polarized under Th1 condition for 5 days as described above. Total proteins were next prepared from cell cultures using the RIPA lysis buffer supplemented with protease inhibitors, and Western blot analyses of targeted proteins were carried out as reported by probing the blots with indicated primary antibodies followed by an HRP-conjugated secondary antibody [18], respectively. The reactive bands were visualized using a Western ECL kit (Bio-Rad, Hercules, CA, USA). GAPDH and β-actin or basal STATs were used for normalization as indicated. Antibody against Jak2, phospho-Jak2, STAT4, phospho-STAT4, STAT1, phospho-STAT1, STAT5, and phospho-STAT5 were purchased from the Cell Signaling Technology (Beverly, MA, USA). The T-bet, Hlx, Runx3, IL-12Rβ2 and GAPDH antibodies were obtained from the Santa Cruz (Santa Cruz, CA, USA).
Histology
Cardiac grafts were harvested and fixed in 4% paraformaldehyde and embedded in paraffin. The sections were stained by hematoxylin and eosin (H&E) as previously described [19,20].
Statistical analysis
Allograft survival curves were generated by the Kaplan and Meier method, and survival differences between groups were determined using the log-rank (Mantel-Cox) test. Comparisons between groups for flow cytometry and real-time PCR data were accomplished by one-way or two-way analysis of variance (ANOVA) and boneferroni’s post hoc test where appropriate. All data are expressed as mean ± standard error (SD) with 3 or 4 independent replications. Graphpad Prism 5 was employed for statistical analysis, and a P value of < 0.05 was considered with statistical significance.
Results
Jak2 deficiency alters Th program and enhances Treg production
Jak2 deficiency was induced in 8 wk old Cre+-Jak2fl/fl mice by i.p. injection of tamoxifen for 5 consecutive days, and Jak2 depletion was firstly confirmed by genotyping of tail blood DNA for the presence of floxed null allele (Figure 1A). Indeed, Jak2 was undetectable in the lysates of spenocytes by Western blot analysis (Figure 1B), demonstrating that tamoxifen efficiently abrogated Jak2 expression. In consistent with our previous studies [13], mice deficient in Jak2 manifested a significant reduction for the total number of splenocytes (Figure 1C). Interestingly, the myeloid lineage cells in Jak2-/- spleens were nearly disappeared on the forward and side scatter image of flow cytometry data along with a significant increase for the proportion of lymphoid lineage (Figure 1D). In line with this observation, the proportion of CD4+ T cells in pan splenic cells was elevated by 1-fold (Figure 1E). The significant elevation for the proportion of lymphoid cells and CD4 T cells were also seen in peripheral blood (Figure 1F and 1G).
Figure 1.

The impact of Jak2 depletion on T cell development. Jak2 deficiency was induced by tamoxifen injection. Four days after last induction, the mice were sacrificed and used for following experiments. A. PCR analysis of tail genomic DNA to check the presence of floxed null allele. B. Western blot analysis to confirm Jak2 depletion in splenic cell lysates. C. Comparison of total splenic cell numbers between Jak2-/- and control mice. Relative cell numbers were normalized by body weight. D. Flow cytometry analysis of splenic cell populations by plots on forward scatter and side scatter. Comparison was carried out for the lymphoid cluster (lower left corner) between Jak2-/- and WT controls. E. Flow cytometry analysis for the percentage of CD3+CD4+ T cells in total splenocytes. F. Flow cytometry analysis of lymphoid cells in peripheral blood. The number of lymphoid cells (lower left corner) was compared between Jak2-/- and WT controls. G. Comparison for the proportion of CD3+CD4+ T cells in peripheral blood mononuclear cells (PBMCs) between Jak2-/- and WT controls. All data were expressed as means ± SD, and four mice were included in each study group.
We next sought to address the impact of Jak2 deficiency on T cell development. Loss of Jak2 did not result in a perceptible change for the CD4+ or CD8+ T cell ratio in total CD3+ splenic cells (Figure 2A), and similar results were obtained in the lymph nodes (Figure 2B) and peripheral blood (Figure 2C). However, Jak2 deficiency resulted in a 1-fold decrease for the CD4+CD44highCD62Llow effector/memory T cells (TEM cells) (Figure 2D) along with a 1.5-fold reduction for the number of CD4+ IFN-γ producing Th1 cells (Figure 2E). On the contrary, the proportion of CD4+Foxp3+ Tregs was noted to be significantly higher in Jak2-/- mice as compared with their counterparts (Figure 2F).
Figure 2.

The impact of Jak2 deficiency on CD4+ T cell development. Loss of Jak2 did not affect the percentage of CD4+ T cells in total CD3+ splenic cells (A), total CD3+ lymph node cells (B), and total CD3+ PBMCs (C). However, the percentage for CD4+CD44highCD62Llow effector/memory T cells (TEM cells) in total CD4+ splenocytes (gated on CD4+ cells) was significantly reduced (D), and similarly, a significant reduction for the percentage of IFN-γ+ Th1 cells was noted (E). On the contrary, a significant increase for the proportion of Foxp3+ Treg cells in CD4+ splenocytes was characterized (F). All flow cytometry data were expressed as mean ± SD, and four mice were analyzed for each group.
Loss of Jak2 attenuates naïve T cells polarizing to IFN-γ producing Th1 cells
To confirm the above data, CD4+CD44lowCD62Lhigh naïve T cells isolated from Jak2-/- and control mice were cultured under Th1 and Treg condition as described, followed by flow cytometry analysis of IFN-γ+ Th1 cells and Foxp3+ Tregs. Indeed, IFN-γ secreting Th1 cells were successfully induced in control cells, while loss of Jak2 almost completely abolished the production of IFN-γ producing Th1 cells. More importantly, addition of IFN-γ (50 ng/ml) markedly augmented Th1 differentiation in control cells, but the repressed Th1 differentiation was not restored by IFN-γ in Jak2-/- cells (Figure 3A). Interestingly, Jak2 deficiency did not affect the production of Foxp3+ Tregs, rather the percentage of Foxp3+ Tregs was even higher than that of control cells (Figure 3B). Particularly, anti-CD28 stimulation substantially increased Foxp3 expression in both Jak2-/- and control cells, while the percentage of Foxp3+ cells was still significantly higher in Jak2-/- cells than that of control cells (Figure 3B). Collectively, these results indicate that loss of Jak2 only selectively impairs Th1 development.
Figure 3.
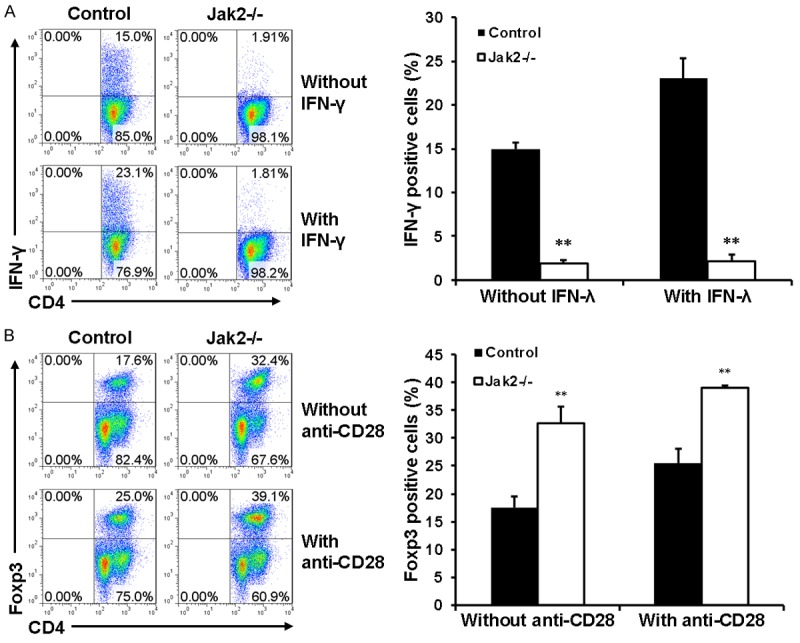
The effect of Jak2 deficiency on Th1 and Treg development. CD4+CD62LhighCD44low naïve CD4+ T cells were purified from Jak2-/- and control mice by magnetic beads as described (cell purity > 85%). A. Loss of Jak2 impaired Th1 development. Naïve CD4+ T cells were cultured under Th1 condition in the presence (lower) or absence (upper) of IFN-γ (50 ng/ml) for five days. The production of IFN-γ secreting Th1 cells were estimated by intracellular staining followed by flow cytometry analysis. B. Loss of Jak2 enhanced Treg production. Naïve CD4+ T cells were induced under Treg condition in the presence (lower) or absence (upper) of anti-CD28 (1 ug/ml) for five days. The production of Foxp3+ Tregs was estimated by flow cytometry as above. Three mice were analyzed in each study group, and the studies were conducted with three replications.
Loss of Jak2 prolongs long-term survival of cardiac allografts
The above findings prompted us to examine the impact of Jak2 deficiency on allograft chronic rejection, a process predominantly mediated by CD4+ T cells. For this purpose, BABL/c (H-2d)-derived cardiac grafts were heterotopically transplanted into the abdomen of Jak2 deficient mice or their control littermates (H-2b). Remarkably, loss of Jak2 significantly prolonged allograft survival [median survival time (MST) 58 ± 30.6 days vs. 7 ± 0.3 days, P < 0.001; Figure 4A]. Particularly, 4 out of 13 Jak2 deficient recipients (30%) showed long-term acceptance of allografts as manifested by the graft survival time > 1 00 days (Figure 4A). To exclude the potential effect of tamoxifen induction on allograft survival, tamoxifen induced Cre-ERT2 transgenic mice (H-2b) were transplanted with BABL/c (H-2d)-derived cardiac grafts as well. No perceptible difference in terms of allograft survival time was noted between vehicle treated control mice and tamoxifen-induced Cre-ERT2 mice (MST 7 ± 0.3 days vs. 7.5 ± 0.4 days).
Figure 4.
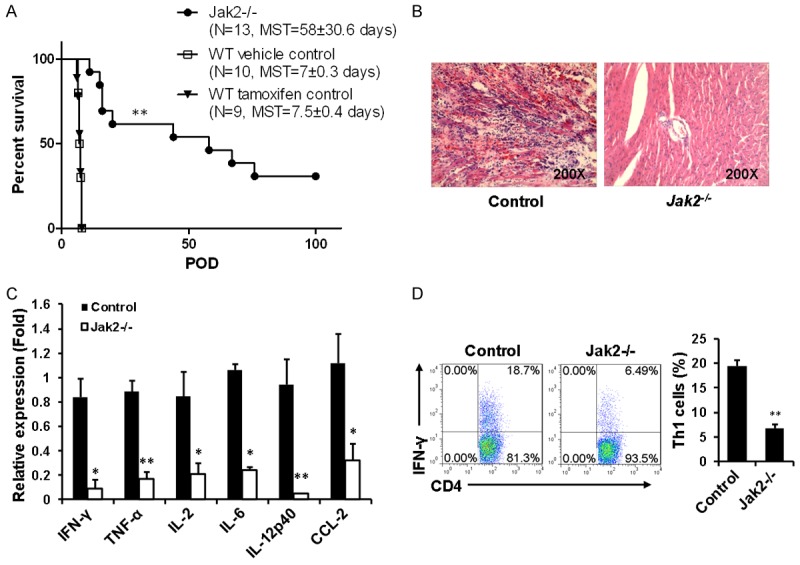
Cardiac allografts were protected from chronic rejection in recipients deficient in Jak2. Allogenic hearts originated from BALB/c (H-2d) mice were implanted into Jak2-/- (H-2b) recipients (n=13), corn oil induced control recipients (H-2b, n=10), and tamoxifen induced Cre-ERT2 recipients (H-2b, n=9) as described, respectively. A. Allograft survival curve generated by the Kaplan and Meier method. B. Histological analysis of allograft sections six days after transplantation. C. Real-time PCR analysis of inflammatory cytokines and chemokines within the grafts six days after transplantation. D. Comparison of splenic IFN-γ+ Th1 cells between Jak2-/- and corn oil induced control recipients six days after transplantation. Four mice were included in each group for the above studies.
Histological analysis was next conducted to further confirm the above data. Indeed, H&E staining of cardiac allograft sections originated control recipients after day 6 of transplantation revealed rigorous inflammatory infiltration along with cardiomyocyte destruction. In sharp contrast, no significant inflammatory infiltration was noted in the graft sections derived from Jak2-/- recipients along with well reserved myocardium (Figure 4B). The attenuated allograft rejection was also confirmed by the differences of mRNA levels for inflammatory cytokines and chemokines. Specifically, allografts originated from control recipients manifested significantly higher levels of IFN-γ, TNF-α, IL-2, IL-6, IL-12p40 and CCL-2 expression as compared with grafts from Jak2-/- recipients. Particularly, IFN-γ and IL-12p40 were almost undetectable in the grafts derived from Jak2-/- recipients (Figure 4C).
Jak2-/- recipients manifest higher proportion of functional Tregs
Since loss of Jak2 altered Th program (Figures 1, 2 and 3), we next compared the difference between Jak2-/- and control recipients for IFN-γ producing Th1 cells after day 6 of transplantation. Flow cytometry analysis of splenic CD4+ T cells revealed that control recipients manifested a 1.9-fold higher IFN-γ+ Th1 cells than that of Jak2-/- recipients (Figure 4D). However, higher proportion of splenic Tregs was noted in Jak2-/- recipients as compared with that of control recipients (Figure 5A). Especially, Jak2-/- recipients manifested a 5% higher proportion of Tregs than that of control recipients in the draining lymph nodes (DLN) (Figure 5B).
Figure 5.

Comparison of Treg suppressive function between Jak2-/- and control recipients six days after transplantation. A. Comparison of splenic Treg numbers between Jak2-/- and control recipients. B. Analysis of Tregs in the draining lymph nodes (DLN) of recipients. C. A representative result for Treg suppressive assays. Proliferation of Tresps upon anti-CD3 stimulation in the presence of accessory cells along with different proportion of Tregs was estimated by flow cytometry based on the halving of CFSE fluorescence after three days of culture. D. Results for Treg suppressive kinetics of all recipients studied for each group. Four recipients were included for each group and the studies were carried out with three replications.
To demonstrate whether Tregs deficient in Jak2 were still functionally competent, Treg suppressive assays were next carried out based on CFSE labeling. CD4+ responders (Tresp) and non-T accessory cells were obtained from WT mice, while Jak2-/- and WT Tregs were added in different proportions into the cultures to repress Tresp proliferation following anti-CD3 stimulation. Interestingly, Jak2-/- Tregs exhibited a comparable suppressive kinetics as that of WT Tregs (Figure 5C, 5D), suggesting that loss of Jak2 does not affect the functionality of Tregs.
Jak2 deficiency does not impact the intrinsic proliferative capability for CD4+ T cells
The next important question is whether Jak2 deficiency impacts the intrinsic capability of CD4+ T cells for proliferation. To address this issue, splenic CD4+ T cells originated from Jak2-/- and control mice were labeled with CFSE and then stimulated with anti-CD3/anti-CD28 antibodies or low dose PMA/Ionomycin as described, respectively. In line with our expectation, Jak2-/- CD4+ T cells manifested similar proliferation potency as that of control CD4+ T cells in response to anti-CD3/anti-CD28 or PMA/Ionomycin stimulation (Figure 6A and 6B), indicating that Jak2 deficiency does not affect their intrinsic proliferative capability. To further demonstrate this question, we conducted mixed lymphocyte reaction (MLR) assays to assess the differences for allo-antigen stimulated proliferation. BALB/c-derived bone marrow dendritic cells (BMDCs) were treated with Mitomycin C and then employed for stimulation of CFSE-labeled Jak2-/- and control CD4+ T cells, respectively. The purity of CD11c positive dendritic cell (DC) was about 95% as manifested by flow cytometric analysis (Figure 6C). Similar as above, Jak2-/- CD4+ T cells displayed comparative proliferative capability as that of their control counterparts in response to allo-antigen stimulation (Figure 6D).
Figure 6.

Analysis of the intrinsic proliferative capability of CD4+ T cells after Jak2 depletion. CD4+ T cells were prepared from Jak2-/- and control mice and labeled with CFSE to serve as responder cells, while T cell depleted control splenocytes were treated with mitomycin C to serve as accessory cells. A. Results for proliferation of responder cells stimulated with PMA (10 ng/ml) and Ionomycin (250 ng/ml). B. Proliferation results for responder cells stimulated by anti-CD3 (0.5 ug/ml) and anti-CD28 (0.5 ug/ml). C. Flow cytometry analysis of allogenic BMDCs prepared from BALB/c mice. D. Proliferation results for responder cells stimulated by BALB/c-derived allogenic BMDCs. Mitomycin C treated BMDCs were co-cultured with CFSE labeled splenic cells originated from Jak2-/- or control mice for 72 h. Cell proliferation was estimated as above based on the halving of CFSE fluorescence intensity. Similarly, four mice were included for each group and the studies were carried out with three replications.
Loss of Jak2 selectively represses IL-12/STAT4 and IFN-γ/STAT1 signaling
To dissect the mechanisms by which Jak2 deficiency impairs the balance between Th1 and Treg program, we first examined cytokine-stimulated Jak2 activity in CD4+ T cells. WT CD4+ T cells were stimulated with IFN-γ, IL-12 and IL-2, followed by analysis of the levels for phosphorylated Jak2 (p-Jak2). High levels of p-Jak2 were detected in both IFN-γ and IL-12 stimulated cells, and in sharp contrast, p-Jak2 was almost undetectable in IL-2 stimulated CD4+ cells (Figure 7A).
Figure 7.
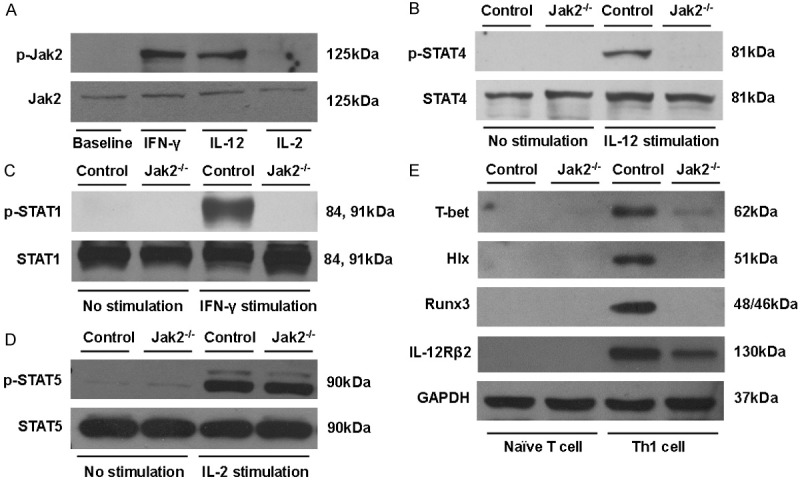
Loss of Jak2 selectively repressed signals essential for Th1 development. A. IFN-γ and IL-12 were potent to stimulate Jak2 phosphorylation (p-Jak2) in CD4+ T cells, while p-Jak2 was undetectable in IL-2 stimulated CD4+ T cells. B. Loss of Jak2 in CD4+ T cells impaired IL-12 induced STAT4 activation (p-STAT4). C. Jak2-/- CD4+ T cells manifested impaired IFN-γ/STAT1 signaling. D. Jak2 deficiency did not impact IL-2/STAT5 signaling in CD4+ T cells. E. Western blot results for analysis of transcription factors relevant to Th1 development. Naïve CD4+ T cells originated from Jak2-/- and control mice were polarized under Th1 condition for five days, followed by analysis of T-bet, Hlx, Runx3 and IL-12Rβ2 expression levels by Western blotting. GAPDH was served as loading controls, and data shown here were a representative of three independent experiments.
The above results prompted us to check the activities of Jak2 downstream signaling molecules following cytokine stimulation. Under steady condition, phosphorylated STAT4 (p-STAT4) was undetectable both in WT and Jak2-/- CD4+ T cells, while high levels of p-STAT4 were noted in WT CD4+ T cells following IL-12 stimulation. However, IL-12 failed to induce the expression of p-STAT4 in Jak2-/- CD4+ T cells (Figure 7B). Similarly, IFN-γ induced high levels of phosphorylated STAT1 (p-STAT1) in WT CD4+ T cells, but p-STAT1 remained undetectable in Jak2-/- CD4+ T cells following IFN-γ stimulation (Figure 7C). Interestingly, unlike STAT4 and STAT1, both WT and Jak2-/- CD4+ T cells manifested low levels of expression for the phosphorylated STAT5 (p-STAT5) under steady condition, and more importantly, IL-2 stimulation induced high levels of p-STAT5 both in WT and Jak2-/- CD4+ T cells (Figure 7D). Taken together, these data suggest that loss of Jak2 selectively repressed IL-12/STAT4 and IFN-γ/STAT1 signaling, which then attenuated naïve CD4+ T cells toward to IFN-γ producing Th1 cells. On the contrary, Jak2 is dispensable for the IL-2/STAT5 signaling, and as a result, Jak2 deficiency did not affect Treg development. Since Jak2 deficiency did not impact IL-2/STAT5 signaling, higher percentage of Tregs noted in Jak2-/- mice is likely caused by the impaired IL-12 and IFN-γ signaling, in which Jak2-/- naïve CD4+ T cells preferentially differentiate into Foxp3+ Tregs in the absence of IFN-γ.
Jak2 is essential for the expression of Th1 transcription factors
To dissect the mechanisms by which loss of Jak2 impairs Th1 development, we first checked transcription factors T box 21 (T-bet) and H 2.0-like homeobox (Hlx), in which T-bet serves as a Th1 “master regulator” [21], while Hlx drives optimal IFN-γ expression to stabilize Th1 phenotype [22]. To this end, naïve T cells isolated from Jak2-/- and control mice were cultured under Th1 condition and then subjected to Western blot analysis of T-bet and Hlx expression. Unlike WT T cells which manifested high levels of T-bet expression, T-bet was hardly detectable in Th1 polarized Jak2-/- T cells, and remarkably, Hlx was completely absent in Th1 polarized Jak2-/- T cells. To further confirm this observation, we examined Runt-Related Transcription Factor 3 (Runx3), another transcription factor that acts in cooperation with T-bet to ensure Th1 polarization [23], and similar results were obtained (Figure 7E). During Th1 development the IL-12Rβ2 subunit for IL-12 receptor is rapidly induced following the expression of T-bet. Unexpectedly, the abrogated T-bet expression did not couple with diminished induction of IL-12Rβ2 as manifested by the detection of relatively lower levels of IL-12Rβ2 in Jak2-/- CD4 T cells (Figure 7E). Collectively, these data suggest that loss of Jak2 attenuates the induction of T-bet/Hlx and the related transcription factor Runx3, which then impairs Th1 polarization.
Discussion
Although Jak2 has been extensively studied for its impact on hematopoietic malignancy, its role in immune cell development, especially in CD4+ T cells, remained poorly understood. Previously, we reported that loss of Jak2 impairs DC development and its maturation [13], and now we demonstrated evidence indicating that Jak2 oriented signaling is essential for Th1 development, but the proportion for CD4/CD8 T cells was unaffected in mice deficient in Jak2. This observation was consistent with previous reports that Jak2-/- fetal liver cells were able to numerically and functionally reconstitute T and B cell lineage in Jak3-/- mice [24,25]. However, the majority of T cells in Jak2-/- mice remained immature and was unable to differentiate into Th1 cells, while Treg development was unexpectedly enhanced. Given the role of Th1 cells played in the setting of allograft rejection [24,25], a cardiac transplantation model was next employed to demonstrate the impact of Jak2 deficiency on cardiac allograft chronic rejection. Remarkably, cardiac allografts were significantly protected from chronic rejection in recipients deficient in Jak2 as manifested by the prolonged median survival time (58 ± 30.6 days vs. 7 ± 0.3 days) and ameliorated allograft inflammatory response. Particularly, four allografts reached long-term acceptance as evidenced by the survival time > 100 days. It was noted that Th1 response was significantly attenuated in Jak2-/- recipients, while Treg proportion was significantly higher in Jak2-/- recipients as compared with that of control recipients. More importantly, Tregs originated from Jak2-/- recipients were functionally intact as evidenced by the comparable capability to suppress effector T cell proliferation.
Of importantly note, in the setting of anti-rejection therapy, combined immunosuppressive drugs are usually employed, while none of our recipient mice following cardiac transplantation administered anti-rejection drugs, and therefore, it is logical that the majority of grafts would be eventually rejected. However, 4 recipient mice reached long-term tolerance without rejection. These results demonstrate that blockade of Jak2 signaling alone may have the potential to reach immune tolerance following transplantation, but the homeostasis of tolerance cannot be achieved in all recipients. This discrepancy could be due to the heterogeneity of immune response following transplantation, which may involve additional mechanisms other than the described imbalance between Th1 and Tregs.
A model for three essential signals has been well established in CD4 T cell activation and differentiation, in which TCR stimulation provides the cardinal signal, followed by the signals generated by B7/CD28 costimulation and inflammatory cytokines [26]. Interestingly, analysis of Treg differentiation revealed that the signals originated from TCR and B7/CD28 were intact after Jak2 depletion, which ensured normal Treg development. However, discrepancies for the signal generated by the extracellular cytokine milieu, which is a fate decision factor for directing CD4 T cell development, were noted under different Th conditions [27]. Particularly, IL-2 elicited strong STAT5 signaling both in WT and Jak2-/- CD4+ T cells, suggesting that Jak2 was not responsible for IL-2 mediated STAT5 activation, which was further confirmed by the observation that Jak2-/- naïve T cells were more prone to differentiate into Tregs with or without anti-CD28. It has been well documented that IL-2/STAT5 pathway plays a non-redundant role for the development, homeostasis and function of Tregs [28], and therefore, mice deficient in IL-2 receptor spontaneously develop autoimmune disease [29]. We postulated that the enhanced Treg production is probably caused by the imbalance and crosstalk between Th1 and Treg after Jak2 depletion [17]. Previously, we demonstrated in DCs that Jak2 plays an essential role for STAT5 signaling as manifested by the impaired development, maturation and secretion of proinflammatory cytokines in Jak2-/- DCs [13]. This discrepant result between our present report might be due to the differences of cell type- and stimulus-specific STAT activation profile between DCs and T cells. Collectively, we have proved that Jak2 is dispensable for IL-2/STAT5 signaling in the setting of Treg development.
Unlike its impact on IL-2/STAT5 signaling, loss of Jak2 almost completely abrogated IL-12/STAT4 and IFN-γ/STAT1 signaling. It was noted that Th1 signature cytokines IL-12 and IFN-γ induced robust Jak2 phosphorylation, which then mediated activation of STAT4 and STAT1, respectively. These results were consistent with previous in vitro studies, in which Jak2 was indispensable for IL-12 [11] and IFN-γ [10] signaling. Activated STAT4 and STAT1 then transcribe the expression of Th1 lineage-defining transcription factor T-bet, which binds to the IFN-γ regulatory elements and promotes IFN-γ transcription. As a result, mice deficient in Stat4 manifest impaired Th1 response along with abrogated IFN-γ secretion [10]. Similarly, STAT1 activation by IFN-γ induces the Th1 transcription factor T-bet [30,32] and act on Th1 cells in an autocrine positive feedback manner to stabilize T-bet and amplify cytokine secretion [31-34]. Other than positive regulation of IFN-γ transcription, another important task for T-bet is to repress the activity of Th2 master transcription factor, GATA-3, by which it attenuates IL-4 transcription and maximizes IFN-γ production [33,35]. Moreover, T-bet is unable to directly repress IL-4 in the absence of Runx3, which is induced during Th1 development in a T-bet dependent manner [23]. T-bet also induces Hlx expression, which coordinates with T-bet to potentiate and stabilize IFN-γ production [22]. However, as described above neither T-bet nor downstream Runx3 and Hlx were present in Jak2-/- cells following Th1 induction. Together, these results provide strong evidence supporting that loss of Jak2 impairs Th1 development by interrupting the IL-12/STAT4, IFN-γ/STAT1, and T-bet/Hlx/Runx3 signaling pathways.
Naïve CD4 T cells do not possess a functional IL-12 receptor as a result of lacking IL-12Rβ2 expression [34]. During Th1 differentiation, IL-12Rβ2 is rapidly induced by TCR stimulation in the presence of IFN-γ [34]. Previous studies suggest that IFN-γ/STAT1 dependent T-bet expression dominants IL-12Rβ2 expression in Th1 cells [35]. Interestingly, despite almost completely abrogated IFN-γ/STAT1 signaling and T-bet expression, induction of IL-12Rβ2 expression was still detected in Jak2-/- CD4 T cells, albeit less than control cells. This discrepant observation suggests that additional signal(s) might be responsible for the induction of IL-12Rβ2 expression once IFN-γ/STAT1 signaling is interrupted. Indeed, a recent study revealed that IL-2 could induce IL-12Rβ2 expression in an IFN-γ independent manner and IL-2 is able to induce IL-12Rβ2 expression in T-bet-/- T cells [36]. Together, our data provided additional evidence that both IFN-γ and IL-2 can induce optimal IL-12Rβ2 expression during Th1 development. It is noteworthy that loss of Jak2 did not affect the proliferative capability of CD4+ T cells in response to non-specific polyclonal anti-CD3/anti-CD28 and PMA/Ionomycin stimulation. Once challenged with allogenic BMDCs, the expansion of Jak2-/- CD4 T cells was unaffected as well. These results suggest that Jak2 is not required for the maintenance of intrinsic proliferative capability in CD4 T cells.
It has been suggested that the number of splenocytes are critical for antibody-mediated rejection (AMR), and splenectomy have been used in the clinical setting of refractory humoral rejection after kidney transplantation of highly sensitized patients [37,38], but it has limited effects on the acute and chronic rejection in other settings. The survival time only last to 27 ± 1.5 days for rat heat transplantation after splenectomy [39]. Interestingly, we also observed reduced number of splenocytes in Jak2 deficient recipients, but our graft survival time is 58 ± 30.6 days, and 4 grafts reached long term survival. These results suggest that Jak2 could be more important in T cell function as compared to its role in maintaining the number of splenocytes during the course cardiac allograft rejection.
Among four Jak family members, Jak1, Jak2 and Tyk2 were ubiquitously expressed, while the expression of Jak3 is limited to lymphocytes, which renders Jak3 to be the best therapeutic target for treatment of immune related diseases in clinical settings. Nevertheless, Jak3 has also been found to function as a key signal transducer downstream of the common gamma chain (γc), through which IL-2 takes effect [40]. Given the essential role of IL-2 played in Treg development, the use of selective Jak3 inhibitors such as CP-690550 also repressed the production of Tregs other than suppressing Th1 functionality [8,41], and indeed, mice deficient in Jak3 manifest significantly reduced Tregs [41]. Unlike Jak3, we herein provided evidence indicating that loss of Jak2 only selectively suppressed Th1 response relevant to cardiac allograft rejection by blocking the IFN-γ-Jak2/STAT1 and IL-12-Jak2/STAT4 pathways, while the signaling pathways relevant to Treg development and functionality were not affected. Our findings raised the feasibility that Jak2 could also be a viable target for prevention and treatment of immune related diseases in clinical settings. However, given that Jak2 is ubiquitously expressed, pan inhibition of Jak2 may cause side effect as well.
In summary, we demonstrated that loss of Jak2 led to diminished Th1 cell development along with a preferential increase of functionally competent Tregs. As a result, cardiac allografts were significantly protected from chronic rejection in recipients deficient in Jak2. Cellular studies revealed that Jak2 deficiency did not impact the intrinsic proliferative capability for CD4+ T cells in response to nonspecific polyclonal and allogenic stimulation. Mechanistic studies documented that the impaired Th1 development was caused by the attenuated IFN-γ/STAT1 and IL-12 /STAT4 signaling along with repressed expression of Th1 transcription factors T-bet, Hlx and Runx3. However, the IL-2/STAT5 signaling remained intact, which ensured normal Treg development in Jak2-/- naïve CD4 T cells. Together, our data support that that suppression of Jak2 may have therapeutic potential for prevention and treatment of allograft rejection in clinical settings.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (81530024, 9174920038, 81770823 and 81470988), the Ministry of Science and Technology (2017ZX09304022 and 2016YFC1305002), the Department of Science and Technology of Hubei State (2017ACA096), and the Integrated Innovative Team for Major Human Disease Programs of Tongji Medical College, Huazhong University of Science and Technology.
Disclosure of conflict of interest
None.
Abbreviations
- ANOVA
analysis of variance
- BMDC
bone marrow dendritic cells
- CFSE
5, 6-carboxyfluorescein diacetate succinimidyl ester
- DC
dendritic cell
- DLN
draining lymph nodes
- GAPDH
Glyceraldehyde-3-phosphate dehydrogenase
- GATA-3
GATA Binding Protein 3
- H&E
hematoxylin and eosin
- Hlx
H 2.0-like homeobox
- Jak
Janus kinase
- Jak2
Janus kinase 2
- IFN-γ
interferon-γ
- MLR
mixed lymphocyte reaction
- MST
median survival time
- PMA
phorbol-12-myristate-13-acetate
- Runx3
Runt-Related Transcription Factor 3
- SD
standard deviation
- T-bet
T box 21
- Teff
effector T
- TEM
effector/memory T cell
- Th1
T helper 1
- Treg
regulatory T cell
- Tyk2
tyrosine kinase 2
- Tresp
T responder
References
- 1.Schliesser U, Streitz M, Sawitzki B. Tregs: application for solid-organ transplantation. Curr Opin Organ Transplant. 2012;17:34–41. doi: 10.1097/MOT.0b013e32834ee69f. [DOI] [PubMed] [Google Scholar]
- 2.Newell KA, Phippard D, Turka LA. Regulatory cells and cell signatures in clinical transplantation tolerance. Curr Opin Immunol. 2011;23:655–659. doi: 10.1016/j.coi.2011.07.008. [DOI] [PubMed] [Google Scholar]
- 3.Bluestone JA. Regulatory T-cell therapy: is it ready for the clinic? Nat Rev Immunol. 2005;5:343–349. doi: 10.1038/nri1574. [DOI] [PubMed] [Google Scholar]
- 4.O’Shea JJ, Plenge R. JAK and STAT signaling molecules in immunoregulation and immune-mediated disease. Immunity. 2012;36:542–550. doi: 10.1016/j.immuni.2012.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kontzias A, Kotlyar A, Laurence A, Changelian P, O’Shea JJ. Jakinibs: a new class of kinase inhibitors in cancer and autoimmune disease. Curr Opin Pharmacol. 2012;12:464–470. doi: 10.1016/j.coph.2012.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wojciechowski D, Vincenti F. Targeting JAK3 in kidney transplantation: current status and future options. Curr Opin Organ Transplant. 2011;16:614–619. doi: 10.1097/MOT.0b013e32834c23ce. [DOI] [PubMed] [Google Scholar]
- 7.Vijayakrishnan L, Venkataramanan R, Gulati P. Treating inflammation with the Janus kinase inhibitor CP-690,550. Trends Pharmacol Sci. 2011;32:25–34. doi: 10.1016/j.tips.2010.10.004. [DOI] [PubMed] [Google Scholar]
- 8.van Gurp EA, Schoordijk-Verschoor W, Klepper M, Korevaar SS, Chan G, Weimar W, Baan CC. The effect of the JAK inhibitor CP-690,550 on peripheral immune parameters in stable kidney allograft patients. Transplantation. 2009;87:79–86. doi: 10.1097/TP.0b013e31818bbea7. [DOI] [PubMed] [Google Scholar]
- 9.Harry BL, Eckhardt SG, Jimeno A. JAK2 inhibition for the treatment of hematologic and solid malignancies. Expert Opin Investig Drugs. 2012;21:637–655. doi: 10.1517/13543784.2012.677432. [DOI] [PubMed] [Google Scholar]
- 10.Watling D, Guschin D, Muller M, Silvennoinen O, Witthuhn BA, Quelle FW, Rogers NC, Schindler C, Stark GR, Ihle JN, et al. Complementation by the protein tyrosine kinase JAK2 of a mutant cell line defective in the interferon-gamma signal transduction pathway. Nature. 1993;366:166–170. doi: 10.1038/366166a0. [DOI] [PubMed] [Google Scholar]
- 11.Bacon CM, McVicar DW, Ortaldo JR, Rees RC, O’Shea JJ, Johnston JA. Interleukin 12 (IL-12) induces tyrosine phosphorylation of JAK2 and TYK2: differential use of Janus family tyrosine kinases by IL-2 and IL-12. J Exp Med. 1995;181:399–404. doi: 10.1084/jem.181.1.399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Silvennoinen O, Witthuhn BA, Quelle FW, Cleveland JL, Yi T, Ihle JN. Structure of the murine Jak2 protein-tyrosine kinase and its role in interleukin 3 signal transduction. Proc Natl Acad Sci U S A. 1993;90:8429–8433. doi: 10.1073/pnas.90.18.8429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhong J, Yang P, Muta K, Dong R, Marrero M, Gong F, Wang CY. Loss of Jak2 selectively suppresses DC-mediated innate immune response and protects mice from lethal dose of LPS-induced septic shock. PLoS One. 2010;5:e9593. doi: 10.1371/journal.pone.0009593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Krempler A, Qi Y, Triplett AA, Zhu J, Rui H, Wagner KU. Generation of a conditional knockout allele for the Janus kinase 2 (Jak2) gene in mice. Genesis. 2004;40:52–57. doi: 10.1002/gene.20063. [DOI] [PubMed] [Google Scholar]
- 15.Fang J, He L, Wang SQ, Ma MJ, Liu HY, Zhu XH, Zhu P, Wei X, Wang CY. A simplified two-stitch sleeve technique for arterial anastomosis of cervical heterotopic cardiac transplantation in mice. Am J Transl Res. 2013;5:521–529. [PMC free article] [PubMed] [Google Scholar]
- 16.Han J, Zhong J, Wei W, Wang Y, Huang Y, Yang P, Purohit S, Dong Z, Wang MH, She JX, Gong F, Stern DM, Wang CY. Extracellular high-mobility group box 1 acts as an innate immune mediator to enhance autoimmune progression and diabetes onset in NOD mice. Diabetes. 2008;57:2118–2127. doi: 10.2337/db07-1499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhong J, Yu Q, Yang P, Rao X, He L, Fang J, Tu Y, Zhang Z, Lai Q, Zhang S, Kuczma M, Kraj P, Xu JF, Gong F, Zhou J, Wen L, Eizirik DL, Du J, Wang W, Wang CY. MBD2 regulates TH17 differentiation and experimental autoimmune encephalomyelitis by controlling the homeostasis of T-bet/Hlx axis. J Autoimmun. 2014;53:95–104. doi: 10.1016/j.jaut.2014.05.006. [DOI] [PubMed] [Google Scholar]
- 18.Yang P, Zhang Y, Pang J, Zhang S, Yu Q, He L, Wagner KU, Zhou Z, Wang CY. Loss of Jak2 impairs endothelial function by attenuating Raf-1/MEK1/Sp-1 signaling along with altered eNOS activities. Am J Pathol. 2013;183:617–25. doi: 10.1016/j.ajpath.2013.04.007. [DOI] [PubMed] [Google Scholar]
- 19.Yang P, Zhang Y, Xu J, Zhang S, Yu Q, Pang J, Rao X, Kuczma M, Marrero MB, Fulton D, Kraj P, Su Y, Wang CY. SUMO1 regulates endothelial function by modulating the overall signals in favor of angiogenesis and homeostatic responses. Am J Transl Res. 2013;5:427–40. [PMC free article] [PubMed] [Google Scholar]
- 20.Ran L, Yu Q, Zhang S, Xiong F, Cheng J, Yang P, Xu JF, Nie H, Zhong Q, Yang X, Yang F, Gong Q, Kuczma M, Kraj P, Gu W, Ren BX, Wang CY. Cx3cr1 deficiency in mice attenuates hepatic granuloma formation during acute schistosomiasis by enhancing the M2-type polarization of macrophages. Dis Model Mech. 2015;8:691–700. doi: 10.1242/dmm.018242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mullen AC, High FA, Hutchins AS, Lee HW, Villarino AV, Livingston DM, Kung AL, Cereb N, Yao TP, Yang SY, Reiner SL. Role of T-bet in commitment of TH1 cells before IL-12-dependent selection. Science. 2001;292:1907–1910. doi: 10.1126/science.1059835. [DOI] [PubMed] [Google Scholar]
- 22.Mullen AC, Hutchins AS, High FA, Lee HW, Sykes KJ, Chodosh LA, Reiner SL. Hlx is induced by and genetically interacts with T-bet to promote heritable T(H)1 gene induction. Nat Immunol. 2002;3:652–658. doi: 10.1038/ni807. [DOI] [PubMed] [Google Scholar]
- 23.Djuretic IM, Levanon D, Negreanu V, Groner Y, Rao A, Ansel KM. Transcription factors T-bet and Runx3 cooperate to activate Ifng and silence Il4 in T helper type 1 cells. Nat Immunol. 2007;8:145–153. doi: 10.1038/ni1424. [DOI] [PubMed] [Google Scholar]
- 24.Wernig G, Kharas MG, Okabe R, Moore SA, Leeman DS, Cullen DE, Gozo M, McDowell EP, Levine RL, Doukas J, Mak CC, Noronha G, Martin M, Ko YD, Lee BH, Soll RM, Tefferi A, Hood JD, Gilliland DG. Efficacy of TG101348, a selective JAK2 inhibitor, in treatment of a murine model of JAK2V617F-induced polycythemia vera. Cancer Cell. 2008;13:311–320. doi: 10.1016/j.ccr.2008.02.009. [DOI] [PubMed] [Google Scholar]
- 25.Parganas E, Wang D, Stravopodis D, Topham DJ, Marine JC, Teglund S, Vanin EF, Bodner S, Colamonici OR, van Deursen JM, Grosveld G, Ihle JN. Jak2 is essential for signaling through a variety of cytokine receptors. Cell. 1998;93:385–395. doi: 10.1016/s0092-8674(00)81167-8. [DOI] [PubMed] [Google Scholar]
- 26.Liu Z, Fan H, Jiang S. CD4(+) T-cell subsets in transplantation. Immunol Rev. 2013;252:183–191. doi: 10.1111/imr.12038. [DOI] [PubMed] [Google Scholar]
- 27.Stepkowski SM, Kirken RA. Janus tyrosine kinases and signal transducers and activators of transcription regulate critical functions of T cells in allograft rejection and transplantation tolerance. Transplantation. 2006;82:295–303. doi: 10.1097/01.tp.0000228903.03118.be. [DOI] [PubMed] [Google Scholar]
- 28.Malek TR, Yu A, Vincek V, Scibelli P, Kong L. CD4 regulatory T cells prevent lethal autoimmunity in IL-2Rbeta-deficient mice. Implications for the nonredundant function of IL-2. Immunity. 2002;17:167–178. doi: 10.1016/s1074-7613(02)00367-9. [DOI] [PubMed] [Google Scholar]
- 29.Suzuki H, Kundig TM, Furlonger C, Wakeham A, Timms E, Matsuyama T, Schmits R, Simard JJ, Ohashi PS, Griesser H, et al. Deregulated T cell activation and autoimmunity in mice lacking interleukin-2 receptor beta. Science. 1995;268:1472–1476. doi: 10.1126/science.7770771. [DOI] [PubMed] [Google Scholar]
- 30.Lighvani AA, Frucht DM, Jankovic D, Yamane H, Aliberti J, Hissong BD, Nguyen BV, Gadina M, Sher A, Paul WE, O’Shea JJ. T-bet is rapidly induced by interferon-gamma in lymphoid and myeloid cells. Proc Natl Acad Sci U S A. 2001;98:15137–15142. doi: 10.1073/pnas.261570598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wenner CA, Guler ML, Macatonia SE, O’Garra A, Murphy KM. Roles of IFN-gamma and IFN-alpha in IL-12-induced T helper cell-1 development. J Immunol. 1996;156:1442–1447. [PubMed] [Google Scholar]
- 32.Schulz EG, Mariani L, Radbruch A, Hofer T. Sequential polarization and imprinting of type 1 T helper lymphocytes by interferon-gamma and interleukin-12. Immunity. 2009;30:673–683. doi: 10.1016/j.immuni.2009.03.013. [DOI] [PubMed] [Google Scholar]
- 33.Usui T, Preiss JC, Kanno Y, Yao ZJ, Bream JH, O’Shea JJ, Strober W. T-bet regulates Th1 responses through essential effects on GATA-3 function rather than on IFNG gene acetylation and transcription. J Exp Med. 2006;203:755–766. doi: 10.1084/jem.20052165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Szabo SJ, Dighe AS, Gubler U, Murphy KM. Regulation of the interleukin (IL)-12R beta 2 subunit expression in developing T helper 1 (Th1) and Th2 cells. J Exp Med. 1997;185:817–824. doi: 10.1084/jem.185.5.817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Afkarian M, Sedy JR, Yang J, Jacobson NG, Cereb N, Yang SY, Murphy TL, Murphy KM. T-bet is a STAT1-induced regulator of IL-12R expression in naive CD4+ T cells. Nat Immunol. 2002;3:549–557. doi: 10.1038/ni794. [DOI] [PubMed] [Google Scholar]
- 36.Liao W, Lin JX, Wang L, Li P, Leonard WJ. Modulation of cytokine receptors by IL-2 broadly regulates differentiation into helper T cell lineages. Nat Immunol. 2011;12:551–559. doi: 10.1038/ni.2030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lipshultz SE, Chandar JJ, Rusconi PG, Fornoni A, Abitbol CL, Burke GW 3rd, Zilleruelo GE, Pham SM, Perez EE, Karnik R, Hunter JA, Dauphin DD, Wilkinson JD. Issues in solid-organ transplantation in children: translational research from bench to bedside. Clinics (Sao Paulo) 2014;69(Suppl 1):55–72. doi: 10.6061/clinics/2014(Sup01)11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tzvetanov I, Spaggiari M, Jeon H, Roca RG, Bhati C, Oberholzer J, Benedetti E. The role of splenectomy in the setting of refractory humoral rejection after kidney transplantation. Transplant Proc. 2012;44:1254–1258. doi: 10.1016/j.transproceed.2012.01.109. [DOI] [PubMed] [Google Scholar]
- 39.Zhu J, Chen S, Wang J, Zhang C, Zhang W, Liu P, Ma R, Chen Y, Yao Z. Splenectomy increases the survival time of heart allograft via developing immune tolerance. J Cardiothorac Surg. 2013;8:129. doi: 10.1186/1749-8090-8-129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Johnston JA, Kawamura M, Kirken RA, Chen YQ, Blake TB, Shibuya K, Ortaldo JR, McVicar DW, O’Shea JJ. Phosphorylation and activation of the Jak-3 Janus kinase in response to interleukin-2. Nature. 1994;370:151–153. doi: 10.1038/370151a0. [DOI] [PubMed] [Google Scholar]
- 41.Sewgobind VD, Quaedackers ME, van der Laan LJ, Kraaijeveld R, Korevaar SS, Chan G, Weimar W, Baan CC. The Jak inhibitor CP-690,550 preserves the function of CD4CD25FoxP3 regulatory T cells and inhibits effector T cells. Am J Transplant. 2010;10:1785–1795. doi: 10.1111/j.1600-6143.2010.03200.x. [DOI] [PubMed] [Google Scholar]