

Abstract
Endometrial carcinoma (EC) is one of the most common malignancies of female reproductive tract in developed countries. MicroRNA is frequently dysregulated in human cancers and acts a key regulator role in tumor cell growth and metastasis. The aims of this study were to investigate the roles of microRNA-184 (miR-184) in EC cells and to identify its potential molecular mechanism. Here, the data revealed that miR-184 was significantly downregulated in human EC tissue compared with normal endometrial tissue, and the level of miR-184 expression was associated with lymph node metastasis and prognosis in patients with EC. In vitro assays, overexpression of miR-184 could suppress the proliferation and invasion of HEC-1B and RL95-2 cells. Moreover, bioinformatics analysis showed that cell division cycle 25A (CDC25A) was a putative target gene of miR-184. Dual luciferase reporter assay confirmed that miR-184 significantly downregulated CDC25A expression via directly interaction with the putative binding site in the 3’-untranslated region (3’-UTR) of CDC25A mRNA. Interestingly, knockdown of CDC25A resulted in inhibition of HEC-1B and RL95-2 cells growth and invasion. Mechanistic investigation revealed that downregulation of the Notch receptors (NOTCH1, NOTCH2, NOTCH3 and NOTCH4) and target gene HES1 by miR-184 could be reversed by CDC25A overexpression. In summary, our data demonstrate that CDC25A is a target gene of miR-184 in EC cells, and decreased expression of miR-184 suppresses the growth and invasion of EC cells via CDC25A-dependent Notch signaling pathway, suggesting that miR-184 may be a promising target for a new therapeutic strategy against EC.
Keywords: Endometrial carcinoma, microRNA-184, CDC25A, target, Notch signaling
Introduction
Endometrial carcinoma (EC) is one of the most common types of gynecologic cancer in developed countries [1,2]. The development of EC is a multistep process with the accumulation of genetic and epigenetic alterations [3]. Approximately 30% patients with EC are diagnosed at a late stage, which is associated with high levels of morbidity and mortality [4]. Despite being a common malignancy, the molecular mechanism underlying the initiation and progression of EC remains poorly understood [5,6]. There is an urgently need to elucidate the molecular mechanisms that occur during EC and to identify new targets for the development of novel therapeutic strategies against EC.
MicroRNAs (miRNAs) are a class of small noncoding RNAs containing ~18-24 nucleotides that silence their target genes by either degrading mRNAs or inhibiting their translation [7,8]. Numerous studies have demonstrated that miRNAs are involved in the regulation of a variety of cellular processes, including proliferation, differentiation, metabolism, and metastasis [9,10]. Emerging evidence suggests that dysregulation of miRNAs is associated with the initiation and progression of many human cancers and that miRNAs function as either oncogene or tumor suppressors [11,12]. For example, Du et al. showed that miR-137 plays a tumor suppressor role in gastric cancer cells by targeting KLF12 and MYO1C [13]. Li et al. reported that miR-20b-5p functions as a tumor suppressor in renal cell carcinoma by regulating cellular proliferation, migration, and apoptosis [14]. Zhang et al. found that miR-19a functions as an oncogene by regulating the PTEN/AKT/pAKT pathway in myeloma [15].
MiR-184 has been identified as a tumor suppressor in renal cell carcinoma [16]. Studies have also indicated that miR-184 overexpression inhibits cell proliferation and metastasis in small cell lung cancer and glioma [17,18]. Another study recently provided evidence that miR-184 is upregulated in patients with osteosarcoma who are treated with doxorubicin and leads to poor response to drug therapy by targeting BCL2L1 [19]. However, little is known about the biological role and molecular mechanism of miR-184 in EC. Notch signaling is a highly conserved pathway in tumor biology, and shows important regulation on cell proliferation, metastasis and differentiation that associated with tumorigenesis [20]. Recent reports have demonstrated the mechanism of EC progression under the Notch signaling regulation [21]. However, the interaction between miR-184 and Notch signaling in EC remains unclear.
In this present study, we explored the expression of miR-184 in 44 EC tissues and normal endometrial tissue through the use of quantitative reverse transcription (qRT)-polymerase chain reaction (PCR) analysis. Subsequently, we completed a series of cellular functional experiments to investigate the roles of miR-184 in HEC-1B and RL95-2 cells. To further identify the mechanism involved in miR-184-mediated biological behavior in EC, miR-184 putative targets were sought and confirmed through the use of bioinformatics analysis and dual luciferase reporter assay. Finally, the effects of target gene on the growth and invasion of EC cells were examined. To better understand its regulatory mechanism, we also evaluated the effects of miR-184 and its target gene on the expression levels of the Notch receptors (NOTCH1, NOTCH2, NOTCH3 and NOTCH4) and target gene HES1 that are documented to be involved in Notch signaling pathway. This finding may provide a clue to the development of novel effective therapies for EC in the future.
Material and methods
Tissue samples
A total of 44 fresh EC tissue specimens and matched normal endometrial tissue specimens (5 cm from the tumor margin) were obtained from patients with EC (aged 27-74 years, with a mean age of 47.51 ± 7.29 years) who underwent abdominal hysterectomy in the Department of General Gynaecology, Tianjin Central Hospital of Gynecology and Obstetrics, between March 2007 and October 2013. The patients EC did not receive any therapy before recruitment into this study. The tissue specimens were further carefully confirmed by two pathologists in our hospital. Specimens were frozen in liquid nitrogen and subsequently stored at -80°C for further RNA extraction. Tumor staging and pathology were based on the FIGO criteria [22]. The ethics committee of Tianjin Central Hospital of Gynecology and Obstetrics approved this research project, and informed consent was obtained from all patients.
Cell culture
Human EC cell lines (HEC-1B and RL95-2) were obtained from the Cell Bank of Shanghai Institute for Biological Sciences, Chinese Academy of Science (Shanghai, China) and cultured in Dulbecco modified Eagle medium (DMEM; Thermo Fisher Scientific; Rockford, IL, USA) containing 10% fetal bovine serum (FBS; Thermo Fisher Scientific) and 1% penicillin/1% streptomycin at 37°C in a humidified atmosphere containing 5% CO2.
MiRNA, siRNA and plasmid transfection
The sequence of miR-184 mimics (mimics) was: 5’-UGGACGGAGAACUGAUAAGGGU-3’, and the sequence for mimic-negative control (mimics NC) was: 5’-UUACUCGACACGUGUCAAGUTT-3’. The CDC25A-specific small interfering RNA (CDC25A siRNA) sequence was synthesized as follows: 5’-CGGUAUGUGAGAGAGAGAG-3’; the following sequence was used as an siRNA-negative control (siRNA NC): 5’-UGCUACGAGCAUGAGUCGUU-3’. All the RNA oligonucleotides were purchased from Cyagen Biosciences (Suzhou, China). The plasmid pCMV/Neo-CDC25A containing transfection-ready CDC25A cDNA (NCBI Reference Sequence: NM_001789.2) and a pure pCMV//Neo were obtained from GeneChem (Shanghai, China). For cell transfection, HEC-1B and RL95-2 cells were seeded at 37°C overnight. After reaching 50-70% confluence, the cells were transfected with either 50 nmol/L mimics or mimics NC and 100 nmol/L CDC25A siRNA or siRNA NC or 2 µg pCMV/Neo-CDC25A and pCMV/Neo with the use of Lipofectamine 2000 reagent (Thermo Fisher Scientific), following the manufacturer’s protocols. The transfection efficiency of RNA oligonucleotides or plasmids in HEC-1B and RL95-2 cells was observed with the use of a Leica TCS-SP confocal scanning laser microscope (Leica Laser-technik GmbH, Heidelberg, Germany) and confirmed by quantitative reverse transcription (qRT)-polymerase chain reaction (PCR) analysis.
3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazoliumbromide assay
HEC-1B and RL95-2 cells were seeded onto 96-well plates at a density of ~5000 cells/well and transfected as described here earlier. At 0-, 24-, 48-, and 72-h time-points after transfection, cell proliferation ability was analyzed by using a 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazoliumbromide (MTT) assay. Briefly, 10 µl of MTT solution (5 mg/ml) was added to each well and incubated at 37°C in a humidified chamber with 5% CO2 for 4 h. Subsequently, 150 µl of DMSO (Sigma, St. Louis, MO, USA) was added to each well and incubated at 37°C for 30 min. The absorbance at 450 nm with 650 nm as the reference was measured by using an ELISA microplate reader (VersaMax; Molecular Devices, Sunnyvale, CA, USA).
Transwell invasion assay
Cells transfected with mimics and mimics NC or CDC25A siRNA and siRNA NC were collected and resuspended in serum-free DMEM. Then, ~60,000 cells were seeded into the upper chamber of Transwell inserts that had been precoated with Matrigel (BD Bioscience, CA, USA). DMEM (500 µl) containing 10% FBS medium was placed into the lower chamber. Cells were allowed to invade for 24 h at 37°C in a humidified chamber with 5% CO2. Finally, the membranes were fixed with 75% methanol and stained with 0.1% crystal violet (Sigma). Cell invasion was measured by counting the number of cells attached to the lower side of the membrane in 10 high-power (200×) fields under a light microscope (Olympus Optical, Tokyo, Japan).
RNA extraction and real-time qPCR assay
Total RNA from EC tissues and cultured EC cells was isolated by using TRIzol solution (Thermo Fisher Scientific; Rockford, IL, USA), following the manufacturer’s protocols. The RNA with 260:280 ratios at 1.8-2.1 was selected for RT. One μg of RNA was reverse transcribed to cDNA by using an miRNA First-Strand cDNA Synthesis kit (Thermo Fisher Scientific) or by M-MLV reverse transcriptase with Oligo (dT18) RT primers. The real-time qPCR assay was conducted by using an SYBR Green Realtime PCR Master Mix (Toyobo, Tokyo, Japan) according to the manufacturer’s instructions. The qPCR primers were shown in Table 1. The condition of qPCR was as follows: incubation at 94°C for 10 min, 94°C for 10 s, 55°C for 30 s, and 72°C for 30 s followed by 40 cycles. Cycle threshold (Ct) values for all samples were determined. To normalize the data for qPCR, U6 and GAPDH was selected as the housekeeping gene. The relative expression of each gene was calculated using the delta-delta Ct method.
Table 1.
The primer sequences for qPCR amplification
| Gene | Forward (5’-3’) | Reverse (5’-3’) |
|---|---|---|
| miR-184 | TGGACGGAGAACTGAUAAGGGT | GTGCAGGGTCCGAGGT |
| U6 | CTCGCTTCGGCAGCACATATACT | ACGCTTCACGAATTTGCGTGTC |
| NOTCH1 | CGAACCCGTGCCAGAA | CAGATGCCCAGTGAAGC |
| NOTCH2 | TGACAGCCTGTATGTGC | CAGTTGTAAGTGTTGACC |
| NOTCH3 | CTGCAAGGACCGAGTCAA | AGCGTGCCCTCAAAGC |
| NOTCH4 | CCCGATGTGAGGAGGA | TGTTTGACAGCGTGGC |
| HES1 | TAAGGTGTTTGGAGGCT | CGCTGTTGCTGGTGTA |
| GAPDH | CCACTCCTCCACCTTTG | ACCACCCTGTTGCTGT |
Dual luciferase reporter assay
Dual luciferase reporter assay was conducted out to confirm the interaction between miR-184 and the 3’-untranslated region (UTR) of CDC25A. Approximately 60,000 cells/well were seeded onto 24-well plate and cotransfected with 50 nmol/L mimics or mimics NC and 2 µg of dual luciferase vector expressing the wild-type (WT) or mutant (MUT) 3’-UTR sequence of CDC25A. After 24 h, luciferase activities were measured with a Dual-Luciferase Reporter Assay System (Promega, WI, USA) according to the manufacturer’s protocols. The ratio of renilla to firefly luciferase signal was used to normalize renilla activity for intraexperimental transfection efficiency.
Western blot analysis
Approximately 106 cultured EC cells were lysed in ice-cold RIPA lysis buffer (Thermo Fisher Scientific), and then the concentration of each sample was measured by using a BCA assay kit (Pierce, Rockford, IL, USA). Approximately 40 μg of protein sample was separated by using 10% sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) gel and transferred onto PVDF membrane (Millipore, Bedford, MA, USA). After blocking with 5% fat-free milk for 2 h at 37°C, the membrane was incubated with primary antibodies against CDC25A (1:1000 dilution; Proteintech, USA) and GAPDH (1:2000 dilution) overnight in 4°C. Next, the membrane was incubated with anti-mouse or anti-rabbit IgG secondary antibody (1:2000; Proteintech) for 1 h at 37°C. Finally, the bands were developed by enhanced chemiluminescence (Pierce) and autoradiography.
Statistical analysis
Statistical analysis of all data was conducted using SPSS statistical software, version 18.0 (SPSS, Chicago, IL, USA). Data are presented as mean ± SD at least three independent experiments. The paired two-tailed Student t-test was used to compare the two groups. Three or more groups were analyzed with the use of one-way ANOVA. A value of P<0.05 was considered statistically significant.
Results
Levels of miR-184 were markedly reduced in EC tissues compared with matched normal endometrial tissues
In an attempt to explore the expression and significance of miR-184 in EC progression, we detected the expression of miR-184 in 44 pairs of EC tissue and matched normal endometrial tissue by using qPCR assay. As shown in Figure 1A, among these EC tissue specimens, 36 specimens exhibited decreased miR-184 expression compared with matched normal endometrial tissue specimens (81.8%, 36/44). Further, statistical analysis demonstrated that the expression of miR-184 was significantly downregulated in EC tissue specimens compared with matched normal endometrial specimens (Figure 1B, P<0.01). We further studied miR-184 expression levels and their association with clinicopathologic parameters in EC. The results revealed that miR-184 expression was associated with lymph node metastasis in patients with EC (Table 2, P = 0.01).
Figure 1.
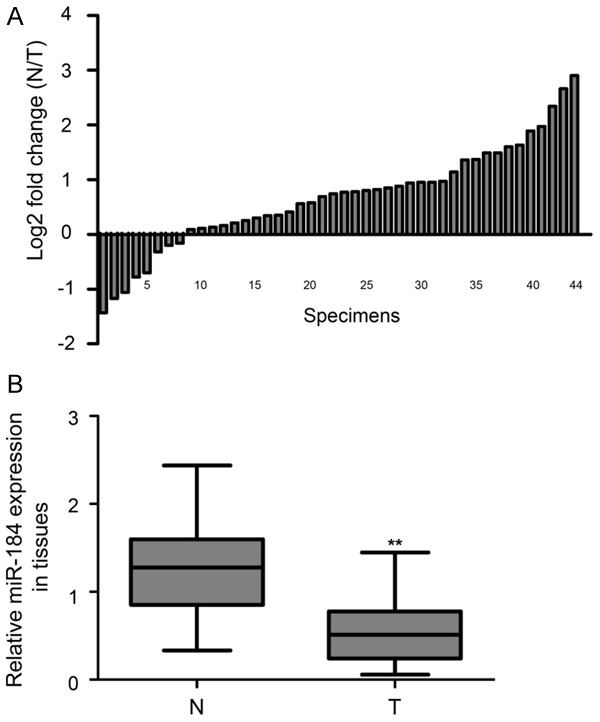
Expression levels of miR-184 in EC specimens. A. The expression levels of miR-184 was normalized relative to the U6 endogenous control in 44 pairs of N and T by real-time qPCR assay, and the data are shown in log2 (N/T). 36 cases of specimens exhibited decreased miR-184 expression. B. Statistical analysis showed that miR-184 was significantly downregulated in T compared with N. EC: endometrial carcinoma; T: EC tissues; N: match normal endometrial tissues. **P<0.01.
Table 2.
Correlation of miR-184 expression with different clinicopathological features of endometrial carcinoma (EC)
| Clinicopathological features | No. of cases | Median expression of miR-184 | p value |
|---|---|---|---|
| Age (years) | |||
| <55 | 15 | 0.59 ± 0.14 | 0.58 |
| ≥55 | 29 | 0.64 ± 0.17 | |
| Pathology | |||
| Endometrioid adenocarcinoma | 37 | 0.60 ± 0.20 | 0.63 |
| Other pathology types | 7 | 0.63 ± 0.21 | |
| FIGO stage | |||
| I-II | 33 | 0.58 ± 0.16 | 0.52 |
| III-IV | 11 | 0.66 ± 0.09 | |
| Pathology classification | |||
| Well + moderate | 30 | 0.54 ± 0.11 | 0.09 |
| Poor | 14 | 0.69 ± 0.13 | |
| Myometrial invasion | |||
| <1/2 | 26 | 0.66 ± 0.14 | 0.15 |
| ≥1/2 | 18 | 0.55 ± 0.10 | |
| Grade | |||
| G1 + G2 | 35 | 0.52 ± 0.07 | 0.07 |
| G3 | 9 | 0.68 ± 0.11 | |
| Lymph node metastasis | |||
| Negative | 32 | 0.71 ± 0.25 | 0.01 |
| Positive | 12 | 0.43 ± 0.06 |
Downregulated miR-184 was associated with unfavorable prognosis in patients with EC
To further validate the prognostic significance of miR-184 expression in EC, Kaplan-Meier survival analysis and log-rank test were performed to assess disease-specific survival in patients with EC. The results revealed that downregulation of miR-184 was significantly correlated with poor disease-specific survival in patients with EC. As shown in Figure 2, patients with low expression of miR-184 had worse survival times than those patients with high expression of miR-184 (P<0.01).
Figure 2.
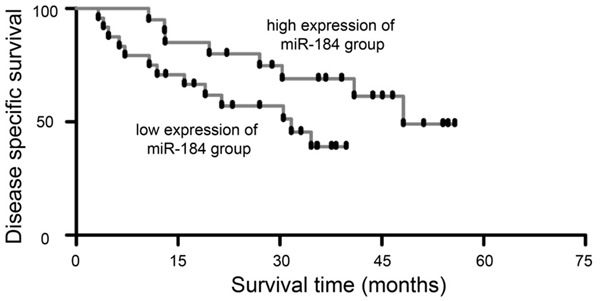
Downregulated expression of miR-184 indicated a poor prognosis in patients with EC. Kaplan-Meier survival curves for 44 EC cases, low expression of miR-184 was defined as short survival and high expression of miR-184 was defined as long survival. Patients with low miR-184 expression had poor survival time than patients with high miR-184 expression.
MiR-184 directly targeted CDC25A and downregulated CDC25A expression in EC cells
We then analyzed the mRNA sequence of CDC25A with use of an miRNA target-detecting software and identified a complementary binding site for miR-184 in the 3’-UTR of CDC25A (Figure 3A). Sequence alignment showed that the binding site was located in conserved regions of the CDC25A 3’-UTR among several vertebrate species (Figure 3B). A dual luciferase reporter assay indicated that miR-184 could directly bind to the 3’-UTR of CDC25A mRNA in HEC-1B and RL95-2 cells (Figure 3C, P<0.01). Furthermore, Western blot analysis confirmed that forced expression of miR-184 significantly reduced the protein levels of CDC25A in HEC-1B and RL95-2 cells (Figure 3D, P<0.01). All these results suggest that CDC25A is a direct target of miR-184.
Figure 3.
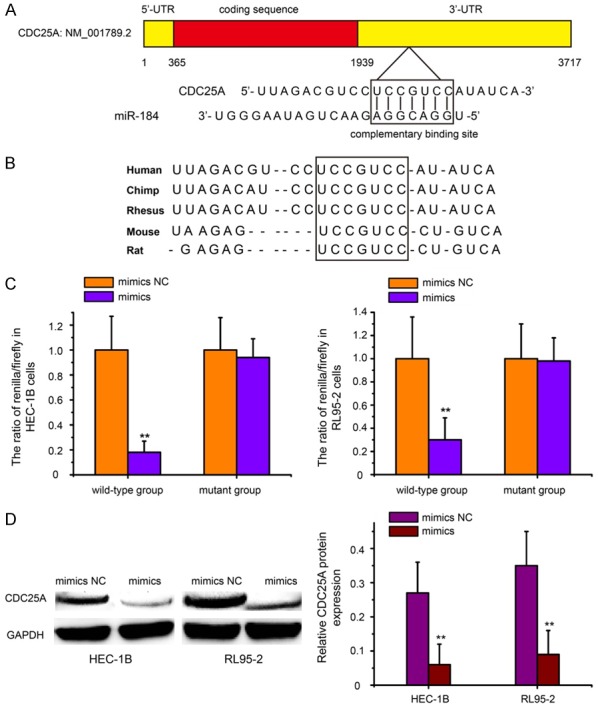
Cell division cycle 25A (CDC25A) is a directly target of miR-184 in EC cells. A. Schematic representation of CDC25A mRNA 3’-UTR showing the putative miR-184 targeting site. The seed-targeting site is framed. B. The targeting site in CDC25A mRNA 3’-UTR was highly conserved among several vertebrate species. C. The luciferase reporter constructs that contained the WT or MUT 3’-UTR of CDC25A, together with mimics or mimics NC, were transfected into HEC-1B and RL95-2 cells. At 48 h after transfection, luciferase activity was detected. Normalized data were calculated as the quotient of renilla/firefly luciferase activity. D. Western blot analysis of CDC25A levels in HEC-1B and RL95-2 cells after transfected with mimics or mimics NC. Forced expression of miR-184 significantly reduced the protein levels of CDC25A in HEC-1B and RL95-2 cells. mimics: miR-184 mimics; mimics NC: mimic negative control. **P<0.01.
Overexpression of miR-184 suppressed cell growth through inhibition of CDC25A
To determine the roles of miR-184 in the progression of EC, we sought to determine whether miR-184 may affect the proliferation of EC cells. The mimics were used to overexpress miR-184 in HEC-1B and RL95-2 cells. As shown in Figure 4A, the relative expression levels of miR-184 were significantly upregulated at 48 hours posttransfection of mimics in HEC-1B (17.92-fold over the mimics NC group, P<0.01) and RL95-2 cells (14.54-fold over the mimics NC group, P<0.01). MTT assay revealed that the proliferation rates of HEC-1B and RL95-2 cells with forced expression of miR-184 were notably decreased compared with cells transfected with mimics NC (Figure 4B, P<0.01).
Figure 4.
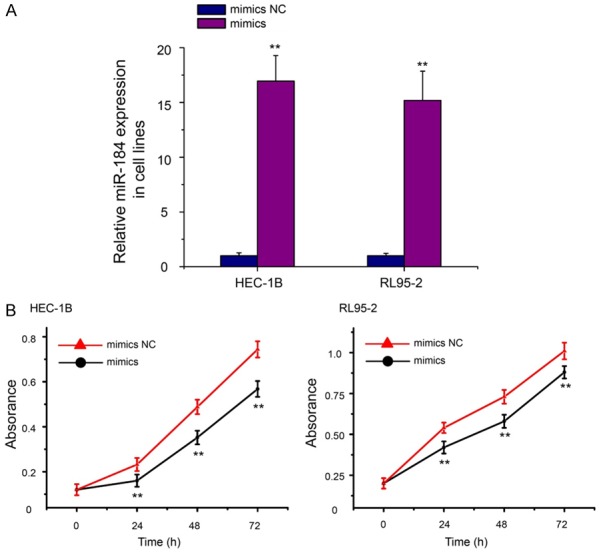
Overexpression of miR-184 inhibited the growth of EC cells. A. Validation of miR-184 expression change after transfection with mimics or mimics NC in HEC-1B and RL95-2 cells. The expression levels of miR-184 were significantly upregulated at 48 hours posttransfection of mimics in HEC-1B and RL95-2 cells. B. Cell growth was measured by MTT assay at different time intervals in both HEC-1B and RL95-2 cells. The growth curve of HEC-1B and RL95-2 cells after mimics transfection compared to mimics NC. MTT: 3-(4,5-dimethylthiazole-2-yl)-2,5-biphenyl tetrazolium bromide. **P<0.01.
Based on these observations, we hypothesized that miR-184 might exert its suppression function by downregulating CDC25A expression. To explore this further, we performed RNA interference directed against the CDC25A gene in HEC-1B and RL95-2 cells and analyzed the effect on cell proliferation. Western blot analysis showed that the protein levels of CDC25A in HEC-1B and RL95-2 cells were substantially restrained after transfection with CDC25A siRNA (Figure 5A, P<0.01). Interestingly, consistent with the effect of miR-184 overexpression, CDC25A knockdown inhibited the growth of HEC-1B and RL95-2 cells (Figure 5B, P<0.01).
Figure 5.
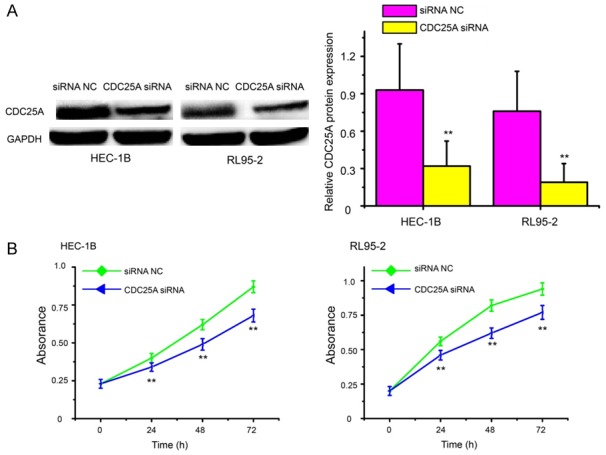
Targeting CDC25A suppressed cell growth in EC. A. Western blot analysis of CDC25A in HEC-1B and RL95-2 cells after transfection of CDC25A siRNA or siRNA NC. The protein levels of CDC25A in HEC-1B and RL95-2 cells were substantially restrained after transfection with CDC25A siRNA. B. The cell proliferative potential was determined in HEC-1B and RL95-2 cells transfected with CDC25A siRNA or siRNA NC. CDC25A knockdown inhibited the growth of HEC-1B and RL95-2 cells. CDC25A siRNA: CDC25A specific siRNA; siRNA NC: siRNA negative control. **P<0.01.
Forced expression of miR-184 repressed cell invasion by targeting CDC25A
To further explore the roles of miR-184 in the progression of EC, we considered whether miR-184 might possess a crucial role in EC cell invasion. Transwell invasion assay demonstrated that increased expression of miR-184 significantly reduced the invasion capacity of both HEC-1B and RL95-2 cells compared with the mimics NC group (Figure 6A, P<0.01). Moreover, the cell invasion assay revealed that CDC25A silencing could inhibit cell invasion in both HEC-1B and RL95-2 cells (Figure 6B, P<0.01), which was similar to the effects of miR-184 overexpression. These results strongly indicate that miR-184 suppresses cell proliferation and invasion in EC via the negative regulation of CDC25A.
Figure 6.
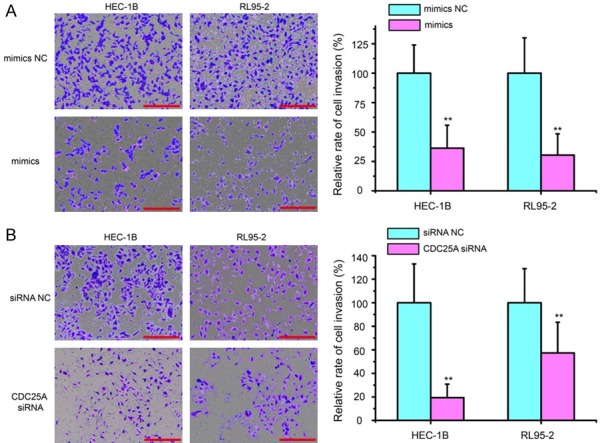
Forced expression of miR-184 repressed cell invasion by targeting CDC25A. A. Cell invasion of HEC-1B and RL95-2 cells was measured by Transwell invasion assay. Increased expression of miR-184 significantly impaired the invasion of EC cells compared with the mimics NC treated cells. B. Cell invasion changes caused by transfection of CDC25A siRNA or siRNA NC in both HEC-1B and RL95-2 cells. CDC25A silencing could inhibit cell invasion in both HEC-1B and RL95-2 cells, which was similar to the effects of miR-184 overexpression. Scar bar = 100 µm. **P<0.01.
CDC25A was required for miR-184-mediated Notch signaling inactivation of EC cells
Notch signaling pathway activation appears to affect tumorigenesis in many cancers including EC [20]. Thus, we speculated that the interaction between miR-184 and CDC25A might be related with the Notch pathway. Then, we used real-time qPCR assay to detect the mRNA levels of Notch receptors (NOTCH1, NOTCH2, NOTCH3 and NOTCH4) and the Notch pathway target protein, HES1, in HEC-1B cells after mimics or mimics NC transfection. The results showed that the expression of Notch receptors and HES1 was significantly reduced by miR-184 over-expression (Figure 7A, P<0.01). Furthermore, cotransfection with pCMV/Neo-CDC25A and mimics reversed the decreased expression of Notch receptors and HES1 caused by miR-184 in HEC-1B cells (Figure 7B, P<0.01). These results suggest a mechanism that AR might be a medium in miR-184-induced Notch pathway inactivation of EC cells.
Figure 7.
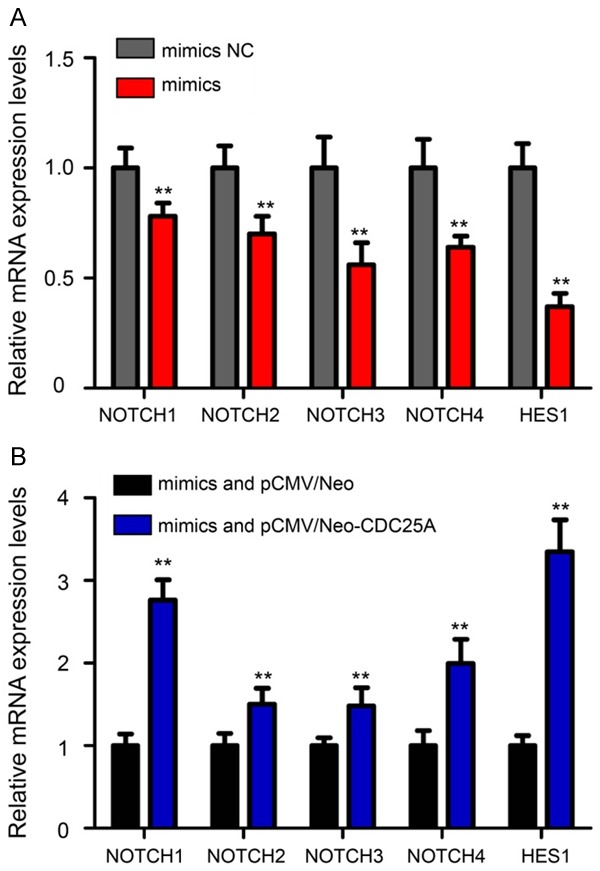
Downregulation of the Notch receptors and target gene HES1 by miR-184 could be reversed by CDC25A overexpression. A. The mRNA levels of NOTCH1, NOTCH2, NOTCH3, NOTCH4 and HES1 in HEC-1B cells after mimics or mimics NC transfection. The expression of Notch receptors and HES1 was significantly reduced by miR-184 over-expression. B. Cotransfection mimics with pCMV/Neo-CDC25A could reverse the decreased expression of Notch receptors and HES1 caused by miR-184 in HEC-1B cells. **P<0.01.
Discussion
Increasing evidence has shown that miRNAs regulate >30-60% of the protein-coding genes in the human genome [23]. Recently, study results have suggested that aberrantly expressed miRNAs plays a crucial role in the tumorigenic process of cancer [24]. Therefore, the identification of key miRNAs and the networks regulated by these miRNAs will provide new insights into the potential molecular mechanisms of EC progression. It has been reported that miR-184 is widely aberrantly expressed in many human cancers, including tongue squamous cell carcinoma [25], nasopharyngeal carcinoma [26], neuroblastoma [27], and hepatocellular carcinoma [28], indicating that miR-184 may play a significant role in tumorigenesis. However, to our knowledge, the functions and potential molecular mechanism of miR-184 are not well understood in EC.
In this study, we provide important evidence in support of miR-184 functioning as a tumor suppressor in EC. We showed the decreased expression of miR-184 in EC tissue relative to adjacent normal tissue. MiR-184 expression was associated with lymph node metastasis and prognosis in patients with EC. Our findings are consistent with studies on miR-184 expression in other types of malignancy [16-18]. The downregulation of miR-184 in EC tissue made us speculate as to its tumor suppressive role in EC. Then, we performed in vitro functional assays to analyze the effects of miR-184 on the biological behaviors of both HEC-1B and RL95-2 cells. Interestingly, we found that overexpression of miR-184 dramatically suppressed cell growth and invasion in HEC-1B and RL95-2 cells. The results provide new insight into the suppressive roles of miR-184 in the progression of EC.
The molecular mechanism underlying the miR-184-mediated inhibition of EC growth and invasion remains inconclusive. Several molecules appear to be the key targets of miR-184, including Bcl-2-like protein 1 (BCL2L1) [19], tumor necrosis factor α-induced protein 2 (TNFAIP2) [18], and MYC proto-oncogene (c-Myc) [29]. Our results demonstrated that CDC25A was a target of miR-184 in EC cells. CDC25A is thought to be oncogenic, and its expression is upregulated in various types of tumor tissue [30]. CDC25A plays an important role in cancer cell cycle progression, overexpression of CDC25A leads to accelerate entry of cells into mitosis [31]. In addition, the expression of CDC25A is regulated at transcriptional [32], translational [33], and posttranslational levels [34]. Here, we found miR-184 inhibited cancer growth and invasion by blocking the expression of CDC25A. The loss of miR-184 leads to the upregulation of CDC25A, consequently resulting in malignant transformation.
The Notch signaling pathway is implicated in the carcinogenesis of various cancers, and blockade of Notch pathway appears to affect cell proliferation in multiple types of cancers [35]. For example, Zang et al. reported Notch pathway inhibition in breast cancer cells induces cell cycle arrest and apoptosis [36]. Cheng et al. found that downregulation of Notch1 contributes to cell growth inhibition in pancreatic cancer [37]. Similarly, Wei et al. showed that abnormal activation of the Notch pathway promotes proliferation in endometrial cancer [38]. In addition, Guo et al. demonstrated that lncRNA-MEG3 inhibits cell proliferation of endometrial carcinoma by repressing Notch signaling [39]. In general, Notch signaling pathway initiates through ligand-receptor interactions, while the intramembranous proteolytic cleavage of Notch receptors contributes to the release of Notch intracellular domain (NICD) in an active form. NICD acts as a transcriptional activator after translocating to the nucleus, showing positive regulation on target genes, including hes family bHLH transcription factor 1 (HES1). HES1 is one HES family members that can function as a transcriptional repressor by negatively regulating genes required for cell proliferation. Based on these observations, we hypothesized that the interaction between miR-184 and CDC25A might be related with the Notch pathway. Our results suggested that downregulation of the Notch receptors (NOTCH1, NOTCH2, NOTCH3 and NOTCH4) and target gene HES1 by miR-184 could be reversed by CDC25A overexpression. These findings indicate that AR might be a medium in miR-184-induced Notch pathway inactivation of EC cells.
In conclusion, our results demonstrate that decreased expression of miR-184 restrains the growth and invasion of EC cells through CDC25A-dependent Notch signaling pathway. This finding not only helps us to understand the molecular mechanism of EC progression but also provides a strong rationale to use miR-184 as a new potential biomarker and therapeutic target for EC in the future.
Disclosure of conflict of interest
None.
References
- 1.Siegel R, Naishadham D, Jemal A. Cancer statistics, 2012. CA Cancer J Clin. 2012;62:10–29. doi: 10.3322/caac.20138. [DOI] [PubMed] [Google Scholar]
- 2.Siegel R, Naishadham D, Jemal A. Cancer statistics, 2013. CA Cancer J Clin. 2013;63:11–30. doi: 10.3322/caac.21166. [DOI] [PubMed] [Google Scholar]
- 3.Amant F, Moerman P, Neven P, Timmerman D, Van Limbergen E, Vergote I. Endometrial cancer. Lancet. 2005;366:491–505. doi: 10.1016/S0140-6736(05)67063-8. [DOI] [PubMed] [Google Scholar]
- 4.Ray M, Fleming G. Management of advanced-stage and recurrent endometrial cancer. Semin Oncol. 2009;36:145–154. doi: 10.1053/j.seminoncol.2008.12.006. [DOI] [PubMed] [Google Scholar]
- 5.Banno K, Nogami Y, Kisu I, Yanokura M, Umene K, Masuda K, Kobayashi Y, Yamagami W, Susumu N, Aoki D. Candidate biomarkers for genetic and clinicopathological diagnosis of endometrial cancer. Int J Mol Sci. 2013;14:12123–12137. doi: 10.3390/ijms140612123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bansal N, Yendluri V, Wenham RM. The molecular biology of endometrial cancers and the implications for pathogenesis, classification, and targeted therapies. Cancer Control. 2009;16:8–13. doi: 10.1177/107327480901600102. [DOI] [PubMed] [Google Scholar]
- 7.Bartel DP. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell. 2004;116:281–297. doi: 10.1016/s0092-8674(04)00045-5. [DOI] [PubMed] [Google Scholar]
- 8.Pasquinelli AE. MicroRNAs and their targets: recognition, regulation and an emerging reciprocal relationship. Nat Rev Genet. 2012;13:271–282. doi: 10.1038/nrg3162. [DOI] [PubMed] [Google Scholar]
- 9.Hwang HW, Mendell JT. MicroRNAs in cell proliferation, cell death, and tumorigenesis. Br J Cancer. 2007;96(Suppl):R40–44. [PubMed] [Google Scholar]
- 10.Zhou X, Yang PC. MicroRNA: a small molecule with a big biological impact. MicroRNA. 2012;1:1. doi: 10.2174/2211536611201010001. [DOI] [PubMed] [Google Scholar]
- 11.Zhang B, Pan X, Cobb GP, Anderson TA. MicroRNAs as oncogenes and tumor suppressors. Dev Biol. 2007;302:1–12. doi: 10.1016/j.ydbio.2006.08.028. [DOI] [PubMed] [Google Scholar]
- 12.Calin GA, Croce CM. MicroRNA signatures in human cancers. Nat Rev Cancer. 2006;6:857–866. doi: 10.1038/nrc1997. [DOI] [PubMed] [Google Scholar]
- 13.Du Y, Chen Y, Wang F, Gu L. miR-137 plays tumor suppressor roles in gastric cancer cell lines by targeting KLF12 and MYO1C. Tumour Biol. 2016;37:13557–13569. doi: 10.1007/s13277-016-5199-3. [DOI] [PubMed] [Google Scholar]
- 14.Li Y, Chen D, Jin L, Liu J, Su Z, Gui Y, Lai Y. MicroRNA-20b-5p functions as a tumor suppressor in renal cell carcinoma by regulating cellular proliferation, migration and apoptosis. Mol Med Rep. 2016;13:1895–1901. doi: 10.3892/mmr.2015.4692. [DOI] [PubMed] [Google Scholar]
- 15.Zhang X, Chen Y, Zhao P, Zang L, Zhang Z, Wang X. MicroRNA-19a functions as an oncogene by regulating PTEN/AKT/pAKT pathway in myeloma. Leuk Lymphoma. 2017;58:932–940. doi: 10.1080/10428194.2016.1213827. [DOI] [PubMed] [Google Scholar]
- 16.Su Z, Chen D, Li Y, Zhang E, Yu Z, Chen T, Jiang Z, Ni L, Yang S, Gui Y, Ye J, Lai Y. microRNA-184 functions as tumor suppressor in renal cell carcinoma. Exp Ther Med. 2015;9:961–966. doi: 10.3892/etm.2015.2199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhou R, Zhou X, Yin Z, Guo J, Hu T, Jiang S, Liu L, Dong X, Zhang S, Wu G. Tumor invasion and metastasis regulated by microRNA-184 and microRNA-574-5p in small-cell lung cancer. Oncotarget. 2015;6:44609–44622. doi: 10.18632/oncotarget.6338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cheng Z, Wang HZ, Li X, Wu Z, Han Y, Li Y, Chen G, Xie X, Huang Y, Du Z, Zhou Y. MicroRNA-184 inhibits cell proliferation and invasion, and specifically targets TNFAIP2 in Glioma. J Exp Clin Cancer Res. 2015;34:27. doi: 10.1186/s13046-015-0142-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lin BC, Huang D, Yu CQ, Mou Y, Liu YH, Zhang DW, Shi FJ. MicroRNA-184 modulates doxorubicin resistance in osteosarcoma cells by targeting BCL2L1. Med Sci Monit. 2016;22:1761–1765. doi: 10.12659/MSM.896451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Previs RA, Coleman RL, Harris AL, Sood AK. Molecular pathways: translational and therapeutic implications of the Notch signaling pathway in cancer. Clin Cancer Res. 2015;21:955–961. doi: 10.1158/1078-0432.CCR-14-0809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jonusiene V, Sasnauskiene A, Lachej N, Kanopiene D, Dabkeviciene D, Sasnauskiene S, Kazbariene B, Didziapetriene J. Down-regulated expression of Notch signaling molecules in human endometrial cancer. Med Oncol. 2013;30:438. doi: 10.1007/s12032-012-0438-y. [DOI] [PubMed] [Google Scholar]
- 22.FIGO (International Federation of Gynecology and Obstetrics) 26th annual report on the results of treatment in gynecological cancer. Int J Gynaecol Obstet. 2006;95(Suppl 1):S1–257. doi: 10.1016/S0020-7292(06)60025-8. [DOI] [PubMed] [Google Scholar]
- 23.Inui M, Martello G, Piccolo S. MicroRNA control of signal transduction. Nat Rev Mol Cell Biol. 2010;11:252–263. doi: 10.1038/nrm2868. [DOI] [PubMed] [Google Scholar]
- 24.Schickel R, Boyerinas B, Park SM, Peter ME. MicroRNAs: key players in the immune system, differentiation, tumorigenesis and cell death. Oncogene. 2008;27:5959–5974. doi: 10.1038/onc.2008.274. [DOI] [PubMed] [Google Scholar]
- 25.Manikandan M, Deva Magendhra Rao AK, Rajkumar KS, Rajaraman R, Munirajan AK. Altered levels of miR-21, miR-125b-2*, miR-138, miR-155, miR-184, and miR-205 in oral squamous cell carcinoma and association with clinicopathological characteristics. J Oral Pathol Med. 2015;44:792–800. doi: 10.1111/jop.12300. [DOI] [PubMed] [Google Scholar]
- 26.Zhen Y, Liu Z, Yang H, Yu X, Wu Q, Hua S, Long X, Jiang Q, Song Y, Cheng C, Wang H, Zhao M, Fu Q, Lyu X, Chen Y, Fan Y, Liu Y, Li X, Fang W. Tumor suppressor PDCD4 modulates miR-184-mediated direct suppression of C-MYC and BCL2 blocking cell growth and survival in nasopharyngeal carcinoma. Cell Death Dis. 2013;4:e872. doi: 10.1038/cddis.2013.376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Foley NH, Bray IM, Tivnan A, Bryan K, Murphy DM, Buckley PG, Ryan J, O’Meara A, O’Sullivan M, Stallings RL. MicroRNA-184 inhibits neuroblastoma cell survival through targeting the serine/threonine kinase AKT2. Mol Cancer. 2010;9:83. doi: 10.1186/1476-4598-9-83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gao B, Gao K, Li L, Huang Z, Lin L. miR-184 functions as an oncogenic regulator in hepatocellular carcinoma (HCC) Biomed Pharmacother. 2014;68:143–148. doi: 10.1016/j.biopha.2013.09.005. [DOI] [PubMed] [Google Scholar]
- 29.Lin TC, Lin PL, Cheng YW, Wu TC, Chou MC, Chen CY, Lee H. MicroRNA-184 deregulated by the microRNA-21 promotes tumor malignancy and poor outcomes in non-small cell lung cancer via targeting CDC25A and c-Myc. Ann Surg Oncol. 2015;22(Suppl 3):S1532–1539. doi: 10.1245/s10434-015-4595-z. [DOI] [PubMed] [Google Scholar]
- 30.Yu YQ, Weng J, Li SQ, Li B, Lv J. MiR-675 promotes the growth of hepatocellular carcinoma cells through the CDC25A pathway. Asian Pac J Cancer Prev. 2016;17:3881–3885. [PubMed] [Google Scholar]
- 31.Blomberg I, Hoffmann I. Ectopic expression of CDC25A accelerates the G(1)/S transition and leads to premature activation of cyclin E- and cyclin A-dependent kinases. Mol Cell Biol. 1999;19:6183–6194. doi: 10.1128/mcb.19.9.6183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Vigo E, Muller H, Prosperini E, Hateboer G, Cartwright P, Moroni MC, Helin K. CDC25A phosphatase is a target of E2F and is required for efficient E2F-induced S phase. Mol Cell Biol. 1999;19:6379–6395. doi: 10.1128/mcb.19.9.6379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lin YM, Chung CL, Cheng YS. Posttranscriptional regulation of CDC25A by BOLL is a conserved fertility mechanism essential for human spermatogenesis. J Clin Endocrinol Metab. 2009;94:2650–2657. doi: 10.1210/jc.2009-0108. [DOI] [PubMed] [Google Scholar]
- 34.Busino L, Chiesa M, Draetta GF, Donzelli M. CDC25A phosphatase: combinatorial phosphorylation, ubiquitylation and proteolysis. Oncogene. 2004;23:2050–2056. doi: 10.1038/sj.onc.1207394. [DOI] [PubMed] [Google Scholar]
- 35.Qiao L, Wong BC. Role of Notch signaling in colorectal cancer. Carcinogenesis. 2009;30:1979–1986. doi: 10.1093/carcin/bgp236. [DOI] [PubMed] [Google Scholar]
- 36.Zang S, Ji C, Qu X, Dong X, Ma D, Ye J, Ma R, Dai J, Guo D. A study on Notch signaling in human breast cancer. Neoplasma. 2007;54:304–310. [PubMed] [Google Scholar]
- 37.Cheng H, Zhu H, Cao M, Lu C, Bao S, Pan Y. HtrA1 suppresses the growth of pancreatic cancer cells by modulating Notch-1 expression. Braz J Med Biol Res. 2018;52:e7718. doi: 10.1590/1414-431X20187718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wei Y, Zhang Z, Liao H, Wu L, Wu X, Zhou D, Xi X, Zhu Y, Feng Y. Nuclear estrogen receptor-mediated Notch signaling and GPR30-mediated PI3K/AKT signaling in the regulation of endometrial cancer cell proliferation. Oncol Rep. 2012;27:504–510. doi: 10.3892/or.2011.1536. [DOI] [PubMed] [Google Scholar]
- 39.Guo Q, Qian Z, Yan D, Li L, Huang L. LncRNA-MEG3 inhibits cell proliferation of endometrial carcinoma by repressing Notch signaling. Biomed Pharmacother. 2016;82:589–594. doi: 10.1016/j.biopha.2016.02.049. [DOI] [PubMed] [Google Scholar]