

Abstract
Hippocampal neuronal death plays a causal role in the cognitive impairment of temporal lobe epilepsy (TLE). Ferroptosis, a novel form of regulated cell death, is strongly linked to cognitive impairment. However, whether ferroptosis is associated with cognitive comorbidities of TLE is unknown. In this study, it was demonstrated that ferroptosis occurs in the hippocampus following kainic acid (KA)-induced TLE in rats. Treatment with ferrostatin-1, a specific inhibitor of ferroptosis, prevented the initiation and progression of ferroptosis in the hippocampus of KA-treated rats. This was through decreased expression of glutathione peroxidase 4, glutathione (GSH) depletion as well as lipid peroxides and iron accumulation. It was also found that ferrostatin-1 prevented hippocampal neuronal loss and rescued cognitive function in KA-induced TLE in rats. These results suggest that ferroptosis is involved in the cognitive impairment of KA-induced TLE in rats, and inhibition of ferroptosis processes ameliorates cognitive impairment in KA-induced TLE in rats.
Keywords: Ferroptosis, cognitive impairment, kainic acid, temporal lobe epilepsy-rats
Introduction
Epilepsy is a common chronic neurological disorder affecting more than 68 million individuals worldwide [1]. It is associated with both physical distress and psychological stress [2]. Up to 50% of patients with epilepsy suffer from psychiatric or cognitive comorbidities. Moreover, the burden of comorbidities severely affects the quality of life [3]. Temporal lobe epilepsy (TLE) is the most prevalent form of epilepsy, accounts for 60% of all epilepsy cases. It is originating primarily from hippocampus and amygdala. Since the hippocampus is the main structure involved in learning and memory, damage to this structure often results in cognitive impairment [4]. Neuroimaging studies show that hippocampal atrophy, neuronal cell loss in the hippocampus and decreased neuronal density in the dentate gyrus positively correlate with memory impairment in patients with TLE [5]. Inhibition of hippocampal neuronal death and oxidative injury improves cognitive dysfunction in temporal lobe epilepsy in rats [3,6]. Although damage to the hippocampal structures is a common cause of seizures and cognitive dysfunction, a growing body of literature suggest that the anesis of cognitive comorbidities of epilepsy is associated with neuroprotection but not with a concomitant decrease in seizure burden. Further, there are few targeted therapies for the management of cognitive comorbidities of epilepsy. In addition, our understanding of the mechanisms of the current drugs is not sufficient [3].
Ferroptosis, a novel form of regulated cell death, was previously been found in an organotypic hippocampal slice culture model of rats with glutamate-induced neurotoxicity [7]. Ferroptosis initiation and execution lies in three critical events: iron accumulation, GSH depletion, and lipid peroxides accumulation [8]. On the other hand, numerous clinicopathological features of dementia are consistent with ferroptosis [9]. Inhibition of hippocampal ferroptosis ameliorates cognitive function in diverse disease models, such as stroke, Alzheimer’s disease [10,11]. It is, however not know if ferroptosis is involved in epilepsy induced cell death and associated cognitive comorbidities of TLE.
Intrahippocampal administration of kainic acid (KA) is particularly useful in studying the behavior and neuropathological characteristics of TLE. Activation of the KA receptor results in the release of excess glutamte caused neuronal death and sustained epileptic activity in the hippocampus [12]. Central KA injections results in progressive increase in iron concentration in the rat hippocampus, leading to accumulation of a large amount of lipid peroxides, accompanied by the depletion of glutathione (GSH), which could promote free radical damage in the lesioned areas [13]. These facts suggest that ferroptosis might occur in KA induced TLE in rats.
In this current, the hypothesis that ferroptosis is involved in epilepsy induced cell death and associated cognitive comorbidities of TLE was tested. Using ferrostatin-1 (Fer-1), a potent and selective inhibitor of ferroptosis [14], it was found that inhibition of hippocampal ferroptosis process prevented hippocampal neuronal loss, and ameliorated cognitive decline in TLE rats.
Materials and methods
Animals
All experiments were conducted on adult male Sprague-Dawley (200 g-220 g, n = 30), obtained from Hunan slake jingda laboratory animal co.,Ltd. (Changsha, China). 3-4 rats/cage were kept in a temperature controlled room under a 12 h-light/dark cycle (lights on at 07:00a.m.) with free access to food and water. Procedures involving animals were approved by the Animal Care and Use Committees of Guangxi Medical University. Rats were randomly divided into three equal-sized groups namely: sham, KA + Fer-1, and KA groups. For intrahippocampal injections, the rats were anesthetized with chloral hydrate (350 mg/kg; i.p.), placed into the stereotaxic frame (Stoelting Co., USA) with the incisor bar set at 3.3 mm below the interaural line. The dorsal surface of the skull was exposed and a burr hole was drilled in the skull using the following stereotaxic coordinates according to the atlas of Paxinos and Watson: anteroposterior, 4.3 mm caudal to bregma, 4.1 mm lateral to the midline (right side), and 4.2 mm ventral to the surface of the skull. A micro syringe filled with 2 μl of normal saline containing 0.5 μg/μl of KA was placed over the burr hole and the kainic acid solution was injected at a rate of 1 μl/min in order to induce an experimental model of status epilepticus (SE) and TLE [15]. All rats showed convulsive SE after 30-60 min following intrahippocampal injection of KA. KA (Sigma-Aldrich, USA) was dissolved in cold normal saline just prior to surgery. The sham group received an equivalent volume of normal saline at the same stereotaxic coordinates. Treatment on rats was commenced 3 h following overt SE [16]. Rats in the Fer-1 group were administered once a day for 2 weeks with an i.p. dose of Fer-1 (2.5 μmol/kg; Selleck, USA), the dose of Fer-1 was selected from previous reports [17] and this pilot study. Fer-1 was dissolved in vehicle. The sham group and KA group were injected with vehicle (200 μl PBS containing 10% DMSO).
Assessment of seizure behavior
From day 30 to day 36 after intrahippocampal injection, all rats were continually monitored via 24/7 video to record the spontaneous recurrent seizures (SRS) and scored according to Racine’s classification [16].
Assessment of cognitive function
Cognitive function of all rats was assessed by a Y-maze test, novel recognition test and morris water maze. If a rat experienced a seizure before testing, they were tested at least 1 h after the seizure. If a rat experienced a seizure during testing, the rat data were excluded at the time of analysis.
Y-maze test
At day 37 after intrahippocampal injection, the Y-maze test, was performed as previously described [18]. The Y-maze consisted of three randomly designated A, B, and C arms (50 cm length,18 cm width, and 18 cm height) with an angle of 120° between each of the two arms. During the study, each rat was first placed at one end of the arm and the total number (N) and the order of the arm entries were recorded by a video camera for every 8 min. Successful spontaneous alternations were defined as consecutive triple entries of different arms choices. The spontaneous alternation response rate was calculated as: spontaneous alternation rate (%) = number of successful alternation/(N - 2) × 100.
Novel object recognition test
From day 38 to day 39 after intrahippocampal injection, the novel object recognition (NOR) test was performed in an open circle arena (50 cm × 50 cm) as previously described [19]. During the training trial, two identical objects were placed along the center line of the arena and the rats were allowed to explore it for 5 min. After each trial, the arena and objects were cleaned with 40% ethanol solution to minimize olfactory cues. Twenty four hours following the training trial, one of the objects was replaced with a novel object and rats were allowed a 5 min exploratory trial. The time exploring familiar object (TF) and exploring novel object (TN) were recorded. Memory was evaluated by discrimination index. Discrimination Index (DI) = (TN - TF)/(TN + TF).
Perls’ staining
The brains were removed from the skulls and immediately put in 4% paraformaldehyde, then dehydrated and embedded in paraffin, and finally sliced into 4 μm thick coronal sections by a microtome. Perls’ staining was used to detect iron accumulation as previously described [20]. After dewaxing, the sections were washed with distilled water and incubated for 20 min in freshly prepared Perls stain solution (Solarbio, Beijing, China). This was followed by washing with distilled water 6 times for 5 min each. Sections were finally counterstained with eosin for 20 s. After rinsing several times in PBS, the sections were clear-mounted. Iron deposition was digitized and analyzed with Image J software.
Nissl staining
After dewaxing, the sections were washed with distilled water and incubated with Nissl Staining Solution (Beyotime Institute of Biotechnology, Nanjing, China) for 10 min at room temperature. The sections were then dehydrated using 95 and 100% ethanol solutions, made transparent using xylene, placed under coverslips and analyzed by microscopy.
Transmission electron microscope (TEM)
Sham and KA group rats were perfused with 2% paraformaldehyde and 2% glutaraldehyde in 0.1 M sodium cacodylate buffer, followed by post fixation in 2% osmium tetroxide with 1.6% potassium ferrocyanide in 0.1 M sodium cacodylate. Samples in CA3 region were then cut and stained en bloc with 2% uranyl acetate (UA), dehydrated in ethanol, and embedded in eponate. The sections (70-90 nm) were then placed on copper slot grids and stained with 2% UA and lead citrate. TEM images were captured with a Hitachi 7560 TEM in the microscopy core of Guangxi Medical University. 10 random micrographs were taken in the hippocampus and measurements made using Image J software for each rat. We quantified the mitochondrial area in neuronal (no morphologic evidence of classic necrosis, apoptosis, and autophagy). The mitochondrial length was determined by measuring the major length from one side to another.
Immumohistochemical staining
After dewaxing, the sections were sequentially treated with 3% hydrogen peroxide for 15 min to block endogenous peroxidase activity, the sections were preincubated with 10% normal goat serum for 10 min in the microwave processor. The sections were incubated overnight at 4°C with primary antibody rabbit-anti- glutathione peroxidase 4 (1:200, Abcam, UK). Following incubation of primary antibody, the sections were washed in PBS and incubated for 10 min with horseradish peroxidase labeled goat anti-rabbit IgG (1:100, Zhongshan Biotechnology, Beijing, China), washed with PBS, and then exposed to diaminobenzidine for 5 min. The sections were washed in distilled water and dehydrated with xylene, mounted, and observed by light microscopy.
Measurement hippocampal lipid peroxidation and GSH
The rats were perfused transcardially with ice-cold physiological saline, hippocampi were quickly excised and stored at -80°C until assayed. Concentrations of MDA, used as a marker of lipid peroxidation, were measured with the thiobarbituric acid (TBA) method spectrophotometric assay kit (Nanjing Jiancheng Bioengineering Institute, China) as previously described [21]. Briefly, this assay was based on the spectrophotometric measurement of the color generated during the reaction to TBA with MDA. MDA concentrations were calculated by reading the absorbance of TBA reactive substances in the supernatant at 532 nm. GSH was determined by a GSH assay kit (Nanjing Jiancheng Bioengineering Institute, China) as commercially recommended by the manufacturer’s protocol. The absorbance of samples were read at 420 nm by spectrophotometer and the values represented the final concentration according to the plotted standard curves.
Statistical analysis
Data were analyzed using the IBM SPSS software 17.0 package and expressed as Mean ± SD. Percentage of rats with spontaneous seizure was examined by χ2 test. The data of mitochondrial area were analyzed by Student’s t test. Data in this study, including spontaneous alternation rates, discrimination indexs, MDA levels and GSH levels, number of nissl positive staining cells, average percentages of iron positive area and number of GPX4 immunoreactive cells/field, were analyzed by one-way ANOVA. Values were considered statistically significant when P < 0.05.
Results
Fer-1 does not attenuate spontaneous seizure of KA induced TLE in rats
Sham group of rats did not show any signs of seizure after 4 weeks. In contrast, 70% in KA and 50% of rats in Fer-1 groups of rats had spontaneous seizures. There was no significant difference between the Fer-1 and sham groups (x2 = 0.833, P = 0.361).
Fer-1 attenuates cognitive impairment of KA-treated rats
Cognitive function was assessed using three hippocampal-dependent learning and memory tasks (Y-Maze test, Novel object recognition test and Morris water maze tests). The data of the Y-Maze test showed that the spontaneous alternation rate of KA group of rats decreased significantly compared with the sham group of rats (P < 0.01).
The spontaneous alternation rate was significantly improved in the Fer-1 group compared to KA group (P < 0.05) (Figure 1A). The Novel object recognition test showed that the discrimination index of KA group decreased significantly compared with the sham group (P < 0.01). The discrimination index was significantly improved in the Fer-1 group compared to KA group (P < 0.05). There were not significantly different between the Fer-1 and sham groups (Figure 1B).
Figure 1.
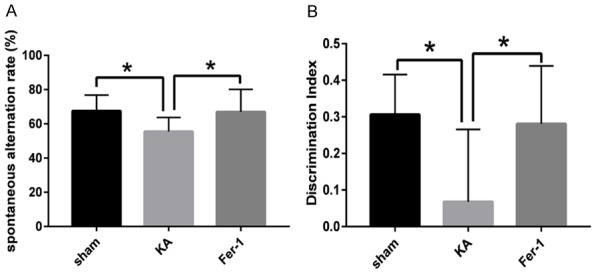
Fer-1 attenuates cognitive impairment of KA-treated rats. A. Spontaneous alternation rate was evaluated in the Y maze test. B. Discrimination index was assessed in the novel object recognition test. *P < 0.05 (vs KA) by one-way ANOVA (n = 10, each group).
Fer-1 attenuates neuron cell death in hippocampus of KA-treated rats
As shown in Figure 2, the results of Nissl staining indicated that the number of Nissl stained cells in CA1 and CA3 regions of the hippocampus decreased remarkably in the KA group as compared to the sham group (P < 0.01). Treatment with Fer-1 resulted in significant attenuation of prominent neuronal loss in CA1 and CA3 regions of hippocampus of KA induced TLE rats (P < 0.01).
Figure 2.
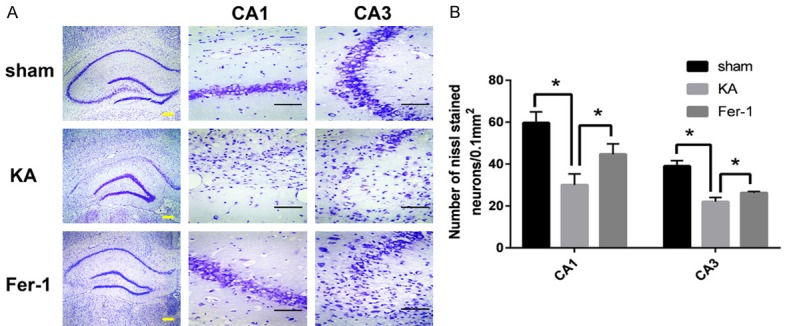
Fer-1 attenuates neuron cell death in hippocampus of KA-treated rats. A. Representative photomicrographs of Nissl-stained neurons in CA1 and CA3 area of hippocampus. B. Quantitative analysis of the number of Nissl-stained neurons. *P < 0.05 (vs KA) by one-way ANOVA (n = 3-5, each group). Scale bars: yellow bar = 200 μm, black bar = 50 μm.
Presence of Ferroptosis in the hippocampus of KA-treated rats
The occurrence of ferroptosis in the hippocampus of KA treated rats was measured by TEM. Our results showed that the average mitochondrial area of the hippocampus neuron of KA group was smaller than that of sham group (P < 0.05), indicating the existence of ferroptosis in the hippocampus following KA treatment in rats (Figure 3).
Figure 3.
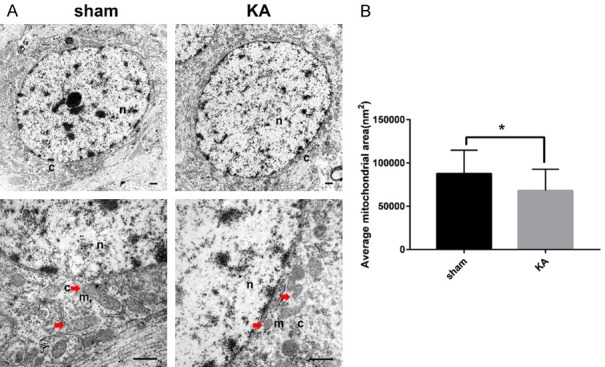
Occurrence of Ferroptosis in the hippocampus of KA-treated rats. A. Ultrastructure of neuron somas. n, nuclei; c, cytoplasm; m, mitochondria. Red arrows show representative mitochondria in somas. B. Quantification of the average mitochondrial area. *P < 0.05 (vs KA) by Student’s t test. Number of mitochondria: sham, n = 60, KA, n = 60. Scale bar: 500 nm. (n = 3 rats each group).
Fer-1 attenuates iron accumulation in the hippocampus of KA-treated rats
As shown in Figure 4, the results of Perls’ staining showed that the average percentages of the iron positive area in CA1 and CA3 regions of the hippocampus increased significantly in the KA group compared with the sham group (P < 0.01). Treatment with Fer-1 deceased prominently the average percentages of the iron positive area in the hippocampus of KA-treated rats (P < 0.01), indicating that Fer-1 attenuates iron accumulation in hippocampus of KA induced TLE rats.
Figure 4.
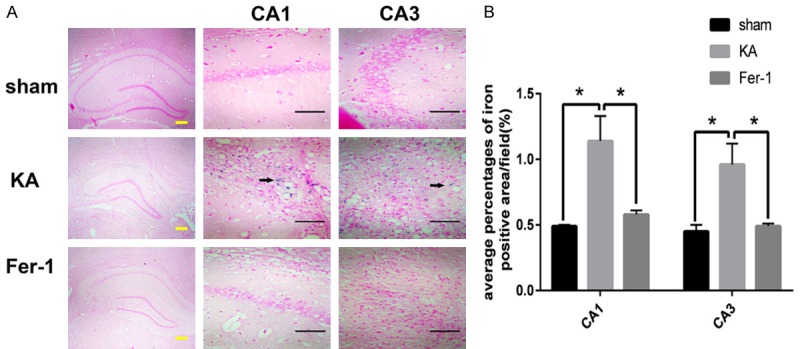
Fer-1 attenuates iron accumulation in hippocampus of KA-treated rats. A. Representative photomicrographs of iron positive area in CA1 and CA3 area of hippocampus. Arrows show representative iron positive area. B. Quantitative analysis of the average percentages of iron positive area. *P < 0.01 (vs KA) by one-way ANOVA (n = 3-5, each group). Scale bars: yellow bar = 200 μm, black bar = 50 μm.
Fer-1 restores GPX4 expression in the hippocampus of KA-treated rats
The number of GPX4 immunoreactive cells in the hippocampus was compared by immunohistochemistry. The results showed that, the number of GPX4 immunoreactive cells in CA1 and CA3 regions of hippocampus decreased significantly in the KA group compared to the sham group, and this effect can be attenuated by Fer-1 (P < 0.05) (Figure 5).
Figure 5.

Fer-1 restores GPX4 expression in the hippocampus of KA-treated rats. A. Representative photomicrographs of the iron positive area in CA1 and CA3 area of the hippocampus. Arrows show representative GPX4 immunoreactive cells. B. Quantitative analysis of the number of GPX4 immunoreactive cells. *P < 0.01 (vs KA) by one-way ANOVA (n = 3-5, each group). Scale bars: yellow bar = 200 μm, black bar = 50 μm.
Fer-1 increases GSH level and attenuates MDA level in the hippocampus of KA-treated rats
Compared to the Sham group, the levels of GSH in the hippocampus decreased significantly in the KA group (P < 0.01). Treatment with Fer-1 attenuated the reduction of GSH in the hippocampus of KA-treated rats significantly (P < 0.05) (Figure 6A). While the levels of MDA in the hippocampus increased significantly in the KA group (P < 0.01). Treatment with Fer-1 attenuated the increases of MDA in the hippocampus of KA-treated rats significantly (P < 0.05) (Figure 6B).
Figure 6.
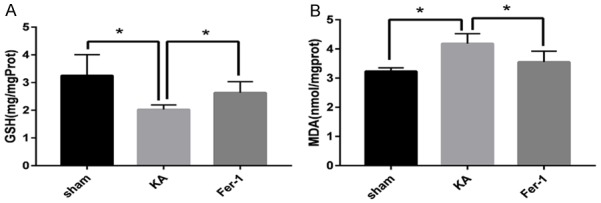
Fer-1 increases GSH level and attenuates MDA level in the hippocampus of KA-treated rats. Quantitative analysis of GSH level and MDA level in the hippocampus show *P < 0.05 (vs KA) by one-way ANOVA (n = 4-5, each group).
Discussion
The hippocampus plays a critical role in memory processes. This is because the spatial and temporal aspects of memory are processed and integrated in the hippocampal circuitry, that is from CA3 to CA1 region [22]. Noteworthy, the hippocampus is more vulnerable than other regions of the brain to insults [23]. The selective neuronal loss in the CA1/CA3 region of the hippocampus is one of the pathological characteristics of hippocampal sclerosis which is the major neuropathological feature of patients with TLE. Accumulating evidence suggests that hippocampal neuronal cell death is one of the principal causes of cognitive comorbidities of TLE [4]. Various forms of cell death have been found out in epilepsy, including apoptosis, autophagy, necrosis, necroptosis, and pyroptosis [24,25]. Inhibiting cell-death processes such as apoptosis, autophagy, pyroptosis, necrosis, and necroptosis, can ameliorate hippocampus damage, and then improve the cognitive function of epilepsy models [26-28].
In the present study, hippocampal dependent learning and memory were assessed using two techniques namely, the novel object recognition test, Y-maze [29-31]. The results demonstrated that ferrostatin-1 (Fer-1) improved cognitive function and mitigated the hippocampal neuronal loss of KA-treated rats. Further, treatment with Fer-1 did not attenuate spontaneous seizure in KA-treated rats, suggesting that spontaneous seizures could not account for differences in learning and memory. It should however be noted that spontaneous seizures were only evaluated by video according to Racine’s classification during the fifth weeks after SE, and its effects on chronic epilepsy could not be ruled out. To our knowledge, Fer-1, a potent and selective inhibitor of ferroptosis, can be used as a probe to study ferroptosis in multiple contexts. Hence, this work speculates that ferroptosis might be involved in the mechanism of cognitive dysfunction of TLE.
Ferroptosis is a novel nonapoptotic form of programmed cell death hallmarked by the accumulation of lethal lipid peroxidation products driven by iron [32]. It is morphologically and biochemically distinguishable from other forms of programmed cell death, including apoptosis, necrosis, and autophagy. The morphology of cells that underwent ferroptosis, exhibited slightly smaller mitochondria, thicker, and denser mitochondrial membranes; lacked rupture and blebbing of the plasma membrane (morphological features of apoptosis) and double-membraned autophagic vacuoles (morphological features of autophagy) [9]. Although ferroptosis has been shon to be involved in various pathological conditions, including neoplastic diseases, glutamate-induced neurotoxicity, neurodegenerative diseases, liver injury, renal failure, cerebral hemorrhage, ischemia/reperfusion injury, it has not been reported in an in vivo epilepsy model to date [11,20,33]. In the present research, using TEM, shrunken mitochondria was observed in the hippocampus of KA-treated rats, confirming the involvement of ferroptosis in the pathological process of TLE.
The initiation and progression of ferroptosis depend on the interactions among amino acids, iron and lipid metabolism [8]. Excessive iron accumulation induces lipid peroxidation, results in accumulation of lipid hydroperoxides and generates free radicals by the Fenton reaction [34,35]. If lipid hydroperoxides could not be scavenged in time before accumulating to toxic levels, the occurrence of ferroptosis is inevitable. Potentially toxic lipid hydroperoxides can be converted to non-toxic lipid alcohols by glutathione (GSH) or activating glutathione peroxidase 4 (GPX4) [36,37]. Since depletion of GSH makes GPX-4 inactivated, GSH metabolism is bound up with ferroptosis [38]. The biosynthesis of glutathione is dependent on the availability of cysteine that is derived from cystine. Because intracellular glutamate and extracellular cystine are exchanged in a 1:1 ratio through system Xc-, extracellular glutamate levels impact cystine import and subsequently impact biosynthesis of glutathione. High concentrations of extracellular glutamate deprive the level of intracellular cysteine and induce ferroptosis [9].
KA is the analog of L-glutamate and the agonist of ionotropic glutamate receptors. After excitotoxic injury induced by KA injections, the massive glutamate released in the rat’s hippocampus decrease the levels of GSH for six weeks and weaken the antioxidant defenses system [39-41]. Accompanied by GSH reduction, GPX-4 expression may be decreased. Further, the expression of divalent metal transporter-1 (DMT1), an iron importer, is elevated in the hippocampus of rats following KA injection [42,43], causing progressive increase in intracellular iron levels [44,45]. High iron content catalyzes lipid peroxidation and leads to the accumulation of lipid hydroperoxides and oxidative stress damage [46,47]. Numerous studies confirmed that the concentrations of malondialdehyde (MDA), a biomarker of lipid hydroperoxides, were increased in the hippocampus of animal models of TLE [48,49]. Similarly this work showed that compared with sham group rats, GSH and GPX-4 levels declined significantly, and the level of both iron and MDA increased markedly in the hippocampus of KA group rats. These pathologic changes are the critical conditions for initiating and executing ferroptosis procedure [50]. Accompanied by these pathologic changes in the hippocampus of KA group rats, hippocampal neurons were obviously lost. This study also revealed that Fer-1 reversed the pathologic changes (reduction of GSH and GPX-4 levels, and elevation of the concentration of both iron and MDA), and alleviated the hippocampal neuronal loss of KA-treated rats.
There were some limitations in this study. Firstly, the effect of Fer-1 on normal rats was not inspected in this study. Fer-1 is served as a potent and selective inhibitor and played an import role in studying ferroptosis in various pathological conditions. In view that the initiation and execution of ferroptosis is influenced by the action of iron accumulation, glutathione depletion, and lipid peroxidation, it is believed that ferroptosis is a form of pathological cell death. To date, the normal physiological function of ferroptosis has not been reported. Further, the dynamic effects of ferroptosis on different phases of TLE rats were not detected in this research. Lastly, the effects of different doses of Fer-1 on TLE rats were not studied. Further studies are recommended to determine the impact of these limitations on our findings.
In conclusion, ferroptosis is involved in the cognitive impairment of KA-induced TLE, and inhibition of ferroptosis processes ameliorates cognitive impairment in kainic acid-induced temporal lobe epilepsy in rats.
Acknowledgements
The present study was supported by the National Natural Science Foundation of China (grant Nos: 81760242).
Disclosure of conflict of interest
None.
References
- 1.Nevalainen O, Ansakorpi H, Simola M, Raitanen J, Isojärvi J, Artama M, Auvinen A. Epilepsy-related clinical characteristics and mortality: a systematic review and meta-analysis. Neurology. 2014;83:1968–1977. doi: 10.1212/WNL.0000000000001005. [DOI] [PubMed] [Google Scholar]
- 2.Sun J, Gao X, Meng D, Xu Y, Wang X, Gu X, Guo M, Shao X, Yan H, Jiang C, Zheng Y. Antagomirs targeting mirorna-134 attenuates epilepsy in rats through regulation of oxidative stress, mitochondrial functions and autophagy. Front Pharmacol. 2017;8:524. doi: 10.3389/fphar.2017.00524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Pearson JN, Rowley S, Liang LP, White AM, Day BJ, Patel M. Reactive oxygen species mediate cognitive deficits in experimental temporal lobe epilepsy. Neurobiol Dis. 2015;82:289–297. doi: 10.1016/j.nbd.2015.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Minjarez B, Camarena HO, Haramati J, Rodríguez-Yañez Y, Mena-Munguía S, Buriticá J, García-Leal O. Behavioral changes in models of chemoconvulsant-induced epilepsy: a review. Neurosci Biobehav Rev. 2017;83:373–380. doi: 10.1016/j.neubiorev.2017.10.016. [DOI] [PubMed] [Google Scholar]
- 5.Holler Y, Trinka E. What do temporal lobe epilepsy and progressive mild cognitive impairment have in common? Front Syst Neurosci. 2014;8:58. doi: 10.3389/fnsys.2014.00058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Falcicchia C, Paolone G, Emerich DF, Lovisari F, Bell WJ, Fradet T, Wahlberg LU, Simonato M. Seizure-suppressant and neuroprotective effects of encapsulated BDNF-producing cells in a rat model of temporal lobe epilepsy. Mol Ther Methods Clin Dev. 2018;9:211–224. doi: 10.1016/j.omtm.2018.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dixon Scott J, Lemberg Kathryn M, Lamprecht Michael R, Skouta R, Zaitsev Eleina M, Gleason Caroline E, Patel Darpan N, Bauer Andras J, Cantley Alexandra M, Yang Wan S, Morrison B, Stockwell Brent R. Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell. 2012;149:1060–1072. doi: 10.1016/j.cell.2012.03.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Xie Y, Hou W, Song X, Yu Y, Huang J, Sun X, Kang R, Tang D. Ferroptosis: process and function. Cell Death Differ. 2016;23:369–379. doi: 10.1038/cdd.2015.158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Stockwell BR, Friedmann Angeli JP, Bayir H, Bush AI, Conrad M, Dixon SJ, Fulda S, Gascón S, Hatzios SK, Kagan VE, Noel K, Jiang X, Linkermann A, Murphy ME, Overholtzer M, Oyagi A, Pagnussat GC, Park J, Ran Q, Rosenfeld CS, Salnikow K, Tang D, Torti FM, Torti SV, Toyokuni S, Woerpel KA, Zhang DD. Ferroptosis: a regulated cell death nexus linking metabolism, redox biology, and disease. Cell. 2017;171:273–285. doi: 10.1016/j.cell.2017.09.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhang YH, Wang DW, Xu SF, Zhang S, Fan YG, Yang YY, Guo SQ, Wang S, Guo T, Wang ZY, Guo C. Alpha-Lipoic acid improves abnormal behavior by mitigation of oxidative stress, inflammation, ferroptosis, and tauopathy in P301S Tau transgenic mice. Redox Biol. 2018;14:535–548. doi: 10.1016/j.redox.2017.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tuo QZ, Lei P, Jackman KA, Li XL, Xiong H, Li XL, Liuyang ZY, Roisman L, Zhang ST, Ayton S, Wang Q, Crouch PJ, Ganio K, Wang XC, Pei L, Adlard PA, Lu YM, Cappai R, Wang JZ, Liu R, Bush AI. Tau-mediated iron export prevents ferroptotic damage after ischemic stroke. Mol Psychiatry. 2017;22:1520–1530. doi: 10.1038/mp.2017.171. [DOI] [PubMed] [Google Scholar]
- 12.Levesque M, Avoli M. The kainic acid model of temporal lobe epilepsy. Neurosci Biobehav Rev. 2013;37:2887–2899. doi: 10.1016/j.neubiorev.2013.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Shin EJ, Jeong JH, Chung CK, Kim DJ, Wie MB, Park ES, Chung YH, Nam Y, Tran TV, Lee SY, Kim HJ, Ong WY, Kim HC. Ceruloplasmin is an endogenous protectant against kainate neurotoxicity. Free Radic Biol Med. 2015;84:355–372. doi: 10.1016/j.freeradbiomed.2015.03.031. [DOI] [PubMed] [Google Scholar]
- 14.Zilka O, Shah R, Li B, Friedmann Angeli JP, Griesser M, Conrad M, Pratt DA. On the mechanism of cytoprotection by ferrostatin-1 and liproxstatin-1 and the role of lipid peroxidation in ferroptotic cell death. ACS Cent Sci. 2017;3:232–243. doi: 10.1021/acscentsci.7b00028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Klee R, Brandt C, Tollner K, Loscher W. Various modifications of the intrahippocampal kainate model of mesial temporal lobe epilepsy in rats fail to resolve the marked rat-to-mouse differences in type and frequency of spontaneous seizures in this model. Epilepsy Behav. 2017;68:129–140. doi: 10.1016/j.yebeh.2016.11.035. [DOI] [PubMed] [Google Scholar]
- 16.Ambrogini P, Minelli A, Galati C, Betti M, Lattanzi D, Ciffolilli S, Piroddi M, Galli F, Cuppini R. Post-seizure alpha-tocopherol treatment decreases neuroinflammation and neuronal degeneration induced by status epilepticus in rat hippocampus. Mol Neurobiol. 2014;50:246–256. doi: 10.1007/s12035-014-8648-2. [DOI] [PubMed] [Google Scholar]
- 17.Wang H, An P, Xie E, Wu Q, Fang X, Gao H, Zhang Z, Li Y, Wang X, Zhang J, Li G, Yang L, Liu W, Min J, Wang F. Characterization of ferroptosis in murine models of hemochromatosis. Hepatology. 2017;66:449–465. doi: 10.1002/hep.29117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rusznak K, Cseko K, Varga Z, Csabai D, Bona A, Mayer M, Kozma Z, Helyes Z, Czeh B. Long-term stress and concomitant marijuana smoke exposure affect physiology, behavior and adult hippocampal neurogenesis. Front Pharmacol. 2018;9:786. doi: 10.3389/fphar.2018.00786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Diaz A, Escobedo C, Trevino S, Chavez R, Lopez-Lopez G, Moran C, Guevara J, Venegas B, Munoz-Arenas G. Metabolic syndrome exacerbates the recognition memory impairment and oxidative-inflammatory response in rats with an intrahippocampal injection of amyloid beta 1-42. Oxid Med Cell Longev. 2018;2018:1358057. doi: 10.1155/2018/1358057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Li Q, Han X, Lan X, Gao Y, Wan J, Durham F, Cheng T, Yang J, Wang Z, Jiang C, Ying M, Koehler RC, Stockwell BR, Wang J. Inhibition of neuronal ferroptosis protects hemorrhagic brain. JCI Insight. 2017;2:e90777. doi: 10.1172/jci.insight.90777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mao GX, Zheng LD, Cao YB, Chen ZM, Lv YD, Wang YZ, Hu XL, Wang GF, Yan J. Antiaging effect of pine pollen in human diploid fibroblasts and in a mouse model induced by D-galactose. Oxid Med Cell Longev. 2012;2012:750963. doi: 10.1155/2012/750963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Khalil A, Kovac S, Morris G, Walker MC. Carvacrol after status epilepticus (SE) prevents recurrent SE, early seizures, cell death, and cognitive decline. Epilepsia. 2017;58:263–273. doi: 10.1111/epi.13645. [DOI] [PubMed] [Google Scholar]
- 23.Fernandez-Fernandez S, Bobo-Jimenez V, Requejo-Aguilar R, Gonzalez-Fernandez S, Resch M, Carabias-Carrasco M, Ros J, Almeida A, Bolanos JP. Hippocampal neurons require a large pool of glutathione to sustain dendrite integrity and cognitive function. Redox Biol. 2018;19:52–61. doi: 10.1016/j.redox.2018.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dingledine R, Varvel NH, Dudek FE. When and how do seizures kill neurons, and is cell death relevant to epileptogenesis? Adv Exp Med Biol. 2014;813:109–122. doi: 10.1007/978-94-017-8914-1_9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Khan MT, Liu J, Nerlich J, Tang Y, Franke H, Illes P. Regulation of P2X7 receptor function of neural progenitor cells in the hippocampal subgranular zone by neuronal activity in the dentate gyrus. Neuropharmacology. 2018;140:139–149. doi: 10.1016/j.neuropharm.2018.08.001. [DOI] [PubMed] [Google Scholar]
- 26.Tan CC, Zhang JG, Tan MS, Chen H, Meng DW, Jiang T, Meng XF, Li Y, Sun Z, Li MM. NLRP1 inflammasome is activated in patients with medial temporal lobe epilepsy and contributes to neuronal pyroptosis in amygdala kindling-induced rat model. J Neuroinflammation. 2015;12:18. doi: 10.1186/s12974-014-0233-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ni H, Zhao DJ, Tian T. Ketogenic diet change cPLA2/clusterin and autophagy related gene expression and correlate with cognitive deficits and hippocampal MFs sprouting following neonatal seizures. Epilepsy Res. 2016;120:13–18. doi: 10.1016/j.eplepsyres.2015.11.021. [DOI] [PubMed] [Google Scholar]
- 28.Wang J, Li Y, Huang WH, Zeng XC, Li XH, Li J, Zhou J, Xiao J, Xiao B, Ouyang DS, Hu K. The protective effect of aucubin from eucommia ulmoides against status epilepticus by inducing autophagy and inhibiting necroptosis. Am J Chin Med. 2017;45:557–573. doi: 10.1142/S0192415X17500331. [DOI] [PubMed] [Google Scholar]
- 29.Motta-Teixeira LC, Machado-Nils AV, Battagello DS, Diniz GB, Andrade-Silva J, Silva S Jr, Matos RA, do Amaral FG, Xavier GF, Bittencourt JC, Reiter RJ, Lucassen PJ, Korosi A, Cipolla-Neto J. The absence of maternal pineal melatonin rhythm during pregnancy and lactation impairs offspring physical growth, neurodevelopment, and behavior. Horm Behav. 2018;105:146–156. doi: 10.1016/j.yhbeh.2018.08.006. [DOI] [PubMed] [Google Scholar]
- 30.Albani SH, Mchail DG, Dumas TC. Developmental studies of the hippocampus and hippocampal-dependent behaviors: insights from interdisciplinary studies and tips for new investigators. Neurosci Biobehav Rev. 2014;43:183–190. doi: 10.1016/j.neubiorev.2014.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Fernandez SP, Muzerelle A, Scottolomassese S, Barik J, Gruart A, Delgadogarcía JM, Gaspar P. Constitutive and acquired serotonin deficiency alters memory and hippocampal synaptic plasticity. Neuropsychopharmacology. 2017;42:512–523. doi: 10.1038/npp.2016.134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cao JY, Dixon SJ. Mechanisms of ferroptosis. Cell Mol Life Sci. 2016;73:2195–2209. doi: 10.1007/s00018-016-2194-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Skouta R, Dixon SJ, Wang J, Dunn DE, Orman M, Shimada K, Rosenberg PA, Lo DC, Weinberg JM, Linkermann A, Stockwell BR. Ferrostatins inhibit oxidative lipid damage and cell death in diverse disease models. J Am Chem Soc. 2014;136:4551–6. doi: 10.1021/ja411006a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Morris G, Berk M, Carvalho AF, Maes M, Walker AJ, Puri BK. Why should neuroscientists worry about iron? The emerging role of ferroptosis in the pathophysiology of neuroprogressive diseases. Behav Brain Res. 2018;341:154–175. doi: 10.1016/j.bbr.2017.12.036. [DOI] [PubMed] [Google Scholar]
- 35.Lewerenz J, Ates G, Methner A, Conrad M, Maher P. Oxytosis/ferroptosis-(Re-) emerging roles for oxidative stress-dependent non-apoptotic cell death in diseases of the central nervous system. Front Neurosci. 2018;12:214. doi: 10.3389/fnins.2018.00214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Shimada K, Skouta R, Kaplan A, Yang WS, Hayano M, Dixon SJ, Brown LM, Valenzuela CA, Wolpaw AJ, Stockwell BR. Global survey of cell death mechanisms reveals metabolic regulation of ferroptosis. Nat Chem Biol. 2016;12:497–503. doi: 10.1038/nchembio.2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cardoso BR, Hare DJ, Bush AI, Roberts BR. Glutathione peroxidase 4: a new player in neurodegeneration? Mol Psychiatry. 2017;22:328–335. doi: 10.1038/mp.2016.196. [DOI] [PubMed] [Google Scholar]
- 38.Gao M, Monian P, Quadri N, Ramasamy R, Jiang X. Glutaminolysis and transferrin regulate ferroptosis. Mol Cell. 2015;59:298–308. doi: 10.1016/j.molcel.2015.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Holmes PV, Reiss JI, Murray PS, Dishman RK, Spradley JM. Chronic exercise dampens hippocampal glutamate overflow induced by kainic acid in rats. Behav Brain Res. 2015;284:19–23. doi: 10.1016/j.bbr.2015.02.002. [DOI] [PubMed] [Google Scholar]
- 40.Baluchnejadmojarad T, Roghani M. Coenzyme q10 ameliorates neurodegeneration, mossy fiber sprouting, and oxidative stress in intrahippocampal kainate model of temporal lobe epilepsy in rat. J Mol Neurosci. 2013;49:194–201. doi: 10.1007/s12031-012-9886-2. [DOI] [PubMed] [Google Scholar]
- 41.Sedaghat R, Taab Y, Kiasalari Z, Afshin-Majd S, Baluchnejadmojarad T, Roghani M. Berberine ameliorates intrahippocampal kainate-induced status epilepticus and consequent epileptogenic process in the rat: Underlying mechanisms. Biomed Pharmacother. 2017;87:200–208. doi: 10.1016/j.biopha.2016.12.109. [DOI] [PubMed] [Google Scholar]
- 42.Loke SY, Siddiqi NJ, Alhomida AS, Kim HC, Ong WY. Expression and localization of duodenal cytochrome b in the rat hippocampus after kainate-induced excitotoxicity. Neuroscience. 2013;245:179–190. doi: 10.1016/j.neuroscience.2013.04.008. [DOI] [PubMed] [Google Scholar]
- 43.Huang E, Ong WY, Go ML, Connor JR. Upregulation of iron regulatory proteins and divalent metal transporter-1 isoforms in the rat hippocampus after kainate induced neuronal injury. Exp Brain Res. 2006;170:376–386. doi: 10.1007/s00221-005-0220-x. [DOI] [PubMed] [Google Scholar]
- 44.Wang XS, Ong WY, Connor JR. Increase in ferric and ferrous iron in the rat hippocampus with time after kainate-induced excitotoxic injury. Exp Brain Res. 2002;143:137–148. doi: 10.1007/s00221-001-0971-y. [DOI] [PubMed] [Google Scholar]
- 45.Liang LP, Jarrett SG, Patel M. Chelation of mitochondrial iron prevents seizure-induced mitochondrial dysfunction and neuronal injury. J Neurosci. 2008;28:11550–11556. doi: 10.1523/JNEUROSCI.3016-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Dixon SJ, Stockwell BR. The role of iron and reactive oxygen species in cell death. Nat Chem Biol. 2014;10:9–17. doi: 10.1038/nchembio.1416. [DOI] [PubMed] [Google Scholar]
- 47.Ashraf A, Clark M, So PW. The aging of iron man. Front Aging Neurosci. 2018;10:65. doi: 10.3389/fnagi.2018.00065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Cardenas-Rodriguez N, Coballase-Urrutia E, Perez-Cruz C, Montesinos-Correa H, Rivera-Espinosa L, Sampieri A 3rd, Carmona-Aparicio L. Relevance of the glutathione system in temporal lobe epilepsy: evidence in human and experimental models. Oxid Med Cell Longev. 2014;2014:759293. doi: 10.1155/2014/759293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Chwiej J, Kutorasinska J, Janeczko K, Gzielo-Jurek K, Uram L, Appel K, Simon R, Setkowicz Z. Progress of elemental anomalies of hippocampal formation in the pilocarpine model of temporal lobe epilepsy--an X-ray fluorescence microscopy study. Anal Bioanal Chem. 2012;404:3071–3080. doi: 10.1007/s00216-012-6425-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bertrand RL. Iron accumulation, glutathione depletion, and lipid peroxidation must occur simultaneously during ferroptosis and are mutually amplifying events. Med Hypotheses. 2017;101:69–74. doi: 10.1016/j.mehy.2017.02.017. [DOI] [PubMed] [Google Scholar]