

Abstract
Brain natriuretic peptide (BNP) has a demonstrable anti-fibrotic effect on diverse organ systems, including the kidney. To understand the molecular mechanism underlying this renoprotective effect, the efficacy of BNP was examined in an in vitro model of glomerular sclerosis by exposing glomerular podocytes to transforming growth factor (TGF)β1-containing media that recapitulates the profibrogenic milieu in chronic glomerular disease. BNP mitigates extracellular matrix (ECM) accumulation in TGFβ1-treated podocytes, as evidenced by Sirius red assay and staining, concomitant with a restoration of the ECM catabolizing activity, as assessed by pulse chase analysis. This effect was in parallel with a mitigating effect on TGFβ1-elicited overexpression of tissue inhibitor of metalloproteinases (TIMP)2, a key inhibitor of a multitude of ECM-degrading metalloproteinases. Mechanistically, glycogen synthase kinase (GSK)3β, a key player in pathogenesis of podocyte injury and glomerulopathies, seems to be involved. BNP treatment considerably induced GSK3β inhibition, marked by inhibitory phosphorylation at the serine 9 residue, and this significantly correlated with the abrogated TIMP2 induction in TGFβ1-injured podocytes. Moreover, genetic knockout of GSK3β in podocytes is sufficient to attenuate the TGFβ1 induced TIMP2 expression and ECM deposition, reminiscent of the effect of BNP. Conversely, ectopic expression of a nonphosphorylatable GSK3β mutant abolished the inhibitory effect of BNP on TGFβ1-elicited TIMP2 overexpression and ECM accumulation, signifying an essential role of GSK3β inhibition in mediating the effect of BNP. Collectively, BNP possesses an anti-fibrotic activity in glomerular epithelial cells. This finding, if validated in vivo, may open a new avenue to the treatment of glomerulosclerosis.
Keywords: Brain natriuretic peptide, podocytes, glomerular disease, fibrosis, transforming growth factor β1, glycogen synthase kinase 3β, extra cellular matrix, cell culture
Introduction
Regardless of the original etiology, diverse chronic kidney diseases (CKD) progress relentlessly via a common pathway to the eventual devastating end stage renal fibrosis, characterized by excessive accumulation of extracellular matrix (ECM) in both glomeruli and tubulointerstitia [1]. Despite numerous pathogenic mechanisms proposed for this process, the primary and direct event of tissue fibrogenesis is the aberration of ECM metabolism that takes place in various parenchymal cells under disease conditions [2]. ECM metabolism involves both matrix synthesis and matrix catabolism or degradation [3]. Theoretically, either increased matrix production or retarded matrix catabolism, or both will result in matrix accumulation. Many cytokines and growth factors have been found to be profibrogenic in the kidney through modulating the matrix metabolism pathways. Among these, TGFβ1 has been widely accepted as a pivotal growth factor contributing to renal fibrosis in various renal diseases [2]. Consistently, in vitro, TGFβ1 was also found to stimulate matrix production as well as retard matrix degradation in cultured renal cells by this [4] and other groups [5]. Many interventions have been attempted; as yet, no one has been completely successful in counteracting the fibrogenic actions of TGFβ1. In the glomerulus, ECM is a major component of supportive scaffolds like glomerular basement membrane (GBM) and mesangium. It exists in a state of dynamic equilibrium between synthesis and degradation that is dictated by all cells constituting the glomerulus, i.e. mesangial cells, glomerular endothelial cells and podocytes. Glomerular podocytes, as the key structural constituent of the glomerular filtration barrier, play a pivotal role in regulating glomerular ECM homeostasis, and thereby affect glomerular permselectivity and contribute to GBM thickening or mesangial expansion, ultimately leading to glomerulosclerosis.
B-type natriuretic peptide (BNP) is a multifunctional peptide, principally produced by ventricular myocytes. Physiologically, BNP exerts vasodilation, natriuresis, and anti-growth activities as well as modulates homeostasis of blood pressure and hemodynamics [6,7]. Distinct from atrial natriuretic peptide (ANP), which is relatively abundant in circulation, recent studies suggest that BNP may also have considerable anti-fibrogenics effect in the heart, which is believed to be achieved via counteraction of TGFβ1’s fibrogenic activity in heart fibroblast cells [8]. Consistent with this view, mice with target deletion of BNP develop cardiac fibrosis [9]. In the kidney, the action of BNP on renal disease has been less investigated. In the only few published studies [10,11], glomerular injury was noted to be attenuated in BNP transgenic mice. This has been attributed to inhibition of mesangial activation. However, it remains unknown if BNP is able to regulate the pathophysiology of other glomerular cells, such as the glomerular podocytes, which play a central role in the development and progression of GBM thickening and glomerular sclerosis, a hallmark of kidney fibrosis [12]. Latest evidence does suggest that glomerular podocytes may also be a target effector of the BNP, because podocyte specific knockout of BNP receptor exacerbates podocyte injury and glomerular sclerosis in a progressive CKD model [13]. Nevertheless, how podocyte specific BNP signaling is involved in glomerular ECM accumulation and glomerular sclerosis remains elusive and was thus examined in this study.
Both BNP and its specific receptor natriuretic peptide receptor-A, also known as membrane-bound guanylyl cyclase receptor A, are expressed in human and rat glomerular and tubular epithelial cells [14], and modulate cellular functions via the intracellular second messenger, cyclic guanosine monophosphate [15], implying the presence of an active autocrine natriuretic peptide system in the kidney and glomerulus. Here, in an effort to examine the effect of BNP on glomerular sclerosis, we employed an in vitro model system of glomerular sclerosis by exposing glomerular podocytes to TGFβ1-containing media that recapitulates the profibrogenic milieu present in the diseased kidney. The efficacy of BNP and the underlying molecular mechanism were tested in this model.
Materials and methods
Cell culture
Conditionally immortalized mouse podocytes, between passages 21 to 25, were cultured under permissive conditions as described previously [16,17]. In brief, podocytes were cultured in RPMI 1640 medium (Life Technologies, Grand Island, NY) supplemented with 10% fetal bovine serum (Life Technologies) and 50 U/mL of recombinant mouse interferon-γ (Millipore, Billerica, MA) at 33°C. Prior to experiments, cells were changed to nonpermissive conditions without interferon-γ and transferred to 37°C incubators to induce differentiation. Differentiated podocytes were treated with BNP peptide (Sigma, St. Louis, MO) and/or recombinant TGFβ1 (R&D Systems Minneapolis, MN) at indicated concentrations. Cells were harvested at the indicated time points.
Sirius red assay and staining
To evaluate the production of fibrotic substances in the cultures, Sirius red assay and staining were applied to assess the extent of ECM collagen accumulation. Concisely, podocytes were grown to subconfluence and treated as stated above. Cells were harvested and sonicated for Sirius red assays. Ammonium sulfate was added to the sonicated cells and the contents were incubated under a slow constant rocking motion at 4°C for 24 h followed by centrifugation. The pellets were resuspended in acetic acid. An aliquot of the resuspended samples was mixed with Sirius red (50 µM) solution (Sigma) made up in 0.5 M acetic acid. After 30 min of constant mixing, the pellets were prepared by centrifuge and resuspended in 1 ml of potassium hydroxide. The optical density of the samples was individually read with a spectrophotometer set to a wavelength of 540 nm. For Sirius red staining, podocytes were grown on chamber slides and received different treatments. At the completion of the experiment, the cells were directly stained with Sirius red and visualized under the light microscope.
Measurement of ECM degrading activity
Matrices of podocytes were prepared as described previously [4,18]. Briefly, differentiated podocytes were grown on 6-well plates at a density of 2×105 cells/well in the presence of ascorbic acid (25 µg/ml, Sigma). The medium was changed every other day with addition of L-[3H]proline (1 µCi/ml). One week after the cells were seeded, cultures were washed with phosphate-buffered saline (PBS), and the cells were removed by addition of 1 ml 2.5 mM NH4OH, 0.1% Triton X-100 per well for 1 min. The matrices left in each well were then washed extensively with PBS and kept covered with sterile distill water at 4°C until further use. Labeled matrices were washed twice with 2 ml of PBS before plating podocytes at 2×105 cells/well under nonpermissive conditions. One week later, cells were washed three times with PBS to remove proteolytic enzyme inhibitors potentially present in the serum. After BNP and/or TGFβ1 treatments for indicated time, cell culture supernatants containing digested matrix were collected. Matrix remaining on the plates was also collected after being digested with 2 ml of 2 N NaOH at 37°C for 18 h. Proteins in all samples were precipitated with cold 10% trichloroacetic acid, washed in cold acetone, and then solubilized and analyzed in a Beckman LS6500 scintillation counter. The values were subtracted by background values obtained with medium in the absence of cells. Counts for both the supernatant and the residual undigested matrix were together considered to be 100%. The percentage of ECM degradation was expressed as the value of supernatant counts divided by that of the sum of counts.
Generation of mice with podocyte-specific GSK3β knockout
Mice with doxycycline inducible podocyte specific knockout of GSK3β (KO) were created as previously elaborated [19,20]. In brief, mice with floxed GSK3β transgene (GSK3βfl/+) were crossed, respectively, with tetO-Cre mice and NPHS2-rtTA mice. Then, Cre/GSK3βfl/+ mice were crossed with rtTA/GSK3βfl/+ mice. All littermates lacking the Cre transgene were designated as control mice (Con). A routine PCR protocol was used for genotyping tail DNA samples. At 8 weeks old, mice received doxycycline hydrochloride (TCI, Tokyo, Japan) treatment via drinking water (2 mg/ml with 5% sucrose, protected from light) for 14 days to induce podocyte-specific GSK3β deletion.
Glomerular isolation and primary culture of control or GSK3β KO podocytes
Isolation of glomeruli from the KO and Con mice was performed as described previously [19,20], with minor modifications. Mice were euthanized and the kidney was perfused with 5 ml of PBS containing 8×107 Dynabeads M-450 (Dynal Biotech ASA, Oslo, Norway). The kidney cortices were minced into 1 mm3 pieces and then digested in a digestion buffer containing 1 mg/ml of collagenase A and 100 U/ml of DNase I for 30 min at 37°C. The tissue was then pressed gently through a 100 μm cell strainer (Falcon, Bedford, MA) and washed with ice-cold sterilized PBS. Washed cells were centrifuged for 5 min at 200 g, and supernatants were aspirated and discarded. Cell pellets were resuspended in ice-cold PBS and glomeruli-containing Dynabeads were gathered using a magnetic particle concentrator. An aliquot (1:1500) of the glomerular isolate was visualized under a microscope to ensure that the samples contained < 5 tubular fragments per ×200 field. The majority of the isolated glomeruli (80%) were decapsulated, which was similar to what had been reported before [19,20]. The enriched glomeruli were plated on collagen type I-coated dishes at 37°C in RPMI 1640 medium (Life Technologies) with 10% fetal bovine serum (Life Technologies), 0.075% sodium pyruvate (Sigma), 100 U/ml penicillin, 100 μg/ml streptomycin (Life Technologies) in a humidified incubator with 5% CO2. Subcultures of primary podocytes were performed by detaching the glomerular cells with 0.25% trypsin-EDTA (Invitrogen, Carlsbad, CA), followed by sieving through a 40 μm cell strainer (Falcon), and culture on collagen type I-coated dishes as reported before [21]. Podocytes of passages 1 or 2 were characterized by the expression of multiple podocyte-specific markers and used in subsequent experiments. Primary podocytes were treated with TGFβ1, BNP or vehicle for indicated time.
Transient transfection
The expression vectors encoding the hemagglutinin (HA) tagged nonphosphorylatable mutant GSK3β (S9A-GSK3β-HA/pcDNA3) was kindly provided by Dr. Gail V.W. Johnson (University of Alabama at Birmingham, Birmingham, AL) [22]. Podocyte transfection was performed using Lipofectamine 2000 (Invitrogen, Carlsbad, CA, USA) as described before [23]. In brief, conditionally immortalized murine podocytes were cultured under permissive conditions at 50% to 70% confluence in the absence of antibiotics. The vector-Lipofectamine 2000 complexes were prepared and applied to proliferating podocytes for transfection. The ratio of Lipofectamine 2000 to vectors was optimized by a series of pilot experiments for each study until the best transfection efficiency was achieved. After transfection, the cells were cultured under nonpermissive conditions in normal growth medium for 48 h before transfection efficiency was assessed by immunocytochemistry staining or by immunoblot analysis for HA. Cells were then subjected to BNP, TGFβ1 or vehicle treatments.
Western immunoblot analysis
Cultured cells were lysed in radioimmunoprecipitation assay buffer supplemented with protease inhibitors (1% Nonidet P-40, 0.1% SDS, 100 mg/ml phenylmethysulfonyl fluoride, 0.5% sodium deoxycholate, 1 mM sodium orthovanadate, 2 mg/ml aprotin, 2 mg/ml leupeptin, 5 mM EDTA in PBS). Protein concentrations were determined using a bicinchoninic acid protein assay kit (Sigma). Samples with equal amounts of total protein (50 µg) were fractionated by 10~15% SDS polyacrylamide gels under reducing conditions and analyzed by western immunoblot as described previously. The antibodies against phosphorylated glycogen synthase kinase (GSK) 3β at serine 9 (pGSK3β), GSK3β, tissue inhibitors of metalloproteinases (TIMP)2, HA and actin were purchased from Santa Cruz Biotechnology (Santa Cruz, CA) or Cell Signaling Technology (Beverly, MA).
Statistical analyses
One investigator in a blinded manner performed computerized morphometric analysis. For immunoblot analysis, bands were scanned and the integrated pixel density was determined using a densitometer and the NIH image analysis program. Unless otherwise indicated, all experimental observations were repeated three times. Statistical analysis of the data from multiple groups was performed by repeated measures ANOVA followed by Fisher’s Least Significant Difference tests. Data from two groups were compared by Student’s t-test. Linear regression analysis was applied to examine possible relationships between two parameters. P < 0.05 was considered significant.
Results
BNP mitigates ECM accumulation in podocytes exposed to a profibrogenic milieu containing TGFβ1, concomitant with a correction of the ECM degrading activity
TGFβ1 is a quintessential profibrotic cytokine and a key player in mediating fibrogenesis in multiple organ systems. It is capable of inducing ECM accumulation in diverse cell types, including podocytes. Indeed, in differentiated immortalized murine podocytes in culture, TGFβ1 treatment for 48 h resulted in massive ECM accumulation, marked by Sirius red staining (Figure 1A). This effect was substantially abrogated by BNP co-treatment. The morphologic findings were corroborated by Sirius red assay of whole cell lysates (Figure 1B), which revealed a dose-dependent anti-fibrotic activity of BNP. ECM accumulation is determined by the activity of ECM metabolism, which is balanced by both ECM synthesis or anabolism and ECM degradation or catabolism. TGFβ1 has been demonstrated to impede ECM catabolism in other kidney cells like renal tubular epithelial cells [4]. However, its effect on podocyte ECM degradation is unknown. To address this issue, pulse chase analysis of ECM pre-labelled with [3H]-proline was performed. Shown in Figure 1C, there was a constitutive ECM degrading activity in podocytes under basal conditions. TGFβ1 drastically retarded ECM degradation, resulting in a lessened level of soluble degraded matrix in the cell culture supernatant and a heightened level of undigested matrix. This effect was markedly abrogated by BNP in a dose dependent mode, though BNP alone did not significantly affect ECM accumulation or degradation. In aggregate, these data suggest that BNP is capable of counteracting the profibrogenic activity of TGFβ1, which suppresses ECM catabolism, resulting in ECM accumulation in podocytes.
Figure 1.
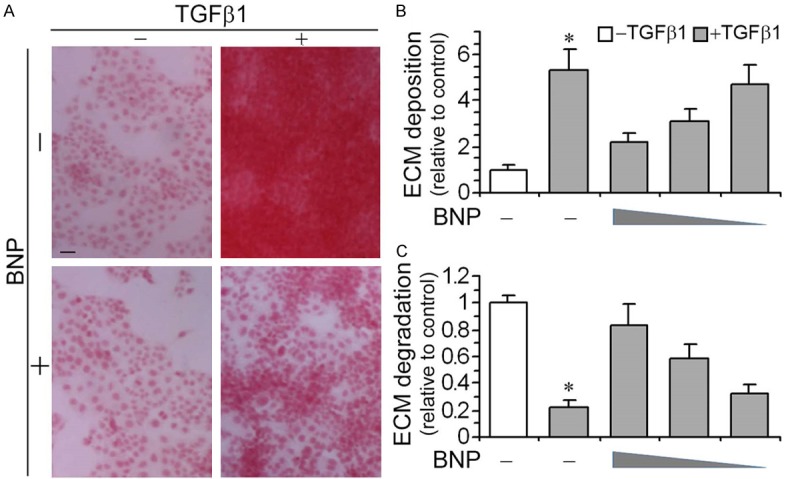
BNP treatment counteracts the profibrogenic effect of TGFβ1 in podocytes, associated with a restored ECM catabolic activity. Differentiated immortalized murine podocytes were treated with vehicle, BNP (10-6 M, 10-7 M, 10-8 M) and/or TGFβ1 (10 ng/ml) for 48 h. A. Cell cultures treated with vehicle, BNP (10-6 M) and/or TGFβ1 (10 ng/ml) for 48 h were stained with Sirius red and visualized under light microscope. Representative microscopic images were shown. Bar =100 µm. B. Cell lysates were collected and processed for Sirius red total collagen assay. Relative abundance of total collagen is shown. *P < 0.05 versus BNP-treated cells (n=3). C. Podocytes were grown on ECM pre-labelled with [3H]proline and differentiated. Cells were then treated with vehicle, BNP (10-6 M, 10-7 M, 10-8 M) and/or TGFβ1 (10 ng/ml) for 48 h followed by pulse-chase analysis of the ECM catabolic activity as specified in the Methods and expressed here as the arbitrary value relative to the vehicle treated control group. *P < 0.05 versus BNP-treated cells (n=3).
BNP overrides TGFβ1-elicited TIMP2 expression, associated with inhibitory phosphorylation of GSK3β
A number of enzymes are involved in regulating ECM degradation, including a number of TIMP, such as TIMP1, 2 and 3. In our study, TIMP2, which plays a critical role in inhibiting the collagen degrading metalloproteinase 2 and 9, rather than TIMP1 and 3 (data not shown) were significantly induced in podocytes by TGFβ1 treatment. BNP co-treatment was able to overcome the TGFβ1 elicited TIMP2 expression in a dose dependent manner, as evidenced by immunoblot analysis of cell lysates (Figure 2A). A growing body of evidence recently suggests that GSK3β is centrally implicated in podocyte injury triggered by various stimuli and in many glomerular diseases [16,17,19-21,23]. To discern if the anti-fibrotic activity of BNP in cultured podocytes is accociated with an effect on GSK3β signaling, immunoblot analysis was conducted to measure the expression of phosphorylated GSK3β at serine 9 residue, which represents the inhibitory phosphorylation of GSK3β. Shown in Figure 2A, BNP treatment augmented this inhibitory phosphorylation of GSK3β in a dose dependent fashion, concomitant with a mitigated TIMP2 induction. Linear regression analysis (Figure 2B) revealed a close negative correlationship between the BNP induced GSK3β phosphorylation and the TIMP2 expression levels in podocytes exposed to TGFβ1.
Figure 2.
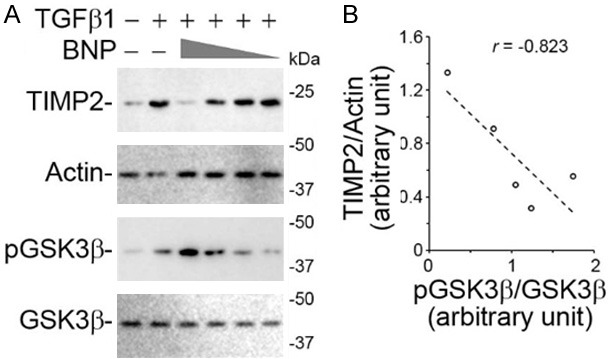
BNP mitigates the TGFβ1 induced expression of TIMP2 in podocytes, correlating with inhibitory phosphorylation of GSK3β, a key player in podocyte injury and glomerular disease. Differentiated immortalized murine podocytes were treated with vehicle, BNP (10-6 M, 10-7 M, 10-8 M) and/or TGFβ1 (10 ng/ml) for 48 h. A. Cells lysates were processed for immunoblot analysis for indicated molecules (full-length blots are included in the Figure S1). B. Densitometric analysis of immunoblots quantified the relative abundance of TIMP2 as TIMP2/actin ratios and that of pGSK3β as pGSK3β/GSK3β ratios. Linear regression analysis showed a negative correlation between inhibitory phosphorylation of GSK3β and TIMP2 in podocytes. The correlation coefficient r was -0.823 (P < 0.05).
GSK3β ablation mimics the BNP effect on diminishing ECM accumulation in TGFβ1-injured podocytes
To further explore a possible role of GSK3β inhibition in mediating the effect of BNP on ECM degradation and accumulation in TGFβ1-treated podocytes, primary cultures of podocytes derived from GSK3β KO mice and control mice were utilized and exposed to TGFβ1 treatment. Shown in Figure 3A, TGFβ1 induced massive ECM accumulation in control podocytes. The morphologic findings were corroborated by Sirius red assay of whole cell lysates (Figure 3B). These effects were largely diminished in KO cells, signifying that GSK3β inhibition is likely sufficient for the suppressive effect of BNP on matrix accumulation.
Figure 3.
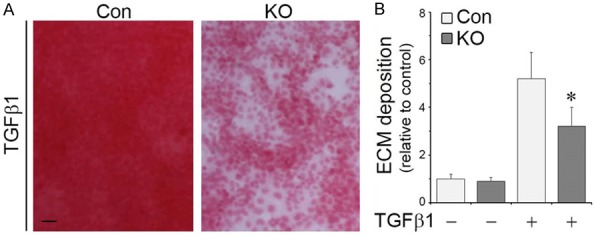
Genetic knockout of GSK3β counteracts the profibrogenic effect of TGFβ1 in primary podocytes, reminiscent of the action of BNP. Primary podocytes were prepared from mice with podocyte specific knockout of GSK3β (KO) or control mice (Con) and treated with vehicle or TGFβ1 (10 ng/ml) for 48 h. A. Cell cultures were stained with Sirius red and visualized under light microscope. Representative microscopic images were shown. Bar =100 µm. B. Cell lysates were collected and processed for Sirius red total collagen assay. Relative abundance of total collagen is shown. *P < 0.05 versus TGFβ1-treated Control cells (n=3).
Loss of GSK3β is sufficient to attenuate the TGFβ1 induced TIMP2 expression in podocytes
To determine if the lessened ECM accumulation in TGFβ1-treated GSK3β KO cells stems from a possible effect on TIMP2 expression, GSK3β KO and control podocytes were exposed to vehicle or TGFβ1 and cell lysates subjected to immunoblot analysis. Shown in Figure 4A, a near complete ablation of GSK3β was noted in KO cells as compared to control cells. Loss of GSK3β seems to barely affect TIMP2 expression in podocytes under basal condition. However, the TGFβ1 elicited TIMP2 overproduction was present in control podocytes but evidently blunted in KO cells (Figure 4A and 4B), denoting that GSK3β inhibition is sufficient for attenuating TIMP2 induction in TGFβ1-treated podocytes.
Figure 4.
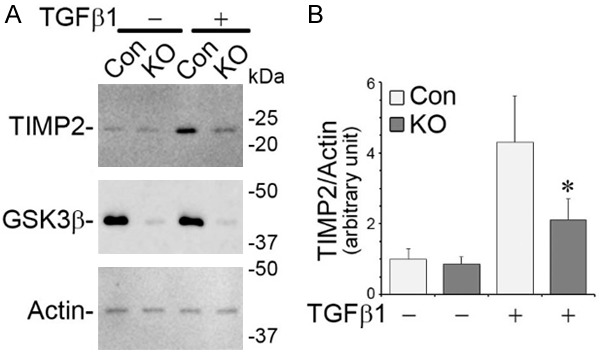
Loss of GSK3β is sufficient for overriding TIMP2 induction in primary podocytes exposed to TGFβ1. Primary podocytes were prepared from mice with podocyte specific knockout of GSK3β (KO) or control mice (Con) and treated with vehicle or TGFβ1 (10 ng/ml) for 48 h. A. Cells lysates were processed for immunoblot analysis for indicated molecules (full-length blots are included in the Figure S2). B. Densitometric analysis of immunoblots quantified the relative abundance of TIMP2 as TIMP2/actin ratios. *P < 0.05 versus TGFβ1-treated Control cells.
Inhibitory phosphorylation of GSK3β at serine 9 is essential for the inhibitory effect of BNP on TIMP2 expression and ECM accumulation in podocytes exposed to TGFβ1
To determine if GSK3β inhibition is essential for the BNP effect on TIMP2 expression and on ECM accumulation, immortalized mouse podocytes were forced to express a mutant GSK3β, in which the serine 9 residue is replaced by alanine (S9A) and thus GSK3β is unable to be phosphorylated and inhibited. Following transient transfection, podocytes exhibited a satisfactory transfection efficiency as shown by immunocytochemistry staining (data not shown) or immunoblot analysis for HA or GSK3β (Figure 5A). Consistent with the findings above, BNP mitigated TIMP2 expression likewise in empty vector (EV)-expressing cells exposed to TGFβ1 (Figure 5A), in parallel with an abrogated ECM accumulation (Figure 5B and 5C). In stark contrast, in podocytes expressing S9A, the efficacy of BNP was largely blunted, suggesting that phosphorylation of GSK3β at serine 9 is required for the BNP effect on TIMP2 expression (Figure 5A) and the ensuing effect on ECM metabolism (Figure 5B and 5C).
Figure 5.
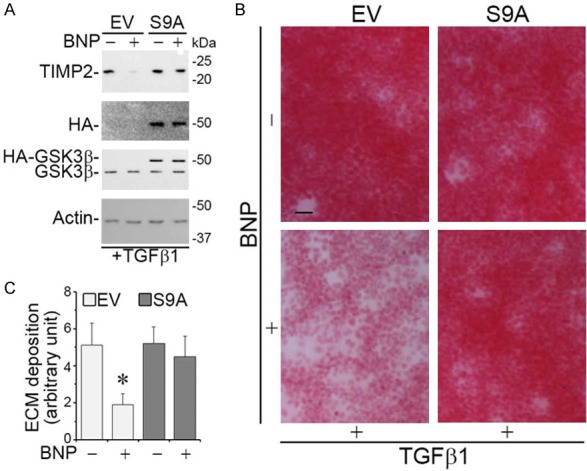
Phosphorylation of GSK3β at serine 9 is required for the inhibitory effect of BNP on TIMP2 expression and the ensuing effect on ECM accumulation. Immortalized murine podocytes were subjected to transient transfection with plasmid vectors encoding the empty vector (EV) or a hemagglutinin (HA)-conjugated nonphosphorylatable GSK3β mutant, in which the serine 9 residue is replaced by alanine (S9A). Cells were treated with TGFβ1 (10 ng/ml) in the presence or absence of BNP (10-6 M) for 48 h. A. Cells lysates were processed for immunoblot analysis for TIMP2, HA, GSK3β and actin (full-length blots are included in the Figure S3). B. Cell cultures were stained with Sirius red and visualized under light microscope. Representative microscopic images were shown. Bar =100 µm. C. Cell lysates were collected and processed for Sirius red total collagen assay. Relative abundance of total collagen is shown. *P < 0.05 versus TGFβ1 alone-treated Control cells (n=3).
Discussion
Podocytes play an important role in maintaining ECM homeostasis in the glomerulus, more specifically in the GBM and mesangial area [24]. Aberration in podocyte ECM metabolism, such as enhanced ECM production or impeded ECM degradation, has been involved in glomerular matrix accumulation, glomerular sclerosis and destruction in various primary and secondary glomerular diseases [25]. For instance, GBM thickening in diabetic nephropathy or in other CKD has been attributable to diminished ECM degradation [26,27]. BNP has been reported to play a beneficial effect in glomerular injury [10,11,13,14]. However, the underlying mechanism remains unknown. Here, we tested the effect of BNP in an in vitro model of glomerular sclerosis by exposing glomerular podocytes to TGFβ1-containing media that recapitulates the profibrogenic milieu in progressive CKD. Our study demonstrated that BNP is capable of suppressing the TGFβ1 induced overexpression of TIMP2 and reinstating ECM degrading activity in podocytes in the profibrogenic milieu. This anti-fibrotic effect of BNP is mediated at least in part by inhibition of the GSK3β signaling. To the best of our knowledge, this is the first report on the regulatory effect of BNP on podocyte ECM catabolism.
Burgeoning evidence suggests that BNP exerts a potent protective effect in animal models of chronic fibrosis in a number of organs, including the heart [9], liver [28] and the kidney [10,11,13]. However, the mechanism responsible for this beneficial anti-fibrotic effect is illusive. As a hormone secreted by cardiomyocytes in the heart ventricles in response to stretching caused by increased ventricular blood volume, BNP has been postulated to attenuate organ fibrosis indirectly via modulating the systemic blood pressure secondary to its powerful control of natriuresis and the circulating volume [7]. However, more recent evidence from transgenic animals with tissue specific knockout of BNP receptors suggests that a tissue specific effect of BNP that is independent of systemic hemodynamics may contribute to its anti-fibrotic activity. To this end, podocyte specific knockout of guanylyl cyclase-A, a cognate receptor for BNP, worsened glomerulosclerosis and proteinuria in mice with progressive CKD elicited by deoxycorticosterone-acetate in combination with high salt intake [13], implying that a podocyte specific BNP signaling confers a mitigating effect on glomerular ECM deposition. TGFβ1 is a critical cytokine that plays a key role in kidney fibrosis and glomerular sclerosis as evidenced by numerous studies that successfully ameliorated renal fibrotic lesions after blocking TGFβ1 signaling [29]. In vivo, the beneficial and anti-fibrotic effect of BNP therapy in diverse disease models has been associated with a mitigating action on TGFβ1 expression [8]. However, whether BNP/guanylyl cyclase-A signaling is able to directly affect any cellular or molecular effect downstream of TGFβ1 pathway in podocytes has not been studied before. To address issue, we tested the effect of BNP in podocytes exposed to TGFβ1. BNP was found to reinstate the ECM degrading activity in TGFβ1-treated podocytes, associated with a diminished induction of TIMP2, a key inhibitor of MMPs and a typical target molecule of TGFβ1. It seems that this effect of BNP is mediated indirectly via inhibition of GSK3β signaling pathway, because loss of GSK3β is sufficient for suppressing TIMP2 expression and mitigating ECM accumulation in TGFβ1-treated podocytes, mimicking the effect of BNP, whereas ectopic expression of a nonphosphorylatable GSK3β mutant abolishes the effect of BNP.
How GSK3β inhibition affects TGFβ1-induced TIMP2 overexpression and aberration in ECM catabolism is still unknown. But there is evidence suggesting that inhibition of GSK3β is able to counteract the signaling activity of TGFβ1 and mitigate diverse TGFβ1-target fibrogenic phenotypes in other cell types. For instance, in cultured renal fibroblast cells, overexpression of constitutively active GSK3β was sufficient to induce myofibroblast activation, marked by α-smooth muscle actin (α-SMA) expression, reminiscent of the effect of TGFβ1 [30]. In consistency, in vivo in mice with ischemia-reperfusion injury, pharmacological inhibition of GSK3 using TDZD-8 significantly suppressed renal fibrosis by reducing the myofibroblast population and deposition of ECM, including collagen I and fibronectin [30]. Similarly in primary pulmonary fibroblasts isolated from patients with chronic obstructive pulmonary disease, blockade of GSK3β by SB216763, a highly selective small molecule inhibitor of GSK3β, dose-dependently attenuated TGFβ1-induced expression of myofibroblast markers like α-SMA and fibronectin [31]. It seems that this effect of GSK3β inhibition on myofibroblast differentiation did not involve Smad, NFκB or ERK1/2 signaling. Rather, GSK3β inhibition increased the phosphorylation of cAMP response element binding protein (CREB), which in its phosphorylated from acts as a functional antagonist of TGFβ/smad signaling [31]. Of course, it merits in-depth investigations to determine whether activation of CREB signaling pathway subsequent to GSK3β inhibiton is also responsible for the TIMP2 suppressing and anti-fibrotic effect of BNP in podocytes.
In summary, BNP is able to restore the ECM degrading activity and reinstate ECM homeostasis in glomerular podocytes exposed to a profibrogenic milieu. This effect is likely attributable to a mitigated TIMP2 induction subsequent to GSK3β inhibition. Our findings suggest that BNP possesses an anti-fibrotic activity in glomerular cells and, if validated in vivo, may become a novel therapeutic option for glomerulosclerosis.
Acknowledgements
The research work of the authors was supported in part by the Natural Science Foundation of China grant 81770672 and 81873612, the U.S. National Institutes of Health grant DK092485 and DK114006. The funders had no role in the design and conduct of this study, collection and interpretation of the data, or preparation and approval of the manuscript.
Disclosure of conflict of interest
None.
Supporting Information
References
- 1.Remuzzi G, Bertani T. Pathophysiology of progressive nephropathies. N Engl J Med. 1998;339:1448–1456. doi: 10.1056/NEJM199811123392007. [DOI] [PubMed] [Google Scholar]
- 2.Eddy AA. Molecular basis of renal fibrosis. Pediatr Nephrol. 2000;15:290–301. doi: 10.1007/s004670000461. [DOI] [PubMed] [Google Scholar]
- 3.Saha AK, Kohles SS. A cell-matrix model of anabolic and catabolic dynamics during cartilage biomolecule regulation. Int J Comput Healthc. 2012;1:214–228. doi: 10.1504/IJCIH.2012.046995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gong R, Rifai A, Tolbert EM, Centracchio JN, Dworkin LD. Hepatocyte growth factor modulates matrix metalloproteinases and plasminogen activator/plasmin proteolytic pathways in progressive renal interstitial fibrosis. J Am Soc Nephrol. 2003;14:3047–3060. doi: 10.1097/01.asn.0000098686.72971.db. [DOI] [PubMed] [Google Scholar]
- 5.Lu P, Takai K, Weaver VM, Werb Z. Extracellular matrix degradation and remodeling in development and disease. Cold Spring Harb Perspect Biol. 2011;3 doi: 10.1101/cshperspect.a005058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Potter LR, Yoder AR, Flora DR, Antos LK, Dickey DM. Natriuretic peptides: their structures, receptors, physiologic functions and therapeutic applications. Handb Exp Pharmacol. 2009:341–366. doi: 10.1007/978-3-540-68964-5_15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kuhn M. Molecular physiology of natriuretic peptide signalling. Basic Res Cardiol. 2004;99:76–82. doi: 10.1007/s00395-004-0460-0. [DOI] [PubMed] [Google Scholar]
- 8.Kapoun AM, Liang F, O’Young G, Damm DL, Quon D, White RT, Munson K, Lam A, Schreiner GF, Protter AA. B-type natriuretic peptide exerts broad functional opposition to transforming growth factor-beta in primary human cardiac fibroblasts: fibrosis, myofibroblast conversion, proliferation, and inflammation. Circ Res. 2004;94:453–461. doi: 10.1161/01.RES.0000117070.86556.9F. [DOI] [PubMed] [Google Scholar]
- 9.Tamura N, Ogawa Y, Chusho H, Nakamura K, Nakao K, Suda M, Kasahara M, Hashimoto R, Katsuura G, Mukoyama M, Itoh H, Saito Y, Tanaka I, Otani H, Katsuki M. Cardiac fibrosis in mice lacking brain natriuretic peptide. Proc Natl Acad Sci U S A. 2000;97:4239–4244. doi: 10.1073/pnas.070371497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kasahara M, Mukoyama M, Sugawara A, Makino H, Suganami T, Ogawa Y, Nakagawa M, Yahata K, Goto M, Ishibashi R, Tamura N, Tanaka I, Nakao K. Ameliorated glomerular injury in mice overexpressing brain natriuretic peptide with renal ablation. J Am Soc Nephrol. 2000;11:1691–1701. doi: 10.1681/ASN.V1191691. [DOI] [PubMed] [Google Scholar]
- 11.Suganami T, Mukoyama M, Sugawara A, Mori K, Nagae T, Kasahara M, Yahata K, Makino H, Fujinaga Y, Ogawa Y, Tanaka I, Nakao K. Overexpression of brain natriuretic peptide in mice ameliorates immune-mediated renal injury. J Am Soc Nephrol. 2001;12:2652–2663. doi: 10.1681/ASN.V12122652. [DOI] [PubMed] [Google Scholar]
- 12.D’Agati VD. Podocyte injury in focal segmental glomerulosclerosis: lessons from animal models (a play in five acts) Kidney Int. 2008;73:399–406. doi: 10.1038/sj.ki.5002655. [DOI] [PubMed] [Google Scholar]
- 13.Staffel J, Valletta D, Federlein A, Ehm K, Volkmann R, Fuchsl AM, Witzgall R, Kuhn M, Schweda F. Natriuretic peptide receptor guanylyl cyclase-a in podocytes is renoprotective but dispensable for physiologic renal function. J Am Soc Nephrol. 2017;28:260–277. doi: 10.1681/ASN.2015070731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kato Y, Mori K, Kasahara M, Osaki K, Ishii A, Mori KP, Toda N, Ohno S, Kuwabara T, Tokudome T, Kishimoto I, Saleem MA, Matsusaka T, Nakao K, Mukoyama M, Yanagita M, Yokoi H. Natriuretic peptide receptor guanylyl cyclase-A pathway counteracts glomerular injury evoked by aldosterone through p38 mitogen-activated protein kinase inhibition. Sci Rep. 2017;7:46624. doi: 10.1038/srep46624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kerkela R, Ulvila J, Magga J. Natriuretic peptides in the regulation of cardiovascular physiology and metabolic events. J Am Heart Assoc. 2015;4:e002423. doi: 10.1161/JAHA.115.002423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bao H, Ge Y, Peng A, Gong R. Fine-tuning of NFkappaB by glycogen synthase kinase 3beta directs the fate of glomerular podocytes upon injury. Kidney Int. 2015;87:1176–1190. doi: 10.1038/ki.2014.428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Xu W, Ge Y, Liu Z, Gong R. Glycogen synthase kinase 3beta orchestrates microtubule remodeling in compensatory glomerular adaptation to podocyte depletion. J Biol Chem. 2015;290:1348–1363. doi: 10.1074/jbc.M114.593830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Brem AS, Morris DJ, Ge Y, Dworkin L, Tolbert E, Gong R. Direct fibrogenic effects of aldosterone on normotensive kidney: an effect modified by 11beta-HSD activity. Am J Physiol Renal Physiol. 2010;298:F1178–1187. doi: 10.1152/ajprenal.00532.2009. [DOI] [PubMed] [Google Scholar]
- 19.Li C, Ge Y, Dworkin L, Peng A, Gong R. The beta isoform of GSK3 mediates podocyte autonomous injury in proteinuric glomerulopathy. J Pathol. 2016;239:23–35. doi: 10.1002/path.4692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhou S, Wang P, Qiao Y, Ge Y, Wang Y, Quan S, Yao R, Zhuang S, Wang LJ, Du Y, Liu Z, Gong R. Genetic and pharmacologic targeting of glycogen synthase kinase 3beta reinforces the Nrf2 antioxidant defense against podocytopathy. J Am Soc Nephrol. 2016;27:2289–2308. doi: 10.1681/ASN.2015050565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Li C, Ge Y, Peng A, Gong R. The redox sensitive glycogen synthase kinase 3beta suppresses the self-protective antioxidant response in podocytes upon oxidative glomerular injury. Oncotarget. 2015;6:39493–39506. doi: 10.18632/oncotarget.6303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gong R, Rifai A, Dworkin LD. Activation of PI3K-Akt-GSK3beta pathway mediates hepatocyte growth factor inhibition of RANTES expression in renal tubular epithelial cells. Biochem Biophys Res Commun. 2005;330:27–33. doi: 10.1016/j.bbrc.2005.02.122. [DOI] [PubMed] [Google Scholar]
- 23.Xu W, Ge Y, Liu Z, Gong R. Glycogen synthase kinase 3beta dictates podocyte motility and focal adhesion turnover by modulating paxillin activity: implications for the protective effect of low-dose lithium in podocytopathy. Am J Pathol. 2014;184:2742–2756. doi: 10.1016/j.ajpath.2014.06.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Abrahamson DR. Role of the podocyte (and glomerular endothelium) in building the GBM. Semin Nephrol. 2012;32:342–349. doi: 10.1016/j.semnephrol.2012.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tufro A. Repairing the GBM step by step. J Am Soc Nephrol. 2018;29:1346–1347. doi: 10.1681/ASN.2018030294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lee HS. Mechanisms and consequences of TGF-ss overexpression by podocytes in progressive podocyte disease. Cell Tissue Res. 2012;347:129–140. doi: 10.1007/s00441-011-1169-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Marshall CB. Rethinking glomerular basement membrane thickening in diabetic nephropathy: adaptive or pathogenic? Am J Physiol Renal Physiol. 2016;311:F831–F843. doi: 10.1152/ajprenal.00313.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sonoyama T, Tamura N, Miyashita K, Park K, Oyamada N, Taura D, Inuzuka M, Fukunaga Y, Sone M, Nakao K. Inhibition of hepatic damage and liver fibrosis by brain natriuretic peptide. FEBS Lett. 2009;583:2067–2070. doi: 10.1016/j.febslet.2009.05.025. [DOI] [PubMed] [Google Scholar]
- 29.Meng XM, Tang PM, Li J, Lan HY. TGF-beta/Smad signaling in renal fibrosis. Front Physiol. 2015;6:82. doi: 10.3389/fphys.2015.00082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Singh SP, Tao S, Fields TA, Webb S, Harris RC, Rao R. Glycogen synthase kinase-3 inhibition attenuates fibroblast activation and development of fibrosis following renal ischemia-reperfusion in mice. Dis Model Mech. 2015;8:931–940. doi: 10.1242/dmm.020511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Baarsma HA, Engelbertink LH, van Hees LJ, Menzen MH, Meurs H, Timens W, Postma DS, Kerstjens HA, Gosens R. Glycogen synthase kinase-3 (GSK-3) regulates TGF-beta(1)-induced differentiation of pulmonary fibroblasts. Br J Pharmacol. 2013;169:590–603. doi: 10.1111/bph.12098. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.